Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2018; 8(12):3380-3391. doi:10.7150/thno.24017 This issue Cite
Research Paper
Somatostatin receptor type 2 as a radiotheranostic PET reporter gene for oncologic interventions
Pedram Heidari*, Anchisa Kunawudhi*, Jordi Martinez-Quintanilla, Alicia Szretter, Khalid Shah, Umar Mahmood ![]()
Department of Radiology, Massachusetts General Hospital, Boston, MA
*Equal contribution
Received 2017-11-25; Accepted 2018-4-8; Published 2018-5-23
Abstract

Reporter gene systems can serve as therapy targets. However, the therapeutic use of reporters has been limited by the challenges of transgene delivery to a majority of cancer cells. This study specifically assesses the efficacy of targeting human somatostatin receptor subtype 2 (hSSTR2) with peptide receptor radionuclide therapy (PRRT) when a small subpopulation of cells bears the transgene.
Methods: The hSSTR2 transgene was delivered to A549 and Panc-1tumors using the lentiviral vector, LV-hSSTR2-IRES-GFP or murine mesenchymal stem cells (mMSC)s using a retroviral vector. SSTR2 expression was assessed using Western blot and correlated to GFP fluorescence and 68Ga-DOTATOC uptake. Wild type (WT), transduced (TD), and mixed population A549 or Panc-1 xenografts were implanted in nude mice. Separate groups with A549WT and Panc-1WT tumors received intratumoral injection of SSTR2-expressing mMSCs. Tumor-bearing mice were treated with 90Y-DOTATOC or saline and evaluated with 68Ga-DOTATOC PET before and after treatment.
Results: Cell studies showed a strong correlation between 68Ga-DOTATOC uptake and SSTR2 expression in A549 (p < 0.004) and Panc-1 cells (p < 0.01). 68Ga-DOTATOC PET SUVmean was 8- and 5-fold higher in TD compared to WT A549 and Panc-1 tumors, respectively (p < 0.001). After 90Y-DOTATOC treatment, 100% TD and mixed population TD xenografts showed growth cessation while the WT xenografts did not. A549WT and Panc-1WT tumors with SSTR2-expressing mMSCs treated with 90Y-DOTATOC showed significantly lower tumor volumes compared to controls (p < 0.05). 68Ga-DOTATOC PET SUVmean of treated TD tumors monotonically declined and was significantly lower than that of non-treated xenografts.
Conclusions: We showed that SSTR2 delivery to a small population of cells in tumor in conjunction with PRRT is effective in tumor growth cessation. The availability of various transgene delivery methods for hSSTR2 and radiotherpaeutic somatostatin analogs highlights the direct translational potential of this paradigm in the treatment of various cancers.
Keywords: somatostatin receptor, theranostic, peptide receptor radionuclide therapy (PRRT), 68Ga-DOTATOC, reporter gene
Introduction
Reporter systems have been employed over the past decades to confirm successful delivery and expression of genes that encode enzymes, transporters, or receptors in vivo and in vitro. These systems work through versatile mechanisms to bind, activate or entrap imaging beacons that act as surrogate markers of gene expression in the target tissue [1]. They can be specifically engineered to express one or multiple reporters, which are detectable using various imaging modalities such as optical imaging including fluorescence and luminescence, magnetic resonance imaging (MRI) and radionuclide imaging with single photon emission computed tomography (SPECT) or positron emission photography (PET) [2-6]. Among these, the radionuclide-based reporter systems are uniquely sensitive and reproducible, while providing a surrogate non-invasive quantitation method of biological molecules or processes. Additionally, the radionuclide-based reporter systems are not limited by the depth of tissue penetration, allowing for imaging deep body structures which facilitates clinical translation [1].
Somatostatin receptors (SSTR) is a superfamily of G-protein coupled receptors with mostly inhibitory downstream signaling that often alters the release of secreted hormones and proteins, decreases cell proliferation and induces apoptosis. SSTRs, particularly somatostatin receptor type 2 (SSTR2), are expressed at very low levels in most organs and tumors, except in differentiated neuroendocrine carcinoma (NECA) [7]. There has been prior successful implementations of hSSTR2 as reporter gene in multiple studies [8]. This paucity of SSTR2 expression in the human body makes this receptor a favorable candidate for a reporter system [8, 9]. Additionally, the overall inhibitory effect of SSTR2 on cell proliferation and endocrine release of active hormones increases the potential of the reporter system for human translation. Also, the availability of clinically used octreotide-based PET probes, makes it feasible to translate the use of human SSTR2 gene (hSSTR2) as a reporter system to quantitate gene delivery in vivo [8-13].
In addition to being a diagnostic reporter gene, hSSTR2, if delivered and expressed in adequate copy numbers, could be used as a therapeutic target in conjunction with somatostatin (SST) analogs labeled with therapeutic radionuclide(s) such as 90Yttrium or 177Lutetium. Local delivery of hSSTR2 to the tumors with minimal or no SSTR expression could be a potential therapeutic option given the vastly improved outcome of patients with SSTR2-positive NECA in multicenter trials including most recently the NETTER-1 trial [14]. Fortunately, there are gene transfer technologies such as viral vectors or mesenchymal stem cells that allow local delivery of hSSTR2 to the target tissue. Previous research demonstrated encouraging results in treating SSTR2-transduced xenografts using peptide receptor radionuclide therapy (PRRT), which resulted in a delay in xenograft growth compared to control groups [15-17]. Practically, however, even with the most robust gene delivery methods only a small fraction of tumor volume is composed of SSTR2-expressing cells.
This study specifically assesses the efficacy of peptide receptor radionuclide radiotherapy (PRRT) in SSTR-lacking tumors following hSSTR2 gene transfer to a small subpopulation of cells; we show a significant therapy response even when a minority of cells express the hSSTR2 transgene. This is especially important since the efficacy of the current local gene transfer technologies is limited and results in stable transgene expression in only a few percent of cells in the tumor 3D matrix; the cross-fire radiation from long-range ß-particles through the SSTR-lacking cells in the tumor enhances therapeutic effect in tumors with low hSSTR2 copy number. There are multiple possible avenues for clinical translation of this paradigm, which most importantly include local gene transfer by interventional procedures through different viral vectors or alternatively through transduced mesenchymal stem cells (MSC). In this study, we demonstrated delivery of hSSTR2 gene to SSTR2-negative tumors using MSCs and showed feasibility for treating the tumors with the transgene with 90Y-DOTATOC as a proof-of-principle for clinical translation, as overviewed in Figure 1.
Conceptual framework of the hSSTR2 theranostic reporter gene system. After packaging of the transgene in viral vectors the transgene may be delivered directly to tumor cells via viral vectors such as lentivirus or adenovirus, or through homing mesenchymal stem cells. Quantitative PET imaging using 68Ga-DOTATOC, or similar radiolabeled SST analogs enables indirect quantitation of the transgene expression in the target after transgene delivery. PRRT using SST analogs labeled with therapeutic radionuclides results in apoptosis in both the TD and WT cells in the tumor due to ß-radiation depositing energy in the TD cells, and adjacent cells up to millimeters distant in each direction in the 3D tumor matrix; this allow for effective tumor control even when a low percentage of cells in the tumor mass express the transgene.
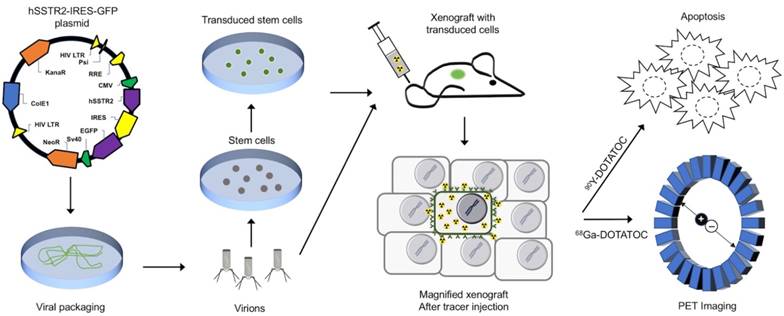
Methods
68Ga radiolabeling of DOTATOC
DOTATOC (Bachem) was labeled according to the procedures previously described in detail [18, 19]. Briefly, 68Ge/68Ga-generator (iThemba Labs) was eluted with 6 mL of 0.6 M HCl (Fluka TraceSELECT®, Sigma Aldrich). The eluate was added to reaction buffer consisting of sodium acetate and 40 µg of DOTATOC and heated at 100 °C for 20 min. The reaction solution was loaded on a reverse phase C18 Sep-Pak mini cartridge (Waters) and eluted with 200 µL of 200 proof ethanol (American Bioanalytical). The final formulation was adjusted to 10% ethanol in saline. The radiochemical purity of 68Ga-DOTATOC was measured through radio-TLC with two mobile phases, 0.1 M sodium citrate (Sigma Aldrich) and 1 M ammonium acetate/methanol (1:1 v/v) (Sigma Aldrich). Plates were analyzed using a radio-TLC plate reader (AR-2000, Bioscan). 68Ga-DOTATOC demonstrated > 95% radiochemical purity.
90Yttrium labelling of DOTATOC
DOTATOC was added to 150 mL of 0.4 M sodium acetate/gentisic acid buffer (1:1 v/v, pH 5.0), followed by the addition of 90Y-chloride (PerkinElmer). Each 1 µg of DOTATOC was mixed with approximately 1 mCi of 90Y and the mixture was heated for 40 min at 92 °C to allow radiometal incorporation to DOTA. Purification and quality control of the products was performed as described above for 68Ga-labeling. The labeling yield was approximately 98 ± 1% and radiochemical purity was > 95%.
Cell culture
All the cell lines were obtained from ATCC. A549, a human alveolar basal epithelial cell carcinoma, was cultured in phenol red free F-12K medium (ATCC), and Panc-1, a human pancreatic adenocarcinoma cell line, and 293T/17 cells were cultured in phenol red free Dulbecco's Modified Eagle's Medium (DMEM) media (ATCC). The media were supplemented by 10% FBS (Atlanta Biologicals) and 1% penicillin (100 U/mL) /streptomycin (100 μg/mL) solution (Gibco). Murine mesenchymal stem cells (mMSC) were grown in DMEM containing 10% FBS, 10% horse serum (Atlanta Biologicals) and 1% penicillin/streptomycin. Cultures were maintained in a humidified incubator at 37 °C and 5% CO2.
Plasmid construct and lentivirus/retrovirus generation
A lentiviral vector (LV), pLV-CSC-IG bearing an IRES-GFP (Internal ribosomal entry site-green fluorescent protein) element was used as a backbone and all restriction sites were engineered into the primers. Retroviral vector (RV), MGRi bearing an IRES-GFP was used as a backbone for RV. hSSTR2 was PCR amplified using pJP1520-hSSTR2 (DNASU Plasmid Repository, Arizona State University) as a template. The resulting hSSTR2 fragment was digested with NheI and XhoI (Promega). This fragment was ligated in-frame into the NheI/XhoI digested pLV-CSC-IG and MGRi vectors. The lentiviral construct was packaged as a LV in 293T/17 cells using a helper virus-free packaging method as previously described [20]. The retroviral construct was packaged as RV by transient transfection of 293T/17 cells. Briefly, cells (15×106) were seeded in 150 mm2 tissue culture plates 24 h and washed with fresh medium 4 h before transfection. Transfection was performed by the calcium phosphate precipitation method using 18 µg of transfer plasmid DNA, the transfer vectors constructed above, and the lentiviral helper plasmids pCMVΔ8.91 (18 µg) and glycoprotein expression plasmid pVSVG (12 µg; Clontech) or the retroviral helper plasmid pCL-Eco (25μg; Addgene). Cells were washed with fresh medium 16-18 h after transfection, and vector supernatants were harvested 48 h after transfection. The supernatants were filtered (0.45 µm) and loaded in a Beckman Quick-Seal ultracentrifuge tube (Beckman Coulter) and centrifuged at 28,000 × g for 90 min. Pellets were resuspended in PBS and stored at -80 °C. Titers were determined by counting fluorescent transduced 293T/17 cells.
Tumor cell transduction and clone selection
hSSTR2 negative cell lines A549 and Panc-1 were transduced with the LV described above, containing both hSSTR2 and GFP reporters (LV-hSSTR2-IRES-GFP), with multiplicity of infection (moi) of 2 in medium containing protamine sulfate (8 μg/mL) (Sigma Aldrich) and selected with puromycin (4 μg/mL) (Sigma Aldrich). mMSCs were transduced with the RV containing either GFP only (RV-GFP) or both hSSTR2 and GFP reporters (RV-GFP-hSSTR2) at moi of 10 by incubating virions in a culture medium containing 4 µg/mL protamine sulfate. Cells were visualized for GFP expression by fluorescence microscopy to confirm transduction yield of at least 50-60%. After 48 h, the cells were sorted by GFP expression with a fluorescence-activated cell sorting (FACSAria Cell-Sorting System, BD Biosciences). The clones were propagated in 96-well plates (Corning) to develop single cell clone populations. The cells were then expanded and select single cell clones with different GFP expression levels by FACS were chosen (i.e., high, medium and low).
Correlation between GFP and hSSTR2 and 68Ga-DOTATOC uptake in vitro
The A549 and Panc-1 cell lines with different expression levels of GFP (low to high) were seeded in 24-well plates at 1×105 cells per well in triplicate and allowed to grow for 48 h to ensure firm attachment to the plates. The GFP fluorescence signal intensity of each well was read using the blue fluorescence module of GloMax®-Multi Detection System (Promega) at 490 nm excitation. After reading the fluorescence signal intensity of wells, approximately 25 µCi of 68Ga-DOTATOC was added to each well and incubated at 37 ℃ for 60 min. The medium was then aspirated, and wells were carefully washed 3 times with Hank's balanced salt solution (HBSS; Fisher Scientific). The cells were harvested using 0.25% Trypsin-EDTA (Gibco). The cells were counted using automated cell counter (Countess™; Invitrogen). The activity of each collected sample was measured using 2480 Wizard™ gamma counter (Perkin Elmer) and decay corrected.
Measuring hSSTR2 expression
A549 and Panc-1 cell lines with different expression levels of GFP (low to high) were seeded in 6-well plates at 5×105 cells per well in triplicate and allowed to grow for 48 h. To quantitate the SSTR2 expression, Western blot assay was performed on the samples as previously described [21]. Briefly, whole protein extract purification was performed on the cell sediments. Protein samples (30 µL) were loaded onto SDS-polyacrylamide gels (Bio-Rad) and run at 120 V and 14 mA for 1.5 h. Gels were blotted on polyvinylidene difluoride (PVDF) membrane and the blots incubated overnight at 4 °C with anti-human SSTR2 monoclonal antibody (Abcam) at 1/500 dilution. ß-actin monoclonal antibody (Santa Cruz) at 1/1000 dilution was used as an internal control. Detection was performed using the BM Chemiluminescence Western Blotting Kit (Roche) and imaged on the in vivo Multispectral FX imaging system (Carestream). The SSTR2 expression corrected for ß-actin was correlated to the 68Ga-DOTATOC uptake per cell.
Binding specificity of 68Ga-DOTATOC to transduced cells
Specificity of the binding of 68Ga-DOTATOC to SSTR2 receptors on TD cells was assessed using a competitive binding assay with octreotide acetate (Abbiotec) acting as the displacement agent. Briefly, the WT and TD A549 and Panc-1 cells with high GFP/SSTR2 expression were seeded in 24-well plates at 1×105 cells and allowed to grow and attach to the plates for 48 h. The TD cells were treated for 60 min with 10 μM concentration of octreotide acetate or vehicle, i.e., 20 µL of phosphate buffered solution (PBS; Gibco), added to the medium. The WT cells were treated with vehicle for 60 min. Approximately 25 µCi of 68Ga-DOTATOC was added to each well and also incubated at 37 °C for 60 min. The wells were washed, and the cells were harvested, as above. The activity of each sample was measured using the 2480 Wizard™ gamma counter and was corrected for cell number.
In vivo tumor implantation
All the experiments and procedures involving the use of small animals were approved by our Institutional Animal Care and Use Committee (IACUC). Nu/nu mice (Cox-7 Laboratories) were randomly divided in 8 groups and were implanted with WT, TD, or a mix of 10% or 50% TD with WT A549 or Panc-1 cells using the methods described previously [22, 23]. Briefly, 5×105 cells were suspended in 100 µL of Matrigel (Corning) and 0.9 N sodium chloride (Hospira) (1:1 / v:v) and implanted subcutaneously to the right upper flank of mice (n = 5 per group).
hSSTR2 delivery to wild type xenografts using mMSCs
We evaluated hSSTR2 delivery to tumors via murine (m)MSCs as delivery vehicle. mMSCs were transduced with either GFP (mMSC-GFP) or hSSTR2-GFP (mMSC-GFP-hSSTR2) and the usefulness of these delivery approaches for receptor radiotherapy using 90Y-DOTATOC was evaluated. Nude mice were injected subcutaneously with either A549WT or Panc-1WT in the upper flanks (n = 5 per group). After one week, 1×106 mMSC-GFP-hSSTR2 or mMSC-GFP (no hSSTR2 gene) cells were injected directly into the WT tumors in each mouse. Tumor-bearing mice with mMSC-GFP-hSSTR2 were randomly selected to receive either 90Y-DOTATOC or saline (vehicle) three days after mMSC injection. The 3-day gap between the mMSC injection and 90Y-DOTATOC administeration was chosen based on our prior experience with mMSCs in multiple tumor models to maximize mMSC migration in the tumor while preserving optimal mMSC viability [20, 24]. All tumor-bearing mice with mMSC-GFP were treated with 90Y-DOTATOC three days after mMSC injection. Tumor volumes were monitored as described above.
90Y-DOTATOC treatment and outcome assessment
All tumor-bearing mice were treated at day 10 (after tumor implantation) with infusion of 0.5 mL isotonic fluid containing 1 mCi of 90Y-DOTATOC over 20 min via the lateral tail vein. The injection was performed slowly using a programmable syringe pump (Harvard Instruments) to avoid volume overload. Static 68Ga-DOTATOC PET was acquired 12 h before and 5, 14 and 18 days after treatment using the procedures previously described [18, 21]. Briefly, tumor size was monitored daily by measuring the largest perpendicular diameters using caliper measurements. Tumor volume (mm3) was estimated using the formula: Volume = 0.52 × (width)2 × length. All mice were euthanized by day 25 of treatment and tumors were extracted for histopathological analysis with hematoxylin and eosin (H&E) staining.
Statistical analysis
For the in vitro studies, unpaired t-test was used to show the difference between the groups. Linear regression was used to establish correlation between the SSTR2 expression and GFP fluorescence signal intensity or 68Ga-DOTATOC uptake. Repeated measure ANOVA was used to evaluate the difference in tumor 68Ga-DOTATOC uptake and tumor volumes among the groups at different time points after 90Y-DOTATOC treatment. A P-value less than 0.05 was considered a statistically significant difference.
Results
SSTR2 expression strongly correlates with 68Ga-DOTATOC uptake and GFP expression in the transduced cells
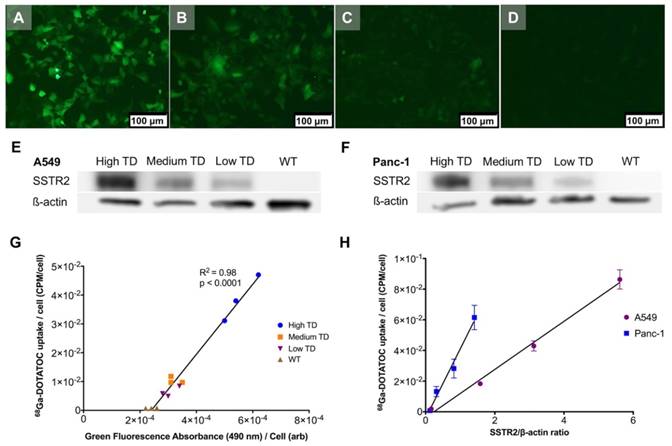
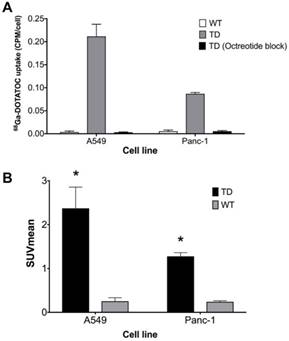
Single cell clones of A549 and Panc-1 cells transduced with the LV-hSSTR2-IRES-GFP vector were selected and expanded based on their GFP expression levels. SSTR2 expression in different clones was studied by GFP epifluorescence, Western blotting for SSTR2 and 68Ga-DOTATOC uptake. A very strong correlation between 68Ga-DOTATOC uptake and GFP fluorescence in A549TD clones (R2 = 0.98, p < 0.0001) was noted. There was also very strong correlation between the corrected SSTR2 protein expression and 68Ga-DOTATOC uptake by TD clones of both A549 (R2 = 0.99, p = 0.0033) and Panc-1 cells (R2 = 0.98, p = 0.0087) (Figure 2). The 68Ga-DOTATOC uptake was 58- and 16- fold higher in the TD clones compared to A549WT and Panc-1WT cells, respectively. Competitive blocking of SSTR2 with 10-fold molar excess octreotide acetate effectively blocked the 68Ga-DOTATOC uptake by the A549TD and Panc-1TD cells to a level similar to A549WT and Panc-1WT cells, demonstrating the specific receptor mediated uptake of 68Ga-DOTATOC after transduction in cells (Figure 3A).
Correlation between transgene expression and radiotracer uptake. (A-D) Representative fluorescence microscopy images of different A549TD cell clones visually expressing high (A), medium (B) and low (C) GFP levels. A549WT cells (D) demonstrate virtually no fluorescence. (E-F) Western blots of SSTR2 and ß-actin in WT and different clones of TD A549 and Panc-1 cells. The transduction levels were designated based on GFP expression level from fluorescence microscopy. (G) Strong correlation between 68Ga-DOTATOC uptake by different clones of A549 and level of GFP expression based on the intensity of the fluorescence signal (R2 = 0.98, p < 0.0001). (H) Strong correlation between 68Ga-DOTATOC uptake by different clones of A549 and Panc-1 and level of corrected SSTR2 expression (R2 = 0.99, p = 0.0033 and R2 = 0.98, p = 0.0087, respectively).
hSSTR2 transduction effect on 68Ga-DOTATOC uptake. (A) A significant increase in 68Ga-DOTATOC uptake in the select TD clones of A549 and Panc-1 cells compared to WT cells was observed. The cellular uptake of 68Ga-DOTATOC was specific and was completely blocked by competitive inhibition from the SST analog, octreotide. (B) In vivo, 68Ga-DOTATOC PET SUVmean was significantly higher in the A549TD and Panc-1TD compared to A549WT and Panc-1WT, respectively.
68Ga-DOTATOC uptake in WT and TD xenografts
In vivo imaging with 68Ga-DOTATOC PET showed significantly higher SUVmean in A549TD compared to A549WT xenografts (2.37 ± 0.48 vs. 0.25 ± 0.08, respectively; p < 0.0001), as well as in Panc-1TD compared to Panc-1WT xenografts (1.28 ± 0.08 vs. 0.24 ± 0.02, respectively; p = 0.001). The SUVmean of A549TD and Panc-1TD were 8 and 5 times higher compared to those of A549WT and Panc-1WT, respectively (Figure 3B).
90Y-DOTATOC treatment and associated changes in xenograft growth curves
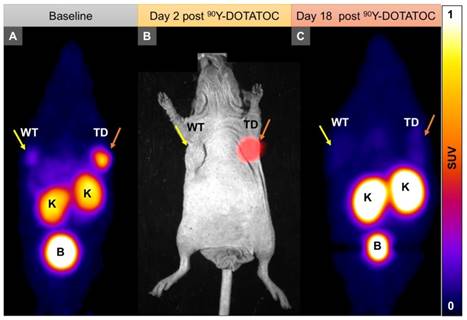
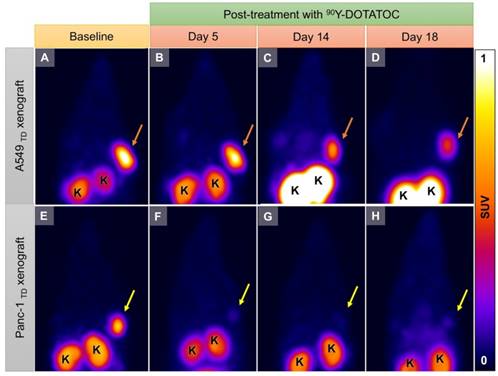
We implanted A549 and Panc-1 xenografts in 4 groups for each cell line using 100% TD, 100% WT as control, or 2 mixed population groups with 50% or 10% TD cells mixed with WT cells. At baseline (12 h before 90Y-DOTATOC injection), mice were imaged using 68Ga-DOTATOC PET to assess SSTR2 expression in the xenografts before 90Y-DOTATOC treatment. Uptake of 90Y-DOTATOC by xenografts was confirmed using radioisotopic fluorescence phosphor imaging. We observed high uptake in the TD tumors including 100%, 50% and 10% TD tumor cell populations, while there was minimal uptake above background in the WT tumor (Figure 4).
Representative imaging of a mouse with Panc-1TD at the left upper flank (orange arrows) and Panc-1WT tumor at the right upper flank (yellow arrows). (A) Baseline 68Ga-DOTATOC PET imaging shows significantly higher tracer uptake in the Panc-1TD compared to Panc-1WT tumor. (B) Multispectral optical imaging with radioisotopic phosphor screen 2 days after injection of 90Y-DOTATOC shows significant uptake in the Panc-1TD tumor while the uptake in Panc-1WT was at the background level. (C) Eighteen days after 90Y-DOTATOC treatment, Panc-1TD tumor completely disappeared from the left upper flank. The tumor bed did not demonstrate 68Ga-DOTATOC uptake above background level. Panc-1WT tumor continued to grow but, as expected, did not demonstrate any uptake above background. K: kidney; B: bladder.
The average volume of TD xenografts decreased significantly after 18 days post treatment in Panc-1TD (p < 0.05) and remained statistically unchanged after 90Y-DOTATOC treatment during the follow-up period in A549TD tumors. The average volume of WT xenografts of both cell lines continued to increase to the date the mice were euthanized at the end of follow up. The 10% and 50% mixed population TD xenografts showed growth curves similar to the 100% A549TD and Panc-1TD tumors (Figure 5A-B).
Tumor growth curves A549 and Panc-1 xenografts before and after PRRT with 90Y-DOTATOC. (A) The average volume of A549TD xenografts did not statistically change after 90Y-DOTATOC treatment during the follow-up while A549WT xenografts continued to grow. 10% and 50% mixed population A549TD xenografts showed growth curves very similar to that of the 100% A549TD tumors. (B) The average volume of Panc-1TD xenografts decreased significantly after day 18 post treatment (p < 0.05), while the average volume of Panc-1WT xenografts continued to increase. 10% and 50% mixed population TD xenografts showed growth curves very similar to that of the 100% Panc-1TD tumors. (C) A549 tumors containing SSTR2-expressing mMSC-GFP-hSSTR2 treated with 90Y-DOTATOC showed significantly lower tumor volumes compared to the control tumors containing mMSC-GFP treated with 90Y-DOTATOC and tumors with mMSC-GFP-hSSTR2 treated with saline starting at day 5 after treatment (p = 0.03). (D) Panc-1 tumors containing mMSC-GFP-hSSTR2 also showed significantly lower tumor volumes compared to control tumors containing mMSC-GFP treated with 90Y-DOTATOC and mMSC-GFP-hSSTR2 treated with saline starting at day 7 after treatment (p < 0.05).
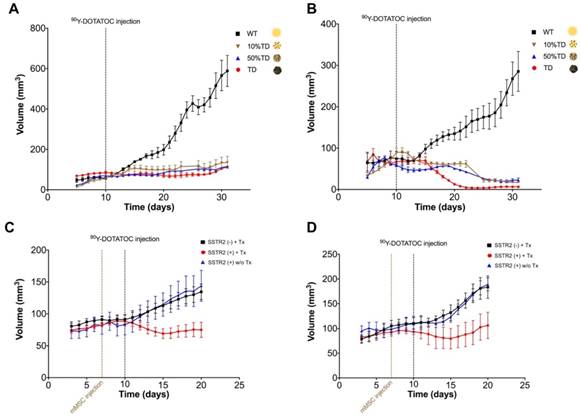
Delivery of 90Y-DOTATOC to tumors using SSTR2-expressing mMSC and alterations in tumor growth
A549WT and Panc-1WT xenografts were injected with selected clones of mMSCs transduced with viral vector containing hSSTR2 (hSSTR2-GFP-mMSC) or lacking hSSTR2 gene (GFP-mMSC). Three days after intratumoral delivery of mMSCs, to allow mMSC engraftment, the mice were treated with 90Y-DOTATOC. A549 tumors containing hSSTR2-GFP-mMSCs showed significantly lower tumor volumes compared to control tumors with GFP-mMSCs starting at day 5 after treatment with 90Y-DOTATOC (69.1 ± 7.7 vs. 103.4 ± 8.3 mm3, respectively; p = 0.03). There was no statistically significant difference between the tumor volumes of the A549 tumors with hSSTR2-GFP-mMSC treated with saline and tumors with GFP-mMSC treated with saline (Figure 5C). Panc-1 tumors containing hSSTR2-GFP-mMSCs showed significantly lower tumor volumes compared to control tumors containing GFP-mMSCs starting at day 7 after 90Y-DOTATOC treatment (88.0 ± 24.0 vs. 156.3 ± 15.4 mm3, respectively; p < 0.05). There was no statistically significant difference between the tumor volumes of the Panc-1 tumors with hSSTR2-GFP-mMSC treated with saline and tumors with GFP-mMSC treated with saline (Figure 5D).
Temporal 68Ga-DOTATOC uptake curves in A549 and Panc-1 xenografts
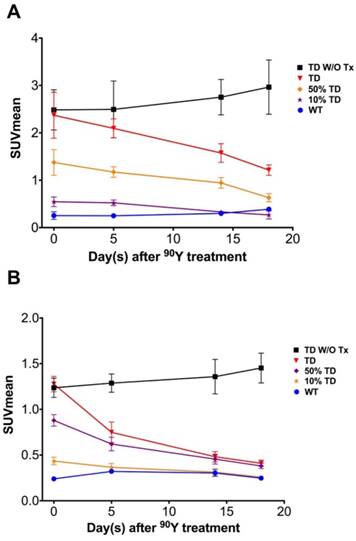
The average 68Ga-DOTATOC uptake in treated WT, TD and mixed-population xenografts as well as non-treated TD xenografts was plotted for both cell lines over 18 days treatment (Figure 6 and Figure 7). We observed a slight increase in the SUVmean of the non-treated TD xenografts although this difference was not statistically significant compared to the baseline. The SUVmean of the treated A549TD tumors, although lower than that of the non-treated A549TD at day 5 after 90Y-DOTATOC treatment, was not significantly different from the baseline (2.1 ± 0.2 vs. 2.5 ± 0.6, respectively; p = 0.54); however, the SUVmean of the treated A549TD tumors was significantly lower than that of the non-treated A549TD xenografts at day 14 (1.6 ± 0.2 vs. 2.8 ± 0.4, respectively; p = 0.023) and 18 (1.2 ± 0.1 vs. 3.0 ± 0.6, respectively; p = 0.017) imaging time points. The SUVmean of the treated A549TD remained consistently higher than that of the A549WT xenografts at all imaging time points. The SUVmean of the 50% TD and 10% TD tumors were between that of 100% A549TD and A549WT at baseline (1.4 ± 0.3 and 0.5 ± 0.1, respectively) and significantly decreased by day 18 (0.6 ± 0.1 and 0.3 ± 0.1, respectively) compared to the baseline (p < 0.03 and p = 0.047, respectively). The SUVmean of the treated Panc-1TD tumors was significantly lower than that of the non-treated group by day 5 after 90Y-DOTATOC treatment (0.7 ± 0.1 vs. 1.3 ± 0.1, respectively; p = 0.007) and further decreased over time to comparable levels as those of the Panc-1WT xenografts by day 18. The SUVmeans of the 50% TD and 10% TD tumors were between those of 100% Panc-1TD and Panc-1WT at baseline (0.9 ± 0.1 and 0.4 ± 0.0, respectively) and significantly decreased by day 18 (0.4 ± 0.0 and 0.3 ± 0.0, respectively) compared to baseline (p < 0.0001 and p = 0.0042, respectively) (Figure 6 and Figure 7).
Representative 68Ga-DOTATOC PET images of A549TD and Panc-1TD tumors at baseline (12 h before 90Y-DOTATOC injection) and days 5, 14 and 18 after 90Y-DOTATOC administration. (A-D) A549TD xenograft showed very high 68Ga-DOTATOC uptake at baseline (A). The 68Ga-DOTATOC uptake gradually decreased over the course of 18 days follow-up but remained above the background level (B-D). (E-H) Panc-1TD xenograft showing very high 68Ga-DOTATOC uptake at baseline (12 h before 90Y-DOTATOC injection) (E). The uptake in the tumor decreased to the background level by day 5 and remained at background level until the last imaging follow-up (F-H). The 68Ga-DOTAOTC uptake difference between Panc-1 and A549 may be related to differences in radiosensitivity of the cells; A549 is known to be a radioresistant cell line. K: kidney.
Average 68Ga-DOTATOC SUVmean of A549 and Panc-1 tumors in different groups. (A) The SUVmean of the A549TD tumors which did not receive PRRT remained high and statistically unchanged compared to baseline after 18 days. The SUVmean of the treated A549TD decreased at every time point until day 18 post treatment and became significantly lower than that of non-treated A549TD xenografts at day 14 (1.6 ± 0.2 vs. 2.8 ± 0.4, respectively; p = 0.023) and 18 (1.2 ± 0.1 vs. 3.0 ± 0.6, respectively; p = 0.017) imaging time points, but remained consistently higher than that of A549WT xenografts at all imaging time points. As expected, the SUVmean of the 50% TD and 10% TD tumors were between those of 100% A549TD and A549WT at baseline and significantly decreased by day 18 compared to baseline (p < 0.03 and p = 0.047, respectively). (B) The SUVmean of the non-treated Panc-1TD tumors remained high and statistically unchanged compared to baseline after 18 days. The SUVmean of the treated Panc-1TD tumors was significantly lower than that of the non-treated group by day 5 after 90Y-DOTATOC treatment (0.7 ± 0.1 vs. 1.3 ± 0.1, respectively; p = 0.007) and further decreased over time to comparable levels compared to Panc-1WT xenografts by day 18. The SUVmean of the 50% TD and 10% TD tumors were between those of 100% Panc-1TD and Panc-1WT at baseline and significantly decreased by day 18 compared to baseline (p < 0.0001 and p = 0.0042, respectively).
Xenograft tissue samples
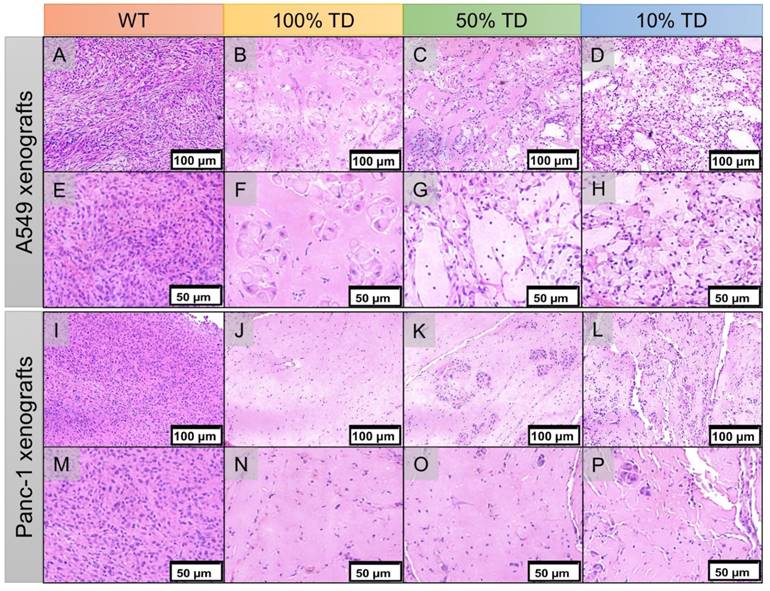
H&E staining of the extracted xenografts 25 days after treatment showed extensive areas of necrosis, cellular ballooning and fragmentation with a small number of remaining tumor cells in the treated 100% TD and mixed population 50% and 10% TD tumors of both A549 and Panc-1 cell lines. As expected, A549WT and Panc-1WT xenografts showed a dense population of cancer cells with high nuclear to cytoplasm ratios (Figure 8).
Hematoxylin and eosin (H&E) staining of A549 (A-H) and Panc1 (I-P) of representative tumor specimens 22 days after PRRT at 100x (A-D, I-L) and 200x (E-H, M-P) magnification. A549WT (A, E) and Panc-1WT (E, M) tumors sections demonstrate high tumor cellularity and high nuclear to cytoplasmic ratio. A549TD (B, F) and Panc-1TD (J, N) tumors showing extensive areas of tumor necrosis with scant cellularity. 50% (C, G) and 10% (D, H) mixed population A549 TD tumors demonstrated much reduced tumor cellularity compared to WT tumors, and large areas of necrosis; there was extensive cellular ballooning and pyknotic cell nuclei. (F-G) 50% (K, O) and 10% (L, P) mixed population Panc-1 TD tumors demonstrate scant cellularity, extensive areas of necrosis and small tumor cell islands with cellular ballooning and pyknotic cell nuclei at the periphery.
Discussion
Transgene delivery using different technologies such as plasmids, viral vectors and MSCs has been safely and effectively tried for treatment of different cancers. The common concern in all gene transfer treatments is to ensure that the therapeutic transgenes reach the target tissue [12, 25-30]. Reporter genes have emerged as a strategy for indirectly monitoring gene- or cell-based therapies including the magnitude, site and timing of transgene delivery and expression [1]. Among the different reporter systems PET-based reporters are particularly interesting due to their quantitative nature and potential for human translation. These reporter genes can be mainly categorized to enzyme-, receptor- or transporter-based systems [1, 6]. Enzyme-based reporters are the most commonly employed and rely on metabolic entrapment of a radiotracer using enzyme(s) not found or abundantly expressed in human cellular machinery. A well know example is herpes simplex virus 1 thymidine kinase (HSV-TK), which has been extensively used as a reporter system in conjunction with radiotracers such as 18F-FHGB [31, 32]. Transporters such as the sodium iodine symporter (NIS) are another category of established but less commonly used reporters [3, 33]. Receptor-based PET reporters employ membrane or intracellular receptors expressed in low copy numbers in physiologic conditions such as SSTR2 [9, 10, 15, 34], dopamine receptor type 2 (D2R) [35] or estrogen receptor (ER) [36]. Of the described PET reporters, hSSTR2, a receptor-based reporter gene, presents several advantages that make it attractive for translation [37]; hSSTR2 is a relatively short sequence, allowing for packaging with other desired therapeutic transgenes even in vectors with limited cargo capacity, such as adeno-associated virus (AAV). hSSTR2 is of human origin and will not elicit an immune response, unlike non-human transgenes such as HSV-TK. Additional advantages of hSSTR2 include direct energy-independent binding of the SST analogs to the receptor, radiolabeled SST analogs for imaging that are approved for human use, and favorable pharmacokinetics of the SST analogs such as rapid urinary clearance and low background tissue retention, which facilitate clinical translation [37].
In this study, we delivered the theranostic gene hSSTR2 into SSTR-negative tumors to achieve different transgene expression levels. Using 68Ga-DOTATOC PET, we confirmed transgene delivery and indirectly quantified hSSTR2 expression in the xenografts. We observed a very strong correlation between the SSTR2 expression and 68Ga-DOTATOC uptake in vitro and in vivo. Both A549TD and Panc-1TD xenografts showed several-fold higher tracer uptake compared to WT tumors. As expected, the 68Ga-DOTATOC uptake in the mixed population A549 and Panc-1 tumors was between the uptake in TD and WT tumors and proportional to the number of TD cells in the tumor. The uptake in the TD cells was specific and paralleled the level of transgene expression in the cancer cells. Our findings are in line with prior reports that demonstrated that hSSTR2-based reporters have the ability to estimate transgene transfer to tumors reported both in vitro and in vivo. For instance, Zhang et al. used an RV to transduce hSSTR2 in several cell lines and demonstrated that hSSTR2 is an effective reporter gene system. They showed that SSTR2 expression can be quantitatively imaged in vivo using 68Ga-DOTATOC PET to provide an accurate surrogate of transgene expression. The authors concluded that hSSTR2 reporter in combination with 68Ga-DOTATOC is promising for translation to clinical studies [8]. Other studies have reported similar results using a variety of different gene transfer technologies for hSSTR2 transduction in the target tissue in conjunction with 99mTc-, 111In- or 188Re-labeled SST-analogs [29, 37-39]. All these studies indicate that hSSTR2 reporter-based imaging is a promising method to track gene delivery and expression of other therapeutic transgenes. hSSTR2 enables observation of the magnitude, duration, and time variation of gene expression, study of the biodistribution of gene transfer vectors, optimization of the administration dose of vector encoding hSSTR2 reporter gene and/or other therapeutic genes, prediction of treatment response of the transduced tumor, and monitoring of antitumor effects of various treatments including gene-based therapies [37].
hSSTR2 can serve as a therapeutic gene alone or in combination with other genes [37]. It has been demonstrated that high SSTR2-expressing tumors show significant response to PRRT with radiotherapeutic SST analogs [14, 40, 41]. In fact, large multicenter trials, such as NETTER-1, have demonstrated marked improvement in the progression-free and overall survival of patients with tumors that highly express SSTR2 who received PRRT [14]. However, these therapeutic benefits do not extend to tumors with no or minimal SSTR expression. Fortunately, it is feasible to deliver hSSTR2 transgene to SSTR-lacking tumors using various gene transfer technologies [15, 17]. Nevertheless, the yield of current gene delivery technologies is generally low and allows for expression of transgene only in a small percentage of the tumor cells. Treatment with ß-emitting radioligands that bind the expressed transgene could potentially extend the therapeutic range of the transgene delivery to the neighboring WT and TD cells. The cross-fire radiation from ß-particles can span through approximately 100-200 cells in each direction and deposit energy in nearby cells that do not necessarily express SSTR2. In this study, we demonstrated delivery of hSSTR2 to neoplasms lacking SSTR expression to enable treatment using PRRT. We showed successful delivery of the hSSTR2 transgene to the cancer cells using 68Ga-DOTATOC PET, and subsequently used 90Y-DOTATOC to treat the xenografts with varying percentages of cells expressing SSTR2 transgene. We specifically explored the therapeutic effects of PRRT in tumors with low SSTR2 copy number and demonstrated significant therapy response. In fact, 90Y-DOTATOC treatment significantly delayed tumor growth even when fewer than 10% of cells in the tumor expressed SSTR2. There was only minimal difference in the tumor growth rate of 100% TD and 50% or 10% mixed population TD xenografts after treatment with 90Y-DOTATOC, while the WT tumors monotonously grew over the course of follow-up. We found significant therapy response with 90Y-DOTATOC therapy in xenografts injected with mMSCs carrying hSSTR2 transgene. The MSCs constitute a potentially interesting and practical transgene delivery method for hSSTR2 since, after local delivery, they preferentially migrate and relocate towards local and disseminated malignant disease and their non-immunogenic nature present them as attractive candidates for cell-based therapies in humans [20]. Our results are concordant with the findings of Zhao et al., who demonstrated the antitumor effect of a 188Re-labeled SST analog (188Re-RC-160) on A549 tumors transfected with plasmid pcDNA3 encoding hSSTR2 reporter gene [42]. The authors reported low-level SSTR2 transduction as the drawback of using a plasmid for transgene delivery but concluded that PRRT, even after hSSTR2 gene transfer with low efficiency, is still effective in reducing tumor growth. In another study, an adenoviral vector with hSSTR2 gene was also utilized in conjunction with 90Y-SMT 487 to treat non-small-cell lung tumors [16]. The median tumor quadrupling time increased to 40-44 days in treated TD tumors compared to untreated TD tumors and treated WT group with median tumor quadrupling times of 16 and 25 days, respectively. Others reported similar findings both in vitro and in vivo [15, 17].
While we demonstrated tumor growth delay in tumors with low copy number hSSTR2 gene after PRRT, this study has several limitations. We used LV in cell lines and mMSCs to deliver the transgene to SSTR-negative tumors. Both delivery mechanisms often result in consistent transgene expression and associated robust radiotracer uptake by the tumor. We did not test our strategy using other transgene delivery vehicles. However, as discussed above, several prior studies have shown successful transduction of SSTR2 using other transgene delivery mechanisms such as plasmids [42], adenovirus [4, 43], adeno-associated virus [25] or even vaccinia virus [28], although some of these have resulted in low or temporary SSTR2 transduction. Another limitation is that only a single dose treatment of 90Y-DOTATOC was used, and therefore conclusions cannot be drawn regarding the effect of the radiation dose fractionation and effect of consecutive 90Y-DOTATOC doses. Although we did not directly address this issue, we observed continuous but decreased SSTR2 expression in the A549TD tumors after PRRT at least until 18 days after therapy, likely allowing dose fractionation and treatment with multiple cycles of PRRT. Lastly, inadequate tissue at the end of follow up period did not allow for quantitative assessment of ex vivo SSTR2 expression by Western blot analysis after treatment, mainly due to the very small tumor size and extensive necrosis. However, we demonstrated reduction of SUV by 68Ga-DOTATOC PET in TD and mixed population tumors after PRRT, which could be indicative of reduction in the number of SSTR2-expressing cells in the tumors.
Conclusions
We have demonstrated that hSSTR2 can serve as an effective target for radiotheranostics when delivered to even a small subpopulation of cells in the tumor matrix. Given the favorable PRRT response in the mixed population tumors and tumors with SSTR2-expressing mMSCs, the therapeutic effect of PRRT might extend beyond the TD cells in the 3D tumor matrix, which is likely explained by cross-fire ß-radiation emitted from the SSTR2-positive cells. The current availability of various transgene delivery methods for hSSTR2 to tumors and clinically used radiolabeled SST analogs highlights the direct translational potential of this paradigm in locoregional control of various human cancers. MSCs may be a particularly attractive gene delivery vehicle for hSSTR2 given the lack of immunogenicity and their ability to migrate and disperse within the tumor matrix.
Abbreviations
111In: 111Indium; 177Lu: 177Lutetium; 188Re: 188Rhenium; 68Ga: 68Gallium; 68Ge: 68Germenium; 90Y: 90Yttrium; 99mTc: 99mTechnesium, D2R: dopamine receptor type 2; GFP: green fluorescent protein; H&E: hematoxylin and eosin; hSSTR2: human somatostatin receptor type 2; HSV-TK: herpes simplex virus 1 thymidine kinase; LV: lentiviral vector; MRI: magnetic resonance imaging; mMSC: murine mesenchymal stem cell; MSC: mesenchymal stem cell; NECA: neuroendocrine carcinoma; NIS: sodium iodine symporter; PCR: polymerase chain reaction; PET: positron emission tomography; PRRT: peptide receptor radionuclide therapy; RV: retroviral vector; SPECT: single photon emission computed tomography; SST: somatostatin; SUV: standardized uptake value; TD: transduced; WT: wild type.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Niu G, Chen X. Molecular imaging with activatable reporter systems. Theranostics. 2012;2:413-23
2. Genove G, DeMarco U, Xu H, Goins WF, Ahrens ET. A new transgene reporter for in vivo magnetic resonance imaging. Nat Med. 2005;11:450-4
3. Ahn BC. Sodium iodide symporter for nuclear molecular imaging and gene therapy: from bedside to bench and back. Theranostics. 2012;2:392-402
4. Ravoori MK, Han L, Singh SP, Dixon K, Duggal J, Liu P. et al. Noninvasive assessment of gene transfer and expression by in vivo functional and morphologic imaging in a rabbit tumor model. PLoS One. 2013;8:e62371
5. van Roessel P, Brand AH. Imaging into the future: visualizing gene expression and protein interactions with fluorescent proteins. Nat Cell Biol. 2002;4:E15-20
6. Hong H, Yang Y, Zhang Y, Cai W. Non-invasive cell tracking in cancer and cancer therapy. Curr Top Med Chem. 2010;10:1237-48
7. Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol. 1999;20:157-98
8. Zhang H, Moroz MA, Serganova I, Ku T, Huang R, Vider J. et al. Imaging expression of the human somatostatin receptor subtype-2 reporter gene with 68Ga-DOTATOC. J Nucl Med. 2011;52:123-31
9. Kundra V, Mannting F, Jones AG, Kassis AI. Noninvasive monitoring of somatostatin receptor type 2 chimeric gene transfer. J Nucl Med. 2002;43:406-12
10. Rogers BE, McLean SF, Kirkman RL, Della Manna D, Bright SJ, Olsen CC. et al. In vivo localization of [(111)In]-DTPA-D-Phe1-octreotide to human ovarian tumor xenografts induced to express the somatostatin receptor subtype 2 using an adenoviral vector. Clin Cancer Res. 1999;5:383-93
11. Yang D, Han L, Kundra V. Exogenous gene expression in tumors: noninvasive quantification with functional and anatomic imaging in a mouse model. Radiology. 2005;235:950-8
12. Singh SP, Yang D, Ravoori M, Han L, Kundra V. In vivo functional and anatomic imaging for assessment of in vivo gene transfer. Radiology. 2009;252:763-71
13. Han L, Ravoori M, Wu G, Sakai R, Yan S, Singh S. et al. Somatostatin receptor type 2-based reporter expression after plasmid-based in vivo gene delivery to non-small cell lung cancer. Mol Imaging. 2013;12:1-10
14. Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B. et al. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N Engl J Med. 2017;376:125-35
15. Verwijnen SM, Sillevis Smith PA, Hoeben RC, Rabelink MJ, Wiebe L, Curiel DT. et al. Molecular imaging and treatment of malignant gliomas following adenoviral transfer of the herpes simplex virus-thymidine kinase gene and the somatostatin receptor subtype 2 gene. Cancer Biother Radiopharm. 2004;19:111-20
16. Rogers BE, Zinn KR, Lin CY, Chaudhuri TR, Buchsbaum DJ. Targeted radiotherapy with [(90)Y]-SMT 487 in mice bearing human nonsmall cell lung tumor xenografts induced to express human somatostatin receptor subtype 2 with an adenoviral vector. Cancer. 2002;94:1298-305
17. Akinlolu O, Ottolino-Perry K, McCart JA, Reilly RM. Antiproliferative effects of 111In- or 177Lu-DOTATOC on cells exposed to low multiplicity-of-infection double-deleted vaccinia virus encoding somatostatin subtype-2 receptor. Cancer Biother Radiopharm. 2010;25:325-33
18. Heidari P, Szretter A, Rushford LE, Stevens M, Collier L, Sore J. et al. Design, construction and testing of a low-cost automated (68)Gallium-labeling synthesis unit for clinical use. Am J Nucl Med Mol Imaging. 2016;6:176-84
19. Leece AK, Heidari P, Yokell DL, Mahmood U. A container closure system that allows for greater recovery of radiolabeled peptide compared to the standard borosilicate glass system. Appl Radiat Isot. 2013;80:99-102
20. Martinez-Quintanilla J, Bhere D, Heidari P, He D, Mahmood U, Shah K. Therapeutic efficacy and fate of bimodal engineered stem cells in malignant brain tumors. Stem Cells. 2013;31:1706-14
21. Heidari P, Wehrenberg-Klee E, Habibollahi P, Yokell D, Kulke M, Mahmood U. Free somatostatin receptor fraction predicts the antiproliferative effect of octreotide in a neuroendocrine tumor model: implications for dose optimization. Cancer Res. 2013;73:6865-73
22. Heidari P, Deng F, Esfahani SA, Leece AK, Shoup TM, Vasdev N. et al. Pharmacodynamic imaging guides dosing of a selective estrogen receptor degrader. Clin Cancer Res. 2015;21:1340-7
23. Heidari P, Esfahani SA, Turker NS, Wong G, Wang TC, Rustgi AK. et al. Imaging of Secreted Extracellular Periostin, an Important Marker of Invasion in the Tumor Microenvironment in Esophageal Cancer. J Nucl Med. 2015;56:1246-51
24. Stuckey DW, Shah K. Stem cell-based therapies for cancer treatment: separating hope from hype. Nat Rev Cancer. 2014;14:683-91
25. Zinn E, Vandenberghe LH. Adeno-associated virus: fit to serve. Curr Opin Virol. 2014;8:90-7
26. Wang N, Zhang H, Zhang BQ, Liu W, Zhang Z, Qiao M. et al. Adenovirus-mediated efficient gene transfer into cultured three-dimensional organoids. PLoS One. 2014;9:e93608
27. ter Horst M, Verwijnen SM, Brouwer E, Hoeben RC, de Jong M, de Leeuw BH. et al. Locoregional delivery of adenoviral vectors. J Nucl Med. 2006;47:1483-9
28. McCart JA, Mehta N, Scollard D, Reilly RM, Carrasquillo JA, Tang N. et al. Oncolytic vaccinia virus expressing the human somatostatin receptor SSTR2: molecular imaging after systemic delivery using 111In-pentetreotide. Mol Ther. 2004;10:553-61
29. Kim KH, Dmitriev I, O'Malley JP, Wang M, Saddekni S, You Z. et al. A phase I clinical trial of Ad5.SSTR/TK.RGD, a novel infectivity-enhanced bicistronic adenovirus, in patients with recurrent gynecologic cancer. Clin Cancer Res. 2012;18:3440-51
30. Matthews K, Noker PE, Tian B, Grimes SD, Fulton R, Schweikart K. et al. Identifying the safety profile of Ad5.SSTR/TK.RGD, a novel infectivity-enhanced bicistronic adenovirus, in anticipation of a phase I clinical trial in patients with recurrent ovarian cancer. Clin Cancer Res. 2009;15:4131-7
31. Yaghoubi SS, Barrio JR, Namavari M, Satyamurthy N, Phelps ME, Herschman HR. et al. Imaging progress of herpes simplex virus type 1 thymidine kinase suicide gene therapy in living subjects with positron emission tomography. Cancer Gene Ther. 2005;12:329-39
32. Liang Q, Nguyen K, Satyamurthy N, Barrio JR, Phelps ME, Gambhir SS. et al. Monitoring adenoviral DNA delivery, using a mutant herpes simplex virus type 1 thymidine kinase gene as a PET reporter gene. Gene Ther. 2002;9:1659-66
33. Niu G, Krager KJ, Graham MM, Hichwa RD, Domann FE. Noninvasive radiological imaging of pulmonary gene transfer and expression using the human sodium iodide symporter. Eur J Nucl Med Mol Imaging. 2005;32:534-40
34. Zinn KR, Chaudhuri TR, Krasnykh VN, Buchsbaum DJ, Belousova N, Grizzle WE. et al. Gamma camera dual imaging with a somatostatin receptor and thymidine kinase after gene transfer with a bicistronic adenovirus in mice. Radiology. 2002;223:417-25
35. MacLaren DC, Gambhir SS, Satyamurthy N, Barrio JR, Sharfstein S, Toyokuni T. et al. Repetitive, non-invasive imaging of the dopamine D2 receptor as a reporter gene in living animals. Gene Ther. 1999;6:785-91
36. Lohith TG, Furukawa T, Mori T, Kobayashi M, Fujibayashi Y. Basic evaluation of FES-hERL PET tracer-reporter gene system for in vivo monitoring of adenoviral-mediated gene therapy. Mol Imaging Biol. 2008;10:245-52
37. Xu C, Zhang H. Somatostatin receptor based imaging and radionuclide therapy. Biomed Res Int. 2015;2015:917968
38. Singh SP, Han L, Murali R, Solis L, Roth J, Ji L. et al. SSTR2-based reporters for assessing gene transfer into non-small cell lung cancer: evaluation using an intrathoracic mouse model. Hum Gene Ther. 2011;22:55-64
39. Zinn KR, Buchsbaum DJ, Chaudhuri TR, Mountz JM, Grizzle WE, Rogers BE. Noninvasive monitoring of gene transfer using a reporter receptor imaged with a high-affinity peptide radiolabeled with 99mTc or 188Re. J Nucl Med. 2000;41:887-95
40. Baum RP, Kulkarni HR. THERANOSTICS: From Molecular Imaging Using Ga-68 Labeled Tracers and PET/CT to Personalized Radionuclide Therapy - The Bad Berka Experience. Theranostics. 2012;2:437-47
41. Vinjamuri S, Gilbert TM, Banks M, McKane G, Maltby P, Poston G. et al. Peptide receptor radionuclide therapy with (90)Y-DOTATATE/(90)Y-DOTATOC in patients with progressive metastatic neuroendocrine tumours: assessment of response, survival and toxicity. Br J Cancer. 2013;108:1440-8
42. Zhao R, Yang W, Wang Z, Li G, Qin W, Wang J. Treatment of transplanted tumor of lung adenocarcinoma A549 transfected by human somatostatin receptor subtype 2 (hsstr2) gene with 188Re-RC-160. Nucl Med Biol. 2010;37:977-87
43. Celinski SA, Fisher WE, Amaya F, Wu YQ, Yao Q, Youker KA. et al. Somatostatin receptor gene transfer inhibits established pancreatic cancer xenografts. J Surg Res. 2003;115:41-7
Author contact
![]() Corresponding author: Umar Mahmood, M.D., Ph.D., Center for Precision Imaging, Department of Radiology, Massachusetts General Hospital, Boston, MA 02114. Tel: 617-726-6477; Fax: 617-726-7422; Email: umahmoodharvard.edu
Corresponding author: Umar Mahmood, M.D., Ph.D., Center for Precision Imaging, Department of Radiology, Massachusetts General Hospital, Boston, MA 02114. Tel: 617-726-6477; Fax: 617-726-7422; Email: umahmoodharvard.edu