Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Acknowledgements
Author contributions
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(12):3416-3436. doi:10.7150/thno.25228 This issue Cite
Research Paper
Antibody affinity and valency impact brain uptake of transferrin receptor-targeted gold nanoparticles
Kasper Bendix Johnsen1,2, Martin Bak2, Paul Joseph Kempen2, Fredrik Melander2, Annette Burkhart1, Maj Schneider Thomsen1, Morten Schallburg Nielsen3, Torben Moos1 ![]() , Thomas Lars Andresen2
, Thomas Lars Andresen2 ![]()
1. Laboratory for Neurobiology, Biomedicine, Department of Health Science and Technology, Aalborg University, Aalborg, Denmark.
2. Center for Nanomedicine and Theranostics, Department of Micro- and Nanotechnology, Technical University of Denmark, Denmark.
3. Department of Biomedicine, Aarhus University, Denmark.
Received 2018-1-30; Accepted 2018-4-17; Published 2018-5-24
Abstract

Rationale: The ability to treat invalidating neurological diseases is impeded by the presence of the blood-brain barrier (BBB), which inhibits the transport of most blood-borne substances into the brain parenchyma. Targeting the transferrin receptor (TfR) on the surface of brain capillaries has been a popular strategy to give a preferential accumulation of drugs or nanomedicines, but several aspects of this targeting strategy remain elusive. Here we report that TfR-targeted gold nanoparticles (AuNPs) can accumulate in brain capillaries and further transport across the BBB to enter the brain parenchyma.
Methods: We characterized our targeting strategy both in vitro using primary models of the BBB and in vivo using quantitative measurements of gold accumulation by inductively-coupled plasma-mass spectrometry together with morphological assessments using light microscopy after silver enhancement and transmission electron microscopy with energy-dispersive X-ray spectroscopy.
Results: We find that the uptake capacity is significantly modulated by the affinity and valency of the AuNP-conjugated antibodies. Specifically, antibodies with high and low affinities mediate a low and intermediate uptake of AuNPs into the brain, respectively, whereas a monovalent (bi-specific) antibody improves the uptake capacity remarkably.
Conclusion: Our findings indicate that monovalent ligands may be beneficial for obtaining transcytosis of TfR-targeted nanomedicines across the BBB, which is relevant for future design of nanomedicines for brain drug delivery.
Keywords: Brain drug delivery, gold nanoparticle, transferrin receptor, targeting, affinity
Introduction
Despite decades of technological advances, delivery of therapeutically active compounds to the brain remains a critical challenge for the treatment of neurological disorders [1, 2]. The low accumulation of drugs in the brain is due to the presence of the blood-brain barrier (BBB), which restricts passage of most compounds into the brain [3]. While the BBB is a major challenge for drug transport into the brain, it contains many receptor and transport proteins that are responsible for nutrient transport into the brain and depict potential targets for brain drug delivery technologies [4, 5]. Among the most exploited target proteins on the luminal membrane of the BBB, the transferrin receptor (TfR) is under normophysiological conditions responsible for binding to and endocytosing iron-transferrin (Tf) complexes for subsequent iron transport into the brain parenchyma [1, 6, 7]. The intracellular sorting mechanism of the TfR in brain capillary endothelial cells (BCECs) is somewhat disputed, but recent evidence indicates that the receptor protein is incapable of transcytosis through the BCEC to the abluminal membrane and brain parenchyma [7-10]. Instead, iron is dissociated during endosomal acidification and in a multistep process pumped into the brain parenchyma independent of TfR and Tf transport [7-10]. Thus, this model does not suggest the TfR to be a relevant target for transport across the BCEC into the brain [1, 6].
While there has been a broad acceptance of the abovementioned role of TfR in physiological iron transport into the brain without transcellular transport of the receptor itself, this role has been continuously challenged by evidence from the brain drug delivery field [1, 6]. For example, antibody constructs efficiently transcytose through the BBB if the binding site of the antibody construct is modified to reduce the overall affinity towards the TfR [11-16]. In addition to the affinity, others have suggested that the valency of the antibody construct against the TfR is critical for antibody transport across the BBB [17-21]. With respect to nanomedicines, the total ligand avidity on nanoparticles is also important for transport into the brain [22, 23], whereas the impact of nanoparticle ligand affinity towards the TfR is less well understood [24]. Importantly, despite critique raised towards the binding kinetics of a nanoparticle to the brain endothelial cell and the ability of nanoparticles to transport into the brain [17], whole TfR-targeted gold nanoparticles (AuNPs) were observed in the brain parenchyma after intravenous administration, suggestive of a functional transport mechanism of this nanoparticle across the BBB mediated by the TfR [22, 23].
The objective of the present study was to investigate the impact of ligand binding mode for nanoparticle transport into the brain. Based on the currently available literature, we expected that modifying the binding affinity or valency of the antibodies targeting the TfR would reduce the brain uptake of the nanoparticle because the strength of the initial interaction between the nanoparticle and the BCEC would be too low to initiate proper endocytosis of the nanoparticle [1, 6]. To study this hypothesis, we decided to examine AuNPs, as they have multifunctionality with respect to the downstream analysis of brain uptake [23]. Concerning the targeting ligands, we gained access to the antibodies developed by Yu et al. [16], which allowed for evaluation of both the difference in affinity (anti-TfRA versus anti-TfRD) and mono- versus bivalency (anti-TfRA/BACE1 versus anti-TfRA) of the TfR-targeting antibody in relation to the subsequent brain uptake of AuNPs. In addition, novel lipoic acid-terminated polyethylene glycol 5000 (PEG5000-LA) constructs were synthesized for polymer surface coating and antibody functionalization of the AuNPs.
We report here that lowering the affinity of the TfR-targeting antibody on the AuNP surface improved the brain uptake modestly but using a monovalent binding antibody increased the brain uptake markedly. This was shown using the bi-specific antibody, anti-TfRA/BACE1. While BACE1 targeting may have a therapeutic potential in relation to the treatment of Alzheimer's disease [25], we could not prove this effect when the anti-TfRA/BACE1 antibody was conjugated to the AuNPs. We quantified the differences in transport both in vitro and in vivo after brain capillary depletion and subsequent inductively-coupled plasma-mass spectrometry (ICP-MS) assessment, and we detected AuNPs in the brain parenchyma using both light microscopy and transmission electron microscopy (TEM) with energy-dispersive X-ray spectroscopy (EDS).
Results
Production of TfR-targeted AuNPs with varying affinities and valencies
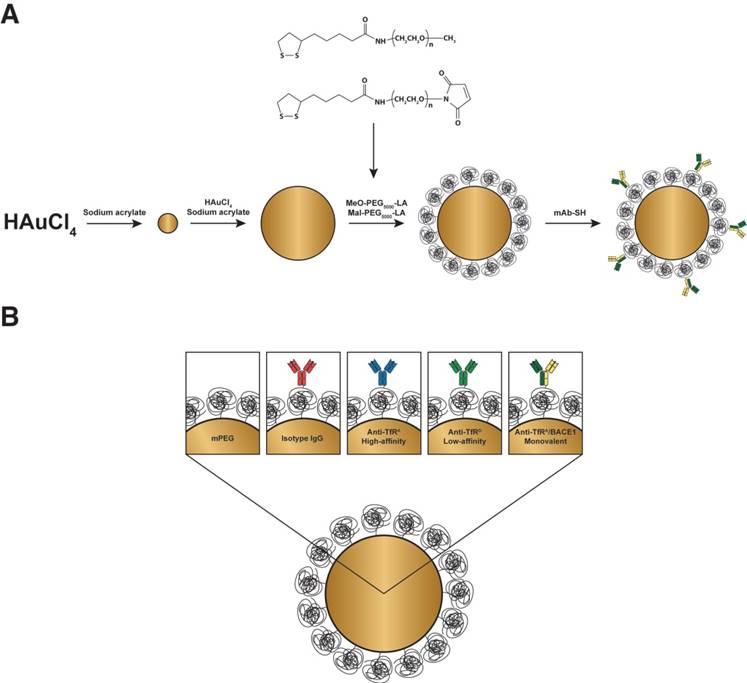
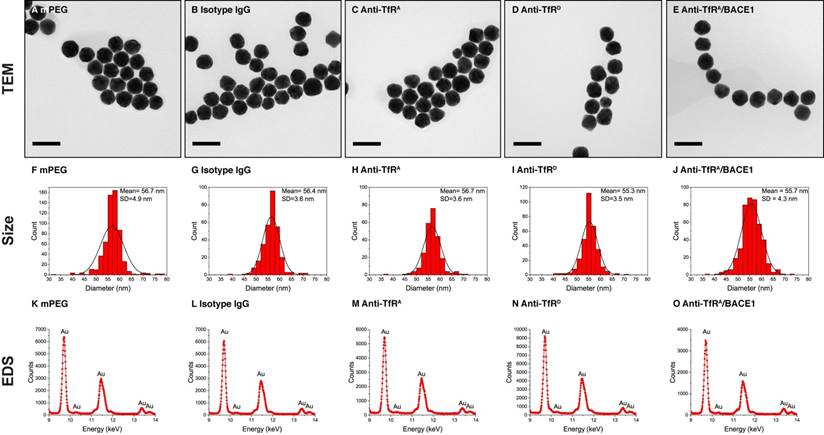
To study the impact of antibody affinity and valency on the brain uptake of nanoparticles, we decided to use AuNPs as our model system, because these nanoparticles allow ample detection with methodologies including ICP-MS, light microscopy after silver enhancement, and TEM with EDS. Uniform AuNPs were produced by a sodium acrylate reduction of a gold salt in solution, done sequentially to obtain a final size of the AuNP core of 52.8 ± 0.7 nm with low polydispersity (Table 1). The AuNPs were subsequently surface-coated with a layer of PEG5000-LA (for schematic, please refer to Figure 1), further increasing the hydrodynamic diameter of the AuNP as determined by dynamic light scattering (DLS; Table 1). Inclusion of a small fraction of 1% maleimide-functionalized PEG5000 (Mal-PEG5000-LA) allowed for conjugation of thiolated anti-TfR antibodies via standard Michael-type addition. The thiolation of the different antibodies was optimized through a screening process to tune the number of reactive thiols group to approximately one per antibody (Figure S1). All antibodies were tested for their reactivity towards the mouse TfR using surface plasmon resonance prior to conjugation to the AuNPs ensuring the desired affinity of the individual antibodies (Table 2 and Figure S2). After the conjugation reaction between the antibodies and AuNPs, all formulations had comparable size distributions and number of antibodies present on the AuNP surface, as directly measured by dot blot (Table 1). Using TEM with EDS, it was possible to validate the homogenous appearance and the presence of gold in all formulations (Figure 2).
DLS, ζ-potential, UVmax, and polydispersity index of the AuNPs
| Group | Hydrodynamic diameter (nm) | ζ-potential (mV) | UVmax (nm) | PDI | Antibodies per AuNP |
|---|---|---|---|---|---|
| AuNPs 1st seed | 35.7 ± 0.2 | -32.8 ± 0.8 | 524 | 0.016 | - |
| AuNPs 2nd seed | 52.8 ± 0.7 | -31.7 ± 5.9 | 534 | 0.082 | - |
| mPEG AuNPs | 92.6 ± 1.5 | -27.2 ± 3.4 | 534 | 0.079 | - |
| mPEG AuNPs + maleimide | 82.0 ± 1.6 | -40.4 ± 1.6 | 536 | 0.206 | - |
| Isotype IgG AuNPs | 75.1 ± 0.6 | -24.9 ± 0.7 | 536 | 0.249 | 8.8 |
| Anti-TfRA AuNPs | 75.2 ± 1.6 | -21.4 ± 1.0 | 536 | 0.217 | 6.5 |
| Anti-TfRD AuNPs | 74.8 ± 1.1 | -16.7 ± 1.4 | 536 | 0.245 | 8.6 |
| Anti-TfRA/BACE1 AuNPs | 77.0 ± 0.3 | -24.1 ± 1.5 | 536 | 0.250 | 7.5 |
Overview of experimental setup. (A) Schematic representation of the synthesis strategy resulting in TfR-targeted AuNPs. (B) Illustration of the different groups compared in subsequent experiments.
Biacore analysis of anti-TfR antibodies against recombinant mouse TfR
| Group | Host | Reactivity | Dissociation constant (nM) |
|---|---|---|---|
| Isotype IgG | Human | - | 3200 |
| Anti-TfRA | Human | Mouse | 21 |
| Anti-TfRD | Human | Mouse | 149 |
| Anti-TfRA/BACE1 | Human | Mouse | 22 |
| RI7217 | Rat | Mouse | 6 |
| OX26 | Mouse | Rat | 1600 |
TEM evaluation of AuNPs. TEM with EDS was performed to characterize the homogeneity, size, and composition of the AuNPs. All groups of AuNPs presented with a homogenous shape (A-E) and size distribution (F-J). A minimum of 250 AuNPs were analyzed in each group to produce the size distribution diagrams. In addition, using EDS, the presence of gold was validated in all groups of AuNPs (K-O). Scale bars depict 100 nm.
AuNP uptake and transport across the blood-brain barrier is increased by TfR-targeting in vitro
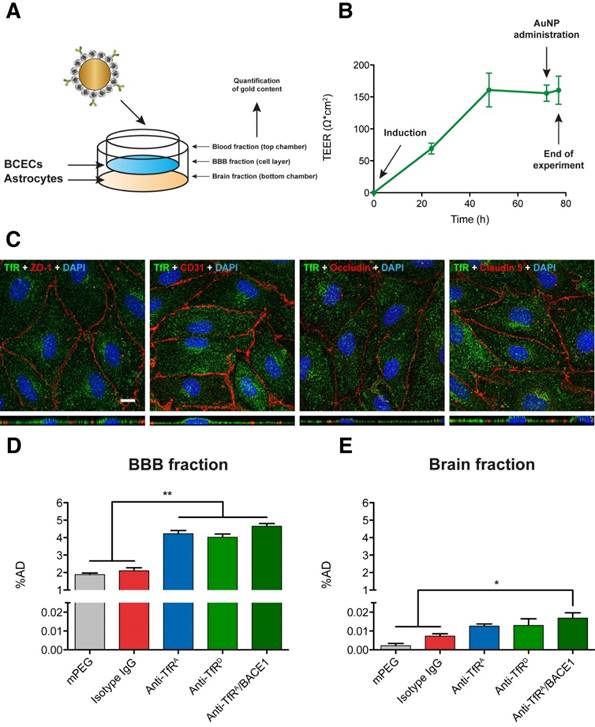
The AuNP formulations were administrated to an in vitro model of the BBB based on freshly isolated mouse BCECs grown in co-culture with primary mouse astrocytes (Figure 3A). The transendothelial electrical resistance (TEER) of the barriers was high (~150 Ω cm2), indicating a tight model of the BBB to study the AuNP transport (Figure 3B) [26]. Importantly, there was no measurable reduction in the barrier integrity during the experiment, as indicated by the TEER being maintained at a high level (Figure 3B). The mouse BCECs of the in vitro models expressed relevant tight junction (TJ) proteins including CD31, ZO-1, occludin, and claudin-5 together with the TfR (Figure 3C and Figure S3). TfR expression could also be confirmed in BCEC lysates by Western blotting (Figure S4). After incubation of the BCECs with the different AuNP formulations, there was an increased uptake of AuNPs into the BCECs as a function of TfR targeting compared to the mPEG and isotype IgG control formulations (Figure 3D, p < 0.01). In general, there was a high baseline level of uptake, which may be due to the high density of the AuNPs, but overall, this level of uptake was doubled by targeting of the TfR (Figure 3D). The transport of AuNPs to the opposite (brain) side of the Transwell culture system was also measured, showing that the transport capacity, especially of anti-TfRA/BACE1 monovalent variant AuNPs, was increased (Figure 3E). This yielded the highest levels of transendothelial transport compared to the mPEG and isotype IgG controls (p < 0.05), although the absolute numbers of AuNPs being transported in this model were disappearingly small (Figure 3E). One reason for the low numbers of AuNP being detected in the bottom chamber of the culture system could be the possibility that the transcytosed AuNPs interacted with the polycarbonate membrane of the Transwell inserts (Figure 3A). In addition, the static conditions of the Transwell setup do not account for the effect of blood flow, and thus, the impact of targeting on both uptake and transport across the BBB may have been underestimated in the in vitro system.
Determination of AuNP transport across the BBB in vitro. (A) Schematic representation of the in vitro model system of the BBB derived from primary mouse BCECs and astrocytes and exposed to BBB induction factors. (B) The in vitro models had a high electrical resistance across the BCEC layer, which did not change over the course of the experiment (n = 24). (C) The in vitro models expressed the TfR together with several TJ-related proteins, including CD31, ZO-1, occludin and claudin-5. XZ planes are presented underneath each image. Single channel images can be viewed in Figure S3. Scale bar depicts 10 µm. (D) BCEC uptake of AuNPs after five hours in the in vitro models. Increased uptake was observed for the TfR-targeted AuNPs compared to the isotype IgG and mPEG controls (p < 0.01, n = 4 - 5, Kruskal-Wallis test with Dunn's multiple comparisons post hoc test). (E) Transport of AuNPs across the in vitro model of the BBB. Only AuNPs functionalized with the monovalent antibody, anti-TfRA/BACE1, were transported to a higher extent across the BCECs with statistical significance (p < 0.05, n = 4 - 5, Kruskal-Wallis test with Dunn's multiple comparisons post hoc test). Data are presented as mean ± SEM (n = 4 - 5) with * p < 0.05 and ** p < 0.01. %AD: percentage of administrated dose; BCECs: brain capillary endothelial cells; TEER: transendothelial electrical resistance; TfR: Transferrin receptor; ZO-1: Zonula occludens 1.
Brain accumulation of AuNPs is improved with lower affinity and monovalency of the antibody
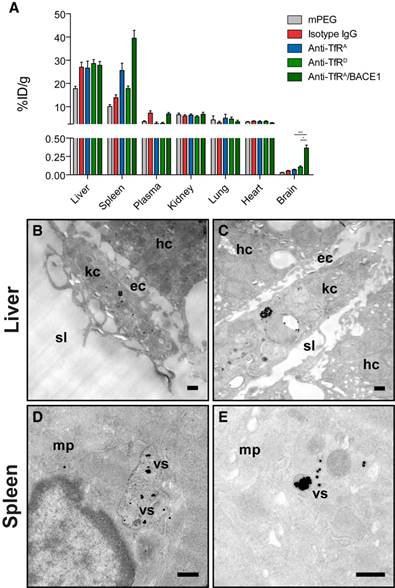
To study the brain targeting abilities of the different AuNP formulations, they were injected via the lateral tail vein into mice and allowed to circulate for 8 h. Lethargy has previously been observed after injection of these humanized anti-TfR antibodies [12, 27], but we saw no signs of this adverse effect, possibly due to the low overall protein concentration of the injected AuNP solution. First, the biodistribution profiles of the AuNP formulations were determined by measuring gold content in resected tissue samples from a range of peripheral organs and the brain using ICP-MS (Figure 4A). The different AuNP formulations primarily accumulated in the liver and the spleen, while approximately 3-7 %ID/g remained in the plasma after 8 h. The low plasma concentration may be a result of the high density of AuNPs, which would allow for pelleting the AuNPs together with red blood cells during plasma isolation. Particularly, attaching an antibody on the surface of the AuNP (regardless of its targeting qualities) seemed to increase the liver uptake of the formulations compared to a non-functionalized (mPEG) control. This was also observed in the spleen, although in this organ the TfR-targeted AuNP formulations with the highest affinity of the conjugated antibody (anti-TfRA and anti-TfRA/ BACE1) seemed to accumulate several-fold more compared to the controls (Figure 4A). TEM analysis confirmed this large accumulation, which was readily observable both in liver (Figure 4B-C) and spleen (Figure 4D-E) tissue samples. Interestingly, we did not observe any AuNP uptake into hepatocytes despite their prominent TfR expression. We found them only to be phagocytosed by resident Kupffer cells (Figure 4B-C). No significant differences were observed with respect to the uptake in other peripheral tissues including the kidney, heart and lungs, whereas the general brain accumulation varied correlating with the binding modus of the TfR-targeting antibody (Figure 4A). Here, the AuNPs functionalized with the low-affinity anti-TfRD variant antibodies accumulated more compared to their high-affinity anti-TfRA counterparts (~1.4-fold increase), although the difference did not reach statistical significance (Figure 4A). However, the AuNPs functionalized with the anti-TfRA/BACE1 monovalent variant antibody seemed to outperform both AuNP formulations functionalized with bivalent antibodies (~4.5-fold and 3.2-fold increase compared to the anti-TfRA and anti-TfRD variants, respectively) with respect to general brain accumulation (p < 0.001 against anti-TfRA, p < 0.05 against anti-TfRD, Figure 4A).
Biodistribution analysis of AuNPs after intravenous injection. (A) AuNPs accumulated to a high extent in both the liver and the spleen, although in the liver the accumulation seemed to scale with the presence of antibodies on the surface of the AuNPs, whereas for the spleen, the accumulation tended to follow the TfR-targeting abilities of the functionalized AuNPs. Large accumulation in the liver (B-C) and spleen (D-E) could be detected using TEM. (B-C) In the liver, AuNPs were seen in Kupffer cells situated in the sinusoidal lumen near the endothelial cell layer and hepatocytes. (D-E) In the spleen, AuNPs were also seen in macrophages with the AuNPs clearly accumulating in vesicular structures. Scale bars depict 200 nm. In other peripheral organs, the general accumulation of the AuNPs was low with no differences between the individual groups. The brain accumulation of AuNPs was improved for the targeted variants compared to the non-targeted variants of AuNPs (statistical significance was found for all TfR-targeted AuNPs compared to both controls, except for anti-TfRA versus isotype IgG (p > 0.05), but not presented in the figure for the sake of a clear overview). The highest brain accumulation was seen for the monovalent anti-TfRA/BACE1 group, which outperformed both the high and low-affinity variants (p < 0.001 and p < 0.05, respectively). Data are presented as mean ± SEM (n = 7-8) with * p < 0.05 and *** p < 0.001. ec: endothelial cell; hc: hepatocyte; kc: Kupffer cell; mp: macrophage; sl: sinusoidal lumen; vs: vesicular structure; %ID/g: Percentage of injected dose per gram.
TfR-targeting with monovalent antibodies significantly improves the transport of AuNPs into the brain parenchyma
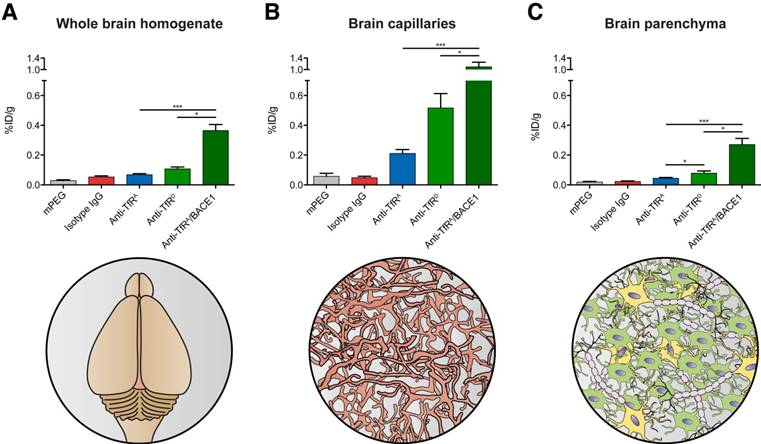
Given the interesting differences in brain accumulation observed between the different AuNP formulations, we chose to investigate further the type of uptake, i.e., whether the increase in brain gold content could be attributed only to an increased brain capillary uptake, or whether it also translated into a measurable transport of AuNPs into the brain parenchyma. Therefore, brain capillary depletion was performed to separate the capillary and parenchymal fractions, and these fractions were subsequently analyzed for their gold content (Figure 5A-C; whole brain homogenate data from Figure 4A re-included as Figure 5A for comparison to the capillary and parenchymal fractions). The purity of each fraction was confirmed by measuring the enzyme activity of γ-glutamyl transferase (Figure S5). As expected, the AuNP uptake into the brain capillaries was high for the TfR-targeted variants (Figure 5B) compared to what was observed in the whole brain homogenates (Figure 5A) and parenchymal fractions (Figure 5C), suggestive of up-concentration at this anatomical site. This also correlated well with the brain capillaries being the entry point to the brain for compounds injected into the systemic circulation. Interestingly, for the capillary fraction, the accumulation of AuNPs was higher for the low-affinity anti-TfRD variant compared to the high-affinity anti-TfRA variant (Figure 5B), but again the difference did not reach statistical significance (p > 0.05). In the parenchymal fraction, the difference between the high-affinity anti-TfRA and the low-affinity anti-TfRD variants was statistically significant (Figure 5C, p < 0.05), suggestive of improved trans-BBB transport. However, these were both inferior to the monovalent anti-TfRA/BACE1 variant (Figure 5C, p < 0.001 against anti-TfRA, p < 0.05 against anti-TfRD for both the capillary and parenchymal fractions). In the capillary fraction, this increased level of AuNP accumulation amounted to more than 1.1 %ID/g, whereas this figure was 0.23 %ID/g in the parenchymal fraction (Figure 5B-C). This indicated that having a monovalent TfR antibody on the AuNP surface was favorable for subsequent transport across the BBB after intravenous administration. Since the monovalent anti-TfRA/ BACE1 variant AuNPs seemed to accumulate to a high degree in the brain parenchyma, we measured the total brain content of β-amyloid after 30 h to evaluate if this level of accumulation allowed for any therapeutic effects of the anti-BACE1 arm. We could not, however, detect any change in the brain β-amyloid load even after doubling the initial anti-TfRA/BACE1 AuNP dose used for the quantitative transport studies (Figure S6).
Accumulation of AuNPs in different brain fractions after capillary depletion. (A) In whole brain homogenates, there was a clear distinction between the different TfR-targeted variants with respect to accumulation of AuNPs (whole brain homogenate data from Figure 3A re-included as Figure 4A for comparison to the capillary and parenchymal fractions). (B) The TfR-targeted AuNPs accumulated in the capillary fraction with 0.2, 0.5, and 1.1 %ID/g for anti-TfRA, anti-TfRD (p > 0.05 against anti-TfRA), and anti-TfRA/BACE1 (p < 0.001 against anti-TfRA, p < 0.05 against anti-TfRD, respectively). (C) In the brain parenchyma, gold could also be detected, indicative of AuNP transport across the BBB. More accumulation was seen for the low-affinity anti-TfRD compared to the high-affinity anti-TfRA variant AuNPs (p < 0.05). Anti-TfRA/BACE1 AuNPs were superior to the other TfR-targeted variant AuNPs with a mean parenchymal accumulation of 0.23 %ID/g compared to 0.04 and 0.08 %ID/g for anti-TfRA and anti-TfRD, respectively (p < 0.001 against anti-TfRA, p < 0.05 against anti-TfRD). Statistical significance was proven for all TfR-targeted AuNPs compared to both controls in brain capillaries and parenchyma after capillary depletion, but not presented in the figure for the sake of a clear overview. Data are presented as mean ± SEM (n = 7-8, Kruskal-Wallis test with Dunn's multiple comparisons post hoc test) with * p < 0.05 and *** p < 0.001. %ID/g: percentage of injected dose per gram.
AuNPs locate to multiple structures inside the brain parenchyma
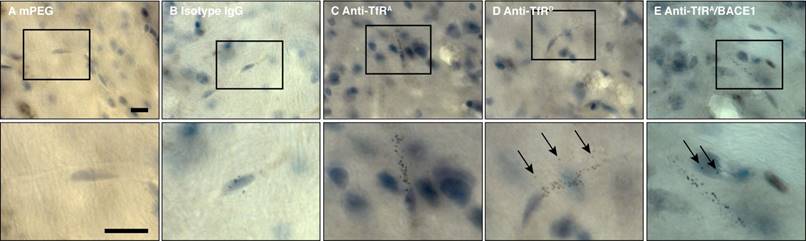
While the brain capillary depletion technique is a suitable tool to indicate the transport of compounds into the parenchymal compartment of the brain (i.e., transported across the BBB), it lacks the robustness of being a standalone analysis. Therefore, we adopted the morphological approach described by Wiley et al. [23] and performed silver enhancement on brain sections from mice having received the different AuNP formulations for subsequent light microscopy (Figure 6). Using this technique, it was apparent that (like when measured using brain capillary depletion) most AuNPs observed in the brain sections were in vessel-like structures, suggestive of capillary uptake and concentration (Figure 6). In mice receiving either the low-affinity anti-TfRD or the monovalent anti-TfRA/BACE1 variants, AuNPs not associated with vessel-like structures could be observed (Figure 6D-E), which indicated that a smaller fraction of these AuNPs had transcytosed into the brain parenchyma. This observation was seldom in mice receiving the high-affinity anti-TfRA variant AuNPs (Figure 6C), and not detected in mice receiving mPEG or isotype IgG-functionalized AuNPs (Figure 6A-B). Capillary accumulation could, however, be observed to a minor extent for the isotype IgG AuNPs (Figure 6A). Immunohistochemical staining against the human Fc region of the AuNP-conjugated antibodies did not reveal any signal indicative of antibody accumulation along the brain capillaries, which was likely due to the low level of accumulation combined with the small number of antibodies per AuNP (data not shown).
Detection of AuNPs in brain tissue after silver enhancement. (A) In mice treated with mPEG AuNPs, no silver grains representing AuNPs could be observed in the brain tissue, (B) whereas some silver grains were observed in capillary structures of mice treated with isotype IgG AuNPs. For all TfR-targeted variants, silver grains representing AuNPs presented as a dotted lining of the capillary structures, indicative of accumulation at this anatomical location (C-E). (C) In mice treated with anti-TfRA AuNPs, there was only little intraparenchymal staining, (D) whereas examples of this could be observed in mice treated with anti-TfRD AuNPs (arrows). (E) In mice treated with anti-TfRA/BACE1 AuNPs, detection of intraparenchymal staining (indicative of AuNP transcytosis) was more apparent (arrows). Quantification was not performed based on the silver enhancement technique. Scale bar depicts 25 µm.
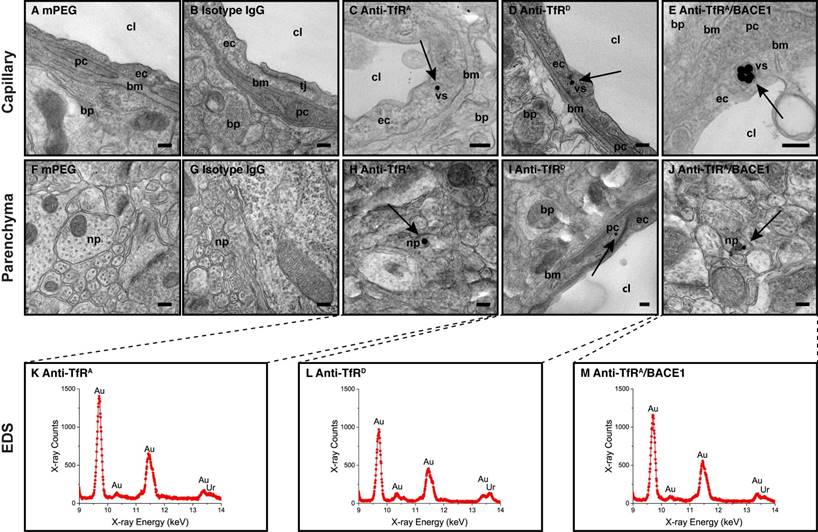
To further substantiate our claim that a fraction of AuNPs had transcytosed into the brain parenchyma, we performed TEM on brains resected from mice having received the different AuNP formulations (Figure 7). The TEM analysis also allowed us to measure the presence of gold by way of EDS (in addition to the direct visualization), which could validate the findings as true AuNPs and not just salt precipitates or other artifacts. Similar to the results of the brain capillary depletion and silver enhancement techniques, the TEM analysis showed the presence of TfR-targeted AuNPs in the capillaries of the brain (Figure 7C-E), whereas no AuNPs could be detected in the brains of mice receiving either mPEG or isotype IgG-functionalized AuNPs (Figure 7A-B). All AuNPs observed in the brain capillaries were associated with intracellular vesicular structures, which correlated with the TfR being an endocytosing receptor (Figure 7C-E and Figure S7). AuNPs were generally located to the larger capillary branches, whereas AuNP uptake in the smallest capillaries was seldom (Figure S8). Interestingly, we also observed a smaller fraction of TfR-targeted AuNPs on the abluminal side of the BCECs (Figure 7H-J), which indicated full transcytosis of the AuNP, especially prominent for the monovalent anti-TfRA/ BACE1 variant (Figure 7J and Figure S9). For example, AuNP could be observed directly below the endothelial basement membrane, in pericytes, and inside neural axons/processes (Figure 7 and Figure S9). All AuNPs present inside brain capillaries and in any region of the brain parenchyma had their gold content validated by EDS (representative spectra for transcytosed AuNPs shown in Figure 7K-M).
TEM detection of AuNPs in brain tissue. Direct visualization of AuNPs in the brain capillaries and parenchyma of treated mice was performed using TEM (n = 2-4). (A-B) No AuNPs could be detected in brain capillaries of mice in the mPEG or isotype IgG groups, whereas the TfR-targeted AuNPs could easily be found at this location (C-E). All AuNPs detected in capillaries were confined to vesicular structures, suggesting receptor-mediated endocytosis as the uptake mechanism. (F-G) No AuNPs could be detected in brain parenchyma of mice in the mPEG or isotype IgG groups. (H-J) AuNPs were observed beyond the BBB or in the brain parenchyma of mice treated with all TfR-targeted variants but were most easily detected in the anti-TfRA/BACE1 group (J). All AuNPs detected in the brain parenchyma were analyzed using EDS to validate the presence of gold in these electron-dense points (K-M). Scale bars depict 200 nm. bp: brain parenchyma; bm: basement membrane; cl: capillary lumen; ec: endothelial cell; np: neural process; pc: pericyte; tj: tight junction; vs: vesicular structure.
Discussion
The use of nanoparticles for drug delivery to the brain has been scrutinized in several studies, however, often only with minor characterization of their mobility at the BBB, as opposed to transport of their encapsulated cargo [1, 6]. To provide more knowledge on this issue, we studied AuNPs as they are stable nanoparticles, their synthesis is easy to control, and they allow for in situ localization using several analysis techniques [28]. The AuNPs were previously used for various brain drug delivery purposes in earlier studies, showing transport through the BBB as a function of both targeting moiety and the diameter of the nanoparticle complex [22, 23, 29-31]. They were also targeted to the TfR on multiple occasions, hence revealing the potential of transport across the BBB, although with varying efficiency [22, 23, 32, 33].
In this present study, we investigated the transport of AuNPs across the BBB in vitro and in vivo both as a function of TfR-targeting and of the binding modus between the anti-TfR antibody and the TfRs expressed on the BCEC surface. Using the antibodies developed by Genentech (anti-TfRA, anti-TfRD, and anti-TfRA/BACE1), we demonstrate both a modest impact of affinity and a marked impact of valency on the absolute transport of AuNPs into the brain. As expected, we found that TfR-targeting increases the uptake of AuNPs into the brain both in vitro and in vivo as compared to non-functionalized (mPEG) and isotype IgG-functionalized AuNPs. This observation is well-described and shows that under normophysiological conditions, the use of active targeting is required for brain accumulation of any drugs or drug delivery vehicles to occur [33, 34]. Whether active targeting is required in the case of manifest neurological disease with possible loss of BBB integrity is widely discussed [35-38]. Recent studies show that for some of the high-incidence, neurodegenerative disorders, the loss of BBB integrity is negligible for transport of antibody-based drugs (150 kDa) [39-45], and hence, will be irrelevant for brain drug delivery using larger nanomedicines like AuNPs.
The increased brain accumulation for TfR- targeted AuNPs also correlated to the binding modus of the surface-bound antibody. Surprisingly, we observed that the low-affinity antibodies allowed for increased accumulation in the brain capillaries compared to those having a high-affinity. This observation did not necessarily follow the expectation that high-affinity would always lead to a higher level of accumulation in the brain capillaries, and it was possibly affected by the time point of analysis (8 h post injection). We have recently found in rats that targeting TfRs with high-affinity antibodies on liposomes leads to high capillary accumulation after 1 h, but that this difference compared to controls was lost over the following 24 h [46]. Hence, what we measure after 8 h in the present study may not encompass the acute increase in accumulation in the first hour after injection of the AuNPs. Supporting this observation, the immediate interaction, uptake, and transcytosis of the AuNPs takes place during the initial 30 min post injection, as described by Cabezón et al. [33], which may indicate that the time point for analysis used in this study is more relevant for studying BBB transport and parenchymal accumulation, instead of the attachment and uptake processes at the luminal surface of the BCEC [33]. We also find that the dissociation constant of the low-affinity antibody remains within the nanomolar range (KD = 149 nM), and accordingly, maintains significant binding potential. Thus, lowering the affinity of the antibodies denotes a potential strategy to make modest increases in the AuNP uptake into the brain. Others found that a high-affinity anti-TfR antibody increased liposome transport across the BBB in vitro compared to an antibody with lower affinity [24]. However, the 'low' affinity antibody (clone RI7217) used in that study has a dissociation constant in the low nanomolar range (KD = 6 nM, see Figure S2), yielding a 25-fold higher affinity than the low-affinity anti-TfRD variant used in this present study [16, 24].
While lowering the affinity could positively impact the transport of AuNPs across the BBB, our data points towards a much larger impact on the transport process imposed by the monovalency of the bi-specific antibody, anti-TfRA/BACE1. Using this antibody as a ligand, we observed a more than three-fold increase in the brain parenchymal uptake of AuNPs compared to the low-affinity anti-TfRD variant. This observation falls in line with prior findings regarding single antibody constructs engineered to be either mono- or bivalent [19, 21]. In the latter studies, injection of bivalent TfR-targeted antibody constructs (brain shuttles) led to only minor transport into the brain parenchyma, and more critically, resulted in a loss of TfR expression on the luminal surface of the BCECs [19]. The loss of TfRs after treatment with bivalent antibody constructs has been known for a long time, and points to the fact that bivalent interactions between the ligand and multiple receptors result in a distorted internalization and sorting process not favoring transendothelial transport into the brain parenchyma [17, 47, 48]. Conversely, engineering the brain shuttles to become monovalent allowed for increased transport into the brain without loss of TfR expression on the BCEC surface [19]. Monovalency was also shown to be important for brain uptake of antibody-based PET tracers for β-amyloid plaque imaging [18, 20]. We found no differences between the dissociation constants of the high-affinity and monovalent variants of antibodies when analyzed using surface plasmon resonance (KD = 21 nM versus 22 nM, respectively, Figure S2), because the setup did not allow for one antibody molecule interacting with several TfRs (anti-TfRA/BACE1 was previously found to have an IC50 = 18 nM [12]). In addition, the level of functionalization of antibodies per AuNP was comparable across the groups. These aspects are important because they allow for interpreting the difference in brain uptake between the anti-TfRA and anti-TfRA/BACE1 variant AuNPs as being caused by the difference in the valency of the antibodies. Others showed that the avidity of AuNPs is important for BBB transport of AuNPs and liposomes [23, 34]. This applied when using endogenous Tf as a ligand, but when performing the studies using comparable levels of functionalization with bivalent antibodies, most of the transport capabilities were lost [22, 23]. Our results underscore this concept, since we found improved uptake using a monovalent antibody and a low level of functionalization (< 10 antibodies per AuNP). This reduces the probability of interacting with multiple TfRs simultaneously, which was previously suggested to reduce the relevance of targeted nanoparticles for brain drug delivery [17]. We cannot, however, rule out the possibility that the higher capillary accumulation resulted in an unspecific mechanism of release of the AuNPs to either the blood or the brain side of the BBB, hereby explaining why the transport into the brain parenchyma scales with the initial capillary accumulation. In addition, we did not observe any therapeutic effects of the anti-TfRA/BACE1 antibody as assessed by measuring the β-amyloid load in the treated brains, as was shown for bi-specific antibody constructs in several studies [16, 49]. This was likely due to the low amount of total protein injected but could also be due to the immobilization of antibodies on AuNPs. This is known to suppress the ability of the antibody to initiate antibody-dependent cellular cytotoxicity because the Fc domains are inaccessible for interaction with immune cells [50].
Transport of TfR-targeted nanoparticles across the BBB is a controversial subject, since the known sorting pathway of the TfR does not include transcytosis through the endothelial layer, and because nanoparticles like the AuNPs used in the present study are of a size thousand-fold larger than that of the Tf protein [1, 7, 9]. The ability of TfR-targeted nanoparticles to pass the BBB and accumulate in the brain parenchyma has been claimed for many years [1, 6, 34, 51-56], although only a few studies have investigated the localization of the nanoparticle itself, and not just the cargo [22, 23, 32, 33, 57]. We demonstrate the presence of AuNPs in the brain parenchyma based on quantitative analysis of the parenchymal fraction of a brain homogenate after capillary depletion. This technique is widely used to estimate any transport of substances across the BBB, and its robustness has been validated in several studies [1, 46, 58, 59]. To ensure the purity of the resulting fractions, we quantified the potential spillover by measuring the enzyme activity of γ-glutamyl transferase in the capillary and parenchymal fractions. In the parenchymal fractions, there was only very little activity of the enzyme, which is suggestive of a capillary depletion with good separation and high purity. To substantiate our claims, we increased the AuNP size in situ using the silver enhancement technique to validate the presence of AuNPs in the brain tissue via light microscopy. The chemistry of this technique is well-known and it has been used quantitatively for studies of AuNP transport into the brain [22, 23, 60, 61]. We found that most silver-enhanced AuNPs located in vessel-like structures with few examples of transcytosis into the brain parenchyma, especially for the anti-TfRA/ BACE1 variant AuNP group. We also visualized the AuNPs directly in the brain cortex using TEM, which showed that AuNPs could indeed be found in the brain parenchymal structures distant from capillaries, although, as depicted by the ICP-MS data, the absolute numbers were low. AuNPs were found beneath the endothelial basement membrane of the BBB, in pericytes, and in neural axons/processes. Whether nanoparticles and other large molecules are mobile in the brain extracellular space after transcytosis from the systemic circulation is still a matter of dispute, and their diffusion properties remain poorly understood [62]. Some suggest the width of the brain extracellular space to be maximum 64 nm [63], whereas others claim that nanoparticles of 100 nm can move in this compartment [64, 65]. The AuNPs used here were > 70 nm, which is above the limit previously described [63], and although our TEM approach was not quantitative, we did not observe any AuNPs in the extracellular space of the brain, but instead they were mostly localized in neural processes. We speculate that the distribution of the minor fraction of AuNPs that had traversed the BBB to the neurons was facilitated by the astrocytes, as has previously been hypothesized, rather than diffusing freely in the brain extracellular space [66].
In conclusion, the findings presented in this study indicate that using monovalent antibodies targeting the TfR expressed by BCECs increases the uptake of AuNPs into the brain parenchyma, whereas lowering the affinity may have a modest impact. Whether these findings will translate into relevance for other types of nanoparticles goes beyond this study, and future studies should seek to validate that an increase in absolute uptake of any type of nanoparticle formulation also leads to increased therapeutic efficacy inside the brain parenchyma. Although the number of AuNPs transported to the brain parenchyma relative to the injected dose remains low, our study shows that shifting from a bivalent to a monovalent binding modus results in a several-fold increase in brain uptake. The improvement of the targeting strategy presented here may thus provide a significant contribution to a future design of a highly efficient nanoparticle formulation for brain drug delivery of otherwise BBB impermeable large molecules.
Methods
Materials
All chemicals were purchased from Sigma-Aldrich Inc. (Brøndby, Denmark) unless otherwise stated. Rabbit anti-claudin 5 (Cat. No. SAB4502981) and γ-Glutamyl Transferase (GGT) Activity Colorimetric Assay kit (Cat. No. MAK089) were also purchased from Sigma-Aldrich Inc. (Brøndby, Denmark). Anti-TfRA, anti-TfRD, anti-TfRA/BACE1 (all human anti-mouse TfR with varying affinities) were obtained from Genentech, Inc. (CA, USA) under MTA A15783. Mal-PEG5000-NH2 (Cat. No. MAL-PEG5000-NH2) was purchased from JenKem technology (Beijing, PRC). MeO-PEG5000-NH2 (Cat. No. PEG1154) was purchased from Iris Biotech GmbH (Marktredwitz, Germany). Tetrachloroauric acid tetrahydrate (Cat. No. 077-00931) was purchased from Wako Chemicals GmbH (Neuss, Germany). LiCor IRDye® 800CW goat anti-human IgG (H + L; Cat. No. 926-32232) and Odessey® Blocking Buffer (in Tris-buffered saline, TBS; Cat. No. 927-50000) was purchased from Li-Cor Biosciences (Køge, DK). DNase I (Cat. No. 1014159001), collagenase/dispase (Cat. No. 109113), basic fibroblast growth factor (bFGF; Cat. No. 1363697), and insulin-transferrin-sodium selenite (Cat. No. 11074547001) were purchased from Roche (Hvidovre, DK). Pierce Traut's reagent (2-iminothiolane; Cat. No. 26101), nitrocellulose blotting membranes (Pre-cut sheets, 80×80 mm, 0.2 µm; Cat. No. LC2000) and 20X TBS buffer (Cat. No. 28358), rabbit anti-ZO-1 (Cat. No. 61-7300), Alexa Fluor 594-conjugated goat anti-mouse IgG (Cat. No. A11032), Alexa Fluor 594-conjugated goat anti-rabbit IgG (Cat. No. A11034), Alexa Fluor 488-conjugated donkey anti-rat IgG (Cat. No. A-21202), human IgG isotype control (Cat. No. 02-7102), collagenase II (Cat. No. 17101105), Dulbecco's Modified Eagle Medium (DMEM; Cat. No. 21885), DMEM-F12 GlutaMAXTM (DMEM-F12; Cat. No. 31331), fetal calf serum (Cat. No. 10270), and Mouse Aβ40 ELISA kit (Cat. No. KMB3481) were purchased from Thermo Scientific (Hvidovre, DK). Bovine serum albumin (BSA; Cat. No. EQBAH62) was purchased at Europa Bioproducts (Cambridge, UK). Plasma-derived bovine serum (Cat. No. 60-00-810) was purchased from First Link (Wolverhampton, UK). Gentamicin sulphate (G418; Cat. No. 17-518Z) was purchased from Lonza Copenhagen (Vallensbæk Strand, DK). Fluorescence mounting medium (Cat. No. S3023) was purchased from DAKO (Glostrup, DK). Hanging cell culture inserts (Transwell; Cat. No. Pirp 15R48), rabbit anti-occludin (Cat. No. ABT146) and Amicon Ultra 4 mL spin filters with 30 kDa cut-off (Cat. No. UFC803096) were purchased from Merck Millipore (Hellerup, DK). Rabbit anti-CD31 (Cat. No. ab28364) was purchased from Abcam (Cambridge, UK). Rat anti-mouse TfR (clone RI7217) was produced in-house using the hybridoma technique. Isoflurane was purchased from Baxter (Søborg, DK). Sodium cacodylate trihydrate (Cat. No. 12300), uranyl acetate (Cat. No. 22400), and nickel TEM grids coated with a carbon-stabilized formvar film (Cat. No. FCF200-Ni) were purchased from Electron Microscopy Sciences (Hatfield, PA, USA). Agar 100 Resin (Cat. No. AGR1031) was purchased from Agar Scientific (Stansted, UK). Absolute ethanol (Cat. No. 20821.310) and acetonitrile (Cat. No. 20066.296) were purchased from VWR International (Søborg, DK). Recombinant mouse TfR protein was purchased from SinoBiological, Inc. (Beijing, PRC).
Instrumentation for chemical synthesis
Reactions were monitored by thin layer chromatography (TLC); visualization was carried out by UV light exposure (254 nm), Cemol and KMnO4-stain. Chromatography refers to open column chromatography on silica gel. NMR spectra were recorded on a Varian Mercury 400 MHz Spectrometer. 1H and 13C NMR were recorded at 400 and 100 MHz, respectively. Chemical shifts (δ) were reported in parts per million (ppm) relative to the solvent signal peak. Mass spectra were recorded on a Bruker AutoflexTM MALDI-TOF MS Spectrometer, using 2,5-dihydroxybenzoic acid (DHB) spiked with sodium trifluoroacetate in CH3OH as matrix. FT-IR was recorded neat on a PerkinElmer Instruments Spectra One FT-IR Spectrometer. Dynamic light scattering (DLS) and ζ-potential measurements were performed on a Brookhaven Instruments Corporation ZetaPALS ζ -potential analyzer. The UV-Vis measurements of the AuNPs were performed on a NanoDrop 2000c spectrophotometer (Thermo Scientific, Waltham, MA, USA). The amount of functionalized antibody per AuNP was quantified by dot blotting and imaged by a Li-Cor OdesseyFC at 800 nm with 5 min exposure time.
Synthesis
N-Hydroxysuccinimide-α-lipoic acid (Figure S10)
α-Lipoic acid (4.85 mmol, 1 equiv.) and N-hydroxysuccinimide (5.82 mmol, 1.2 equiv.) were dissolved in dry tetrahydrofuran (THF; 24 mL, 0.2 M) in a flame-dried flask, fitted with a magnet and N2-atmosphere. The mixture was cooled to 0 ºC. DCC (5.82 mmol, 1.2 equiv.) was dissolved in dry THF (4 mL, 1.4 M) and added dropwise. The yellow reaction mixture was left to react and reach room temperature overnight. The reaction was stopped by removing the precipitate by filtration through Celite filter, and the filtrate concentrated in vacuo. The crude product was washed with cold ether and set to precipitate in the freezer before decanting. The product was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ (ppm): 3.57 (dq, J = 8.2, 6.4 Hz, 1H), 3.21 - 3.07 (m, 2H), 2.83 (bs, 4H), 2.62 (t, J = 7.3 Hz, 2H), 2.50 - 2.42 (m, 1H), 1.92 (dq, J = 13.5, 6.9 Hz, 1H), 1.82 - 1.66 (m, 4H), 1.61 - 1.48 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 169.24, 168.52, 56.21, 40.28, 38.64, 34.53, 30.91, 28.44, 25.72, 24.48. MALDI-TOF MS, m/z: Calc. mass [M+Na]+ = 326.0; found mass [M+H]+ = 325.8.
MeO-PEG5000-LA (Figure S10)
MeO-PEG5000-NH2 (0.052 mmol, 1 equiv.) was dissolved in dry CH2Cl2 (5 mL, 0.01 M, dried with 4 Å molecular sieves) in a 10 mL flame dried flask fitted with a magnet and N2-atmosphere. Dry Et3N (0.36 mmol, 7 equiv.) was added and let to react for 15 min. NHS-LA (0.27 mmol, 5 equiv.) was dissolved in dry CH2Cl2, transferred to the reaction mixture and left to react for 2 days. The reaction was stopped by concentrating in vacuo. The crude MeO-PEG-LA was purified by flash column chromatography (FCC; eluent: CHCl3:CH3OH; gradient: 1-20% over 20 fractions; fraction volume: 20 mL). The pure fractions were collected and concentrated in vacuo, dissolved in CH3OH:MilliQ water (1:4) and lyophilized to a white powder (75% yield). TLC (CHCl3:CH3OH (9:1), KMnO4 stain). 1H NMR (CDCl3, 400 MHz), δ (ppm): 6.29 (1H, bs, NH); 3.82-3.79 (2H, m, O-CH2-CH2-NH-); 3.69-3.53 (m, -CH2(OCH2CH2)115-OCH3 and CH-S); 3.48-3.43 (2H, m, NH-CH2); 3.37 (3H, s, OCH3); 3.20-3.06 (4H, m, S-CH2-, O-CH2-CH2-NH); 2.50-2.41 (1H, m, S-CH2-HCH-CH-S); 2.29 (2H, t, CH2-C(O)NH); 1.96-1.85 (1H, m, S-CH2-HCH-CH-S); 1.79-1.60 (4H, m, CH2-CH2-CH2-CH2-C(O)-NH); 1.53-1.41 (2H, m,-CH2-CH2-CH2-C(O)-NH). 13C-NMR (100 MHz, CDCl3), δ (ppm): 172.8 (-C(O)-); 71.9; 70.8-69.9 (-CH2(OCH2CH2)115- OCH3); 59.3 (O-CH3); 56.4 (CH); 40.3 (S-CH2-CH2-CH-S); 39.1 (NH-CH2); 38.5 (S- CH2); 36.3 (CH2-CO); 34.7 (CH2-CH2-CH2-CO); 28.9 (-CH2-CH2-CH2-CO); 25.4 (CH2-CH2-CH2-CH2-CO). FT-IR: ν (cm-1): 2883, 2745, 2165, 1670, 1467, 1360, 1341, 1097, 1060, 958, 841. MALDI-TOF MS, m/z: Calc. mass [M+Na]+ = 5217.04±nx44; found mass [M+H]+ = 5220.06±nx44.
Mal-PEG5000-LA (Figure S10)
Mal-PEG5000-NH2 (0.023 mmol, 1 equiv.) was dissolved in dry CHCl3 (3 mL, 38 mM, dried with 4 Å molecular sieves) in a 10 mL flame dried flask fitted with a magnet and N2-atmosphere. Dry Et3N (0.05 mmol, 2 equiv.) was added and left to react for 15 min. NHS-LA (0.114 mmol, 5 equiv.) was dissolved in dry CH2Cl2, transferred to the reaction mixture and set to react for 2 days. The reaction was stopped by concentrating in vacuo. The crude MeO-PEG-LA was purified by FCC (Eluent: CHCl3:CH3OH; gradient: 1-20% over 20 fractions; fraction volume: 20 mL). The pure fractions were collected and concentrated in vacuo, dissolved in CH3OH:MilliQ water (1:4) and lyophilized to a white powder (30% yield). TLC (CHCl3:CH3OH (9:1), KMnO4 stain). 1H NMR (400 MHz, CDCl3) δ (ppm): 6.68 (2H, s, -CH=CH-); 6.24 (1H, bs, NH); 3.85-3.77 (2H, m, O-CH2-CH2-NH-); 3.50-3.70 (m and br s, -CH2(OCH2CH2)115-OCH3 and CH-S); 3.46-3.36 (5H, m, OCH3 and O-CH2-CH2-NH); 3.19-3.04 (2H, m, S-CH2-); 2.51-2.42 (1H, m, S-CH2-HCH-CH-S); 2.18 (2H, t, CH2-C(O)NH); 1.93-1.84 (1H, m, S-CH2-HCH-CH-S); 1.74-1.56 (m, 4 H, CH2-CH2-CH2-CH2-C(O)-NH); 1.49-1.36 (2H, m, -CH2-CH2-CH2-C(O)-NH). 13C NMR (100 MHz, CDCl3) δ (ppm): 172.9 (-C(O)-); 170.6 (-C(O)-, maleimide); 134.3 (-CH=CH-); 70.8-69.8 (-CH2(OCH2CH2)115- OCH3); 56.5 (CH); 40.3 (S-CH2-CH2-CH-S); 39.3 (NH-CH2); 39.3 (CH2-NH, maleimide) 38.5 (S- CH2); 36.4 (CH2-CO); 34.8 (CH2-CH2-CH2-CO); 34.6 (CH2-CO, maleimide); 29.0 (-CH2-CH2-CH2-CO); 25.5 (CH2-CH2-CH2-CH2-CO). FT-IR: ν (cm-1): 2883; 1708; 1667; 1545; 1467; 1360; 1343; 1280; 1241; 1146; 1097; 961; 841. MALDI-TOF MS, m/z: Calc. mass [M+Na]+ = 5217.04±nx44; found mass [M+H]+ = 5220.06±nx44.
Synthesis of AuNP seeds
All glassware for the AuNP seed synthesis was cleaned with freshly made aqua regia, rinsed with MilliQ water and dried at 150 ºC before seeding the AuNPs. All the AuNPs were characterized by ICP-MS, DLS, ζ-potential and UV-Vis spectrophotometry. The final formulation was characterized by TEM and the Au concentration determined by ICP-MS. First seeding of AuNPs, d = 30 nm: A solution of HAuCl4·4H2O (0.23 mM, 1000 mL) was adjusted to pH 7.0 by addition of NaOH (1 M). Sodium acrylate (40.9 mM, 332 mL) was added and the reaction flask gently swirled by hand. The reaction was left at room temperature without stirring for 5 days. The combined solution was concentrated 2.6X by centrifugation (12,000 ×g, 10 min/cycle). Second seeding of AuNP, d = 40 nm: A solution of HAuCl4·4H2O (0.29 mM, 1871 mL) was adjusted to pH 7.0 by addition of NaOH (1 M). The concentrated acrylate stabilized AuNPs (d = 30 nm, 504 mL) were added followed by sodium acrylate (51.2 mM, 625 mL). The reaction flask was gently swirled by hand and left at room temperature without stirring for 3 days. The mPEG AuNPs were concentrated 60X to a final volume of 100 mL by centrifugation (12,000 ×g, 10 min/cycle). The final AuNP solution was characterized by UV-Vis, DLS, ζ-potential (see Table 1). ICP-MS was used to determine the Au concentration before PEGylation.
Surface coating of AuNPs with polyethylene glycol 5000
Previous experiments have justified the use of a surplus of PEG (8 PEGs/nm2) for the surface coating of the AuNPs [1, 6, 34, 51-56, 67, 68]. A 100 mL of AuNP solution was diluted 3 times with MilliQ water and divided into 2 portions of sterile 300 mL plastic vessels fitted with a magnet stir-bar (75 mL for control AuNP and 225 mL for the maleimide functionalized AuNPs). The PEG-mixtures were pre-mixed before adding them to the AuNP solution. The control AuNPs were PEGylated with MeO-PEG5000-LA (8 PEGs/nm2, 2.9 µmol) and the Ab-functionalized AuNPs were PEGylated with Mal-PEG5000-LA:MeO-PEG5000-LA (1:99, 8 PEGs/nm2, 0.087 µmol : 8.6 µmol). The PEG mixtures were added to the AuNPs and vortexed. The reaction was left to react overnight at room temperature. UV-Vis, DLS and ζ-potential were measured to characterize the AuNPs (Table 1). A substantial change in morphology was observed between the non-PEGylated versus the PEGylated AuNPs. The AuNPs were subsequently washed 3 times with sterile water and 3 times with sterile-filtered sodium borate buffer (0.2 M, pH 8.5) using centrifugation (6,500 ×g for 10 min/cycle). The Au concentration was determined by ICP-MS prior to antibody functionalization.
Thiolation of antibodies and functionalization of AuNPs
Anti-TfRA, anti-TfRD, anti-TfRA/BACE1 (all human anti-mouse TfR with varying affinities, kindly provided by Genentech, Inc., CA, USA under MTA A15783) and isotype IgG (human IgG, Thermo Fisher Scientific, Nærum, DK) antibodies were diluted in 0.2 M sodium borate buffer (pH 8.5) to a final protein concentration of 2.5 mg/mL. The antibodies were mixed in the thiolating reagent, 2-iminothiolane (Traut's reagent), in a molar ratio of 1:15 to obtain approximately 1 thiol group per antibody. The molar ratio of 2-iminothiolane was chosen based on an optimization of thiolation of the specific antibodies (Figure S2). The antibody-2-iminothiolane reagent mixture was reacted for 60 min at room temperature, and the thiolated antibodies transferred to Amicon Ultra 4 mL spin filters (MW cut-off: 30 kDa) with an additional volume of 0.2 M sodium borate buffer (pH 8.5). The spin filters were centrifuged at 2,200 ×g for 30 min at 4 ºC, and the filtrate discarded. This washing step was repeated twice to clear the thiolated antibody solution from unreacted 2-iminothiolane. Finally, the rinsed, thiolated antibodies were immediately mixed with maleimide-functionalized AuNPs and allowed to conjugate.
The maleimide preparation of AuNPs was divided into four fractions for isotype IgG, anti-TfRA, anti-TfRD, anti-TfRA/BACE1 functionalization in 50 mL Falcon tubes, fitted a magnetic stir-bar and diluted with sterile sodium borate buffer (0.2 M, pH 8.5, C(Au) = 3.5 mM). An excess (approximately 3X molar excess to the added maleimide) of the individual thiolated antibodies were added to the respective AuNP fraction and allowed to react at room temperature overnight with light stirring. The reaction was stopped by washing the AuNPs with sterile-filtered sodium borate buffer (0.2 M, pH 8.5) 3 times, and 3 times with sterile-filtered, isotonic HEPES buffer (10 mM, 150 mM NaCl, pH 7.4) using centrifugation (6,500 ×g for 15 min/cycle). Each fraction was concentrated to approx. 35 mM Au prior to determining the Au concentration by ICP-MS. The fractions were subsequently diluted with sterile-filtered, isotonic HEPES buffer (10 mM, 150 mM NaCl, pH 7.4) to a particle concentration of 3.33×1013 AuNP/mL and aliquoted. The final AuNPs were characterized by UV-Vis, TEM, EDS, DLS, ζ-potential, and dot blot.
Determination of AuNP size by dynamic light scattering
DLS measurements were performed on a ZetaPALS instrument (Brookhaven, Holtsville, USA) equipped with a 35 mV solid-state laser (659 nm). Samples were diluted in MilliQ water followed by equilibration to 25 °C before a minimum of five measurements was made of each sample.
ζ-Potential measurements
ζ-Potential measurements were performed on a ZetaPALS instrument (Brookhaven, Holtsville, USA) equipped with a 35 mV solid-state laser (659 nm). Subsequently, the ζ-Potential measurements were made on the samples prepared for DLS. The samples were loaded to a pre-conditioned capillary cell and the ζ-potential was measured with an applied voltage of 4 V. Ten measurements were made per sample.
Determination of antibody functionalization on AuNPs
The degree of antibody functionalization was determined by dot blot. One milliliter of the final AuNP samples was added 4 µL of 2-mercaptoethanol and heated to 70 ºC with 500 rpm shaking for 30 min. The reduced AuNPs were spun down with 7,000 ×g for 5 min, and the supernatant isolated. The supernatant (10 µL) was then spotted onto a nitrocellulose blotting membrane (8×8 cm, 0.2 µm, Thermo Scientific), dried at 37 ºC for 5 min and blocked with blocking buffer (TBS (20X dilution in MilliQ water):Odessey Blocking Buffer, 1:1, 10 mL) for 1 h. The blotting membrane was washed twice with MilliQ water, followed by blotting with a secondary antibody (LiCor IRDye® 800CW goat anti-human IgG (H + L)) in the blocking buffer (10 mL, 5,000X dilution). The blotting membrane was then washed with TBS (20X dilution in MilliQ water) with 0.1% Tween20 (10 mL, 3 × 10 min), with TBS (20X dilution in MilliQ water) (10 mL, 2 × 2 min) and then dried at 37 ºC in the dark. Finally, the blotting membrane was imaged at 800 nm with 5 min exposure time. The amount of Ab in the samples was then correlated to an isotype IgG standard curve.
The number of AuNPs per volume (NAuNP) was determined by knowing the Au concentration and the diameter of the gold core measured by DLS. NAuNP was calculated by the two following equations:
mAuNP = VAuNP × ρ(fcc) = (4/3) × π × r3 × ρ(fcc) (1)
NAuNP = mAuNP / CAu(ICP-MS) = (4/3) × ((π × r3 × ρ(fcc)) / CAu(ICP-MS)) (2)
where VAuNP is volume per AuNP, r is the radius of the AuNP, ρ(fcc) is the density of face-centered cubic (fcc) gold (19.3 g/cm3) and CAu (ICP-MS) is the concentration of Au determined by ICP-MS. Thus, the number of antibodies per AuNP was determined to be approximately 0, 9, 7, 9 and 8 for the mPEG, isotype IgG, anti-TfRA, anti-TfRD, and anti-TfRA/BACE1 groups, respectively (Table 1).
Inductively coupled plasma mass spectrometry of AuNPs
Au concentration of the pure AuNPs was determined by ICP-MS by dissolving 20 µL concentrated AuNP sample in fresh aqua regia (380 µL) and diluting with MilliQ water (9.60 mL) containing 2% HCl and an Ir-standard (0.5 PPB). The prepared samples were directly analyzed by iCAP Q ICP-MS (Thermo Scientific, Waltham, MA) together with an Au calibration curve. The Au-concentration was determined from plotting the calibration curve from an ICP-MS Au standard (intensity of 197Au) with a known concentration.
Transmission electron microscopy characterization of AuNPs
Three microliters of AuNP solution were deposited onto freshly glow discharged 200 mesh nickel TEM grids coated with a carbon-stabilized formvar film (Electron Microscopy Sciences, Hatfield, USA). The AuNPs adsorbed onto the film surface for 5 min after which the excess solution was wicked away using filter paper. The grids were then washed in quick succession in three droplets of MilliQ water. Excess water was removed using filter paper and the grids allowed to air dry.
TEM was performed utilizing a FEI Tecnai T20 G2 (Thermo Fisher Scientific, Waltham, USA) operating at 200 kV located at the Center for Electron Nanoscopy at DTU. Both low and high magnification images, as well as energy dispersive X-ray spectra (EDS), were acquired for each AuNP sample. Images were acquired using a CCD camera equipped with Digital Micrograph (Gatan, Pleasanton, USA), and EDS spectra were acquired using a X-maxN Silicon Drift Detector (Oxford Instruments, Abingdon, UK). Image analysis was performed using Fiji analysis software [69]. Nanoparticle diameter was determined using the 'analyze particle function' of Fiji to obtain an area for each nanoparticle, making sure not to include nanoparticles that were overlapping in the measurements, and converting this value to a diameter assuming a spherical nanoparticle. A minimum of 250 nanoparticles were measured for each sample.
Surface plasmon resonance analysis
The binding affinities of the different anti-TfR antibodies were evaluated by surface plasmon resonance (SPR) analysis performed using a Biacore 3000 instrument (Biacore, Sweden) equipped with an activated CM5 sensor chip. Recombinant mouse TfR protein (SinoBiological, Inc., Beijing, PRC) was purchased with a His-tag and immobilized on the sensor chip gold surface to densities of 58 fmol/mm2. Samples (human isotype IgG, anti-TfRA, anti-TfRD, anti-TfRA/BACE1) were injected for binding to the immobilized TfR protein at 5 μL/min at 25 °C in 10 mM HEPES, 150 mM NaCl, 1.5 mM CaCl2, 1 mM EGTA, and 0.005% Tween 20 (pH 7.4). Antibody binding to the immobilized TfR protein was expressed as the response difference between the protein-coupled and an uncoupled, but activated, sensor chip. The dissociation constant (KD) values were calculated using the BIAevaluation 4.1 software using a Langmuir 1:1 binding model, using a drifting baseline with global fitting to the curves of the considered concentration range. The anti-TfR antibody clones RI7217 (rat anti-mouse) and OX26 (mouse anti-rat) were also analyzed as positive and negative control, respectively.
Cell culture
Mouse BCECs were isolated from 6-10-week-old C57BL/6 mice according to the procedure described in Thomsen et al. [26]. All experiments and handling of animals were approved by the Danish National Council for Animal Welfare (License no. 2013-15-2934-00893). The experiments were performed in accordance with relevant guidelines and regulations (EU directive 2010/63/EU and relevant local additions to this directive).
The mice were anesthetized with isoflurane (Baxter, Søborg, DK), and their heads submerged in 70% ethanol before decapitation. The mouse brains were carefully removed from the skull, followed by dissection and removal of meninges and white matter using tweezers and filter paper. The gray matter was cut into small pieces using sterile scalpel blades while kept in ice-cold DMEM-F12 GlutaMAXTM (Thermo Scientific, Nærum, DK). The resulting suspension of gray matter was digested with collagenase II and DNase I in DMEM-F12 GlutaMAXTM at 37 ºC for 75 min in an Incubating Mini Shaker (VWR, Søborg, DK), after which the enzymes were inhibited by dilution of the tissue solution in additional DMEM-F12 GlutaMAXTM. Afterwards, the solution was centrifuged at 1,000 ×g for 8 min. The different cell types were then separated by mixing and centrifugation in 20% bovine serum albumin (BSA) at 1,000 ×g for 20 min, followed by digestion of the microvessel-containing pellet with collagenase/ dispase and DNase I in DMEM-F12 GlutaMAXTM at 37 ºC for 50 min. The microvessels were further separated from other cell types on a continuous Percoll gradient (33%). The microvessel fraction was then collected and seeded onto plastic dishes coated with collagen IV (0.15 mg/mL) and fibronectin (0.05 mg/mL), where the BCECs could detach from the microvessels and grow on the culture dish surface. The primary BCECs were cultured in DMEM-F12 GlutaMAXTM supplemented with 10% plasma-derived serum (First Link, Wolverhampton, UK), bFGF (1 ng/mL, Roche), heparin (100 µg/mL, Sigma-Aldrich), insulin-transferrin-sodium selenite (100 µg/mL, Roche), and gentamicin sulphate (10 µg/mL, Lonza Copenhagen, Vallensbæk Strand, DK) in an incubator with 5% CO2 at 37 ºC. Puromycin (an efflux transporter substrate, 4 µg/mL, Sigma-Aldrich) was added to the culture medium to remove pericytes present in the BCEC preparation.
Mixed glial cells were isolated from the brains of 2-day-old C57BL/6 mouse pups (following the procedure of Thomsen et al. [26]). The pups were decapitated, the brains removed from the skull, and subsequently resuspended in DMEM-F12 GlutaMAXTM using a Hypodermic 21G needle (Sterica, B. Braun Medical A/S, Frederiksberg, DK) followed by filtering through a Corning 40 µm nylon cell strainer (Sigma-Aldrich, Brøndby, DK). The filtered cells were then seeded into T75 culture flasks coated with poly-L-lysine (0.1 mg/mL, Sigma-Aldrich, Brøndby, DK), and cultured in an incubator at 5% CO2 at 37 ºC. After 5 days of culture, the cells were shaken and thoroughly washed in PBS to release and remove some of the contaminating microglia cells. The growth medium was replenished, and the cells grown for another 14 days until confluency. The mixed glial cells (mostly astrocytes) were then seeded in poly-L-lysine-coated 12-well plates to be used for construction of the primary mouse in vitro co-culture models of the BBB.
Construction of in vitro BBB models
To construct in vitro co-culture models of the BBB, BCECs were detached from the culture dish and seeded onto the porous membrane of hanging cell culture inserts fitted for 12-well plates (Transwell, polyethylene terephthalate porous membrane, pore size: 1 µm). All culture inserts were coated with collagen IV and fibronectin. To increase the tightness of the BCECs, the hanging cell culture inserts were transferred to a 12-well plate containing a confluent layer of mixed glial cells (mostly astrocytes) in the bottom of each well, and treated with hydrocortisone, cAMP and RO-201724 at concentrations of 550 nM, 250 μM and 17.5 μM, respectively.
The integrity of the in vitro BBB was evaluated by measuring the transendothelial electrical resistance (TEER) using the Millicell ERS-2 Epithelial Volt-Ohm meter and STX01 Chopstick Electrodes (Millipore, Hellerup, DK). The corresponding permeability parameters to the TEER values of the in vitro BBB models were as presented in Thomsen et al. [26]. The TEER value of a coated insert without any cells were subtracted from all TEER measurements as normalization, and all values multiplied by the area of the culture insert (1.12 cm2). All data were analyzed using GraphPad 5.0 (GraphPad Software, Inc., CA, USA), and the TEER values presented as Ω cm2.
In vitro uptake and transcytosis studies of the AuNPs
To evaluate the uptake and transcytosis potential of the different AuNP formulations, the primary in vitro models of the BBB were treated with 100 µg/mL AuNPs in the Transwell insert culture medium. The cells and AuNPs were incubated together for 5 h to obtain sufficient quantities of gold in the bottom chamber of the Transwell co-culture system for ICP-MS detection. All experiments were performed after reaching high TEER (regarded as > 130 Ω cm2 as suggested by Burkhart et al. [26] and Thomsen et al. [70]). No reduction in the TEER values were detected during the course of the experiment, indicating that the integrity of the in vitro BBB models was maintained (Figure 2B). After the incubation, samples were taken from the culture medium present in the Transwell culture insert ('blood fraction') and the bottom well ('brain fraction'). The BCECs attached to the filter membrane of the Transwell culture insert were washed 3 × 5 min in 0.1 mg/mL heparin in 0.1 M PBS, followed by detachment of the BCECs by trypsinization ('BCEC fraction'). The gold content was measured in the different fractions using ICP-MS (see below). The data was analyzed in GraphPad Prism 5.0 (GraphPad Software, Inc., CA, USA), and results for gold uptake and transport were plotted as percentage of administrated dose (%AD) relative to samples taken from the Transwell culture insert immediately after AuNP administration.
Immunocytochemistry
Immunocytochemistry was performed on the primary in vitro models of the BBB to validate the expression of important markers for BBB integrity as well as the TfR. BCECs were grown to confluence on glass cover slips in the bottom of a 12-well cell culture plate. After reaching full confluency, the BCECs were washed 3 × 5 min in 0.1 M PBS (pH 7.4) and fixed in 4% paraformaldehyde in 0.1 M PBS (pH 7.4) for 15 min. After additional washing in 0.1 M PBS (pH 7.4, 3 × 5 min), the BCECs were permeabilized and blocked in 0.1% Triton X-100 and 3% BSA in 0.1 M PBS (pH 7.4) for 30 min. BCECs were incubated with antibodies against different TJ protein markers, including ZO-1, claudin-5, occludin, and CD31 in a co-staining setup with a primary antibody against the TfR (all primary antibodies were diluted 1:200 in 0.1% Triton X-100 and 3% BSA in 0.1 M PBS (pH 7.4)). Surface plasmon resonance with recombinant mouse TfR protein functionalized on the gold surface was used to ensure the specificity of the anti-TfR antibody (clone RI7217, KD = 6 nM, Figure S2). Incubation with primary antibodies proceeded for 60 min at room temperature, and was followed by washing in a 1:50 dilution of the incubation buffer (0.1% Triton X-100 and 3% BSA in 0.1 M PBS (pH 7.4)). After washing, Alexa Fluor 594-conjugated goat anti-rabbit, Alexa Fluor 594-conjugated goat anti-mouse, or Alexa Fluor 488-conjugated donkey anti-rat secondary antibodies were administrated in a 1:200 dilution in incubation buffer (0.1% Triton X-100 and 3% BSA in 0.1 M PBS (pH 7.4) and incubated for 60 min. Lastly, the BCECs were washed thoroughly in 0.1 M PBS (pH 7.4)), the nuclei were stained with 4,6-diamino-2-phenylindole (DAPI), and the glass cover slips were mounted on glass slides with fluorescence mounting medium (DAKO, Glostrup, DK). Confocal microscopy was performed using a ZEISS LSM 800 confocal microscope (Carl Zeiss, Jena, DE), and images were taken using a Zeiss 63X magnification Plan Apochromat Oil DIC objective lens (NA 1.40). All images were processed using Fiji software [69].
Western blotting
Additional validation of TfR expression in isolated BCECs was done by Western blot analysis. The BCECs were harvested using a cell scraper and PBS, and transferred into a tube and centrifuged at 16,000 ×g for 5 min at 4 ºC followed by discarding of the supernatant. One hundred microliters of lysis buffer (150 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 1% Triton X-100, and Complete Protease Inhibitor) was added to the cell pellet, and the cell pellet was dissolved on ice for 20 min with vortexing every 3-5 min. After dissolving, the solution was centrifuged at 16,000 ×g for 10 min at 4 ºC and the supernatant was transferred into a clean tube. The protein concentration was determined using the BCA assay kit (Sigma-Aldrich, Brøndby, DK). The samples were prepared by mixing with NuPAGE sample buffer and 0.02 M DTT before loading onto a precast Novex NuPAGE 4-12% Bis-Tris Gel with a total sample protein input of 3 µg. SeeBlueTM Plus2 Pre-stained Protein Standard was loaded onto the gel for size reference, and the gel was run at 200 V for 40 min. The gel was then transferred to a dry blot cassette on top of a nitrocellulose membrane, and dry blotting was performed for 7 min on an Invitrogen Novex iBlot Dry Blotting system. The membrane was then blocked in TBST containing 5% skim milk for 1 h at 4 ºC before incubation overnight with a 1:1000 dilution of primary antibodies against the mouse TfR (Abcam, ab84036, rabbit polyclonal) or β-actin (Sigma-Aldrich, A5441, mouse monoclonal) in TBST containing 5% skim milk. The next day, the membrane was washed 4 × 5 min in TBST containing 1% skim milk at room temperature, and a 1:2000 dilution of secondary antibodies (horse anti-mouse HRP-linked, Cell Signaling, 7076s, or goat anti-rabbit HRP-linked, Cell Signaling, 7074s) in TBST containing 5% skim milk were applied to the membrane and allowed to incubate for 3 h at 4 ºC. After extensive washing in TBST containing 1% skim milk, the membrane was developed using Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare Life Sciences, Brøndby, DK) and imaged using ImageQuant LAS 4000 (GE Healthcare Life Sciences, Brøndby, DK).
In vivo studies and tissue isolation
All procedures and handling of animals were approved by the Danish National Council for Animal Welfare (License no. 2013-15-2934-00893) and planning was made using the Experimental Design Assistant (National Centre for the Replacement, Refinement & Reduction of Animals in Research, UK). The experiments were all performed in accordance with relevant guidelines and regulations (EU directive 2010/63/EU and relevant local additions to this directive). To study the uptake and trans-BBB transport capacity of the different TfR-targeted AuNP formulations plus controls in vivo, the AuNPs were injected via the lateral tail vein into 8-week-old female Balb/c mice (n = 8 per group) and allowed to circulate for 8 h [23]. The administrated dose of the individual AuNP formulations (mPEG, isotype IgG, anti-TfRA, anti-TfRD, or anti-TfRA/BACE1) was 5×1011 AuNPs/ mouse as previously described in Wiley et al. (2013)[23].
After systemic circulation for 8 h, the mice were deeply anesthetized by a subcutaneous injection of 0.5 mL/10 g body weight of Hypnorm/Dormicum (fentanyl/fluanisone mixed with midazolam and sterile water in a ratio of 1:1:2). The chest cage of the mouse was opened, and the heart was exposed for transcardial perfusion of 0.1 M PBS (pH 7.4). After perfusion, the skull was opened, and the brain removed for subsequent brain capillary depletion (one hemisphere, see below) or sampling for biodistribution analysis (one hemisphere). Furthermore, tissue samples were taken from the liver, spleen, kidney, heart, and lungs were resected for biodistribution analysis as well. All biodistribution tissue samples were quickly frozen on dry ice and stored until further processing for ICP-MS quantification of the gold content.
Before perfusion of the animals, a blood sample was drawn from the left ventricle of the heart into a tube containing heparin. All blood samples were centrifuged at 2,000 ×g for 15 min to pellet cells and platelets contained in the sample, and the resulting plasma was transferred into a clean tube and stored until further processing for ICP-MS. The gold content in all blood and tissue samples were measured using ICP-MS (see below) and the resulting data plotted as %ID/g in GraphPad Prism 5.0 (GraphPad Software, Inc., CA, USA).
A subset of mice (n = 4 per group) was dedicated to morphological analyses using either light microscopy or TEM. After circulation of the AuNPs, the mice were deeply anesthetized by a subcutaneous injection of 0.5 mL/10 g body weight of Hypnorm/Dormicum (fentanyl/fluanisone mixed with midazolam and sterile water in a ratio of 1:1:2). Once full anaesthesia was reached, the chest cage was opened, and the mice were transcardially perfused with 0.1 M PBS (pH 7.4). Afterwards, the mice were re-perfused with either 4% paraformaldehyde in 0.1 M PBS (pH 7.4) for subsequent cryosectioning and light microscopy, or 4% paraformaldehyde, 2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for subsequent processing for TEM. The brains dedicated to light microscopy were transferred into a vial containing 4% paraformaldehyde in 0.1 M PBS (pH 7.4) to continue the fixation process overnight. After fixation, the brains were moved to a 30% sucrose solution and allowed to settle, before they were cryosectioned into 40 µm thick slices and stored at -20 ºC in antifreeze solution until further processing. The brains dedicated to TEM were transferred into a vial containing 4% paraformaldehyde, 2% glutaraldehyde in 0.1 M sodium cacodylate buffer and allowed to fixate until further processing for TEM (see below).
Another subset of mice (n = 4 per group, only groups isotype IgG, anti-TfRA/BACE1, and untreated control) received a dose of 1×1012 AuNPs/mouse (corresponding to a doubling of the dose used for quantitative studies), which circulated for 30 h before termination. The mice were then anesthetized and perfused with 0.1 M PBS (pH 7.4) as described above, before their skulls were opened and their brains removed. The brains were quickly frozen on dry ice and stored at -80 ºC before processing for subsequent ELISA-based quantification of their β-amyloid protein load (see below).
Brain capillary depletion
To indicate the presence of AuNPs in the brain parenchyma, the brain capillary depletion technique was employed to separate the brain capillaries from the rest of the brain cells according to the protocol by Triguero et al. with minor modifications [46, 59]. One brain hemisphere was sampled from each mouse and homogenized by four strokes in a dounce homogenizer containing ice-cold homogenization buffer (10 mM HEPES, 141 mM NaCl, 4 mM KCl, 2.8 mM CaCl2, 1 mM MgSO4, 1 mM NaH2PO4, and 10 mM glucose; pH 7.4), followed by addition of ice-cold 30% dextran (MW: 60,000, Sigma-Aldrich) and further homogenization by four strokes in the dounce homogenizer. The brain homogenate was transferred into a 15 mL Falcon tube, and subsequently centrifuged at 3,500 ×g for 40 min at 4 ºC with slow deceleration. After centrifugation, the brain homogenate had separated into a brain capillary-containing pellet, and a supernatant containing the brain parenchymal fraction. Samples were taken from both the capillary and parenchymal fractions, and the gold content was measured by ICP-MS (see below).
Measurement of γ-glutamyl transferase activity
The purity of the depleted brain capillaries and parenchyma was analyzed by measuring the activity of γ-glutamyl transferase in the sampled fractions using the γ-Glutamyl Transferase (GGT) Activity Colorimetric Assay kit (Sigma-Aldrich, Brøndby, DK) according to the manufacturer's protocol. Briefly, sample volumes corresponding to a total of 10 mg tissue were taken from each fraction and homogenized in ice-cold GGT Assay buffer. The homogenized tissue samples were then centrifuged at 13,000 ×g for 10 min at 4 ºC to pellet any insoluble material. Ten microliters of the supernatant were transferred into a flat-bottom 96-well plate together with positive controls and standard curve samples. Additional 90 µL GGT Substrate Solution were added to each well (except for the standard curve samples), and the mixture was allowed to incubate for 3 min at 37 ºC before taking an initial measurement of the absorbance at 418 nm using an EnSpire Multimode Plate Reader (Perkin-Elmer, Waltham, MA, USA). The samples continued to incubate at 37 ºC and the absorbance at 418 nm was measured every 5 min to follow the conversion process. The initial and final absorbance measurements were used for calculating the enzyme activity presented as nmol/min/mL.
Inductively-coupled plasma mass spectrometry of tissue and cell samples
Inductively-coupled plasma mass spectrometry (ICP-MS) was used to analyze the gold content of the extracted tissue samples. A maximum of 100 mg tissue per sample were digested in aqua regia overnight at 65 ºC, and after digestion, the samples were diluted in pure MilliQ water containing 0.5 ppb iridium (Fluka, Sigma-Aldrich, Brøndby, DK). Just prior to ICP-MS analysis, the samples were further diluted in 2% HCl containing 0.5 ppb iridium, and the samples were analyzed on a iCAP Q ICP-MS system (Thermo Scientific, Hvidovre, DK) fitted with an ASX-520 AutoSampler and a Neclar ThermoFlex 2500 chiller. The system was calibrated using TUNE B iCAP Q element mixture (Thermo Scientific, Hvidovre, DK) prior to analysis. To obtain the gold concentration of the samples, a standard curve was generated based on serial dilution of an analytical standard gold solution (Fluka, Sigma-Aldrich, Brøndby, DK) to obtain data points ranging from 0.08-5 ppb. The iridium concentration was also measured during the analysis as an internal standard, showing that all samples were analyzed similarly. The resulting data was plotted as %ID/g in GraphPad Prism 5.0 (GraphPad Software, Inc., CA, USA).
Transmission electron microscopy of AuNPs in brain tissue
The cortices from the tissue set aside for TEM analysis were carefully excised from the remaining brain tissue and cut into 1 mm3 pieces for further processing. The tissue pieces were washed 2 × 10 min in 0.1 M Na cacodylate buffer to remove residual fixative and suspended in 1% OsO4 in 0.1 M sodium cacodylate buffer for 1 h at 4 ºC. The samples were then washed twice in MilliQ water followed by resuspension in 1% uranyl acetate in water overnight. The next day, the samples were washed twice in MilliQ water and re-suspended in increasing concentrations of ethanol; 50%, 70%, 95%, 100% and 100% for 15 min each. The samples were then washed 3 × 5 min in acetonitrile and infiltrated with a 1:1 solution of Agar 100 resin:acetonitrile for 1 hour (Agar 100 resin is a replacement resin for Epon 812). Next, the samples were infiltrated with a 2:1 solution of Agar 100 resin:acetonitrile overnight. The following day, the samples were infiltrated with pure Agar 100 resin for 1 h. The tissue pieces were then placed in BEEM capsules (Agar Scientific, Stansted, UK) and covered with resin. Then, the samples were placed in a curing oven at 60 °C overnight to polymerize. After polymerization, the samples were removed from the BEEM capsules, trimmed using a razor blade and thin sections (200 nm thick) were cut using a RMC MT-7 Ultramicrotome (Boeckeler Instruments, Tucson, USA) equipped with a glass knife. These thin sections were then placed on 200 mesh nickel TEM grids coated with a carbon stabilized formvar film (Electron Microscopy Sciences, Hatfield, USA) with a hole length of 95 µm.
TEM was performed utilizing a FEI Tecnai T20 G2 (Thermo Fisher Scientific, Waltham, USA) operating at 200 kV located at the Center for Electron Nanoscopy at DTU. Images were acquired using a CCD camera equipped with Digital Micrograph (Gatan, Pleasanton, USA) and EDS spectra were acquired using a X-maxN Silicon Drift Detector (Oxford Instruments, Abingdon, UK). Brain tissue was imaged in a systematic manner by scanning entire grid sections at low magnification, 2500x. The contrast was optimized by condensing the beam such that the top left corner of the CCD camera was not illuminated. This allowed for straight forward identification of AuNPs versus salt crystals and other sample preparation artifacts. Once AuNPs were identified higher magnification images were acquired, and for AuNPs located within the brain parenchyma, EDS spectra were acquired. Processing and TEM imaging of liver and spleen tissues were performed using the same methodology.
Quantification of brain β-amyloid protein load
To study the potential therapeutic efficacy of the anti-TfRA/BACE1 antibody after AuNP transport into the brain, the brain β-amyloid protein load was measured using enzyme-linked immunosorbent assay (ELISA)[16]. Approximately 100 mg brain tissue was weighed and homogenized in 5 M guanidine-HCl/50 mM Tris. The mixture was incubated at room temperature for four hours under constant shaking. After incubation, the mixture was diluted in cold BSAT-PBS containing 1X protease inhibitor cocktail to reduce the guanidine concentration to less than 0.1 M. The diluted samples were then centrifuged at 16,000 ×g for 20 min at 4 ºC, and the supernatant was transferred to a clean tube and stored at -80 ºC until β-amyloid quantification by ELISA.
The β-amyloid load was measured using the Mouse Aβ40 ELISA kit (Thermo Fisher Scientific, Naerum, DK) according to manufacturer's protocol. Briefly, the mouse brain protein samples were administrated to individual wells in a 96-well plate containing surface-bound capture antibodies against mouse Aβ40 and incubated for two hours. A detection antibody solution was then applied and incubated for another hour, followed by incubation with HRP-labelled secondary antibody solution for 30 min. After washing, stabilized chromogen solution (HRP substrate) was administrated and incubated for 30 min to induce a color change correlating with the Aβ40 concentration. Lastly, a stop solution was applied to halt the reaction, and the absorbance at 450 nm was read immediately on an EnSpire Multimode Plate Reader (Perkin-Elmer, Waltham, MA, USA).
Silver enhancement on brain tissue sections
Silver enhancement was performed on fixed brain tissue sections to develop the AuNPs to a size adequate for the resolution of the light microscope [22, 23, 60]. The silver enhancement was made using the Silver Enhancement Kit (Sigma-Aldrich, Brøndby, DK) according to the manufacturer's instructions. Brain cryosections (40 µm) from animals treated with the different AuNP formulations were thoroughly washed in KPBS three times before fixation in 2.5% glutaraldehyde for 15 min at room temperature. After fixation, the brain sections were rinsed three times in MilliQ water to remove traces of buffer salts etc., followed by removal of excess fluid before the silver enhancement process. The Silver Enhancer Mixture was applied to the brain sections and allowed to react for 15 min before rinsing in MilliQ water. Finally, the brain sections were fixed in 2.5% sodium thiosulfate for 3 min before additional washing with MilliQ water. Counterstaining with hematoxylin was performed for 5 min with thorough rinsing in tap (lukewarm) and MilliQ water afterwards. The mounted brain sections were then dried, covered in mounting medium and protected by glass coverslips. Liver sections were treated as explained above and served as a positive control for the specific development of AuNP size. Images were acquired on an Axioplan 2 Imaging Microscope (Carl Zeiss, Jena, DE) using a Zeiss 100X magnification Plan Neofluar Oil objective lens (NA 1.30), and brightness and contrast were adjusted using Fiji software [69].
Statistical analysis
All data were analysed in GraphPad Prism 5.0 (GraphPad Software, Inc., CA, USA) and presented as the arithmetic mean ± standard error of the mean (SEM). The statistical significances of intergroup differences were calculated using the parametric unpaired T-test or one-way ANOVA with Tukey's multiple comparisons post hoc test for normally distributed data/data with equal variances, or the non-parametric Mann-Whitney's U Test or Kruskal-Wallis test with Dunn's multiple comparisons post hoc test for non-normally distributed data/data with unequal variances. Potential outliers in a dataset were tested using Grubbs' test in GraphPad QuickCalcs (GraphPad Software, Inc., CA, USA). A p-value < 0.05 was considered statistically significant. * p < 0.05, ** p < 0.01, *** p < 0.001.
Abbreviations
AuNP: Gold nanoparticle; BACE1: Beta-secretase 1; BBB: Blood-brain barrier; BCEC: Brain capillary endothelial cell; bFGF: Basic fibroblast growth factor; BSA: Bovine serum albumin; cAMP: Cyclic adenosine monophosphate; CCD: Charge-coupled device; CD: Cluster of differentiation; DAPI: 4,6-diamino-2-phenylindole; DHB: 2,5-dihydroxybenzoic acid; DLS: Dynamic light scattering; DMEM: Dulbecco's modified Eagle's medium; EDS: Energy-dispersive X-ray spectroscopy; ELISA: Enzyme-linked immunesorbent assay; Fc: Fragment crystallizable; FT-IR: Fourier-transform infrared; GGT: Gamma-glutamyl transferase; h: hour; HRP: Horseradish peroxidase; IC50: Half maximal inhibitory concentration; ICP-MS: Inductively-coupled plasma-mass spectrometry; IgG: Immunoglobulin; KD: Dissociation constant; kDa: kilo Dalton; LA: Lipoic acid; Mal: Maleimide; MALDI-TOF: Matrix-assisted laser desorption/ionisation-time of flight; mPEG: Methoxy polyethylene glycol; MS: Mass spectrometry; MTA: Material transfer agreement; MW: Molecular weight; NHS: N-hydroxysuccinimide; NMR: Nuclear magnetic resonance; PBS: Phosphate-buffered saline; PDI: Polydispersity index; PEG: Polyethylene glycol; SPR: Surface plasmon resonance; TBS: Tris-buffered saline; TEER: Transendothelial electrical resistance; TEM: Transmission electron microscopy; Tf: Transferrin; TfR: Transferrin receptor; TJ: Tight junction; TLC: Thin-layer chromatography; UV: Ultraviolet; ZO: Zonula occludens; %AD: Percentage of administrated dose; %ID/g: Percentage of injected dose per gram.
Acknowledgements
The work presented in this study was funded by generous grants from the Lundbeck Foundation Research Initiative on Brain Barriers and Drug Delivery (Grant no. R155-2013-14113), Fonden til Lægevidenskabens Fremme (Grant no. 14-191), and Kong Christian den Tiendes Fond (Grant no. 2016-851/10-0748). The authors wish to acknowledge Hanne Krone Nielsen, Laboratory for Neurobiology, Aalborg University for her invaluable technical assistance. Sarah Christine Christensen, Department of Biomedicine, Aarhus University, is acknowledged for her assistance with Western blot analysis of TfR expression. Anne Marie Bundsgaard, Department of Biomedicine, Aarhus University, is acknowledged for her technical expertise related to surface plasmon resonance experiments. Genentech, Inc. is acknowledged for providing the anti-TfRA, anti-TfRD, and anti-TfRA/BACE1 antibodies used in the study.
Author contributions
K.B.J. contributed to the experimental design and analysis, conducted experiments related to AuNP synthesis and in vitro/in vivo assessments with subsequent ICP-MS and light microscopy analysis, and wrote the manuscript. M.B. conducted experiments related to chemical and AuNP synthesis, and contributed to the data analysis and manuscript preparation. P.J.K. contributed to morphological analysis using TEM. T.L.A. and T.M. contributed to the experimental design and analysis, and supervised the manuscript preparation. F.M., A.B., M.S.T., and M.S.N. conducted and analyzed experiments. All authors read and approved the final manuscript.
Competing interests
All authors have declared no conflicting interest in relation to the theme and contents of the manuscript. The anti-TfRA, anti-TfRD, and anti-TfRA/BACE1 antibodies were obtained from Genentech, Inc. via a Material Transfer Agreement (MTA A15783). Genentech, Inc. had no influence on study design, experimental procedures, data analysis, or preparation of the manuscript.
Supplementary Material
Supplementary figures.
References
1. Johnsen KB, Moos T. Revisiting nanoparticle technology for blood-brain barrier transport: unfolding at the endothelial gate improves the fate of transferrin receptor-targeted liposomes. J Control Release. 2016;222:32-46
2. Saraiva C, Praça C, Ferreira R, Santos T, Ferreira L, Bernardino L. Nanoparticle-mediated brain drug delivery: overcoming blood-brain barrier to treat neurodegenerative diseases. J Control Release. 2016;235:34-47
3. Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178-201
4. Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41-53
5. Zuchero YJ, Chen X, Bien-Ly N, Bumbaca D, Tong RK, Gao X. et al. Discovery of novel blood-brain barrier targets to enhance brain uptake of therapeutic antibodies. Neuron. 2016;89:70-82
6. Johnsen KB, Burkhart A, Andresen TL, Moos T, Thomsen LB. The use of the transferrin receptor as a target for brain drug delivery using nanomedicines. In: (ed.) Morales J, Gaillard P. Nanomedicines for Brain Drug Delivery. Springer Nature Neuromethods Series: Springer Nature. 2018
7. Skjørringe T, Burkhart A, Johnsen KB, Moos T. Divalent metal transporter 1 (DMT1) in the brain: implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front Mol Neurosci. 2015;8:19
8. Burkhart A, Skjørringe T, Johnsen KB, Siupka P, Thomsen LB, Nielsen MS. et al. Expression of iron-related proteins at the neurovascular unit supports reduction and reoxidation of iron for transport through the blood-brain barrier. Mol Neurobiol. 2016;53:7237-53
9. Duck KA, Connor JR. Iron uptake and transport across physiological barriers. Biometals. 2016;29:573-91
10. Pardridge WM. Targeted delivery of protein and gene medicines through the blood-brain barrier. Clin Pharmacol Ther. 2015;97:347-61
11. Bien-Ly N, Yu YJ, Bumbaca D, Elstrott J, Boswell CA, Zhang Y. et al. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J Exp Med. 2014;211:233-44
12. Couch JA, Yu YJ, Zhang Y, Tarrant JM, Fuji RN, Meilandt WJ. et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci Transl Med. 2013;5:183ra57 1-12
13. Thom G, Burrell M, Haqqani AS, Yogi A, Lessard E, Brunette E. et al. Enhanced delivery of galanin conjugates to the brain through bioengineering of the anti-transferrin receptor antibody OX26. Mol Pharm. 2018;15:1420-31
14. Webster CI, Hatcher J, Burrell M, Thom G, Thornton P, Gurrell I. et al. Enhanced delivery of IL-1 receptor antagonist to the central nervous system as a novel anti-transferrin receptor-IL-1RA fusion reverses neuropathic mechanical hypersensitivity. Pain. 2017;158:660-8
15. Yu YJ, Atwal JK, Zhang Y, Tong RK, Wildsmith KR, Tan C. et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med. 2014;6:261ra154
16. Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y. et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med. 2011;3:84ra44
17. Freskgård PO, Urich E. Antibody therapies in CNS diseases. Neuropharmacology. 2017;120:38-55
18. Hultqvist G, Syvänen S, Fang XT, Lannfelt L, Sehlin D. Bivalent brain shuttle increases antibody uptake by monovalent binding to the transferrin receptor. Theranostics. 2017;7:308-18
19. Niewoehner J, Bohrmann B, Collin L, Urich E, Sade H, Maier P. et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron. 2014;81:49-60
20. Syvänen S, Fang XT, Hultqvist G, Meier SR, Lannfelt L, Sehlin D. A bispecific Tribody PET radioligand for visualization of amyloid-beta protofibrils - a new concept for neuroimaging. Neuroimage. 2017;148:55-63
21. Villaseñor R, Ozmen L, Messaddeq N, Grüninger F, Loetscher H, Keller A. et al. Trafficking of endogenous immunoglobulins by endothelial cells at the blood-brain barrier. Sci Rep. 2016;6:25658
22. Clark AJ, Davis ME. Increased brain uptake of targeted nanoparticles by adding an acid-cleavable linkage between transferrin and the nanoparticle core. Proc Natl Acad Sci USA. 2015;112:12486-91
23. Wiley DT, Webster P, Gale A, Davis ME. Transcytosis and brain uptake of transferrin-containing nanoparticles by tuning avidity to transferrin receptor. Proc Natl Acad Sci USA. 2013;110:8662-7
24. Gregori M, Orlando A, Re F, Sesana S, Nardo L, Salerno D. et al. Novel antitransferrin receptor antibodies improve the blood-brain barrier crossing efficacy of immunoliposomes. J Pharm Sci. 2016;105:276-83
25. Atwal JK, Chen Y, Chiu C, Mortensen DL, Meilandt WJ, Liu Y. et al. A therapeutic antibody targeting BACE1 inhibits amyloid-β production in vivo. Sci Transl Med. 2011;3:84ra43
26. Thomsen MS, Birkelund S, Burkhart A, Stensballe A, Moos T. Synthesis and deposition of basement membrane proteins by primary brain capillary endothelial cells in a murine model of the blood-brain barrier. J Neurochem. 2017;140:741-54
27. Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes JM, Zhang Y. et al. Effector-attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J Biol Chem. 2017;292:3900-8
28. Male D, Gromnicova R, McQuaid C. Gold Nanoparticles for imaging and drug transport to the CNS. Int Rev Neurobiol. 2016;130:155-98
29. Gromnicova R, Davies HA, Sreekanthreddy P, Romero IA, Lund T, Roitt IM. et al. Glucose-coated gold nanoparticles transfer across human brain endothelium and enter astrocytes in vitro. PLoS One. 2013;8:e81043
30. Gromnicova R, Kaya M, Romero IA, Williams P, Satchell S, Sharrack B. et al. Transport of gold nanoparticles by vascular endothelium from different human tissues. PLoS One. 2016;11:e0161610
31. Gromnicova R, Yilmaz CU, Orhan N, Kaya M, Davies H, Williams P. et al. Localization and mobility of glucose-coated gold nanoparticles within the brain. Nanomedicine (Lond). 2016;11:617-25
32. Cabezón I, Augé E, Bosch M, Beckett AJ, Prior IA, Pelegrí C. et al. Serial block-face scanning electron microscopy applied to study the trafficking of 8D3-coated gold nanoparticles at the blood-brain barrier. Histochem Cell Biol. 2017;148:3-12
33. Cabezón I, Manich G, Martín-Venegas R, Camins A, Pelegrí C, Vilaplana J. Trafficking of gold nanoparticles coated with the 8D3 anti-transferrin receptor antibody at the mouse blood-brain barrier. Mol Pharm. 2015;12:4137-45
34. Huwyler J, Wu D, Pardridge WM. Brain drug delivery of small molecules using immunoliposomes. Proc Natl Acad Sci USA. 1996;93:14164-9
35. Avnir Y, Turjeman K, Tulchinsky D, Sigal A, Kizelsztein P, Tzemach D. et al. Fabrication principles and their contribution to the superior in vivo therapeutic efficacy of nano-liposomes remote loaded with glucocorticoids. PLoS One. 2011;6:e25721
36. Carvey PM, Zhao CH, Hendey B, Lum H, Trachtenberg J, Desai BS. et al. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur J Neurosci. 2005;22:1158-68
37. Kizelsztein P, Ovadia H, Garbuzenko O, Sigal A, Barenholz Y. Pegylated nanoliposomes remote-loaded with the antioxidant tempamine ameliorate experimental autoimmune encephalomyelitis. J Neuroimmunol. 2009;213:20-5
38. Turjeman K, Bavli Y, Kizelsztein P, Schilt Y, Allon N, Katzir TB. et al. Nano-drugs based on nano sterically stabilized liposomes for the treatment of inflammatory neurodegenerative diseases. PLoS One. 2015;10:e0130442
39. Alata W, Ye Y, St-Amour I, Vandal M, Calon F. Human apolipoprotein E ɛ4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J Cereb Blood Flow Metab. 2015;35:86-94
40. Bien-Ly N, Boswell CA, Jeet S, Beach TG, Hoyte K, Luk W. et al. Lack of widespread BBB disruption in Alzheimer's Disease models: focus on therapeutic antibodies. Neuron. 2015;88:289-97
41. Cheng Z, Zhang J, Liu H, Li Y, Zhao Y, Yang E. Central nervous system penetration for small molecule therapeutic agents does not increase in multiple sclerosis- and Alzheimer's disease-related animal models despite reported blood-brain barrier disruption. Drug Metab Dispos. 2010;38:1355-61
42. Do TM, Alata W, Dodacki A, Traversy MT, Chacun H, Pradier L. et al. Altered cerebral vascular volumes and solute transport at the blood-brain barriers of two transgenic mouse models of Alzheimer's disease. Neuropharmacology. 2014;81:311-7
43. Do TM, Dodacki A, Alata W, Calon F, Nicolic S, Scherrmann JM. et al. Age-dependent regulation of the blood-brain barrier influx/efflux equilibrium of amyloid-β peptide in a mouse model of Alzheimer's disease (3xTg-AD). J Alzheimers Dis. 2016;49:287-300
44. Gustafsson S, Lindström V, Ingelsson M, Hammarlund-Udenaes M, Syvänen S. Intact blood-brain barrier transport of small molecular drugs in animal models of amyloid beta and alpha-synuclein pathology. Neuropharmacology. 2018;128:482-91
45. St-Amour I, Paré I, Alata W, Coulombe K, Ringuette-Goulet C, Drouin-Ouellet J. et al. Brain bioavailability of human intravenous immunoglobulin and its transport through the murine blood-brain barrier. J Cereb Blood Flow Metab. 2013;33:1983-92
46. Johnsen KB, Burkhart A, Melander F, Kempen PJ, Vejlebo JB, Siupka P. et al. Targeting transferrin receptors at the blood-brain barrier improves the uptake of immunoliposomes and subsequent cargo transport into the brain parenchyma. Sci Rep. 2017;7:10396
47. Crépin R, Goenaga AL, Jullienne B, Bougherara H, Legay C, Benihoud K. et al. Development of human single-chain antibodies to the transferrin receptor that effectively antagonize the growth of leukemias and lymphomas. Cancer Res. 2010;70:5497-506
48. Lesley J, Schulte R, Woods J. Modulation of transferrin receptor expression and function by anti-transferrin receptor antibodies and antibody fragments. Exp Cell Res. 1989;182:215-33
49. Ruderisch N, Schlatter D, Kuglstatter A, Guba W, Huber S, Cusulin C. et al. Potent and selective BACE-1 peptide inhibitors lower brain Aβ levels mediated by brain shuttle transport. EBioMedicine. 2017;24:76-92
50. Ahmed M, Pan DW, Davis ME. Lack of in vivo antibody dependent cellular cytotoxicity with antibody containing gold nanoparticles. Bioconjug Chem. 2015;26:812-6
51. Pardridge WM. Tyrosine hydroxylase replacement in experimental Parkinson's disease with transvascular gene therapy. NeuroRx. 2005;2:129-38
52. Shi N, Boado RJ, Pardridge WM. Antisense imaging of gene expression in the brain in vivo. Proc Natl Acad Sci USA. 2000;97:14709-14
53. Shi N, Pardridge WM. Noninvasive gene targeting to the brain. Proc Natl Acad Sci USA. 2000;97:7567-72
54. Zhang Y, Boado RJ, Pardridge WM. In vivo knockdown of gene expression in brain cancer with intravenous RNAi in adult rats. J Gene Med. 2003;5:1039-45
55. Zhang Y, Calon F, Zhu C, Boado RJ, Pardridge WM. Intravenous nonviral gene therapy causes normalization of striatal tyrosine hydroxylase and reversal of motor impairment in experimental parkinsonism. Hum Gene Ther. 2003;14:1-12
56. Zhang Y, Schlachetzki F, Pardridge WM. Global non-viral gene transfer to the primate brain following intravenous administration. Mol Ther. 2003;7:11-8
57. Prades R, Guerrero S, Araya E, Molina C, Salas E, Zurita E. et al. Delivery of gold nanoparticles to the brain by conjugation with a peptide that recognizes the transferrin receptor. Biomaterials. 2012;33:7194-205
58. Paris-Robidas S, Brouard D, Emond V, Parent M, Calon F. Internalization of targeted quantum dots by brain capillary endothelial cells in vivo. J Cereb Blood Flow Metab. 2016;36:731-42
59. Triguero D, Buciak J, Pardridge WM. Capillary depletion method for quantification of blood-brain barrier transport of circulating peptides and plasma proteins. J Neurochem. 1990;54:1882-8
60. Choi CH, Alabi CA, Webster P, Davis ME. Mechanism of active targeting in solid tumors with transferrin-containing gold nanoparticles. Proc Natl Acad Sci USA. 2010;107:1235-40
61. Danscher G. Localization of gold in biological tissue: a photochemical method for light and electronmicroscopy. Histochemistry. 1981;71:81-8
62. Pizzo ME, Wolak DJ, Kumar NN, Brunette E, Brunnquell CL, Hannocks MJ. et al. Intrathecal antibody distribution in the rat brain: surface diffusion, perivascular transport and osmotic enhancement of delivery. J Physiol. 2018;596:445-75
63. Thorne RG, Nicholson C. In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc Natl Acad Sci USA. 2006;103:5567-72
64. Nance EA, Woodworth GF, Sailor KA, Shih TY, Xu Q, Swaminathan G. et al. A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci Transl Med. 2012;4:149ra19
65. Reimold I, Domke D, Bender J, Seyfried CA, Radunz HE, Fricker G. Delivery of nanoparticles to the brain detected by fluorescence microscopy. Eur J Pharm Biopharm. 2008;70:627-32
66. Begley DJ. Brain superhighways. Sci Transl Med. 2012;4:147fs29
67. Jølck RI, Binderup T, Hansen AE, Scherman JB, Munch af Rosenschold P, Kjaer A. et al. Injectable colloidal gold in a sucrose acetate isobutyrate gelating matrix with potential use in radiation therapy. Adv Healthc Mater. 2014;3:1680-7
68. Jølck RI, Rydhög JS, Christensen AN, Hansen AE, Bruun LM, Schaarup-Jensen H. et al. Injectable colloidal gold for use in intrafractional 2D image-guided radiation therapy. Adv Healthc Mater. 2015;4:856-63
69. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T. et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676-82
70. Burkhart A, Thomsen LB, Thomsen MS, Lichota J, Fazakas C, Krizbai I. et al. Transfection of brain capillary endothelial cells in primary culture with defined blood-brain barrier properties. Fluids Barriers CNS. 2015;12:19
Author contact
![]() Corresponding authors: Thomas Lars Andresen, PhD, Center for Nanomedicine and Theranostics, DTU Nanotech, Technical University of Denmark, Ørsteds Plads, Building 345B, Room 050, 2800 Kongens Lyngby, Denmark. E-mail: tlandtu.dk; Phone number: 0045 2537 4486 and Torben Moos, MD, PhD, DMSc, Laboratory for Neurobiology, Biomedicine, Department of Health Science and Technology, Aalborg University, Fredrik Bajers Vej 3B, 1.216, 9220 Aalborg Ø, Denmark. E-mail: tmoosaau.dk; Phone number: 0045 9940 2420
Corresponding authors: Thomas Lars Andresen, PhD, Center for Nanomedicine and Theranostics, DTU Nanotech, Technical University of Denmark, Ørsteds Plads, Building 345B, Room 050, 2800 Kongens Lyngby, Denmark. E-mail: tlandtu.dk; Phone number: 0045 2537 4486 and Torben Moos, MD, PhD, DMSc, Laboratory for Neurobiology, Biomedicine, Department of Health Science and Technology, Aalborg University, Fredrik Bajers Vej 3B, 1.216, 9220 Aalborg Ø, Denmark. E-mail: tmoosaau.dk; Phone number: 0045 9940 2420