Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(12):3256-3267. doi:10.7150/thno.23964 This issue Cite
Research Paper
A tightly controlled Src-YAP signaling axis determines therapeutic response to dasatinib in renal cell carcinoma
Jingya Sun1,2, Xin Wang1,2, Boyun Tang3, Hongchun Liu1,2, Minmin Zhang1,2, Yueqin Wang1,2, Fangfang Ping1,2, Jian Ding1,2 ![]() , Aijun Shen1,2
, Aijun Shen1,2 ![]() , Meiyu Geng1,2
, Meiyu Geng1,2 ![]()
1. Division of Anti-tumor Pharmacology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;
2. University of Chinese Academy of Sciences, Beijing 100049, China;
3. Gene Company Ltd, Shanghai 200233, China.
Received 2017-11-21; Accepted 2018-3-27; Published 2018-5-11
Abstract

Over the past decade, therapies targeting the VEGF/VEGFR and mTOR pathways have served as the standard of care for the clinical management of renal cell carcinoma (RCC) patients. Albeit promising, these targeted drugs have attained only modest clinical benefits with limited prolonged progression-free survival. Therefore, alternative reasonable and applicable therapeutic approaches should be introduced to improve the clinical outcome of RCC patients.
Methods: FDA approved kinase inhibitors were screened to evaluate their abilities to suppress the proliferation of RCC cells. Then, the downstream effector, therapeutic target and signaling pathway of the selected drug were identified by gene expression array, RNAi, kinase profile and rescue verification. Finally, the in vivo effectiveness of the drug was assessed in cell line-based xenograft models and patient-derived xenograft models.
Results: In this study, we discovered that dasatinib is a potent agent that can impair RCC cell viability in vitro and decrease tumor growth in vivo. Mechanistically, we improved the understanding of the precise mechanistic role of YAP as a pivotal effector of dasatinib-induced anti-proliferation through Src-JNK-LIMD1-LATS signaling cascade in RCC cells. Meanwhile, our results indicated that the alteration of p-YAP is closely correlated to the growth inhibition caused by dasatinib in sensitive RCC models.
Conclusion: Our findings provide evidence that dasatinib may serve as a powerful drug candidate to treat subgroups of RCC patients with hyper-activated Src-YAP signaling axis, and the alteration of p-YAP could serve as a functional response biomarker of dasatinib in RCC.
Keywords: dasatinib, YAP, Src, renal cell carcinoma
Introduction
Renal cell carcinoma (RCC) is one of the most frequently occurring malignant tumors worldwide. Distinct from other solid malignancies, RCC is highly vascularized and refractory to conventional chemotherapy and radiotherapy. Nevertheless, an improved understanding of the underlying biology of RCC over the past decades has led to the clinical implementation of novel therapeutic strategies. Notably, RCC, especially clear-cell renal cell carcinoma, is characterized by various hotpot inactive mutations of the VHL gene in up to 70% of cases. These mutations result in the persistent accumulation and activation of HIF-1α, which increases the production of pro-angiogenic factors such as VEGF [1]. In addition, the transcription and translation of HIF-1α can be regulated by the mTOR pathway in a VHL-independent fashion [2, 3]. These insights provide a sound basis for the perturbation of the critical HIF-1α/VEGF axis in RCC through the blockade of VEGF/VEGFR signaling in endothelial cells or mTOR signaling in cancer cells, respectively [4, 5].
Indeed, the current targeted therapies aimed at VEGF/VEGFR or mTOR inhibition have witnessed more considerable advances in the clinical treatment of RCC patients than former standard cytokine-based immunotherapies [6-8]. However, the overall clinical results are modest because only prolonged progression-free survival, but not complete or sustained responses was demonstrated [9-12]. Furthermore, the inherent occurrence of primary and acquired resistance has been observed in clinical settings [13-15]. Therefore, to compensate for these limitations, the identification of new subtypes and alternative therapeutic options for RCC is urgently needed. To this end, comprehensive genomic and epigenomic aberrations in RCC patients were investigated to identify potential driving targets for further therapy [16, 17]. Intriguingly, several unusual somatic mutations in PBRM1 (~50%), BAP1 (~15%) and SETD2 (~15%), which rarely occur in other solid tumors, were recently reported in RCC patients [18-20]. However, tremendous effort may be required to validate their biological contributions to RCC and develop specific inhibitors that can be translated into clinical applications.
In this study, to bridge the gap between clinical requirements and drug development, we directly profiled the efficacy of available clinical agents in RCC cells to seek candidates that might be suitable for clinical practice. Strikingly, we discovered that dasatinib, a multi-target kinase inhibitor, is a potent agent that suppressed the proliferation of all tested RCC cells. Interestingly, we found that YAP served as a key downstream effector of dasatinib to control cell viability through the inhibition of Src and subsequently through the JNK-LIMD1-LATS pathway. In addition, we found that the alteration of p-YAP could be considered as a potential responsive biomarker to dictate the pharmacological effects of dasatinib in sensitive RCC cells.
Methods
Cell lines and reagents
Caki-1, 769-P, ACHN, 786-O, A498 and OS-RC-2 cells used in this study were obtained from American Type Culture Collection (ATCC) and authenticated by short tandem repeat (STR) testing at Genesky Biopharma Technology (Shanghai, China). Cells were maintained in appropriate culture medium, as suggested by the suppliers.
Dasatinib was obtained from LC Laboratories (Woburn, MA), and the other inhibitors were obtained from Selleck Chemicals (Shanghai, China). All reagents were dissolved in DMSO for in vitro studies. Dasatinib was dissolved in 0.5% CMC-Na for in vivo studies.
DNA plasmid construction, virus production and infection
DNA plasmids pDONR223-Src (#23934), pDONR223-Yes (#23938), pBABE-YAP (#15682) and pcDNA3-HA-TAZ (#32839) were obtained from Addgene (Cambridge, MA). The retroviral constructs pBABE-Src, pBABE-Yes and pBABE-TAZ were constructed using recombinant polymerase chain reaction and subsequently subcloned into the pBABE-puro vector. pBABE-Src-T341I, pBABE-Yes-T348I, pBABE-YAP-5SA and pBABE-TAZ-4SA were constructed with a site-directed mutagenesis kit (Sbsbio, Shanghai, China).
For virus production and infection, the plasmids were transfected into amphotropic Phoenix 293T packaging cells at 60% confluence using Lipofectamine 2000 (Invitrogen, Grand Island, NY) according to the manufacturer's instructions. After an additional 48 h incubation, the supernatant was collected, filtered using a 0.45 μm filter (Millipore, Cork, Ireland), and used to infect host Caki-1 or 769-P cells in the presence of 6 μg/mL polybrene (Millipore). The resultant stable polyclonal populations of infected cells were then selected with 1 μg/mL puromycin (Sigma, St. Louis, MO) for two weeks, followed by validation by immunoblotting.
RNA interference
Cells were seeded in 6-well or 96-well plates at 30% confluence. After 24 h, cells were transfected with the indicated siRNA oligonucleotides using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. Then, the cells were cultured for 72 h and harvested either for cell viability assays or for immunoblotting analysis. The target sequences of siRNA oligonucleotides are as follows: siSrc#1: 5'-GAGAACCUGGUGUGCAAAG-3', siSrc#2: 5'-CAGUUGUAUGCUGUGGUUU-3', siYAP#1: 5'-GGTCCTCTTCCTGATGGAT-3', siYAP#2: 5'-GACCAATAGCTCAGATCCTTT-3', siTAZ#1: 5'-GACAUGAGAUCCAUCACUA-3', siTAZ#2: 5'-GGACAAACACCCAUGAACA-3'. The customized pre-designed genOFF™ RNAi library used in this study was obtained from RiboBio (Guangzhou, China).
Luciferase reporter assay
Caki-1 cells were seeded in 96-well plates at 60% confluence. After 24 h, cells were co-transfected with 0.2 μg of 8×GTIIC-Luciferase (#34615, Addgene) and 2 ng of Renilla luciferase per well using Lipofectamine 2000 (Invitrogen). Compounds were dispensed into each well 6 h after transfection. Then, the cells were cultured for 24 h, and cell lysates were examined for firefly luciferase activity and Renilla luciferase activity with the Dual-Luciferase Reporter Assay Kit (Promega, Beijing, China). Relative transcriptional activity = Firefly Luciferase / Renilla Luciferase.
Animal studies
Four- to six-week-old nu/nu female athymic BALB/c mice were obtained from the Shanghai Laboratory Animal Center, Chinese Academy of Sciences (Shanghai, China). All studies were performed in compliance with the guidelines set forth by the Institutional Animal Care and Use Committee of Shanghai Institute of Materia Medica. Tumors were generated by transplanting 5×106 Caki-1 cells re-suspended in PBS (200 μL/mouse) into the right flank. Prior to treatment initiation, mice were randomized among control and treated groups (n = 6 per group). Mice bearing Caki-1 cells were administered the indicated doses of dasatinib once a day for 21 days, and their body weights and tumor volumes were measured every three days. To prepare lysates, the mice were sacrificed, and tumor tissues were resected and homogenized in cold RIPA lysis buffer (Beyotime) supplemented with protease and phosphatase inhibitors (Merck) and processed for immunoblotting analysis.
Animal studies using patient-derived xenograft (PDX) models were conducted by WuXi AppTec and Crown Bioscience and in strict accordance with the Guide for the Care and Use of Laboratory Animals set forth by the National Institutes of Health (NIH).
Statistics
The data are presented as the mean ± SD. Differences between experimental groups were analyzed using the unpaired two-tailed Student's t test analysis. P < 0.05 was considered significant. *P < 0.05, **P < 0.01, ***P < 0.001. All statistical analyses were performed using GraphPad Prism software (San Diego, CA).
Results
Dasatinib specifically inhibits RCC cell proliferation
To initiate this study, we profiled the sensitivity of a focused panel of Food and Drug Administration (FDA)-approved kinase inhibitors across 6 RCC cell lines, aiming to identify novel therapeutic options for RCC treatment. Intriguingly, dasatinib, an orally available multi-target kinase inhibitor originally used in chronic myelogenous leukemia (CML) patients, emerged as the most potent agent capable of suppressing the proliferation of all RCC cells tested, with the IC50 values ranging from 40 nM to 1 μM (Figure 1A). Likewise, dasatinib treatment strongly inhibited the colony formation of RCC cells (Figure S1A). Interestingly, when expanding such analysis to a collection of cell lines originated from different source of solid tumors, RCC still represented as the relatively most sensitive tumor type among them (Figure 1B). These data implied the potential clinical application of dasatinib in RCC.
Dasatinib notably suppresses RCC cell survival by inducing G1/S cell cycle arrest. (A) IC50 values of FDA-approved kinase inhibitors in 6 RCC cell lines were assessed using an SRB assay. The data are represented as a heatmap drawn using R. (B) IC50 values of dasatinib in different solid tumor types were assessed using an SRB assay. Bars, mean ± SD. (C-E) Caki-1 and 769-P cells were treated with DMSO or dasatinib for 24 h. The gene expression array was subjected to KEGG pathway analysis (C). Cell cycle distribution was analyzed by flow cytometry. Bars, mean ± SD (D). DNA replication capability was evaluated by EdU incorporation. Scale bar, 300 μm (E).
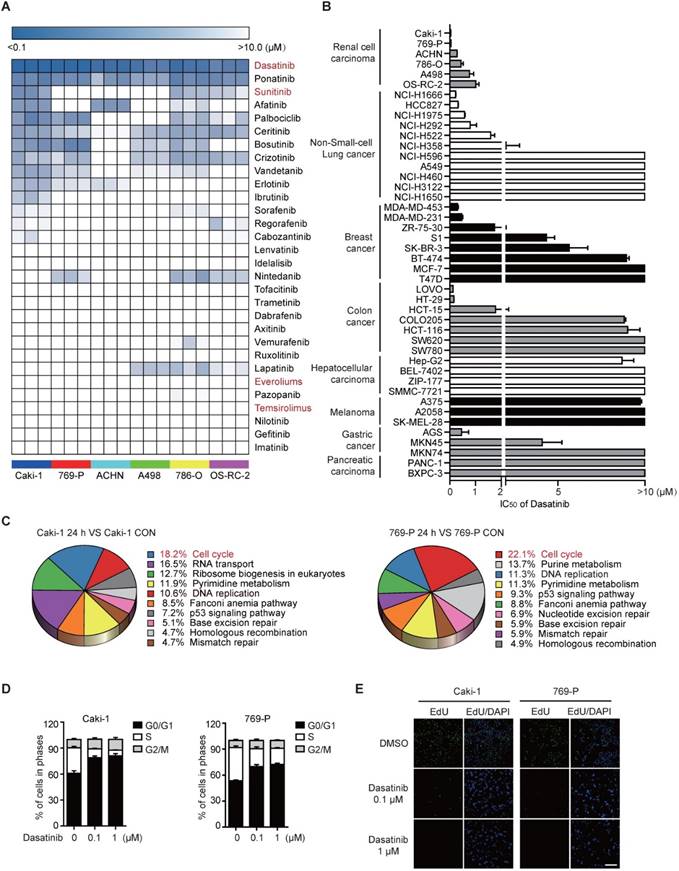
Then, two most sensitive RCC cell lines (Caki-1 and 769-P) were selected to investigate the biological effects of dasatinib induced proliferation inhibition. To this end, we treated Caki-1 and 769-P cells with dasatinib for 24 h and then performed global transcriptional gene expression analysis using microarray. Bioinformatics analysis demonstrated that cell cycle progression was the most down-regulated pathway modulated by dasatinib (Figure 1C). Therefore, we measured cell cycle distribution and found that 0.1 μM and 1 μM dasatinib significantly induced G1-S phase arrest in both cell lines, as determined by propidium iodide (PI) staining and evidenced by the decreased expression of phospho-RB and Cyclin D1 (Figure 1D and Figure S1B). In line with this observation, DNA synthesis was largely reduced following dasatinib treatment as determined by in situ EdU incorporation staining (Figure 1E). In addition, no accumulation of apoptosis was observed (data not shown), suggesting the cytostatic role of dasatinib in RCC cells.
Taken together, these data revealed that RCC is sensitive to dasatinib treatment through proliferation inhibition triggered by obvious G1-S phase arrest.
Dasatinib exerts its anti-proliferative effects through the inhibition of YAP activity
Over the past decade, multiple clinical trials have been carried out to evaluate the application of dasatinib in solid tumors. However, the overall clinical outcome of dasatinib has been quite disappointing, which might stem from a lack of sensitivity, as we observed in this study (Figure 1B). By contrast, most of the RCC cells we tested were relatively sensitive to dasatinib, which spurred us to explore the underlying molecular mechanisms in this setting. To do so, we treated Caki-1 and 769-P cells with dasatinib for 6, 12 and 24 h and used a microarray to analyze gene expression changes. Surprisingly, we noticed that several known targets of the Hippo pathway, such as CTGF, Cyr61, and AJUBA [21, 22], ranked among the top 10 most down-regulated genes modulated by dasatinib (Figure 2A). Meanwhile, gene set enrichment analysis (GSEA) annotation further confirmed the down-regulation of conserved YAP target genes in dasatinib-treated cells (Figure 2B), indicating the involvement of the Hippo pathway in mediating growth arrest after dasatinib treatment.
Dasatinib impairs YAP transcriptional activity in sensitive RCC cell lines. (A) Heatmap representation of the top 20 down- and up-regulated genes (P < 0.001) following dasatinib treatment for 6, 12 and 24 h in Caki-1 and 769-P cells. (B) GSEA enrichment plots showing the down-regulation of the YAP_Conserved_Signature gene set in Caki-1 and 769-P cells treated with dasatinib. (C) Caki-1 and 769-P cells were treated with dasatinib at the indicated concentration, and YAP/TAZ levels were assessed by immunoblotting. (D) Caki-1 cells were treated with dasatinib for 1 h. The cytoplasm and nucleus were separated and subjected to immunoblotting analysis of YAP/TAZ. (E) Caki-1 cells were treated with dasatinib for 3 h. Cell lysates were precipitated with YAP or IgG antibodies for co-immunoprecipitation. YAP and TEAD were detected by immunoblotting. (F) Caki-1 cells were transfected with either TEAD-Luciferase plasmid (8×GTIIC) or empty vector and co-transfected with Renilla Luciferase as a reference. Then, cells were treated with dasatinib for 24 h. Cell lysates were assayed for luciferase activity. Bars, mean ± SD. ***P < 0.001. (G) Caki-1 and 769-P cells were transfected with scramble or YAP siRNAs for 72 h. Cell viability was measured using an SRB assay. Bars, mean ± SD. **P < 0.01, ***P < 0.001. (H-I) Caki-1 cells stably expressing either empty vector, YAP-WT or YAP-5SA. Cells were treated with dasatinib at 1 μM for 3 h for subsequent immunoblotting (H). Cell viability after dasatinib treatment for 72 h was assessed by SRB assay. Bars, mean ± SD. ***P < 0.001 (I). (J) RCC cells were treated with dasatinib at 1 μM for 3 h followed by immunoblotting analysis.
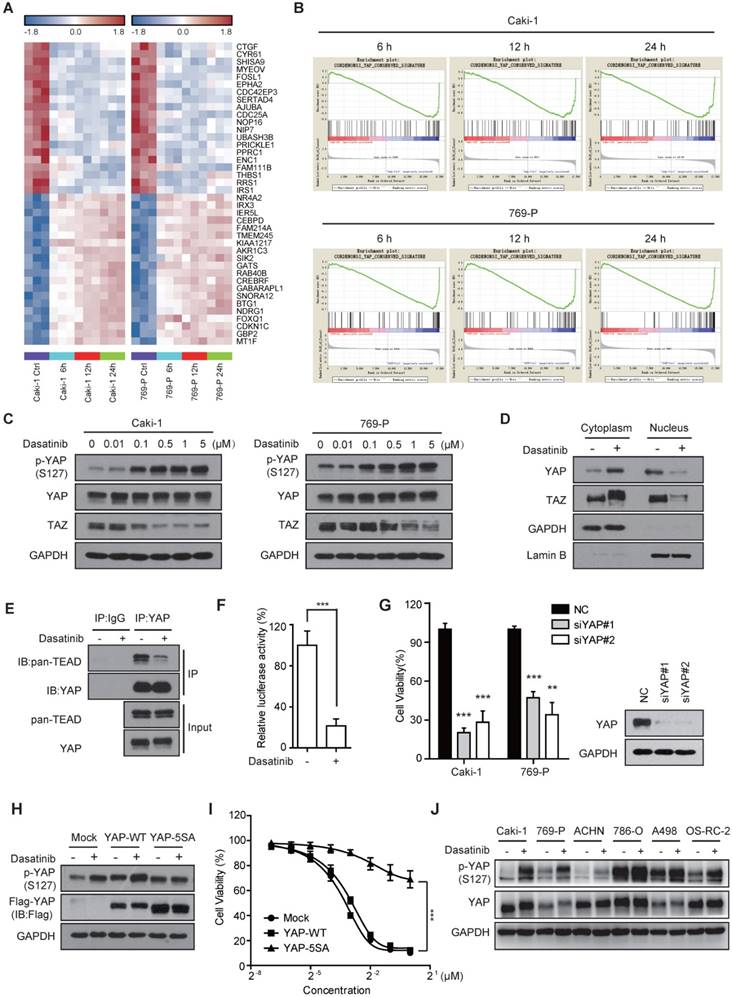
To test this hypothesis, we first examined whether dasatinib inhibited the activity of YAP/TAZ, two homologous transcriptional co-activators that are the key mediators of the Hippo pathway [23]. As expected, dasatinib treatment significantly up-regulated the canonical serine phosphorylation of YAP (Ser127) and down-regulated the protein expression of TAZ in RCC cells in a dose-dependent manner (Figure 2C). Of note, the phosphorylation of YAP promoted its sequestration in the cytoplasm and decreased its binding to TEAD, which subsequently attenuated TEAD-dependent transcriptional activity (Figure 2D-F and Figure S2A). In accordance, the expression of YAP target genes, such as Cyr61, was inhibited at both the mRNA and protein levels (Figure S2B-C). These results demonstrated that the YAP/TAZ-dependent transcriptional program was dramatically blocked by dasatinib treatment.
Then, we proceed to ascertain whether YAP/TAZ determines the therapeutic response of RCC cells to dasatinib. To this end, we first directly depleted YAP and TAZ expression using siRNAs to test whether their inhibition can mimic the effect of dasatinib. Obviously, either disruption of YAP or TAZ yielded a significant decrease in cell survival (Figure 2G and Figure S2D), along with reduced EdU incorporation (Figure S2E). Based on this, we further constructed YAP-5SA and TAZ-4SA mutants, the constitutively activated forms of YAP and TAZ, respectively, in Caki-1 cells. We found that dasatinib-induced p-YAP alteration, downstream target gene expression and the final growth inhibition were largely rescued in YAP-5SA mutant cells (Figure 2H-I and Figure S2F). Strikingly, however, similar results were not obtained in TAZ-4SA mutant cells, suggesting its redundancy with respect to dasatinib sensitivity (Figure S2G-I). In agreement with this observation, p-YAP was consistently up-regulated in almost all RCC cell lines we tested (Figure 2J). In addition, we also excluded the engagement of tyrosine phosphorylation of YAP in the therapeutic determination of dasatinib in RCC cells because the YAP-3YE mutant didn't rescue the effect of dasatinib despite its uncertain down-regulation observed upon dasatinib treatment (Figure S2J-L).
Taken together, these findings clearly clarified that the canonical transcriptional activity of YAP is essential to determine the therapeutic response of dasatinib in RCC cells.
Dasatinib inhibits Src kinase to impair YAP activity in RCC cells
Given that dasatinib is a multi-target kinase inhibitor, we thus sought to identify its predominant target in RCC cells and the corresponding converged signal pathway that regulates YAP activity, which will be helpful for selecting appropriate patients in the clinic in future. To address this, we conducted a KinaseProfilerTM screening analysis (Table S1, 316 human kinases were involved) and mined the literature (including phosphoproteomics data to classify dasatinib binding kinases) to find the converged spectrum of kinases that are both targeted and inhibited by dasatinib [24-27]. As a result, a total of 41 selected kinases were evaluated for target identification in RCC by treating Caki-1 and 769-P cells with a customized pre-designed genOFF™ RNAi library. The results of RNAi screening demonstrated that the inhibition of Src showed the strongest ability to decrease cell viability and inhibit DNA replication in RCC cell lines, similar to that of dasatinib treatment (Figure 3A-B and Figure S3A). Strong proliferation inhibition was also observed when treating Caki-1 and 769-P cells with AZD0530, another putative Src kinase inhibitor (Figure S3B). In addition, the phosphorylation of Src kinase was consistently inhibited by dasatinib in all tested RCC cell lines (Figure 3C). These data suggested that Src might be the most relevant target of dasatinib in RCC cells.
Src kinase is the most dominant therapeutic target of dasatinib that regulates YAP activity in RCC cells. (A) Caki-1 and 769-P cells were treated with a customized pre-designed genOFF™ RNAi library for 72 h. Cell viability was assessed by SRB assay. The mean viability of cells receiving different siRNAs for the same gene was evaluated by Log2 calculation. (B) Caki-1 and 769-P cells were transfected with scramble or Src siRNAs. Cell viability was measured using SRB assay. Bars, mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. (C) RCC cells were treated with dasatinib at 1 μM for 3 h followed by immunoblotting analysis. (D-E) Caki-1 and 769-P cells were stably transfected with retrovirus expressing either empty vector, Src-WT or Src-T341I. The resultant cells were treated with dasatinib (0.1 and 1 μM) for 3 h and the phosphorylation of Src was measured by immunoblotting (D). Cell viability after dasatinib treatment for 72 h was assessed by SRB assay. Bars, mean ± SD. ***P < 0.001 (E). (F-G) Caki-1 cells were treated with scramble or Src siRNAs for 72 h and then subjected to immunoblotting with the indicated antibodies (F). The subcellular location of YAP was determined by immunofluorescence. Scale bar, 50 μm (G). (H) Caki-1 cells stably expressing either empty vector, Src-WT or Src-T341I were treated with dasatinib for 3 h, and YAP phosphorylation was measured by immunoblotting.
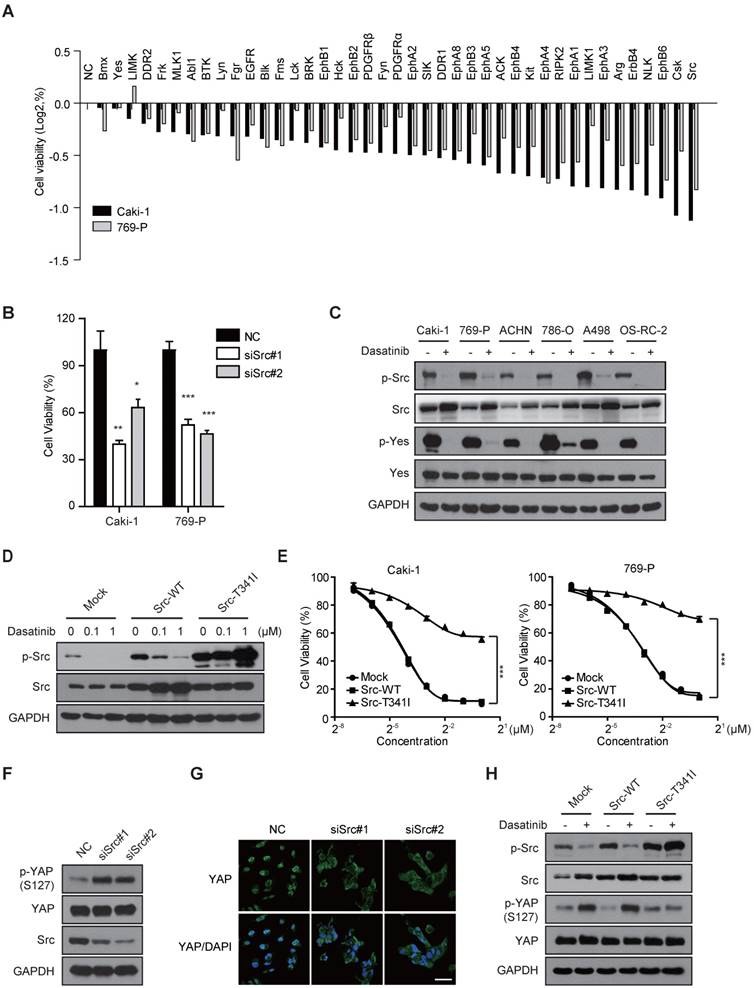
To test this possibility, we further confirmed the role of Src kinase in governing cell survival and therapeutic response to dasatinib. We found that the ability of dasatinib to decrease Src phosphorylation and, accordingly, cell viability in Caki-1 and 769-P cells was largely relieved by the generation of a Src-T341I gatekeeper mutant, which abolished the ATP binding pocket of Src (Figure 3D-E). Nevertheless, the modulation of Yes, the closest homolog to Src, had only marginal effects on cell survival and sensitivity to dasatinib (Figure S3C-E).
Next, we sought to determine whether dasatinib-induced inhibition of YAP activity was directly mediated by Src kinase inhibition. Indeed, siRNA-mediated knockdown of Src expression resulted in significant induction of serine phosphorylation and cytoplasmic sequestration of YAP (Figure 3F-G). Meanwhile, the dasatinib-induced phosphorylation of YAP was reversed in cells harboring the Src-T341I mutant kinase (Figure 3H). However, in HCC827 and H1975 cell lines, which were sensitive to dasatinib via targeting of EGFR instead of Src, we did not observe an up-regulation of p-YAP after dasatinib treatment or even EGFR disruption (Figure S3F-H). This observation indicated a tightly controlled Src-YAP signaling axis impacted by dasatinib in RCC.
Together, these data implied that Src kinase is the bona fide target of dasatinib in RCC cells, whose inhibition contributes to the silencing of YAP transcriptional activity.
Dasatinib inhibits YAP activity via a Src-JNK-LIMD1-LATS-dependent pathway
Next, we attempted to elucidate how dasatinib modulates YAP activity through Src inhibition. It was recently reported that Src can bind to YAP and regulate its activity directly [28]. Intriguingly, the co-immunoprecipitation results demonstrated that YAP was not regulated by direct Src binding (Figure S4A). Thereafter, we treated Caki-1 cells with dasatinib to test whether the up-stream regulators of YAP in the conventional Hippo pathway were involved. Interestingly, we found that the phosphorylation of LATS1 (Thr1079) was significantly up-regulated and YAP phosphorylation was dramatically relieved by the disruption of LATS1/2 before dasatinib treatment (Figure 4A-B). However, similar results were not observed for MST1/2 (Figure S4B), indicating that the regulation of YAP activity is LATS, but not MST, dependent. These findings, together with the identification of Src kinase, raised a possibility that Src, instead of the typical MST1/2, is the specific up-stream regulator of LATS-YAP signaling in RCC cells. In line with this speculation, a recent study reported that Src regulates the serine phosphorylation of YAP via direct tyrosine phosphorylation of LATS [29]. However, our data showed that, although dasatinib somehow inhibited the tyrosine phosphorylation of LATS1, the up-regulation of p-YAP could not be abolished by transfection of LATS1-2YF into Caki-1 cells, excluding its involvement (Figure S4C-D).
The JNK-LIMD1-LATS1 signaling cascade mediates the modulation of YAP following Src inhibition in RCC cells. (A) Caki-1 cells were treated with dasatinib for 3 h and the regulators of the Hippo pathway were measured by immunoblotting. (B) Caki-1 cells were treated with scramble or LATS1/2 siRNAs for 72 h; then, the cells were exposed to DMSO or dasatinib at 1 μM for 1 h and subjected to immunoblotting analysis. (C) RCC cells were treated with DMSO or dasatinib at 1 μM for 3 h followed by immunoblotting analysis. (D-E) Caki-1 cells were treated with DMSO, dasatinib (0.1 μM), MK2206 (1 μM), AZD6244 (1 μM), JNK-IN-8 (5 μM), and Skepinone-L (10 μM) for 3 h followed by immunoblotting analysis (D) or for 72 h followed by SRB assay to measure cell viability (E). Bars, mean ± SD. ***P < 0.001. (F) Caki-1 cells were treated with 1 μM anisomycin for 1 h and then treated with 1 μM dasatinib for 1 h, then subjected to immunoblotting. (G) Caki-1 cells stably expressing either empty vector, Src-WT or Src-T341I were treated with dasatinib at 1 μM for 3 h for subsequent immunoblotting. (H) Caki-1 cells were treated with DMSO or dasatinib at 1 μM for 3 h. Cells lysates were precipitated with LATS1 or IgG antibodies for co-immunoprecipitation. LIMD1 and LATS1 were detected by immunoblotting.
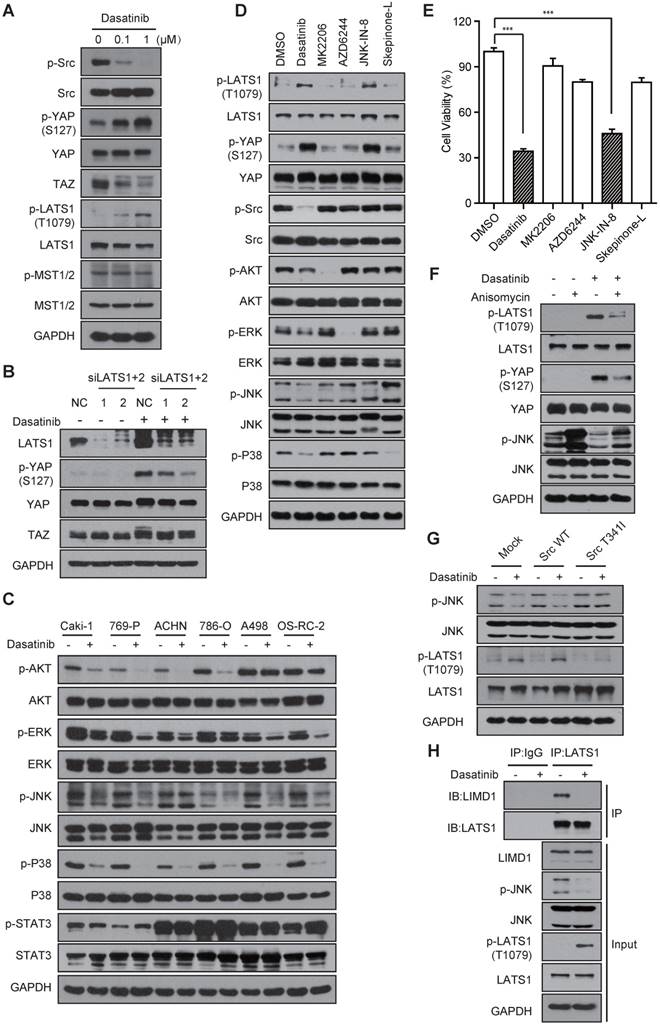
We then sought to examine other signal molecules that are involved in the modulation of LATS and YAP phosphorylation by Src kinase in RCC cells. Intriguingly, the previously reported Src-PI3K-PDK1 signaling did not participate as their inhibition failed to induce upregulated phosphorylation of LATS and YAP (Figure S4E) [30]. The AKT, MAPK and STAT pathways are the other well-documented down-stream components that are regulated by Src signaling to control cell proliferation [31, 32]. To test their involvement, we firstly treated RCC cells with dasatinib and evaluated the inhibition of these pathways. The results demonstrated that treatment with dasatinib caused marked down-regulation of the AKT and MAPK pathways (including ERK, JNK and P38), while the STAT pathway remained intact (Figure 4C). Then, we introduced inhibitors of the AKT (MK2206), MEK (AZD6244), JNK (JNK-IN-8, a covalent inhibitor of JNK) and P38 (Skepinone-L) pathways in Caki-1 cells and investigated which of these contributed to the activation of LATS and YAP phosphorylation. Strikingly, only inhibition of the JNK pathway caused an obvious up-regulation in LATS and YAP phosphorylation, similar to the effects observed after Src inhibition (Figure 4D). Meanwhile, JNK inhibition significantly suppressed cell growth, as expected (Figure 4E).
In addition, the up-regulation of LATS and YAP phosphorylation by JNK inhibition was effectively reversed by pretreatment with anisomycin, an activator of JNK signaling (Figure 4F). The marked reduction of JNK phosphorylation and subsequent up-regulation of LATS phosphorylation caused by dasatinib treatment were largely abolished in Caki-1 cells harboring the Src-T341I mutation (Figure 4G), suggesting that the JNK pathway is a downstream mediator of Src kinase that regulates LATS and YAP. It was reported that JNK signaling inhibits the tumor-suppressive activity of LATS via promoting the binding of LATS to its negative regulator LIMD1. In agreement with these findings, a significant diminishment in the binding of LATS and LIMID1 was observed in dasatinib-treated Caki-1 cells (Figure 4H).
In conclusion, these data revealed that the activity of YAP is mainly modulated through a Src-JNK-LIMD1-LATS signaling cascade in dasatinib-treated RCC cells.
YAP activity indicates the therapeutic response to dasatinib in RCC cells in vivo
The above results have demonstrated the critical role of YAP in mediating growth inhibition by dasatinib through a Src-JNK-LIMD1-LATS-dependent pathway in RCC cells. Functional downstream effectors that determine cell survival, such as BIM and c-Myc, have been considered effective biomarkers to indicate therapeutic response and resistance of kinase inhibitors [33-35]. Thus, we attempted to determine whether modulation of YAP activity is indicative of the biological effects of dasatinib in RCC cells, in vivo in particular.
We first tested this possibility using cell line-based xenograft models. Mice bearing Caki-1 xenografts were treated with dasatinib at 25, 50 or 100 mg/kg once a day for 21 consecutive days. Tumor volume was examined every 3 days, and intratumoral expression of the Src-YAP signaling axis was detected after the mice were sacrificed. As expected, dasatinib treatment significantly suppressed tumor growth in vivo at all doses tested (Figure 5A). In agreement with the observations obtained in RCC cells, intratumoral phosphorylation of Src and YAP was profoundly affected after dasatinib treatment (Figure 5B), indicating the ability of dasatinib to combat tumor growth via modulation of the Src-YAP signaling axis.
YAP alteration correlates with the sensitivity of RCC cells to dasatinib. (A-B) Caki-1 xenograft-bearing nude mice were given dasatinib at 25, 50 and 100 mg/kg or vehicle once a day for 21 consecutive days. Tumor volumes were measured every three days and presented as the average tumor volume ± SD (n = 6). ***P < 0.001 (A). Tumor lysates were prepared and subjected to immunoblotting analysis with the indicated antibodies (B). (C-D) Nude mice bearing patient-derived KI0047 or KI0060 xenografts were administered either vehicle control or dasatinib (50 mg/kg) once daily for 28 consecutive days (n = 6 for each group). Tumor volumes were measured every three days and presented as the average tumor volume ± SD. **P < 0.01, ***P < 0.001 (C). Tumor lysates were prepared and subjected to immunoblotting analysis with the indicated antibodies (D). (E-F) Nude mice bearing patient-derived KI0025 or KI0326 xenografts were administered either vehicle or dasatinib (50 mg/kg) once daily for 23 or 21 consecutive days (n = 6 for each group). Tumor volumes were measured every three days and are presented as the average tumor volume ± SD (E). Tumor lysates were prepared and subjected to immunoblotting analysis with the indicated antibodies (F).
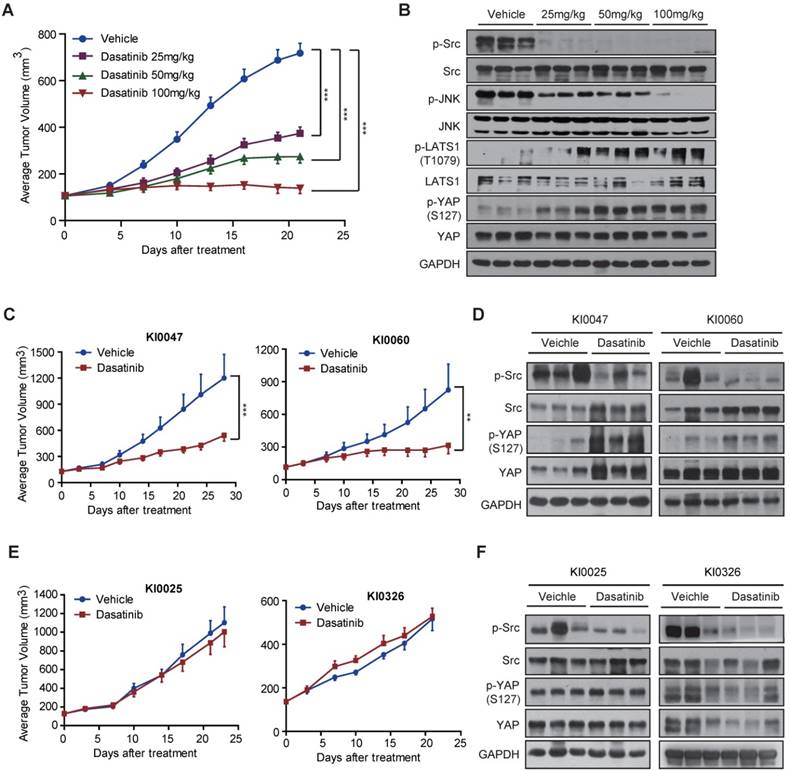
Next, we moved to patient-derived xenograft (PDX) models, which highly mirror the clinical conditions of patients, especially the genetic heterogeneity and tumor microenvironment [36]. We selected 4 RCC PDX models and treated with dasatinib at 50 mg/kg once a day for 21-28 consecutive days. Among them, we found that two models, namely KI0047 and KI0060, were sensitive to dasatinib treatment. Similar to the observations obtained in the Caki-1 model, dasatinib treatment significantly affected the phosphorylation of Src and YAP, as expected (Figure 5C-D). Conversely, we observed loss of up-regulation of YAP phosphorylation in another two models (KI0025 and KI0326) that showed de novo resistance to dasatinib treatment, despite the conserved blockade of Src signaling (Figure 5E-F). The distinct role of YAP activity was further confirmed by immunohistochemistry staining (Figure S5). Collectively, these findings strongly support that p-YAP alteration might indicate the therapeutic response/resistance to dasatinib, which could serve as a response biomarker for RCC patients who receive dasatinib treatment in the clinic.
Discussion
To obtain a deeper insight into the response toward kinase inhibitors in molecular targeted therapies, a thorough map elucidating the mechanism of kinase inhibition-induced growth arrest might be necessary. Our study revealed that dasatinib targeted Src kinase directly and thereby triggered a JNK-LIMD1-LATS signaling cascade to down-regulate a YAP-mediated transcriptional program responsible for the viability of RCC cells (Figure 6). Our findings thus paved the way for the application of dasatinib as a novel therapeutic option for RCC patients, which should be taken into consideration for further clinical investigation.
A proposed model illustrating the mechanism whereby dasatinib impacts survival in sensitive or resistant RCC.
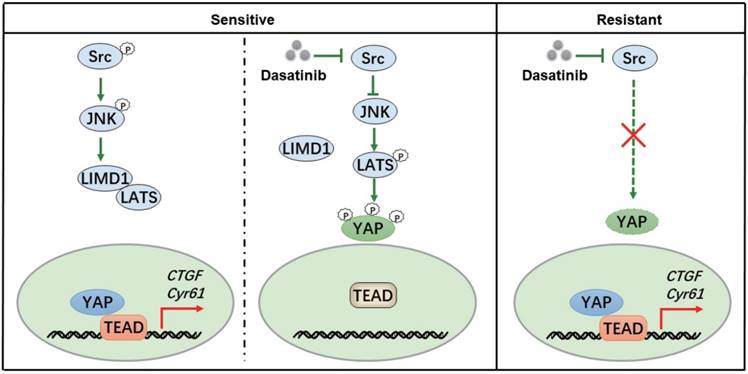
Alternatively, YAP has been regarded as an appealing target for cancer therapy thanks to its critical role in tumor development and progression. However, as YAP is a transcriptional coactivator without known catalytic activity, direct therapeutic intervention against YAP activity appears unachievable now [37]. In contrast, our findings in this study offer an alternative approach to disrupt YAP activity through the indirect modulation of its upstream regulators, such as Src kinase. Indeed, if the corresponding kinase-YAP axis paradigm is established, it will help us discover more inhibitors that block YAP activity and better understand the biological mechanisms of kinases in cancer biology in subgroups of YAP-dominated tumors.
In summary, we propose that cells that rely on the tightly controlled Src-YAP axis for survival may confer sensitivity to dasatinib and that this axis should be considered a powerful predictive biomarker for the enrollment of patients for treatment. In addition, we suggest that the induction of YAP phosphorylation might be regarded as a potential response biomarker to indicate and monitor the clinical effectiveness of dasatinib. Hence, the coming challenge lies in two aspects. First is the clinical stratification of patient subgroups, of RCC patients in particular, that are co-addicted to the Src-YAP axis. To address this issue, we believe that the hyper-activation of the Src-YAP axis, where p-Src is high while p-YAP is low, should be first detected and confirmed in clinical samples. The second issue is how to facilitate the detection of YAP phosphorylation in the clinic. While the serial collection of patient biopsies appears unreasonable, it is imperative to develop alternative strategies to aid clinical detection. Considering that YAP phosphorylation status is closely correlated to its transcription activity and subsequent target gene expression, we suppose that monitoring its down-stream cytokines, such as CTGF or Cyr61, through blood sampling might be an alternative option in the clinic.
Abbreviations
ATCC: American Type Culture Collection. CML: chronic myelogenous leukemia. DMSO: dimethyl sulfoxide. EdU: 5-ethynyl-2'-deoxyuridine. FDA: Food and Drug Administration. GSEA: gene set enrichment analysis. NIH: National Institutes of Health. PDX: patient-derived xenograft. PI: propidium iodide. RCC: renal cell carcinoma. SRB: Sulforhodamine B. STR: short tandem repeat.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No.81402966, No.81673472 and No.91229205), the National Program on Key Basic Research Project of China (No.2012CB910704), the Natural Science Foundation of China for Innovation Research Group (No. 81321092), and Personalized Medicines-Molecular Signature-based Drug Discovery and Development, the Strategic Priority Research Program of the Chinese Academy of Sciences (No.XDA12020101 and No.XDA12020105).
Contributions
AJS, JYS and MYG designed the study. JYS, XW, HCL, MMZ, YQW and FFP performed the experiments. JYS, AJS and BYT analyzed data. JYS, AJS and MYG wrote the manuscript. AJS, JD and MYG supervised the research.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nat Rev Cancer. 2015;15:55-64
2. Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B. et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122-7
3. Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8:851-64
4. Motzer RJ, Jonasch E, Agarwal N, Beard C, Bhayani S, Bolger GB. et al. Kidney cancer, version 3.2015. J Natl Compr Canc Netw. 2015;13:151-9
5. Coppin C, Kollmannsberger C, Le L, Porzsolt F, Wilt TJ. Targeted therapy for advanced renal cell cancer (RCC): a Cochrane systematic review of published randomised trials. BJU Int. 2011;108:1556-63
6. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O. et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115-24
7. Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C. et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370:2103-11
8. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S. et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449-56
9. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S. et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584-90
10. Escudier B, Bellmunt J, Negrier S, Bajetta E, Melichar B, Bracarda S. et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol. 2010;28:2144-50
11. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M. et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol. 2009;27:3312-8
12. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S. et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma: final results and analysis of prognostic factors. Cancer. 2010;116:4256-65
13. Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009;10:992-1000
14. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592-603
15. Bottsford-Miller JN, Coleman RL, Sood AK. Resistance and escape from antiangiogenesis therapy: clinical implications and future strategies. J Clin Oncol. 2012;30:4026-34
16. Rydzanicz M, Wrzesinski T, Bluyssen HAR, Wesoly J. Genomics and epigenomics of clear cell renal cell carcinoma: recent developments and potential applications. Cancer Lett. 2013;341:111-26
17. Jones J, Libermann TA. Genomics of renal cell cancer: the biology behind and the therapy ahead. Clin Cancer Res. 2007;13:685s-92s
18. Creighton CJ, Morgan M, Gunaratne PH, Wheeler DA, Gibbs RA, Robertson AG. et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43-49
19. Pena-Llopis S, Christie A, Xie XJ, Brugarolas J. Cooperation and antagonism among cancer genes: the renal cancer paradigm. Cancer Res. 2013;73:4173-9
20. Randall JM, Millard F, Kurzrock R. Molecular aberrations, targeted therapy, and renal cell carcinoma: current state-of-the-art. Cancer Metastasis Rev. 2014;33:1109-24
21. Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2014;13:63-79
22. Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13:246-57
23. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287-312
24. Shi HB, Zhang CJ, Chen GYJ, Yao SQ. Cell-based proteome profiling of potential dasatinib targets by use of affinity-based probes. J Am Chem Soc. 2012;134:3001-14
25. Li J, Rix U, Fang B, Bai Y, Edwards A, Colinge J. et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat Chem Biol. 2010;6:291-9
26. Rix U, Hantschel O, Durnberger G, Remsing Rix LL, Planyavsky M, Fernbach NV. et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood. 2007;110:4055-63
27. Hantschel O, Rix U, Superti-Furga G. Target spectrum of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib. Leuk Lymphoma. 2008;49:615-9
28. Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ. et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151:1457-73
29. Si Y, Ji X, Cao X, Dai X, Xu L, Zhao H. et al. Src inhibits the Hippo tumor suppressor pathway through tyrosine phosphorylation of Lats1. Cancer Res. 2017;77:4868-80
30. Kim NG, Gumbiner BM. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J Cell Biol. 2015;210:503-15
31. Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4:470-80
32. Kim LC, Song LX, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6:587-95
33. Shen A, Wang L, Huang M, Sun J, Chen Y, Shen YY. et al. c-Myc alterations confer therapeutic response and acquired resistance to c-Met inhibitors in MET-addicted cancers. Cancer Res. 2015;75:4548-59
34. Liu HY, Ai J, Shen AJ, Chen Y, Wang XY, Peng X. et al. c-Myc alteration determines the therapeutic response to FGFR inhibitors. Clin Cancer Res. 2017;23:974-84
35. Bean GR, Ganesan YT, Dong YY, Takeda S, Liu H, Chan PM. et al. PUMA and BIM are required for oncogene inactivation-induced apoptosis. Sci Signal. 2013;6:ra20
36. Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C. et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998-1013
37. Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015;15:73-79
Author contact
![]() Corresponding authors: Meiyu Geng, Aijun Shen and Jian Ding; Division of Anti-tumor Pharmacology; State Key Laboratory of Drug Research; Shanghai Institute of Materia Medica; Chinese Academy of Sciences; 555 Zuchongzhi Road, Shanghai 201203, China; Tel.: (86) 21-50806072; Fax: (86) 21-50806072; E-mail: Meiyu Geng, mygengac.cn; Aijun Shen, shenajac.cn; Jian Ding, jdingac.cn
Corresponding authors: Meiyu Geng, Aijun Shen and Jian Ding; Division of Anti-tumor Pharmacology; State Key Laboratory of Drug Research; Shanghai Institute of Materia Medica; Chinese Academy of Sciences; 555 Zuchongzhi Road, Shanghai 201203, China; Tel.: (86) 21-50806072; Fax: (86) 21-50806072; E-mail: Meiyu Geng, mygengac.cn; Aijun Shen, shenajac.cn; Jian Ding, jdingac.cn