Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2018; 8(13):3530-3543. doi:10.7150/thno.24475 This issue Cite
Research Paper
CCR2-dependent monocytes/macrophages exacerbate acute brain injury but promote functional recovery after ischemic stroke in mice
Weirong Fang1,3, Xuan Zhai1,4, Dong Han1, Xiaoxing Xiong1, Tao Wang1, Xun Zeng5, Shucheng He3, Rui Liu3, Masaaki Miyata6, Baohui Xu2, Heng Zhao1 ![]()
1. Department of Neurosurgery, Stanford University School of Medicine, Stanford, CA 94305, USA
2. Department of Surgery, Stanford University School of Medicine, Stanford, CA 94305, USA
3. Department of Physiology, China Pharmaceutical University, Nanjing 210009, China
4. Children's Hospital of Chongqing Medical University, Chongqing 400014, China
5. The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310003, China
6. Department of Cardiovascular Medicine and Hypertension, Graduate School of Medical and Dental Sciences, Kagoshima University, Japan
Received 2017-12-21; Accepted 2018-4-21; Published 2018-6-6
Abstract

Rationale: Peripheral blood monocytes are recruited into the ischemic brain and transform into macrophages after stroke. Nevertheless, the exact role of CCR2-dependent monocytes/macrophages in brain injury after stroke remains elusive.
Methods: We used CCR2 knockout (KO) mice and the CCR2 pharmacological inhibitor, propagermanium (PG), to address the role of CCR2-dependent monocytes/macrophages in the acute stage and neurological functional recovery after middle cerebral artery (MCA) occlusion and reperfusion.
Results: CCR2 KO resulted in smaller infarct size and lower mortality than in wild type (WT) mice, when measured 3 days after stroke. However, from 5 to 28 days after stroke, the KO mice had higher mortality and showed no obvious neurological functional recovery. In addition, WT mice treated with PG had similar stroke outcomes compared with CCR2 KO, as measured by T2 weighted MRI. Flow cytometry and real-time PCR analyses suggest that monocyte-derived macrophages (MoDMs) in the stroke brains mainly polarized to pro-inflammatory macrophages at the early stage, but gradually switched to anti-inflammatory macrophages at 7 days after stroke. In addition, adoptive transfer of anti-inflammatory macrophages into CCR2 KO mice at 4 and 6 days after stroke alleviated mortality and promoted neurological recovery.
Conclusion: CCR2-dependent monocytes/macrophages are a double-edged sword; they worsen acute brain injury, but are essential for neurological recovery by promoting anti-inflammatory macrophage polarization.
Keywords: CCR2, monocytes, functional recovery, ischemic stroke
Introduction
Acute ischemic brain injury and functional recovery after stroke are mediated by neuroinflammation [1]. Macrophages are a major modulator of this neuroinflammation, which include monocyte-derived macrophages (MoDMs) and their brain resident counterparts, microglia-derived macrophages (MiDMs) [2-3]. Microglia contribute to brain homeostasis, which are immediately activated after ischemic stroke [4-7]. In addition to microglia, blood monocytes are recruited into the ischemic brain a few hours after stroke, where they transform into macrophages, and may modulate both acute brain injury and functional recovery.
The chemokine receptor CCR2, when activated by the ligand CCL2 and expressed on monocytes, is responsible for monocyte emissions from the bone marrow, and migration into the ischemic brain [8-9]. A number of studies have explored the role of CCR2-dependent monocytes/macrophages in acute brain injury after stroke, but the results have been controversial. Some studies have shown that CCR2 KO and pharmacological inhibition of CCR2 attenuated acute brain injury [10], while others have shown that a selective CCR2 antagonist resulted in a more extensive lesion in a mouse ischemic model [11]. Therefore, the exact role of CCR2-dependent MoDMs remains elusive. Furthermore, how MoDMs regulate brain functional recovery after stroke remains largely unexplored. Only recently, Wattananit et al. reported that the anti-CCR2 antibody, MC-21, abolished long-term behavioral recovery. They reported that MC-21 inhibited the anti-inflammatory genes TGF-β, CD163, and Ym-1 expression, but the underlying protective mechanisms are unknown [12]. Therefore, whether monocytes are detrimental or beneficial factors in stroke remains controversial, and whether they have distinctive effects in the acute and delayed phase are not known. In this study, we used CCR2 KO mice and the CCR2 pharmacological inhibitor, propagermanium (PG) to clarify the role of monocytes/macrophages in both acute brain injury and delayed functional recovery in a mouse MCA occlusion model.
Methods
Animals
C57BL/6J wild-type (WT) and CCR2-/- (B6.129(Cg)-CCR2<tm2.1Ifc>/J) (CCR2 KO) mice from Jackson Laboratory (Bar Harbor, ME, USA) were bred and housed in the Stanford Medical School Animal Care Facility. Experimental protocols were approved by the Stanford University Administrative Panel on Laboratory Animal Care, and experiments were conducted in accordance with the guidelines of Animal Use and Care of the National Institutes of Health and Stanford University.
A total of 167 out of 237 WT mice, and 153 out of 207 CCR2 KO mice were included for data analyses. The causes of animal exclusion include: 1) Mice that showed no neurological deficits. 2) Brains with evidence of surgical subarachnoid hemorrhage. Only 0 to 2 mice were excluded in each group due to surgical subarachnoid hemorrhage. Mice with secondary hemorrhage after stroke were not excluded. 3) Mice that died 1-3 d after MCAO.
In the long-term behavior and Nissl staining study, there were some mortalities, especially in CCR2 KO mice and PG treated WT mice. 30 WT mice and 10 CCR2 KO mice were used to culture bone marrow derived macrophage (BMDM) for the adoptive transfer experiment. Monocytes were separated from the spleens and bone marrow from these same animals for the migration assays.
All animal experiments were conducted in a blinded fashion. The researcher performing the surgeries was unaware of the animal groups, and the researcher analyzing the data was unaware of the group conditions.
Focal cerebral ischemia
Male mice aged 8 to 10 weeks, weighing 24-28 g, were randomly assigned to experimental groups. Mice were anesthetized with 5% isoflurane and maintained at 1% to 2% isoflurane during surgery. Body temperature was maintained at 37 ± 0.5 °C with a surface heating pad during the entire procedure. Focal cerebral ischemia was induced by transient MCA occlusion for 45 min by inserting a silicone-coated 6-0 monofilament (Doccol Corp, Redlands, CA, USA) into the left CCA to block the MCA, as we have reported previously [13-14]. For the sham group, surgeries were conducted, but without monofilament insertion into the left CCA.
Infarction size was detected by using the 2, 3, 5-triphenyltetrazolium chloride (TTC) staining. At 72 h after ischemia, the mice were deeply anesthetized with isoflurane and euthanized. The brains were rapidly removed and coronally sectioned into 5 slices with 2 mm thickness, stained in 2% TTC (Sigma, St. Louis, MO) at 37 °C for 15 min, and fixed in 4% paraformaldehyde for 24 h. The brain slices were scanned and infarctions were measured using NIH ImageJ software. To avoid the influence of cerebral edema, the infarction in each section was normalized to the non-ischemic contralateral side and expressed as a percentage of the contralateral hemisphere with the following formula:
Infarct size = (contralateral area - ipsilateral non-infarct area) / contralateral area × 100%.
Brain atrophy at 14 or 28 days after stroke was measured with Nissl staining. Mice were euthanized at 14 or 28 days after ischemia, and then perfused with ice-cold PBS followed with 4% (w/v) paraformaldehyde in PBS [13]. Using a vibratome (Leica VT1000, Wetzlar, Germany), 50 μm coronal brain slices were cut after post-fixation with 4% (w/v) paraformaldehyde for 72 h. Brain slices were stained in 0.1% cresyl violet solution for 30 min, and then rinsed in distilled water. Stained sections were fixed by serial dehydration in ethanol (70%, 90%, 100%) and xylene [15]. Fixed slices were then scanned and quantified with NIH ImageJ software, as mentioned above. Tissue loss in each section was expressed as a percentage of the contralateral hemisphere with the following formula:
Tissue loss = (contralateral area - ipsilateral non-infarct area) / contralateral area × 100%.
Magnetic resonance imaging (MRI)
MRI was performed repeatedly at 3 and 14 days after stroke on a dedicated small animal 7 T MRI system (PharmaScan, Bruker, Ettlingen, Germany) [16]. The image protocol consisted of a T2-weighted sequence to assess the location and extent of infarction. T2w TSE sequence parameters were: TE = 36.8 ms, TR = 5000 ms, FA = 132.6°, FOV = 1.34/1.62 cm, 30 slices, and 0.4 mm slice thickness with 0.4 mm gap. MRI pictures were then scanned and quantified with NIH ImageJ software, and hemisphere volume was calculated as: ∑hemisphere area × slice thickness (0.4 mm).
Infarct or atrophy size = (contralateral area - ipsilateral non-infarct area) / contralateral area × 100%.
Primary culture of bone marrow-derived macrophages (BMDMs) and adoptive transfer of anti-inflammatory macrophages
Bone marrow cells were collected by flushing out the femoral and tibial bone cavities of male C57BL/6J mice, aged 8-10 weeks [17]. The cells were treated with ACK lysing buffer (GIBCO; Invitrogen, Carlsbad, CA, USA) to eliminate red blood cells. The remaining cells were cultured at 37 °C in 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM, GIBCO), supplemented with 10% fetal calf serum (GIBCO), and 20 ng/mL murine macrophage-colony stimulating factor (M-CSF) (PeproTech Inc., Rocky Hill, NJ, USA) [18]. Seven days after plating, the BMDM were stimulated with murine IL-4 (20 ng/mL) (PeproTech Inc., Rocky Hill, NJ, USA) for 48 h to polarize the macrophages into anti-inflammatory phenotypes. To prove that the anti-inflammatory macrophages might be essential for neurological recovery, the in vitro polarized anti-inflammatory macrophages or vehicle PBS were adoptively transferred into CCR2 KO mice (2×106 cells/mouse, 0.2 mL via retro-orbital injection) at 4 and 6 days after ischemic stroke, respectively.
Behavioral tests
To evaluate the role of CCR2 on neurological deficits after stroke, we used three behavioral tests to measure neurological function, as described below:
Rotating beam walking test
The rotating beam walking test was used to evaluate neurological deficits in coordination and integration of movement in mice after ischemic stroke [19]. The mice were trained to walk along a 100 cm rotating wood beam (80 mm in diameter, approximately 80 cm above the floor, at 3 rpm rotation) for 7 days (3 trials per day), then tested before and at 2, 4, 7, 10, 14, 21, and 28 days after stroke. The walking time at each time point for each mouse was then recorded.
Pole test
The pole test was conducted by placing a 50 cm vertical pole (1 cm diameter) in a mouse cage. A mouse is placed, with its head upward, so that it climbs to the top of the pole, then it turns around and descends to the cage floor. “Time to turn around” and “time to reach the floor” are recorded [20]. Mice were trained for 3 days (5 trials per day) and then tested before and 2, 4, 7, 10, 14, 21, and 28 days after stroke.
Flow cytometry analysis for functional macrophage phenotypes
To analyze pro- and anti- inflammatory functional macrophage phenotypes from monocytes, we conducted FACS analysis for cell surface (PD-L1 and PD-L2) and intracellular markers (iNOS and Arg-1) to identify pro- and anti- inflammatory phenotypes. All antibodies were purchased from BioLegend (San Diego, CA, USA), unless stated otherwise. Immune cells were isolated from the brain tissues, as described previously [21-22]. In brief, mice were deeply anesthetized with isoflurane, euthanized, and then transcardially perfused with cold PBS. The ischemic hemispheres were collected in flow cytometry staining buffer (PBS with 1% FBS and 0.1% sodium azide), mildly homogenized on ice, and centrifuged (514 rcf) for 10 min. 6 mL of 37% Percoll was then added to the cells, vortexed, and loaded with 2 mL of 70% Percoll under the cell suspension. The samples were then centrifuged (514 rcf) for 30 min at 4 °C. The mononuclear cells at the interphase were isolated, washed 3 times in the flow cytometry staining buffer, filtered through a 70 μm strainer, re-suspended, and counted.
For cell surface marker staining, the cells were blocked with serum, Fc Block, and then stained on ice for 30 min in the dark with FITC-anti-CD45, PE/Cy7-anti-CD11b, Pacific blue-anti-Ly6C, APC/Cy7 anti-Ly6G, APC-anti-PD-L1, and PE-anti-PD-L2 antibodies, respectively. For intracellular iNOS and arginase 1 (Arg-1) staining, after staining with anti-CD45, anti-11b, anti-Ly6C, and anti-Ly6G, cells were fixed and permeabilized with BD Cytofix/Cytoperm solution (BD Biosciences, No. 554714, San Jose, CA, USA) for 20 min on ice, followed by staining with APC-conjugated arginase 1 (R&D system, No. IC5868A, Minneapolis, MN, USA,) and FITC-conjugated iNOS (BD Biosciences, No. 610330) antibodies, or isotype, as controls [23]. Samples were run on a BD LSR II flow cytometer using Diva software (v6.1.2; Becton Dickinson, San Jose, CA, USA) and data were analyzed using FlowJo software (v7.6.2; Tree Star, Ashland, OR).
In vivo monocyte migration assay in stroke mice
To analyze how CCR2 affects monocyte migration after stroke in mice, we conducted a post-stroke, in vivo monocyte migration assay by flow cytometry. Monocytes (2×108 cells/mL) extracted from bone marrows and spleens of 8-10 week old CCR2 KO mice were incubated with the red fluorescent dye, TRITC, (0.8 μg/mL) in labeling buffer (50% RPMI, 48.5% HBSS without calcium and magnesium, 1.5% bovine calf serum, and 10 mM HEPES) at 37 °C for 15 min. Monocytes from bone marrows and spleens of WT mice were labeled with green fluorescent dye CFSE (3 μM) under the same conditions [24]. Then 1×108 mixed labeled monocytes were transferred via retro-orbital injection in each WT mouse at 3 or 7 days after ischemic stroke, respectively. Two hours after adoptive transfer, monocytes isolated from brain and blood suspensions in host mice, as well as donor monocytes used for the transfer (input cells), were incubated for 30 min at 4 °C in 100 μL of 0.5 μg/mL PE/Cy7-anti-CD11b, APC/Cy7-anti-CD45, and Pacific blue-anti-Ly6C. FACS assay was conducted to identify CFSE+ and TRITC+ MoDMs, and data was analyzed, as described, in the flow cytometry analysis section. Data are expressed as the ratio of the TRITC+ cell number from CCR2 KO divided by the FITC+ cell number from WT monocytes in the blood and ischemic brain.
Quantitative real-time PCR
To evaluate the pro- and anti- inflammatory macrophage functional gene phenotypes from monocytes and microglia, we conducted real-time PCR to identify the mRNA gene expressions of pro- and anti- inflammatory phenotypes. The ischemic cortex of mice were collected after transcardial perfusion with cold PBS, and immediately placed in RNAlater (Qiagen, No. 76104) [25]. Homogenates and RNA were purified using the RNeasy Plus Mini Kit (Qiagen, No. 74134, USA). Reverse transcription was performed using the Superscript III First-Strand Synthesis System for PCR (Invitrogen Life Technologies, No. 18080-051, Grand Island, NY, USA). Real-time PCR was performed with SYBR GreenER qPCR SuperMix Universal (Invitrogen, No. 11762-100) on a Stratagene Mx3000p QPCR System (Stratagene, La Jolla, CA, USA). PCR primer probe sets were predeveloped and produced by BIO-RAD (PrimePCR™ PreAmp for SYBR® Green Assay: TNF-α, mouse; IFN-γ, mouse; IL-4, mouse; TGF-β1, mouse). The expression levels of target genes were normalized to that of the β-actin. Quantities of products were determined by the comparative threshold cycle (CT) method using the equation, 2-ΔΔ CT, to determine the fold change (as relative to sham controls).
To further determine the cytokine gene expressions in MoDMs in the ischemic brain after stroke, immune cells were collected and incubated with FITC-anti-CD45, PE/Cy7-anti-CD11b, Alexa Fluor 647-anti-Ly6G, and Pacific blue-anti-Ly6C, as described in the flow cytometry section. MoDMs (CD11b+CD45highLy6G-Ly6Chigh) were then sorted into tubes by FACSAria (BD Biosciences) for analysis, as described above. The purity of CD11b+CD45highLy6G-Ly6Chigh sorted monocytes/macrophages was >97% [26]. For sham mice, MiDMs (CD11b+CD45int) were sorted into tubes as control. The target gene expression levels were normalized to the β-actin endogenous control. Quantities of products were determined by the comparative threshold cycle (CT) method using the equation, 2-ΔΔ CT, to determine fold changes. The expression levels are displayed as relative to that of microglia (CD45intCD11b+) from the sham group.
Immunostaining
To study macrophage expressions after stroke, immunohistochemistry and immunofluorescence staining were performed. Mice were transcardially perfused with ice-cold PBS and PBS containing 4% (w/v) paraformaldehyde, respectively, after being fully euthanized [13]. Tissues were post-fixed with 4% (w/v) paraformaldehyde for 72 h and snap-frozen in OCT-freeze medium, after which a freezing microtome (Leica 3050, Wetzlar, Germany) was used to cut 10 μm coronal brain slices.
For immunochemistry staining, acetone-fixed frozen sections were stained using a three-stage immunoperoxidase system, as previously described [27]. Briefly, the brain slices were incubated at room temperature for 1 h with primary anti CD68 antibody (BIO-RAD, No. 0713R Hercules, CA, USA), or Iso type control antibody (BioLegend, Biotin Rat IgG2a). Then, the brain slices were incubated at room temperature for 30 min with biotin-conjugated anti-rat IgG secondary antibody. Slices were then incubated with Peroxidase-conjugated Streptavidin (Jackson ImmunoResearch, No. 016-030-084, Suffolk CB8 7SY, UK) and Peroxidase substrate kit AEC (Vector laboratories, SK-4200, Burlingame, CA, USA) sequence with 3 PBS washes between each step. Slides were examined by light microscopy. The number of positive cells in the ischemic area were counted using ImageJ software. Counts were performed on coded sections by an investigator who did not know the codes.
For immunofluorescence staining, brain slices were incubated for 1 h with blocking solution (0.1 M PBS, 0.3% Triton X-100, and 5% normal goat serum), and then incubated at 4 °C for 24 h with anti CD68 antibody, anti Ym-1 antibody (Stemcell Technologies, No. 01404. Vancouver, Canada). Brain slices were washed 3 times with PBS, and incubated at room temperature for 2 h with goat anti-mouse Ym-1, or Alexa Fluor 633-conjugated goat anti-rat (CD68) antibodies, all diluted to 1:400 (Invitrogen, Carlsbad, CA, USA). Brain slices were then washed and mounted on glass slides with 4′,6-diamidino- 2-phenylindole (DAPI, Vector Laboratories, Burlingame, CA). Negative controls without primary antibodies were performed in parallel and showed no staining. Fluorescent imagines were taken by a confocal microscope (Zeiss LSM510, Jena, Germany). Three sections (-1.70 to -2.18 relative to Bregma) / mouse were randomly chosen and the number of positive cells in the core were counted using ImageJ software. Counts were performed on coded sections by a blinded investigator.
Drug administration
For MRI assay, the CCR2 inhibitor, propagermanium (PG, 50 mg/kg/day) or the vehicle PBS was administered, via oral gavage, to WT mice one day before MCAO surgery, until 14 days after stroke. Freshly prepared PG in PBS was administered three times a day (50 mg/kg/day). In the vehicle control group, an equal volume of PBS was administered using the same regimen. For behavioral tests, PG (50mg/kg/day) or the vehicle PBS was administered, via oral gavage, to WT mice from 4 days until 14 days after stroke.
Statistics
Results are shown as mean ± SD. The specific statistical methods are described in each figure legend. One-way ANOVA was performed when comparing different groups, and the Mann-Whitney U test was used for analysis on real-time PCR data. P values of ≤ 0.05 denote significant changes.
Results
CCR2 KO protects against acute brain injury, but worsens delayed damage after stroke
The TTC results showed that infarct sizes measured 3 days after stroke in CCR2 KO mice were significantly smaller than in WT mice (Figure 1A), which suggests that CCR2 KO is neuroprotective at the acute stage after stroke.
The effect of CCR2 KO on stroke outcomes in mice. (A) Representative infarction by TTC staining. Bar graph showing infarct size expressed as percentages measured at 3 days after ischemic stroke. n=6. (B) Representative coronal brain sections with cresyl/violet staining at 28 days after stroke. The bar graph shows quantification of tissue loss expressed as percentage of hemispheric volume in WT and CCR2 KO mice. n=6. (C) Survival curve of mice from 0 to 28 days after MCAO in WT and CCR2 KO mice. n=12. (D) Behavioral tests. a. Performance was calculated as the walking time (seconds) along the rotating beam (100 cm long, 3 rpm rotation). b. Pole test. Performance calculated by turnaround time. c. Pole test. Performance calculated by walking down time. n=6. *, ** vs. WT stroke, P<0.05, 0.01; one-way ANOVA performed to compare the WT stroke and CCR2 KO stroke groups.
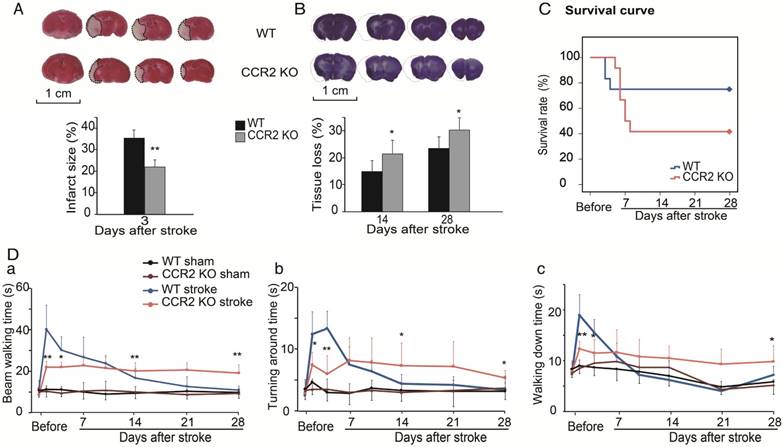
To examine the effect of CCR2 KO on brain injury at later time points, animals were euthanized at 14 and 28 days after stroke, and the brain injury sizes were measured by cresyl/violet staining. In contrast to the acute phase, a significantly larger atrophy in CCR2 KO mice was observed compared to WT mice (Figure 1B), suggesting that CCR2 depletion resulted in brain injury deterioration in a delayed pattern.
We then compared the mortality rate of the CCR2 KO and WT stroke mice. The results suggest that CCR2 KO resulted in lower mortality at the early stage (1 to 4 days) but higher mortality at the delayed stage (after 5 days) compared with WT mice (Figure 1C).
The behavioral results showed that the WT mice walked a significantly longer time on the rotating beam than CCR2 KO mice measured at 2 and 4 days after MCAO, but showed a quicker recovery time thereafter (Figure 1Da). For the pole test, the results showed similar patterns to the beam test (Figure 1Db-c), suggesting that CCR2 depletion resulted in neurological protection at the early time points after stroke but did not offer recovery at the delayed time points.
Inflammatory monocytes migrating into the ischemic brain are CCR2 dependent
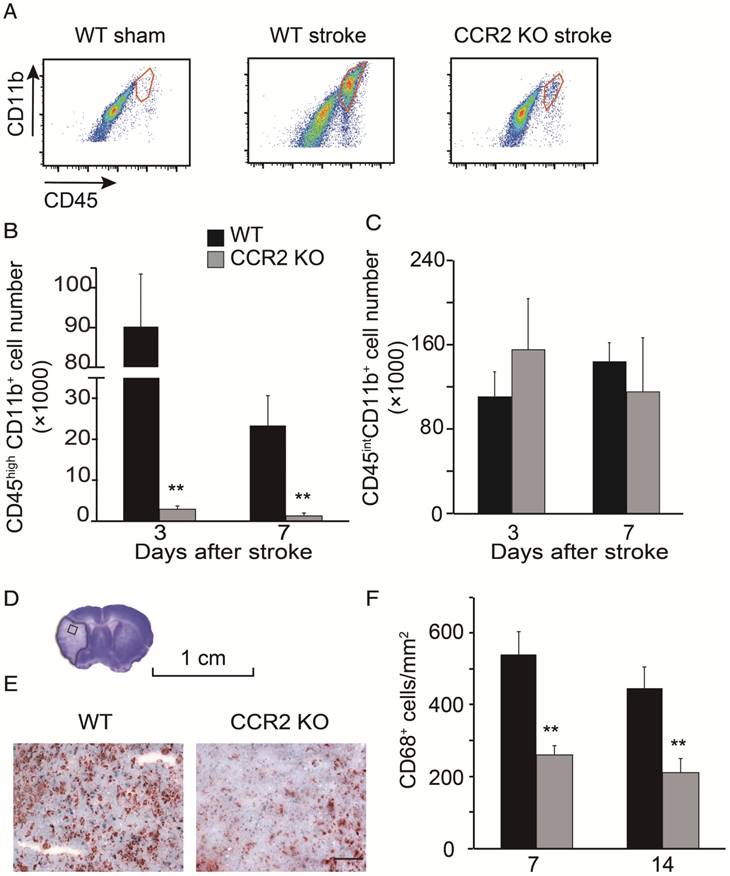
By using flow cytometry, MoDMs and MiDMs were identified and quantified as CD45hiCD11b+ and CD45intCD11b+, respectively [28-29]. The MoDM numbers in CCR2 KO mice measured at 3 and 7 days after stroke were significantly smaller than those in WT mice (Figure 2B). However, the MiDM numbers did not differ significantly between CCR2 KO mice and WT mice (Figure 2C). The results of the immunohistochemical staining indicate that the numbers of CD68 positive cells at 7 and 14 days was significantly lower in CCR2 KO mice than in WT mice (Figure 2F).
Monocyte infiltration into the ischemic brain is CCR2 dependent. (A) CD45hiCD11b+ and CD45intCD11b+ in the ischemic brain of WT and CCR2 KO mice. (B) Bar graph shows the quantification of CD45hiCD11b+ at 3 and 7 days after stroke in the ischemic hemisphere of WT and CCR2 KO mice. n=6. (C) Bar graph shows the quantification of CD45intCD11b+ at 3 and 7 days after stroke in the ischemic hemisphere of WT and CCR2 KO mice. n=6. (D) The box in the cresyl/violet stained section indicates the ischemic core region where photomicrographs were taken to quantify cell numbers. (E) Representative images of CD68 staining from the ischemic brains of WT and CCR2 KO mice. Scale bar 100 μm. (F) Bar graph shows the statistical results of CD68+ cells at 7 and 14 days after stroke. n=6. **, vs. WT, P<0.01; one-way ANOVA was performed to compare the WT stroke and CCR2 KO stroke groups.
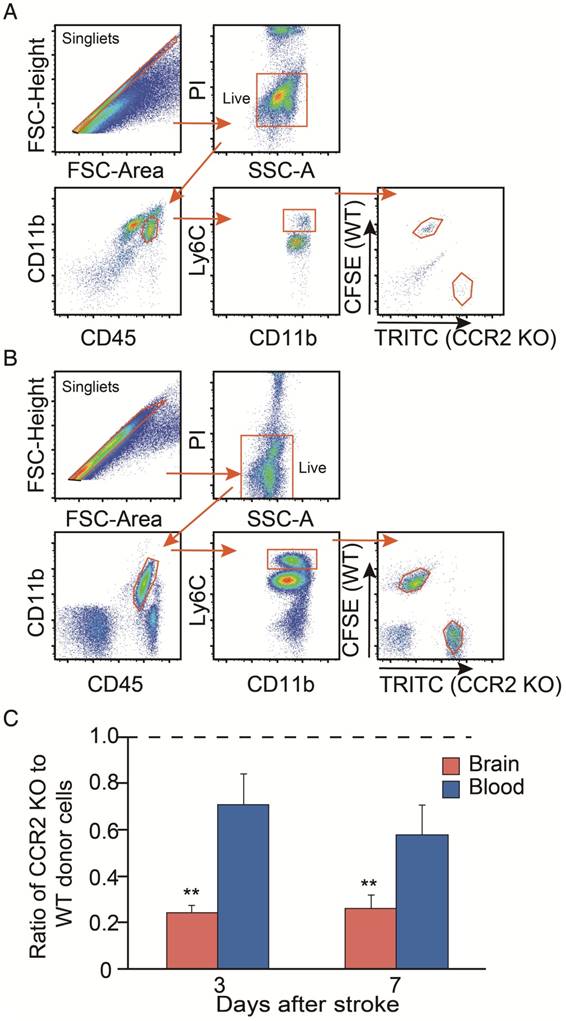
We then compared the migrating ability of WT monocytes vs. CCR2 KO monocytes from the blood into the ischemic brain after stroke. The CCR2 KO monocytes stained with TRITC fluorescent dye were mixed with the WT monocytes labeled with CFSE fluorescent dye, and then adoptively transferred into the animals after stroke. We gated TRITC vs. CFSE positive cells from CD45hiCD11b+Ly6Chi and CD45+CD11b+Ly6Chi cell populations in the ischemic brain and peripheral blood, respectively (Figure 3A-B). The results showed that the monocyte migrating ratios of CCR2 KO monocytes compared with WT monocytes in the ischemic brain were only 0.24 and 0.25 at 3 and 7 days, respectively (Figure 3C), indicating that monocyte migration is CCR2 dependent.
Analyses of in vivo CCR2-dependent monocyte migration into the ischemic brain. (A) Representative flow cytometry gating strategy for the mixed WT and CCR2 KO brain monocytes. (B) Representative flow cytometry gating strategy for the mixed WT and CCR2 KO blood monocytes. (C) Quantification of CCR2 KO monocytes vs. WT monocytes in both the ischemic brain and blood at 3 and 7 days after stroke. The ratio was calculated by the WT number divided by the CCR2 KO number. n=3; ** P<0.01 vs. input ratio where the value was set to 1 (one-sample t-test).
PG shows similar effects as CCR2 KO in stroke
We used MRI to measure infarction in WT mice treated with PG [30]. In order to compare it with CCR2 KO mice, PG or PBS was administered one day before MCAO surgery until 14 days after ischemic stroke. The results show that, compared with PBS treated animals, PG had smaller infarction at 3 days, but larger infarction at 14 days, suggesting that PG has similar effects as CCR2 KO (Figure 4A).
Effects of the CCR2 inhibitor, propagermanium (PG), on infarction, mortality and behavior after ischemic stroke in WT mice. (A) Representative MRI T2 images are shown. PG (50 mg/kg/day, three times a day) or vehicle PBS was administered via oral gavage to WT mice one day before MCAO surgery until 14 days after ischemic stroke. The bar graph shows the statistical results of atrophy size measured at 3 and 14 days after stroke. n=3. (B) PG (50 mg/kg/day, three times a day) or vehicle PBS was administered via oral gavage to WT mice from 4 days until 14 days after stroke. Curve showing the mortality ratio from 0 to 14 days after stroke. n=12. (C) Behavioral tests. PG (50 mg/kg/day, three times a day) or vehicle PBS was administered via oral gavage to WT mice from 4 days until 14 days after stroke. a. Rotating beam walking test. b. Pole test shows the average time walking to the top of the vertical pole. c. Pole test shows the average time walking from the top to the bottom of the pole. n=6. (D) Representative CD68 immunohistochemistry staining. Scale bar, 100 μm. Bar graph shows the quantification of CD68+ macrophages at 7 and 14 days, treated with PBS or PG after stroke. n=6. *, ** vs. vehicle, P<0.05, P<0.01, respectively; one-way ANOVA was performed to compare the PG- and PBS-treated groups.
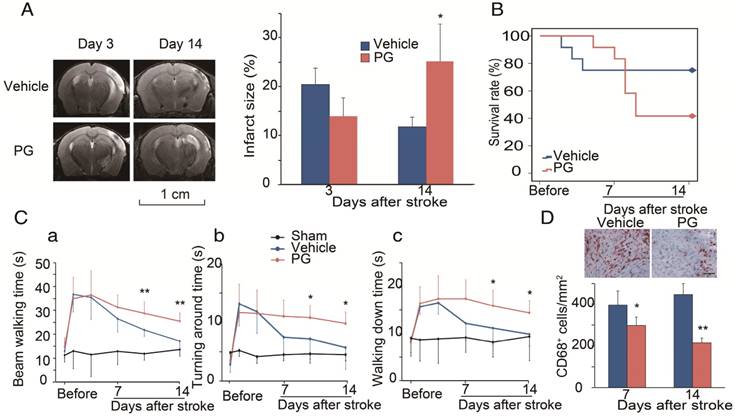
To test whether CCR2 inhibition alters animal behaviors, PG was administered from 4 days until 14 days after stroke. Like CCR2 KO, PG treatment resulted in a significantly higher mortality, starting from 5 days to 12 days after stroke (Figure 4B), and impaired behavioral recovery (Figure 4Ca-c). PG also dramatically inhibited CD68 expression at 7 and 14 days (Figure 4D).
Macrophages convert from the pro-inflammatory to anti-inflammatory phenotype at recovery stage after stroke in WT mice
The results showed that in WT mice, the mRNA levels of the pro-inflammatory macrophage genes, TNF-α and IFN-γ, dramatically increased at 3 and 5 days, but then declined at 7 and 14 days. In contrast, the anti-inflammatory macrophage genes, TGF-β1 and IL-4, in WT mice gradually increased up to 7 days, and then decreased by 14 days. Conversely, the pro-inflammatory macrophage genes increased up to 7 days, while the anti-inflammatory macrophage genes decreased from 3 to 14 days after stroke in the CCR2 KO mice (Figure 5A).
Because the mRNA levels in the ischemic tissues may not precisely reflect the gene expressions in MoDMs, we used a flow cytometry sorter to isolate MoDMs for gene expression quantification. TNF-α mRNA was significantly elevated at 3 to 5 days, and then decreased significantly at 7 days in the purified MoDMs. In contrast, a significant increase of TGF-β1 mRNA expression was observed from 3 days onward, and remained high until 7 days (Figure 5B).
Analyses of cytokine gene expressions and macrophage phenotypes. (A) Real-time PCR analysis of Pro-inflammatory and anti-inflammatory gene expressions in the ischemic brains of WT and CCR2 KO mice at different time points after ischemic stroke. n=6. (B) Real-time PCR analyses of TNF-α and TGF-β1 gene expressions in sorted MoDMs. CD45hiCD11b+ cells were first gated, and then Ly6G-Ly6Chi (CCR2-dependent inflammatory MoDMs) were gated and sorted (~50,000 cells in a total of 6 mice) for real-time PCR analysis. n=4. (C) Flow cytometry analysis of pro-inflammatory and anti-inflammatory MoDMs phenotypes in the ischemic brain. Single cells were obtained by gating with FSC-height vs. FSC-area, then live/dead cells were gated by using propidium iodide (PI) or Aqua to exclude dead cells; PI and Aqua were used for surface cell markers and intracellular staining antibodies, respectively. Microglia/macrophages were first gated as CD45intCD11b and CD45hiCD11b+, respectively. Neutrophils were excluded from MoDMs by gating with Ly6G-. The leftover MoDMs were further gated with Ly6C, and analyzed with the pro- inflammatory markers, PD-L1 or iNOS, and the anti- inflammatory markers, PD-L2 or Arg-1. n=6; *P<0.05, **P<0.01; Mann-Whitney U test used for analysis of real-time PCR data.
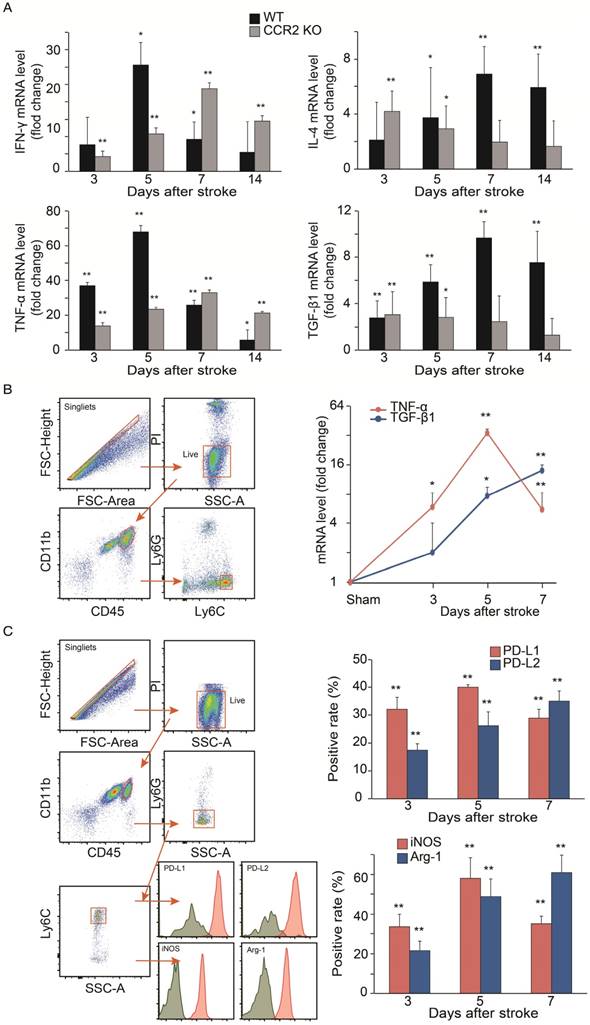
We further investigated protein expressions by flow cytometry to support the result of pro-inflammatory vs. anti-inflammatory status in the ischemic brain. We gated CD45hiCD11b+Ly6G-Ly6Chi cell populations as MoDMs in the ischemic brain (Figure 5C). Consistent with the real-time PCR results, both the pro-inflammatory macrophage markers (PD-L1 and iNOS) were elevated significantly at 3 days, and then declined at 7 days, while the anti-inflammatory macrophage markers (PD-L2 and Arg-1) gradually increased at 3 to 7 days after ischemic stroke (Figure 5C).
Adoptive transfer of the anti-inflammatory macrophages promoted functional recovery in CCR2 KO mice after stroke
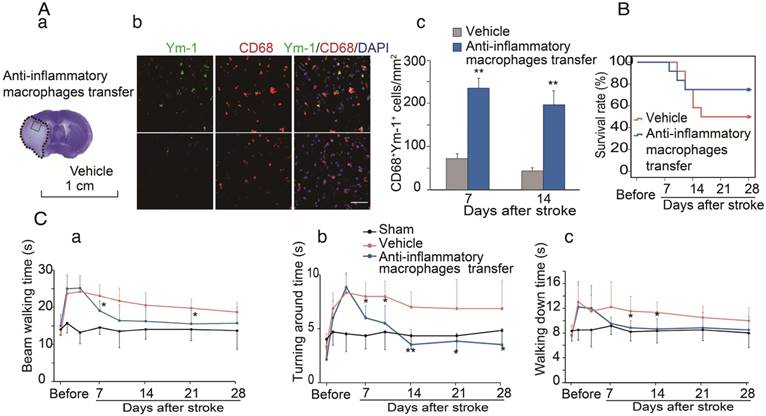
We have provided evidence that CCR2-dependent MoDMs are vital for the neuroinflammatory responses of pro-inflammatory vs. anti-inflammatory macrophages, and we speculated that the anti-inflammatory macrophages might be essential for neurological recovery. To prove this hypothesis, anti-inflammatory macrophages or vehicle PBS were adoptively transferred into CCR2 KO mice at 4 and 6 days after ischemic stroke. The double immunostaining suggests that the adoptive transfer of anti-inflammatory macrophages resulted in increases of the anti-inflammatory macrophage phenotype (Ym-1) in the ischemic brain (Figure 6A). The increased anti-inflammatory microglia/macrophages lead to a lower mortality and improvement in neurological recovery (Figure 6B-C).
The protective effects of adoptive transfer of anti-inflammatory macrophages to CCR2 KO mice. Bone marrow derived monocytes were cultured and stimulated with murine IL-4 to polarize into the anti-inflammatory phenotypes. The anti-inflammatory macrophages or vehicle PBS were adoptively transferred into CCR2 KO mice (2×106 cells/mouse, 0.2 mL via retro-orbital injection) at 4 and 6 days after ischemic stroke, respectively. (A) a. A representative brain section with cresyl violet staining indicates the ischemic region from which the immunostaining pictures were taken. b. Representative images of double immunofluorescence staining with Ym-1 (green), CD68 (red), and DAPI counterstain (blue). Scale bar, 50 μm. c. Quantification of double-positive CD68 and YM-1 macrophage staining at 7 and 14 days after stroke in CCR2 KO mice receiving adoptive transfer of anti-inflammatory macrophages or vehicle. n=6 mice/group, 3 sections/mouse. (B) The curves represent the survival ratio from 0 to 28 days after MCAO in CCR2 KO mice receiving adoptive transfer of anti-inflammatory macrophages and vehicle. n=12. (C) Behavioral tests. a. Rotating beam test showing the average walking time (seconds). b. Pole test showing the climbing time to the top of the vertical pole. c. Pole test showing the time to climb from the top to the bottom of the vertical pole. n=6; *, ** vs. vehicle, P<0.05, 0.01, respectively; one-way ANOVA was performed for the study of adoptive transfer of anti-inflammatory macrophages.
Discussion
We unprecedentedly compared the effects of monocytes in the acute and delayed phase after stroke, and found that CCR2-dependent monocytes are a double-edged sword: they exacerbate acute brain injury but promote neurological functions at the delayed phase after stroke. We showed that the migration and infiltration of MoDMs into the ischemic brain is CCR2 dependent, which has distinctive roles in the acute and delayed phase after stroke. First, both CCR2 KO and the administration of the CCR2 inhibitor, PG, resulted in larger injury and higher mortality at the delayed stage after stroke compared with WT mice. Second, MoDMs migration is CCR2 dependent, and functional recovery after stroke required the infiltration of MoDMs into the ischemic brain. Third, the role of MoDMs in promoting functional recovery is anti-inflammatory macrophage dependent, as the majority of macrophages converted into the anti-inflammatory phenotype at the recovery stage. In addition, the adoptive transfer of anti-inflammatory macrophages promoted functional recovery.
Macrophages play an important role in brain injury after stroke, but whether macrophage-mediated neuroinflammation has distinctive roles in the acute and delayed phases of brain injury remains elusive. Because MoDMs enter the ischemic brain in a delayed pattern, it has been assumed that MoDMs are more important in functional recovery rather than in acute stroke. Our results suggest that MoDMs play a detrimental role in acute brain injury measured at 3 days after stroke, but plays a beneficial role in functional recovery measured 7 to 28 days after stroke. The results refute the assumption that MoDMs are not important for acute brain injury.
After showing that monocyte migration is CCR2 dependent, we then examined the effects of MoDMs on stroke outcomes. We show that MoDMs depletion in the ischemic brain in CCR2 KO mice resulted in a smaller infarct measured 3 days after stroke, but with larger injuries, higher mortality, and less functional recovery measured at 14 and 28 days. This phenomenon is further strengthened by the use of the CCR2 inhibitor, PG, which has relatively specific effects on monocytes, rather than on other cell types. Our collaborator, Dr. Baohui Xu, and colleagues, have shown that PG treatment decreased monocytes solely in bone marrow, but it did not affect the relative numbers of leukocyte subsets in the blood, bone marrow, lymph nodes, and spleen in normal mice or with LPS stimulation. They concluded that PG treatment produced similar effects as CCR2 KO [30]; thus, PG was used to verify our study of CCR2 KO mice. Our results are supported by Wattananit et al., who recently reported that the anti-CCR2 antibody, MC-21, abolished long-term behavioral recovery after ischemic stroke [12], suggesting that MoDMs contribute to spontaneous, long-term functional recovery after stroke. Our results in the acute stage after stroke conflicts with the findings of Chu and colleagues [11], who showed that monocytes are neuroprotective against acute infarction. The discrepancy between our study and Chu et al. might be due to differences in experimental designs, animal strains and models, and the CCR2 inhibitors. We used both WT and CCR2 KO mice, but Chu et al., only used WT mice, which showed that CCR2 inhibition did not affect the expressions of the CCR2 ligand, CCL2. In addition, they used the CCR2 inhibitor, INCB3344, which was administered 1 day before stroke. But in our study, the CCR2 inhibitor, PG, was administered 1 day before MCAO surgery to 14 days after ischemic stroke. INCB3344 differs from PG, but how these two inhibitors differ in their pharmacological effects is not known. Furthermore, in their study, the infarction was measured only at 1 day after stroke, while we measured infarct size at 3 and 7 days, and up to 1 month after stroke.
Macrophages can polarize into pro-inflammatory and anti-inflammatory phenotypes [31]. Macrophage functional phenotypes include a wide spectrum between the two extremes of M1 and M2 phenotypes, which might be over-simplified, as pointed out by Murray et al. [32]. Therefore, we refer to the M1 and M2 phenotypes as pro- and anti-inflammatory phenotypes, respectively. Macrophages may convert from one functional phenotype into another in response to the local microenvironment [33], and in response to the different time courses of tissue injuries [34-35]. We found that the pro-inflammatory phenotypes were dominant in the acute phase after stroke, while the anti-inflammatory phenotypes accumulated in the delayed phase. First, in WT mice, the pro-inflammatory genes, TNF-α and INF-γ, were robustly increased in the acute phase, but declined at 7 days, while the anti-inflammatory genes, IL-4 and TGF-β1, gradually increased and remained high until 7 days. Conversely, the pro-inflammatory genes increased up to 7 days, while the anti-inflammatory genes decreased from 3 to 14 days after stroke in the CCR2 KO mice, indicating that the anti-inflammatory macrophages are linked with functional recovery after stroke. Therefore, macrophages may convert from pro-inflammatory into anti-inflammatory phenotypes along the time course after stroke. Second, the results of TNF-α and TGF-β1 gene expressions in the sorted MoDMs further confirmed that the pro-inflammatory gene expressions declined at 7 days while the anti-inflammatory gene expressions remained high. Third, the flow cytometry results showed a similar change pattern in the cell surface and intracellular markers for both pro- and anti-inflammatory phenotypes. Therefore, we conducted the experiment by adoptively transferring the anti-inflammatory macrophages into CCR2 KO mice at 4 and 6 days after MCAO, in order to supply more anti-inflammatory macrophages at a later period. The results indicated that the adoptive transfer of the anti-inflammatory macrophages increased the anti-inflammatory protein, Ym-1, in both MiDMs and MoDMs, improved functional outcomes, and attenuated mortality in CCR2 KO stroke mice.
The different gene expressions in the ischemic brains between the WT and CCR2 KO mice may be due to different cell compositions, as the CCR2 mice have fewer macrophages in their ischemic brain. Therefore, gene expressions measured in the CCR2 KO mice may reflect gene expression changes in other cell types, such as microglia or astrocytes, which also take part in neuroinflammation after stroke. Indeed, A1 astrocytes, induced by activated microglia, might be harmful, by secreting IL-1α, TNF and C1q [36]. In addition, macrophage induced osteopontin, which further activates astrocytes to repair ischemic neurovascular units, suggests an interaction between macrophages and astrocytes [37]. Therefore, the reduced number of MoDMs would affect microglia or astrocyte phenotypes.
Nevertheless, there was still about 25% recruitment of MoDMs in the CCR2 KO mice, suggesting that there are other receptors involved in monocyte recruitment besides the CCL2/CCR2 axis. Although we do not exactly know the non-CCR2 mechanisms, our previous works and others have shown that CCR5 (CCL5/CCR5) are also expressed in CD45hiCD11b+ MoDMs, both in the experimental ischemic brain and aneurysm tissue [22, 27]. Therefore, CCL5, the CCR5 ligand, also recruits inflammatory monocytes into target tissues, and promotes macrophage induced inflammatory conditions.
Our results on the early pro-inflammatory and late anti-inflammatory activities after stroke are similar to other non-stroke diseases, such as musculoskeletal regeneration, spinal cord injury and myocardial infarction [38-40]. Nevertheless, a report from Chen et al., using reverse transcriptase PCR and immunohistochemical staining methods, suggests that microglia/macrophages primarily exhibit the anti-inflammatory phenotype at an early phase after stroke, and then convert to the pro-inflammatory phenotype at a later phase [41], which differs from our results. This may be due to the different models and techniques used to identify the functional macrophage phenotypes. For example, in our study, we used the ischemic area and not the entire hemisphere for real-time PCR. We used flow cytometry to distinguish microglia from MoDMs, and used cytometry sorting to purify inflammatory monocytes to verify our real-time PCR results. Taken together, our results suggest that MoDMs are the main effectors of pro- and anti-inflammatory pattern changes in the ischemic brain at the early and recovery stages after stroke.
There are a number of limitations in our current study. First, MiDMs and MoDMs are identified as CD45intCD11b+ and CD45hiCD11b+ cell populations, but the CD45hiCD11b+ cell population in the ischemic brain may also include neutrophils. Nevertheless, the method of isolating leukocytes excludes most neutrophils in the samples, thus the majority of isolated cells are mononuclear cells. Although this cell population mainly contains monocytes, we cannot completely exclude the fact that the MoDMs population is contaminated by some neutrophils. Second, CD68 staining was used to verify the effect of CCR2 KO on MoDMs recruitment in the ischemic brain. However, CD68 is highly expressed in both MoDMs and MiDMs after stroke, thus CD68 staining cannot distinguish between MoDMs and MiDMs. Third, we reported that the adoptive transfer of anti-inflammatory macrophages improves the long term behavior performance and animal survival ratio, but brain atrophy sizes were not measured. Fourth, we showed that the adoptive transfer of anti-inflammatory macrophages promotes functional recovery by enhancing Ym-1 positive macrophages. Nevertheless, this treatment may also affect microglia, but we did not distinguish Ym-1 expressions between MiDMs and MoDMs.
Summary
In conclusion, CCR2-dependent MoDMs play a double-edged role in ischemic stroke, causing more injury in the early phase after stroke, while promoting functional recovery at the late stage. These MoDMs may achieve their effects via a conversion of pro-inflammatory to anti-inflammatory phenotypes.
Acknowledgements
This study was supported by R01NS06413606 (HZ), National Natural Science Foundation of China (Program No. 81673442, WF), China Scholarship Council (File No. 201307060033), and Qing Lan Project (2016).
Abbreviations
BMDM: bone marrow-derived macrophages; CCA: common carotid artery; CCR2: C-C chemokine receptor 2; CNS: central nervous system; DAPI: 4′, 6-diamidino- 2-phenylindole; IL-4: interleukin-4; KO: knockout; MCAO: middle cerebral artery occlusion; M-CSF: macrophage-colony stimulating factor; MiDMs: microglia-derived macrophages; MoDMs: monocyte-derived macrophages; MRI: Magnetic Resonance Imaging; PG: propagermanium; TGF-β: transforming growth factor β; TNF-α: tumor necrosis factor-α; TTC: 2,3,5-triphenyltetrazolium chloride; WT: wild type.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Petrovic-Djergovic D, Goonewardena SN, Pinsky DJ. Inflammatory disequilibrium in stroke. Circ Res. 2016;119:142-58
2. Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P. et al. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. 2015;11:56-64
3. Kim E, Cho S. Microglia and monocyte-derived macrophages in stroke. Neurotherapeutics. 2016;13:702-18
4. Kreutzberg GW. Microglia: A sensor for pathological events in the cns. Trends Neurosci. 1996;19:312-8
5. Li T, Pang S, Yu Y, Wu X, Guo J, Zhang S. Proliferation of parenchymal microglia is the main source of microgliosis after ischaemic stroke. Brain. 2013;136:3578-88
6. Umekawa T, Osman AM, Han W, Ikeda T, Blomgren K. Resident microglia, rather than blood-derived macrophages, contribute to the earlier and more pronounced inflammatory reaction in the immature compared with the adult hippocampus after hypoxia-ischemia. Glia. 2015;63:2220-30
7. Xiong XY, Liu L, Yang QW. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog Neurobiol. 2016;142:23-44
8. Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV Jr, Broxmeyer HE. et al. Impaired monocyte migration and reduced type 1 (th1) cytokine responses in c-c chemokine receptor 2 knockout mice. J Clin Invest. 1997;100:2552-61
9. Kurihara T, Warr G, Loy J, Bravo R. Defects in macrophage recruitment and host defense in mice lacking the ccr2 chemokine receptor. J Exp Med. 1997;186:1757-62
10. Chu HX, Arumugam TV, Gelderblom M, Magnus T, Drummond GR, Sobey CG. Role of ccr2 in inflammatory conditions of the central nervous system. J Cereb Blood Flow Metab. 2014;34:1425-9
11. Chu HX, Broughton BR, Kim HA, Lee S, Drummond GR, Sobey CG. Evidence that ly6c(hi) monocytes are protective in acute ischemic stroke by promoting m2 macrophage polarization. Stroke. 2015;46:1929-37
12. Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J. et al. Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci. 2016;36:4182-95
13. Xiong X, Xu L, Wei L, White RE, Ouyang YB, Giffard RG. Il-4 is required for sex differences in vulnerability to focal ischemia in mice. Stroke. 2015;46:2271-6
14. Joo SP, Xie W, Xiong X, Xu B, Zhao H. Ischemic postconditioning protects against focal cerebral ischemia by inhibiting brain inflammation while attenuating peripheral lymphopenia in mice. Neuroscience. 2013;243:149-57
15. Teng F, Zhu L, Su J, Zhang X, Li N, Nie Z. et al. Neuroprotective effects of poly(adp-ribose)polymerase inhibitor olaparib in transient cerebral ischemia. Neurochem Res. 2016;41:1516-26
16. Ludewig P, Sedlacik J, Gelderblom M, Bernreuther C, Korkusuz Y, Wagener C. et al. Carcinoembryonic antigen-related cell adhesion molecule 1 inhibits mmp-9-mediated blood-brain-barrier breakdown in a mouse model for ischemic stroke. Circulation research. 2013;113:1013-22
17. Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008 Chapter 14: Unit 14 11
18. Koronyo Y, Salumbides BC, Sheyn J, Pelissier L, Li S, Ljubimov V. et al. Therapeutic effects of glatiramer acetate and grafted cd115(+) monocytes in a mouse model of alzheimer's disease. Brain. 2015;138:2399-422
19. Linden AM, Sandu C, Aller MI, Vekovischeva OY, Rosenberg PH, Wisden W. et al. Task-3 knockout mice exhibit exaggerated nocturnal activity, impairments in cognitive functions, and reduced sensitivity to inhalation anesthetics. J Pharmacol Exp Ther. 2007;323:924-34
20. Balkaya M, Endres M. Behavioral testing in mouse models of stroke. Rodent Models of Stroke. Neuromethods. 2010;47:182-184
21. Xiong X, Gu L, Zhang H, Xu B, Zhu S, Zhao H. The protective effects of t cell deficiency against brain injury are ischemic model-dependent in rats. Neurochem Int. 2013;62:265-70
22. Fan Y, Xiong X, Zhang Y, Yan D, Jian Z, Xu B. et al. Mkey, a peptide inhibitor of cxcl4-ccl5 heterodimer formation, protects against stroke in mice. Journal of the American Heart Association. 2016;5(9):e003615
23. Zeng X, Wei YL, Huang J, Newell EW, Yu H, Kidd BA. et al. Gammadelta t cells recognize a microbial encoded b cell antigen to initiate a rapid antigen-specific interleukin-17 response. Immunity. 2012;37:524-34
24. Xu B, Wagner N, Pham LN, Magno V, Shan Z, Butcher EC. et al. Lymphocyte homing to bronchus-associated lymphoid tissue (balt) is mediated by l-selectin/pnad, alpha4beta1 integrin/vcam-1, and lfa-1 adhesion pathways. J Exp Med. 2003;197:1255-67
25. Anderson CF, Lira R, Kamhawi S, Belkaid Y, Wynn TA, Sacks D. Il-10 and tgf-beta control the establishment of persistent and transmissible infections produced by leishmania tropica in c57bl/6 mice. J Immunol. 2008;180:4090-7
26. Valaperti A, Nishii M, Liu Y, Naito K, Chan M, Zhang L. et al. Innate immune interleukin-1 receptor-associated kinase 4 exacerbates viral myocarditis by reducing ccr5(+) cd11b(+) monocyte migration and impairing interferon production. Circulation. 2013;128:1542-54
27. Iida Y, Xu B, Xuan H, Glover KJ, Tanaka H, Hu X. et al. Peptide inhibitor of cxcl4-ccl5 heterodimer formation, mkey, inhibits experimental aortic aneurysm initiation and progression. Arterioscler Thromb Vasc Biol. 2013;33:718-26
28. Prinz M, Priller J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15:300-12
29. Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA. 1991;88:7438-42
30. Fujimura N, Xu B, Dalman J, Deng H, Aoyama K, Dalman RL. Ccr2 inhibition sequesters multiple subsets of leukocytes in the bone marrow. Sci Rep. 2015;5:11664
31. Gliem M, Schwaninger M, Jander S. Protective features of peripheral monocytes/macrophages in stroke. Biochim Biophys Acta. 2016;1862(3):329-38
32. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S. et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity. 2014;41:14-20
33. Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. 2005;175:342-9
34. Mylonas KJ, Nair MG, Prieto-Lafuente L, Paape D, Allen JE. Alternatively activated macrophages elicited by helminth infection can be reprogrammed to enable microbial killing. J Immunol. 2009;182:3084-94
35. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723-37
36. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481-7
37. Gliem M, Krammes K, Liaw L, van Rooijen N, Hartung HP, Jander S. Macrophage-derived osteopontin induces reactive astrocyte polarization and promotes re-establishment of the blood brain barrier after ischemic stroke. Glia. 2015;63:2198-207
38. Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A. et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. 2007;204:1057-69
39. Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G. et al. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6:e1000113
40. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL. et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037-47
41. Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S. et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063-70
Author contact
![]() Corresponding author: Heng Zhao, Department of Neurosurgery, Stanford University School of Medicine, 1201 Welch Rd, MSLS Bldg., Room 306, Stanford, CA 94305-5327, USA. Phone: 1 (650) 725-7723; email: hzhaoedu
Corresponding author: Heng Zhao, Department of Neurosurgery, Stanford University School of Medicine, 1201 Welch Rd, MSLS Bldg., Room 306, Stanford, CA 94305-5327, USA. Phone: 1 (650) 725-7723; email: hzhaoedu