Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(12):3176-3188. doi:10.7150/thno.23207 This issue Cite
Research Paper
The HER2 inhibitor lapatinib potentiates doxorubicin-induced cardiotoxicity through iNOS signaling
Wan-Tseng Hsu1,2, Ching-Ying Huang1, Christopher Y.T. Yen1, Ann-Lii Cheng3, Patrick C.H. Hsieh1,4,5,6 ![]()
1. Institute of Biomedical Sciences, Academia Sinica, Taiwan;
2. School of Pharmacy, National Taiwan University, Taiwan;
3. Department of Oncology, National Taiwan University and Hospital, Taiwan;
4. Institute of Medical Genomics and Proteomics, National Taiwan University College of Medicine, Taiwan;
5. Institute of Clinical Medicine, National Taiwan University College of Medicine, Taiwan;
6. Division of Cardiovascular Surgery, Department of Surgery, National Taiwan University Hospital, Taiwan.
Received 2017-10-7; Accepted 2018-2-20; Published 2018-5-9
Abstract

Rationale: Lapatinib (LAP) is a crucial alternative to trastuzumab upon the onset of drug resistance during treatment of metastatic human epidermal growth factor receptor 2-positive breast cancer. Like trastuzumab, LAP is commonly used alongside anthracyclines as a combination therapy, due to enhanced anti-cancer efficacy. However, this is notably associated with cardiotoxicity so it is imperative to understand the mechanisms driving this cardiotoxicity and develop cardioprotective strategies. To this end, here we utilize human pluripotent stem cell-derived cardiomyocytes (hPSC-CMs), which exhibit several characteristics representative of in vivo cardiomyocytes that make them breakthrough models to study drug toxicity.
Methods: We investigated LAP- and doxorubicin (DOX)-induced toxicity in hPSC-CMs and evaluated the involvement of inducible nitric oxide (NO) synthase (iNOS). The significance of iNOS-mediated cardiotoxicity was furthermore evaluated in animal studies.
Results: LAP synergistically increased DOX toxicity in hPSC-CMs in a dose- and time-dependent manner. At concentrations that were otherwise non-apoptotic when administered separately, LAP significantly potentiated DOX-induced hPSC-CM apoptosis. This was accompanied by increased iNOS expression and pronounced production of NO. iNOS inhibition significantly reduced hPSC-CM sensitivity to LAP and DOX co-treatment (LAP-plus-DOX), leading to reduced apoptosis. Consistent with our observations in vitro, delivery of an iNOS inhibitor in mice treated with LAP-plus-DOX attenuated myocardial apoptosis and systolic dysfunction. Moreover, inhibition of iNOS did not compromise the anti-cancer potency of LAP-plus-DOX in a murine breast cancer xenograft model.
Conclusions: Our findings suggest that iNOS inhibition is a promising cardioprotective strategy to accompany HER2-inhibitor/anthracycline combination therapies. Furthermore, these results support the promise of hPSC-CMs as a platform for investigating cardiotoxicity and developing cardioprotectants as a whole.
Keywords: HER2 inhibitor, cardiotoxicity, doxorubicin, iPSC, iNOS
Introduction
Cardiotoxicity is a prevalent side-effect following anti-cancer therapy, and is notoriously associated with the use of anthracyclines (ANTs) and human epidermal growth factor receptor 2 (HER2)-inhibitors. In recent years, HER2 has attracted great attention as a drug discovery target for breast and lung cancers due to the association of its overexpression with progression of malignancy and poor prognosis [1,2]. Amongst the many HER2-targeted therapies that have successfully reached the market, trastuzumab (TraZ) and lapatinib (LAP) are two of the most studied and clinically applied [3,4]. Furthermore, it has been shown that significantly better therapeutic outcomes can be achieved when HER2-inhibitors are administered in combination with ANT-based chemotherapy, such as doxorubicin (DOX) [5]. Importantly, combining HER2-inhibitors with DOX may be particularly useful in patients with previous ANT exposure to reduce the cumulative dose of DOX. However, for both TraZ [5] and LAP [6], this combination unfortunately leads to an increased risk of cardiotoxicity.
In this study, we focus specifically on the interaction between LAP and DOX for several reasons. First, LAP, which blocks both HER1 and HER2 [7], can potentially be used for other types of cancer as opposed to TraZ, which only targets HER2. Second, LAP works by reversible binding to the intracellular ATP-binding site of HER1 and HER2 tyrosine kinases, thus enabling it to target TraZ-resistant tumors that express truncated versions of HER2 [8]. Indeed, cancer patients at this stage often will have already undergone multiple courses of treatment, making them more prone to incidental adverse effects. Third, LAP is an oral medication that negates the need for frequent clinical visits over the course of a treatment regimen. However, reduced monitoring by medical professionals subsequently makes it difficult to catch cardiotoxicity early on, and prevent unnecessary patient risk exposure. Hence, cardioprotective strategies for use prior to and early in the administration of LAP, especially in conjunction with DOX, need to be developed.
It is well established that development of oxidative stress is a major mechanism of DOX-induced cell death in the myocardium [9]. This results from the formation of reactive nitrogen- and oxygen- species (RNS and ROS, respectively) [10], most notably peroxynitrite formed from nitric oxide (NO) and superoxide (O2-). NO is a negative regulator of β-adrenergic chronotropic and inotropic effects through the elevation of intracellular cGMP [11], and can also directly trigger cardiomyocyte apoptosis—a pivotal component of heart failure [12]. On the other hand, HER2 plays a crucial role during protection against oxidative stress and, accordingly, HER2 can protect cancer cells from apoptosis by suppressing iNOS expression and NO production [13]. In congruence with this mechanism, patients treated with an antibody against HER2 display increased cardiomyocyte death through a ROS-dependent mechanism [14]. Interestingly, it has been shown that DOX treatment actually induces the expression of HER2 in the rat heart, implying that HER2 upregulation might serve to counteract oxidative stress induced by DOX [15]. Indeed, activation of HER2 signaling by the ligand neuregulin-1β has been shown to protect against DOX-induced cardiotoxicity [16]. These lines of evidence suggest that drug therapies targeting HER2 may therefore be inhibiting the endogenous antioxidant systems necessary for maintaining normal heart function, especially in conditions of oxidative stress triggered by DOX. Since iNOS signaling is a major inducer of the ROS and RNS cascades, it is postulated that iNOS could represent the basis by which HER2 inhibitors can potentiate DOX-induced cardiotoxicity.
Cardiotoxicity can manifest at the level of acute mechanical dysfunction down to changes to intracellular organelles, cumulatively leading to loss of cardiomyocyte viability and the onset of cardiomyopathy. As such, choice of model system when probing the many causes and mechanisms of cardiotoxicity becomes crucial. Recent advances in large-scale cardiomyocyte (CM) production from human embryonic stem cells (hESC) and induced pluripotent stem cells (hiPSC), together termed human pluripotent stem cell-derived cardiomyocytes (hPSC-CMs), gives rise to the opportunity to establish a human in vitro model system capable of recapitulating drug-induced toxicities observed in patients [10,17,18]. This is because hPSC-CMs exhibit many characteristics of in vivo cardiomyocytes including appropriate ion channel, receptor and transporter expression, syncytial and contractile activities, and physiologically relevant electrophysiological and biochemical responses upon exposure to environmental stimuli. Indeed, Burridge and colleagues demonstrated the fidelity of hPSC-CMs to stratify patient susceptibility to DOX-induced cardiomyopathy [10]. Ultimately, it may be possible to generate safety indices for specific drugs by combining insights gained from in vitro assays utilizing patient-specific hiPSC-CMs with clinical and FDA-documented data [19]. In this study, we investigated the ability of LAP-plus-DOX to trigger apoptosis in hPSC-CMs, and the role of iNOS in this effect. We then explored cardiomyocyte apoptosis as a driver of cardiotoxicity following drug treatment in mice. Furthermore, we examined whether inhibition of iNOS could attenuate cardiotoxicity induced by LAP-plus-DOX without compromising the anti-cancer potency of the treatment in a murine SK-BR-3 breast cancer xenograft model.
Methods
Human induced pluripotent stem cell generation
All protocols were approved by the Institutional Review Board of Biomedical Science Research at Academia Sinica (approval number AS-IRB01-104027). All human iPSC cell lines were generated by the Taiwan iPSC Consortium. Blood samples from healthy donors were obtained, cryopreserved and reprogrammed after informed written consent. iPSCs were generated from cryopreserved peripheral blood mononuclear cells with OKSM (Oct4, Klf4, Sox2 and c-Myc) factors delivered using Sendai virus. Upon successful reprogramming on mouse embryonic fibroblasts, human iPSCs were maintained on hESC-qualified Matrigel (BD Biosciences)-coated plates in E8 medium (STEMCELL Technologies) until they reached 80-90% confluency.
Cardiomyocyte differentiation of human ESC and iPSC
A hESC cell line, RUES2, was obtained under license from the Rockefeller University. Undifferentiated hESCs and hiPSCs were grown in E8 (Stem cell Technologies) on Matrigel-coated plates and passaged every 4 days using Accutase (Innovative Cell Technologies). Cells were differentiated into CMs by sequential targeting of the Wnt pathway [20]. Following differentiation, CM purity was determined by flow cytometry analysis (>90% cTnI+).
Compound exposure and toxicity evaluation
hPSC-CMs were treated with either LAP (Selleckchem) or DOX (Selleckchem) alone, or in combination with each other, in the presence or absence of N6- (1-iminoethyl)-L-lysine (L-NIL; Cayman) for 24, 48 or 72 h as indicated. In some experiments, LAP was replaced with another HER2 inhibitor, either TraZ (Genentech) or afatinib (Selleckchem). In addition, human umbilical vein endothelial cells (HUVECs, Food Industry Research and Development Institute, Taiwan) and non-cardiac human embryonic kidney cells 293 (HEK293, ATCC) were used to evaluate cell specificity of LAP-plus-DOX toxicity. Cell viability was assessed by Trypan Blue (Bio-Rad) exclusion assay and TetraZ cell counting kits (BioLegend).
Annexin V and 7-AAD staining
After drug treatment, cells were dissociated with TrypLE express (Gibco). For detection of apoptosis, cells were stained with 7-aminoactinomycin D (7-AAD) and annexin V (both from BioLegend) following the manufacturer's instruction. Stained cells were then washed twice, resuspended in cold buffer, and analyzed using a FACSCalibur cell analyzer. The results were processed using CellQuest software (BD Biosciences).
Measurement of iNOS activity
iNOS activity in cell culture supernatants and cardiac tissue was determined as a function of total nitric oxide product formation (nitrate + nitrite), using a colorimetric enzymatic nitric oxide assay kit (Cayman).
Intracellular staining of inducible nitric oxide synthase (iNOS)
To stain for iNOS expression, cells were fixed with 4% paraformaldehyde (eBioscience) followed by permeabilization with 0.1% saponin (eBioscience) and subsequent incubation with anti-iNOS (Abcam) and corresponding isotype control.
Mitochondrial bioenergetics analysis
Oxygen consumption rate (OCR) was assessed using a XF96 Extracellular Flux Analyzer (Seahorse Bioscience, Agilent Technologies). hPSC-CMs were seeded at densities of 1,000, 5,000, or 10,000 iPSC-CMs per well to determine the optimal cell number. A seeding density of 10,000 hPSC-CMs per well was chosen to conduct the experiments as this gave rise to a uniform monolayer with linear OCR. Prior to analysis, cells were incubated for 1 h in unbuffered RPMI (Life Technologies). OCR was obtained by plotting change in oxygen concentration over time and obtaining the gradient. After establishing baseline OCR, additional measurements were made following sequential automatic injections of a final concentration of 1 μM oligomycin (an ATP synthase inhibitor), 0.5 μM carbonyl cyanide p-[trifluoromethoxy]-ohenylhydrazone (FCCP, a mitochondrial uncoupler), and 0.5 μM rotenone/antimycin A (mitochondrial electron transport chain complex III inhibitor). Spare respiratory capacity was determined as maximal OCR minus baseline OCR.
LAP and DOX treatment protocol in mice
All mice were housed in individually ventilated cages (IVCs) at the animal core facility at the Institute of Biomedical Sciences, Academia Sinica. All procedures involving animals were carried out in accordance with the Guides for the Use and Care of Laboratory Animals (ARRIVE guidelines) and all animal protocols were approved by the Institutional Animal Care and Use Committee of Academia Sinica, Taiwan. Male C57BL/6 mice (~8 weeks) were injected with a cumulative dose of up to 10 mg/kg LAP and/or DOX, or equivalent volume of vehicle control via intraperitoneal (i.p.) injections. Subsequent analyses were performed 7 days after drug injection. No mortality was associated with this dose regimen. For ex vivo analyses, mice were sacrificed by Zoletil (Virbac) overdose and the hearts were excised and either frozen in liquid nitrogen and stored at -80 °C or fixed in 10% neutral-buffered formalin solution for further studies. A separate cohort of mice was treated with L-NIL (1, 5, or 10 mg/kg i.p.) on top of LAP-plus-DOX.
Heart extraction and cell dissociation
The hearts were excised upon sacrifice from mice administered with heparin (100 IU/kg, i.p.) and anaesthetized with Zoletil (80 mg/kg, i.p.). The continually beating hearts were rinsed by immersion in ice-cold Hank's Balanced Salt Solution (HBSS, Gibco) immediately after removal. Freshly isolated whole left-ventricles were minced and dissociated into single-cell suspension with a cocktail of collagenase II (600 U/mL, Sigma-Aldrich) and DNase I (60 U/mL, AppliChem) in HBSS. After incubation at 37 °C for 60 min, with mechanical dissociation applied every 15 min, the cell-suspension was centrifuged, resuspended in cold HBSS and passed through cell strainers (70 μm mesh size, BD Falcon) to remove non-dissociated clumps. The cells were then pelleted by centrifugation at 300 ×g for 10 min and the red blood cells were removed from the sample using ACK lysing buffer (NH4Cl 150 mmol/L, Wako Pure Chemical Industries, Ltd.; KHCO3 10 mM, J.T. Baker; EDTA 0.01 mmol/L, Sigma-Aldrich).
Measurement of caspase-3 activity
Cardiac lysates were centrifuged at 10,000 ×g and protein concentrations were determined. Protein extracts were used for each assay reaction using a caspase-3 colorimetric assay kit (BioVison). The assay is based on spectrophotometric detection of the chromophore p-nitroaniline (pNA) after cleavage from the labeled substrate DEVD-pNA. pNA light emission, which indicates caspase-3 activity, was quantified using a microtiter plate reader at 405 nm.
Echocardiographic assessment of cardiac function
Cardiac function for all treatment groups was assessed using noninvasive transthoracic echocardiography at 7 days after drug administration. Mice were anesthetized with 2% isoflurane, placed on a temperature-controlled heating pad, and had their chest shaved. Echocardiography was performed using a GE LOGIQ Ultrasound system with an i12L-RS linear array transducer probe. Left-ventricular internal dimension-diastole (LVIDd), left-ventricular internal dimension-systole (LVIDs), fractional shortening (LVIDd - dLVIDs / LVIDd × 100), and ejection fraction (left-ventricular volume at end diastole (EDV) - left-ventricular volume at end systole (ESV) / EDV) were calculated using the Teichholz formula at the mid- papillary muscle level in the short-axis view.
Detection of apoptosis by TUNEL assay
Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) was performed using ApopTag® Fluorescein in situ apoptosis detection kit according to the manufacturer's instructions (Millipore). The percentage of TUNEL-positive cells was calculated as follows: (number of TUNEL-positive cells / total number of nuclei) × 100.
Western blot
Cytoplasmic and nuclear protein were extracted using the Nuclear and Cytoplasmic Protein Extraction Kit (Thermo Scientific). Myocardial protein extracts were resolved by SDS/PAGE and transferred onto a polyvinylidene difluoride membrane (Millipore). Membranes were blocked with 5% milk in Tris-buffered saline with Tween 20 for 2 h at 37 °C and then incubated overnight at 4 °C with primary antibodies. The primary antibodies used for all Western blots were ErbB2/HER2 (Cell signaling), phosphatidylinositol 3 kinase (PI3K, Cell signaling), phospho-PI3K (Cell signaling), protein kinase B (Akt, Cell signaling), phospho-Akt (Ser473) (Cell signaling), phospho-IκBα (Cell signaling), nuclear factor kappa B (NF-κB, Cell signaling), iNOS (Abcam), histone (Cell signaling), or β-actin (GTX-109639, GeneTex).
Immunohistochemistry
Paraformaldehyde-fixed tissue sections were subjected to antigen retrieval by heating in 10 mM sodium citrate buffer (pH 7.2) for 10 min in a microwave, then treated for 5 min in 3% hydrogen peroxide to block endogenous peroxidase activity. Tissue sections were incubated with antibodies against iNOS (Abcam) or Collagen I (Abcam) overnight at 4 °C. Isotype control antibodies were used in negative controls. Sections were subsequently developed with a 30 min incubation step with a polymer-HRP detection kit QD420-YIK (BioGenex) followed by 3,3'-diaminobenzidine detection.
Murine SK-BR-3 breast cancer xenograft model
Human breast cancer cell-line, SK-BR-3, was obtained from ATCC and maintained in Dulbecco's Modified Eagle's Medium (DMEM) with 10% FBS. For in vivo experiments, 6-8-week-old female BALB/c-nu mice were purchased from the Taiwan National Laboratory Animal Center. To inoculate the mice, a cell suspension of 100 μL of PBS containing 5x106 luciferase-transfected SK-BR-3 cells was subcutaneously injected into the bilateral fat pads surrounding the fourth mammary glands of the nude mice. Serial measurements of tumor photon flux from all SK-BR3-bearing nude mice were taken using an IVIS® Spectrum In Vivo Imaging System (PerkinElmer) at weekly intervals, starting from time of inoculation until sacrifice 42 days later. At each imaging time-point, Luciferin (150 mg/kg body weight) was administered into the peritoneal cavity.
Cardiac catheterization
Hemodynamics measurements were taken by catheterization with a pressure-volume catheter with a diameter of 1.4F and 0.8F along the catheter body (Millar Instruments, Houston, SPR‐839) at 42 days after inoculation with SK-BR-3 cells. The catheter was inserted into the right carotid artery and advanced to the left ventricle (LV). One milliliter of blood was drawn from the heart to calibrate electrical conductivity for volume conversion. Baseline LV pressure‐volume loops were recorded after stabilization, and the inferior vena cava was transiently compressed through an incision in the upper abdomen to obtain occlusion loops. The pressure-volume loops with volume calibration were analyzed with commercial software (PVAN3.2; Millar Instruments).
Qualification of cardiac troponin I concentration
For analysis of cardiac troponin I levels, blood was drawn from the anesthetized mouse, after which animals were sacrificed by cervical dislocation. Samples were centrifuged at 1700 ×g for 10 min at 4 °C and stored at -80 °C. Levels of cardiac troponin I were measured using the High Sensitive Mouse Cardiac Troponin I ELISA Kit (Life Diagnostics).
Gene expression analyses via real-time RT-PCR
Total RNA was purified from myocardial tissue using the RNeasy Mini Kit (Qiagen) and reverse transcribed to cDNA using MMLV reverse transcriptase (Promega). Real-time PCR reactions were performed in triplicate on a StepOne® Real-Time PCR System (Applied Biosystems) using the LightCycler® FastStart DNA MasterPLUS SYBR Green I kit (Roche) and oligonucleotide primers specific to target genes. The mixture was incubated at 95 °C for 10 min followed by 40 cycles of 95 °C for 10 s, 60 °C for 15 s, and 72 °C for 10 s. Quantitative data was obtained by calculating the crossing point (CP) value, or the turning point that corresponded to the first maximum of the second derivative curve, using the LightCycler3 data analysis software. The following primers were used: β-actin, f: 5'-GCATTGCTGACAGGATGCAG-3', r: 5'-CCTGCTTGCTGATCCACATC-3'; atrial natriuretic peptide (ANP): f: 5'-CCTAAGCCCTTGTGGTGTGT-3', r: 5'-CAGAGTGGGAGAGGCAAGAC-3'; brain natriuretic peptide (BNP): f: 5'-CTGAAGGTGCTGTCCCAGAT-3', r: 5'-CCTTGGTCCTTCAAGAGCTG-3'. mRNA levels of β-actin were taken for normalization. Relative mRNA levels in untreated controls were set to 1.0.
Statistical analysis
All data are presented as mean ± SD. Group differences were assessed by t-test, and three or more data sets were compared using one-way ANOVAs with Bonferroni's multiple comparison post-hoc test for measuring significance. P-values of P < 0.05 were considered statistically significant.
Results
LAP-plus-DOX combined treatment results in synergistically increased toxicity in hPSC-CMs, in a dose- and time-dependent manner
hPSC-CMs were generated from four independent cell-lines and demonstrated expression of sarcomeric proteins such as α-actinin (Figure 1A) and cardiac troponin I (cTnI; Figure 1B). In addition, HER2 expression was confirmed in these cardiomyocytes using flow cytometry (Figure 1C). Exposure of hPSC-CMs to DOX for 24 h resulted in a dose-dependent decrease in cell viability compared to vehicle-treated control cells, with an IC50 of 4.3 and 58.2 μM for iPSC-CM and RUES2-CM, respectively (Figure 1D). Based on the pharmacokinetic characteristics of LAP in humans, i.e., steady-state plasma peak (Cmax) and trough (Cmin) concentrations for a daily dose of 1200 mg being 2.7 and 0.8 μM, respectively [21], we treated hPSC-CMs with LAP at concentrations of 0.05-5 μM in conjunction with DOX at 3 different concentration (0.5, 1.0, and 2.5 μM) to model combined treatment (LAP-plus-DOX). In line with a previous study which examined cytotoxicity after 72 h of LAP treatment in five healthy control hiPSC-CM lines [19], LAP alone was not highly cytotoxic in hPSC-CMs at concentrations of 0.05-5 μM (Figure 1E). At concentrations of DOX above 1.0 μM, combined treatment with LAP at concentrations above 2.5 μM significantly reduced the viability of hPSC-CMs compared to DOX alone (Figure 1E). Therefore, we selected LAP (2.5 μM) plus DOX (1.0 μM) to investigate the toxic effects on hPSC-CMs over time. A treatment duration of 24 to 72 h was chosen based on the half-life of these drugs in humans being 14-24 h for LAP and 20-48 h for DOX. For each time-point, treatment of hPSC-CMs with LAP alone, i.e., in the absence of DOX, did not have a significant impact on the percentage of cell death (Figure 1F). However, LAP-plus-DOX synergistically decreased the viability of hPSC-CMs (Figure 1F), indicating that LAP potentiated DOX-induced toxicity. We next substituted LAP with another HER2 inhibitor, either TraZ or afatinib, to investigate whether synergistic toxicity was a drug-class effect. At concentrations of DOX above 1.0 μM, combined treatment with TraZ at concentrations above 100 μg/mL significantly reduced the viability of hPSC-CMs compared to DOX alone (Figure 1E). However, neither afatinib alone nor afatinib-plus-DOX induced significant toxicity (Figure 1E). Interestingly, LAP potentiation of DOX-induced toxicity appeared to be cell-type specific as combined treatment did not appear to synergistically decrease the viability of HUVEC or HEK293 cells (Figure 1F).
Human iPSC-derived cardiomyocytes are capable of modeling the synergistic cardiotoxicity of lapatinib (LAP)-plus-doxorubicin (DOX) combination treatment. (A) Representative phase contrast (left) and sarcomeric protein immunostaining (right) images of human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs). ACTN1: actinin alpha 1. Scale bars: 100 μm and 20 μm. (B-C) Flow cytometry analysis of cardiac troponin I (cTnI) and human epidermal growth factor receptor-2 (HER2) expression in hiPSC-CMs. (D) Effect of DOX treatment (after 24 h) on viability of hiPSC-CM and RUES2-CM using a TetraZ-based dye assay. (E) Quantification of cell viability in a representative hiPSC-CM line treated with LAP-plus-DOX, LAP-plus-trastuzumab, or LAP-plus-afatinib at different pharmacological plasma level concentrations. (F) Effects of LAP (2.5 μM) on DOX (1 μM)-induced hiPSC-CM, RUES2-CM, HUVEC, and HEK293 toxicity at different time-points based on quantification of cell viability. (G) Representative images of early (annexin V+) and late (annexin V+7-AAD+) apoptotic hiPSC-CMs 24 h after treatment with LAP-plus-DOX using image-in-flow cytometry. (H) Pro-apoptotic effects of LAP-plus-DOX on hPSC-CMs based on flow cytometry analysis of annexin V+ and 7-AAD+ cells. All data are presented as mean ± SD (n≥3). *P < 0.05 vs. control; † P < 0.05 vs. DOX.
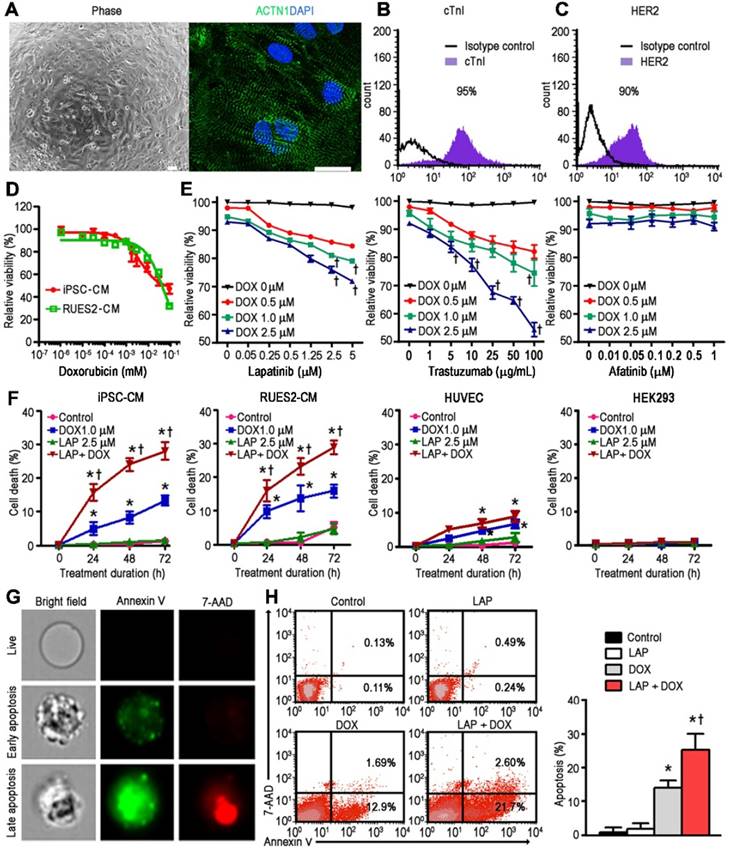
A non-apoptotic concentration of LAP potentiates DOX-induced hPSC-CM apoptosis
Next, we selected the 24 h treatment time-point to elucidate the mechanism behind the synergistic toxicity of LAP-plus-DOX combined treatment. Positive Annexin V staining precedes the loss of membrane integrity during the early stages of programmed cell death (Figure 1G). Viable and early-apoptotic cells with intact membranes exclude 7-AAD, whereas the membranes of late-apoptotic cells are permeable to both Annexin V and 7-AAD (Figure 1G). Annexin V-FITC and 7-AAD double-positive staining showed that DOX treatment could induce apoptosis in hPSC-CMs, as opposed to vehicle control and the LAP-treated groups (Figure 1H). Notably, whilst LAP by itself induced very little toxicity, the rate of apoptosis in the LAP-plus-DOX group was significantly higher than in the DOX alone-treated group (Figure 1H). These findings suggest that LAP treatment may enhance the sensitivity of hPSC-CMs to DOX-induced toxicity, mainly through synergistically increased apoptosis.
L-NIL-mediated inhibition of iNOS in hPSC-CMs reduces synergistic apoptosis induced by LAP-plus-DOX combined treatment
With previous studies demonstrating that DOX-induced cardiomyocyte death is mediated by NO production [22], we next aimed to clarify whether LAP-plus-DOX combined treatment had an effect on pathways related to these findings. Using flow cytometry, cTnI+-gated cells were subsequently assessed for iNOS-expression and a synergistic effect of LAP-plus-DOX on iNOS expression was observed (Figure 2A). Whilst LAP alone slightly increased NO production, the addition of LAP significantly increased DOX-induced NO production (Figure 2B). Thus, we further investigated whether L-N6-(1-iminoethyl)-lysine (L-NIL), a selective iNOS inhibitor that competes with arginine for the active site of iNOS, could concomitantly suppress apoptosis and NO production in LAP-plus-DOX-treated cardiomyocytes. At a dose range of 5 to 25 μM, L-NIL significantly attenuated apoptosis in the LAP-plus-DOX group compared to the DOX-alone group (Figure 2C-D). This effect was consistent with lower iNOS activity, as indicated by reduced production of NO (Figure 2E), and could also be observed in TraZ-plus-DOX-treated cardiomyocytes (Figure S1). Intriguingly, L-NIL-mediated cardioprotection still occurred in hiPSC-CMs treated with DOX alone, suggesting that L-NIL protects against apoptosis at the level of NO (Figure 2C-E). As one of the most potent regulatory effects of NO is the slowing of respiration through cytochrome c oxidase inhibition [23], we subsequently investigated the role of iNOS inhibition on mitochondrial oxygen consumption in vitro. We found that LAP-plus-DOX caused a decrease in respiration rate, which could be reversed upon supplementation with L-NIL (Figure 2F). Taken together, these findings suggest that LAP may potentiate DOX-induced apoptosis and mitochondrial dysfunction through iNOS-dependent nitrosative stress, and that NOS inhibitors can be used to restore mitochondrial respiration and cell viability.
N6-(1-iminoethyl)-L-lysine (L-NIL)-mediated inhibition of inducible nitric oxide synthase (iNOS) in hPSC-derived cardiomyocytes decreases LAP-plus-DOX-induced toxicity. (A) Cytoplasmic iNOS protein expression as determined in hiPSC-CMs treated with vehicle (Control), LAP alone (2.5 μM), DOX alone (1.0 μM), or both LAP-plus-DOX. Cells were labeled with anti-iNOS, or an isotype-matched antibody. Percentage of iNOS-expressing cells are displayed as representative flow cytometry histogram plots (left) and quantitative analysis (right). (B) iNOS activity, as a function of total nitric oxide product formation (nitrate + nitrite), was measured using a colorimetric assay. (C) Proportion of viable, apoptotic and necrotic cells, based on flow cytometry analysis of 7-AAD and Annexin V co-labeling. (D) Quantification of fold changes in apoptosis upon L-NIL co-administration. (E) Quantification of L-NIL-mediated iNOS inhibition based on total nitric oxide product formation. (F) Mitochondrial function of hiPSC-CMs cultured in each experimental group. Oxygen consumption rate (OCR) of hiPSC-CMs demonstrating basal OCR and spare OCR respiratory capacity measured after consecutive addition of oligomycin, FCCP, and antimycin. (G) Flow cytometry analysis of HER2 expression in hiPSC-CMs. (H) Effects of L-NIL on LAP-plus-DOX-induced HER2 activity, PI3K/Akt activation, IκBα phosphorylation, and nuclear translocation of NF-κB p65 subunit. All data are presented as mean ± SD (n≥3). *P < 0.05 vs. control; † P < 0.05 vs. DOX; ‡ by P < 0.05 vs. LAP + DOX.
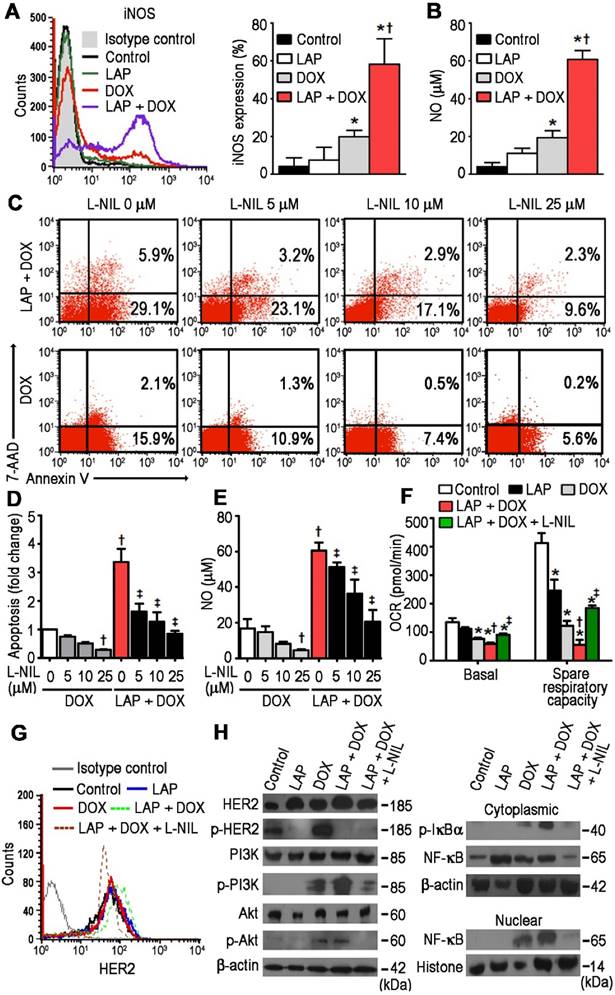
L-NIL-mediated inhibition of iNOS in hPSC-CMs abolishes activation of PI3K/Akt and NF-κB signaling pathways induced by LAP-plus-DOX combined treatment
To further investigate the mechanism behind iNOS inhibition in the context of LAP-plus-DOX cardiotoxicity, HER2 and its downstream signaling were investigated. L-NIL inhibition of iNOS did not affect the expression (Figure 2G) or phosphorylation (Figure 2H) of HER2. Next, we examined PI3K/Akt signaling, which is mutually regulated by iNOS and HER2 signaling. We found that LAP-plus-DOX promoted PI3K and Akt phosphorylation whereas iNOS inhibition downregulated PI3K/Akt activity (Figure 2H). Given that PI3K/Akt signaling is a critical regulator of NF-κB activation [24], the possible inhibitory effects of L-NIL on IκB phosphorylation were further investigated. Whilst LAP-plus-DOX augmented phosphorylation of IκB, this effect was repressed by L-NIL (Figure 2H). To further confirm whether PI3K/Akt signaling contributes to LAP-plus-DOX-induced NF-κB activity, we also examined NF-κB nuclear translocation and found that LAP-plus-DOX-induced NF-κB p65 nuclear translocation was inhibited by L-NIL. Taken together, our results show that the PI3K/Akt/NF-κB signaling axis is involved in LAP-plus-DOX-mediated cardiotoxicity and can be modulated by iNOS inhibition.
L-NIL-mediated inhibition of iNOS attenuates myocardial apoptosis and systolic dysfunction in mice administered with LAP-plus-DOX combined treatment
To confirm whether our findings of increased apoptosis upon LAP-plus-DOX treatment in hPSC-CMs could be applied in vivo, mice were divided to receive i.p. injections of either LAP (LAP1: 1 mg/kg, LAP5: 5 mg/kg, or LAP10: 10 mg/kg), DOX (DOX2: 2 mg/kg, DOX5: 5 mg/kg, or DOX10: 10mg/kg), or LAP-plus-DOX (LAP1 + DOX 5, LAP5 + DOX5, or LAP10 + DOX5). Preliminary studies in wild-type mice indicated that the Cmax concentration after a single 10 mg/kg i.p. bolus dose ranged from 1000 to 4000 ng/mL (Figure S2), which is comparable with human plasma Cmax values (4.2 μM or 2432 ng/mL) after a daily oral 1250 mg dose. At 72 h after treatment, hearts were harvested, digested and the isolated cardiomyocytes were stained with annexin V and 7-AAD (Figure 3A). We found that co-treatment of mice with LAP10 plus DOX5 (combo) significantly increased the percentage of apoptotic cardiomyocytes compared to those treated with DOX5 alone (Figure 3B). This dose-regimen was therefore selected for following experiments investigating the cardioprotective potential of iNOS inhibition on LAP-plus-DOX-mediated cardiotoxicity. We found that treatment with L-NIL could attenuate combo-mediated apoptosis in mice, also in a dose-dependent manner (Figure 3B). Out of three dose-regimens tested, a 10 mg/kg dose of L-NIL exhibited the strongest response. Moreover, a time-course study revealed that activation of caspase-3 by DOX, which was further augmented by co-treatment with LAP, was attenuated in response to L-NIL rescue (Figure 3C). In parallel, we observed a downregulation in the number of TUNEL-positive cells at 7 days in combo treated mice receiving L-NIL (Figure 3D), which was associated with preserved left-ventricular function (Figure 3E). Hematoxylin and eosin staining evaluation of myocardial immune infiltration showed that DOX induced interstitial mononuclear infiltration, which was further enhanced when DOX was combined with LAP (Figure 3F). L-NIL reduced LAP-plus-DOX-stimulated cell infiltration, suggesting that iNOS inhibition counteracted the inflammatory cellular infiltrates evoked by combined treatment. Immunostaining revealed prominent iNOS expression in the hearts of combo-treated mice, whereas expression was weaker in LAP- or DOX alone-treated hearts (Figure 3F). Interestingly, immunostaining of iNOS showed that expression was localized not only in cardiomyocytes but could also be observed in infiltrating cells in the myocardium (Figure S3). Notably, no iNOS expression was observed in control hearts (Figure 3F). Western blots similarly showed significantly increased levels of iNOS protein in combo-treated mice, with a lesser extent in LAP- or DOX alone-treated mice (Figure 3G). Consistently, LAP or DOX alone induced NO generation in the heart, which was potentiated upon combining the drugs (Figure 3H). As expected, treatment with L-NIL did not alter expression of iNOS protein levels in the myocardium (Figure 3F-G) but instead decreased NO production, thus confirming inhibition of iNOS activity (Figure 3H).
L-NIL-mediated iNOS inhibition attenuates myocardial apoptosis and systolic dysfunction in mice treated with LAP-plus-DOX. (A) Representative flow cytometry scatter plots and (B) statistical analysis of annexin V+ and 7-AAD+ cells from digested hearts of mice treated with different dose-regimens of LAP-plus-DOX. (C) Caspase-3 activity assay performed on ventricular extracts from hearts of mice treated with different drug-regimens for 0 to 7 days as indicated. (D) TUNEL immunostaining of left-ventricular sections at 7 days after drug administration. Percentages of TUNEL-expressing cTnI+ cells (white arrows) are displayed as representative images and cumulative data. TUNEL, red; cTnI, green; DAPI, blue. Scale bar: 10 μm. (E) Echocardiographic measurements of mice treated with different drug-regimens taken 7 days after commencement of treatment. (F) Hematoxylin and eosin-staining (left) and immunohistochemical localization of iNOS expression (right) in myocardial sections. Scale bar: 50 μm. (G) Western blots showing iNOS expression and (H) NO levels in the myocardium. In Figures (D-H): Control = vehicle; LAP = lapatinib 10 mg/kg; DOX = doxorubicin 5 mg/kg; Combo = LAP 10 mg/kg + DOX 5 mg/kg; L-NIL = vehicle + L-NIL 10 mg/kg; Combo + L-NIL = LAP 10 mg/kg + DOX 5 mg/kg + L-NIL 10 mg/kg. LVEF: left-ventricular ejection fraction; FS: fractional shortening; EDV: end-diastolic volume; ESV: end-systolic volume. All data are presented as mean ± SD (n≥4). *P < 0.05 vs. control; † P < 0.05 vs. DOX; ‡ by P < 0.05 vs. Combo.
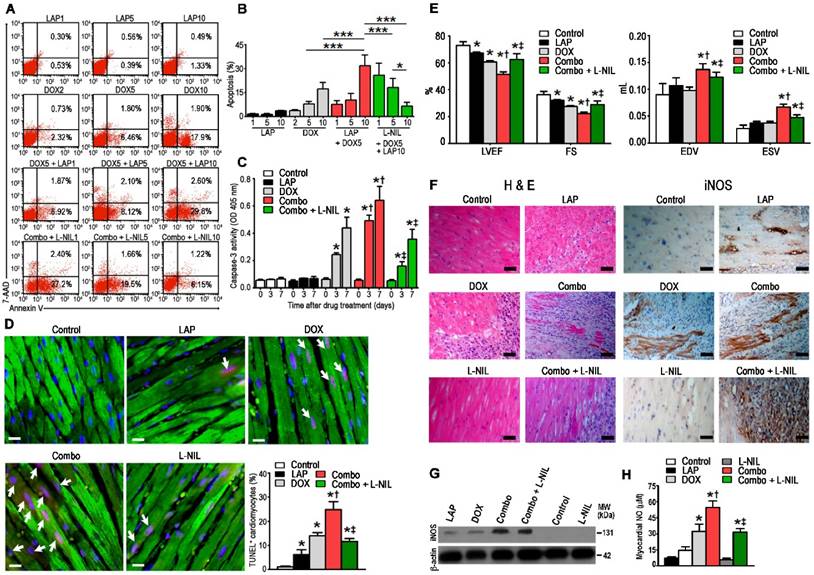
L-NIL-mediated cardioprotection does not compromise the anti-cancer potency of LAP-plus-DOX combined treatment
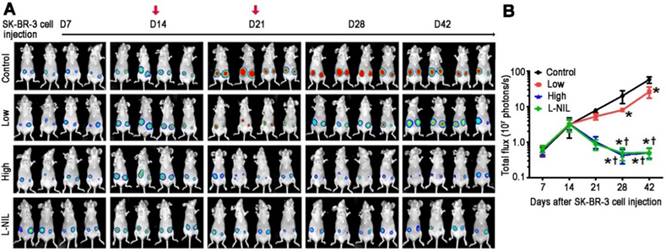
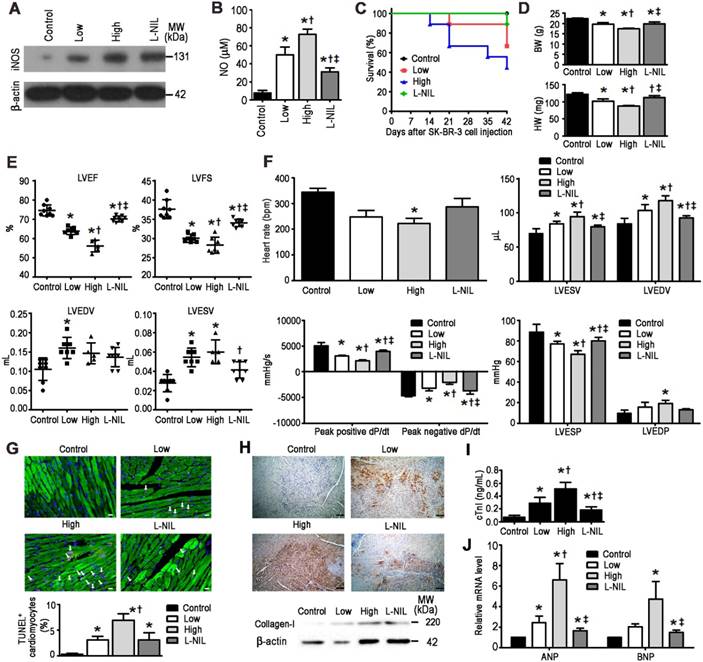
To investigate the possibility that L-NIL-mediated cardioprotection may compromise the anti-cancer potency of LAP-plus-DOX combined treatment, we administered the drugs in a murine xenograft model of SK-BR-3 breast cancer. A non-invasive In Vivo Imaging System (IVIS) was used to follow the induction of tumorigenesis and treatment effects in the xenograft model (Figure 4A). Serial images were taken from each experimental group after SK-BR-3 cell inoculation until sacrifice at 42 days after inoculation. In the control group, increases in bioluminescence were observed in the weeks following inoculation, indicating successful tumorigenesis. As expected, this was significantly attenuated in the LAP-plus-DOX group (Figure 4B), with high dose LAP-plus-DOX (LAP 10 mg/kg + DOX 10 mg/kg) exerting stronger inhibitory effects on tumor progression than low dose LAP-plus-DOX (LAP 5 mg/kg + DOX 5 mg/kg). This therapeutic effect lasted the entire 42 days after induction of tumorigenesis, thus validating the anti-cancer efficacy of LAP-plus-DOX in our xenograft model. We then co-administered L-NIL with high dose LAP-plus-DOX to investigate whether there would be any impact on LAP-plus-DOX anti-cancer efficacy. Crucially, L-NIL did not appear to compromise the effectiveness of LAP-plus-DOX combined treatment, based on comparing the inhibition of tumor progression between high dose LAP-plus-DOX in the presence (L-NIL group) or absence of L-NIL co-administration. This suggests that iNOS inhibition does not interfere with the efficacy of high dose LAP-plus-DOX (Figure 4B). Furthermore, we evaluated the degree of cardiotoxicity in the tumor-bearing mice based on animal survival rate (Figure 5C), plasma levels of cTnI (Figure 5D), echocardiography (Figure 5E), and catheter hemodynamic measurements (Figure 5F). As expected, although high dose LAP-plus-DOX exerted better anti-cancer effects, this was associated with marked cardiotoxicity. Increased mortality was observed in the LAP-plus-DOX-treated groups, which was associated with reduced body and heart weight (Figure 5D), as well as systolic dysfunction as measured by echocardiography (Figure 5E). Catheterization prior to sacrifice on day 42 similarly showed depressed LV contractility and relaxation in the LAP-plus-DOX groups, as evidenced by measurements of ±dP/dt and LV end-systolic and -diastolic pressure (LVESP/LVEDP) (Figure 5F). LV functional impairment was supported by observations of increased pathological severity in LAP-plus-DOX groups, such as increased cardiomyocyte apoptosis (Figure 5G), myocardial fibrosis (Figure 5H), plasma cTnI (Figure 5I) and gene expression of stress markers such as ANP and BNP (Figure 5J). Crucially, co-administration of L-NIL could distinctly alleviate the cardiotoxic effects of high dose LAP-plus-DOX treatment, as evidenced by significantly improved survival (Figure 5C) and protection against cardiac dysfunction (Figure 5E-F). Consistently, severity of myocardial apoptosis and fibrosis was attenuated upon L-NIL administration (Figure 5G-H), as well as the increases in plasma cTnI and gene expression levels of ANP and BNP (Figure 5I-J). Mechanistically, we observed that LAP-plus-DOX induced cardiotoxicity was associated with similar dose-dependent increases in iNOS expression and NO production, as confirmed by immunoblotting (Figure 5A) and colorimetric assay (Figure 5B), respectively. Co-administration of L-NIL resulted in a significant decrease in NO production, reflecting the cardioprotective effect observed. As expected, iNOS expression remained unaffected by L-NIL due to its mechanism of action in targeting the active site of iNOS.
L-NIL-mediated iNOS inhibition maintains anti-cancer potency of LAP-plus-DOX combined treatment in a murine SK-BR-3 breast cancer xenograft model. (A) Tumor progression displayed as representative serial, non-invasive In Vivo Imaging System images. (B) Time-course chart showing tumor size as determined by bioluminescent signal intensity in the region of interest, expressed in photons per second. Control = vehicle; Low = low dose of LAP (5 mg/kg) + DOX (5 mg/kg); High = high dose of LAP (10 mg/kg) + DOX (10 mg/kg); L-NIL = high dose of LAP + DOX + L-NIL (10 mg/kg). In (A), red arrows indicate the time points of drug administration. All data are presented as mean ± SD (n≥4). *P < 0.05 vs. Ctl; † P < 0.05 vs. Low; ‡ P < 0.05 vs. High.
L-NIL-mediated iNOS inhibition alleviates LAP-plus-DOX-induced cardiotoxicity in a murine SK-BR-3 breast cancer xenograft model. (A) Immunoblots indicating iNOS and (B) NO levels in the myocardium. Cardiotoxicity was evaluated by (C) animal survival rate, (D) body weight and heart weight, (E) echocardiography, (F) catheter-derived hemodynamic measurements, (G) TUNEL-expressing cTnI+ cells (white arrows), (H) collagen I expression, (I) plasma levels of cTnI, and (J) gene expression of stress markers. Scale bars indicate 10 μm in (G) and 50 μm in (H), respectively. Control = vehicle; Low = low dose of LAP (5 mg/kg) + DOX (5 mg/kg); High = high dose of LAP (10 mg/kg) + DOX (10 mg/kg); L-NIL = high dose of LAP + DOX + L-NIL (10 mg/kg). LVEF: left-ventricular ejection fraction; LVFS: left ventricular fractional shortening; LVEDV: left ventricular end-diastolic volume; LVESV: left ventricular end-systolic volume; LVESP: left ventricular end-systolic pressure; LVEDP: left ventricular end-diastolic pressure; ANP: atrial natriuretic peptide; BNP: brain natriuretic peptide. All data are presented as mean ± SD (n≥4). *P < 0.05 vs. Ctl; † P < 0.05 vs. Low; ‡ P < 0.05 vs. High.
Discussion
Our results demonstrate that hPSC-CMs can consistently recapitulate the synergistic cardiotoxicity of LAP-plus-DOX combination therapy. Whilst LAP alone, at its pharmacological plasma concentration (2.5 μM), displayed minimal effects on hPSC-CM cell viability, this non-toxic concentration of LAP significantly augmented DOX-induced apoptosis. Mechanistically, this was accompanied by up-regulation of iNOS and NO production. Following these human in vitro findings, we further showed that prophylactic therapy with an iNOS inhibitor, L-NIL, significantly reduced cardiotoxicity in a murine model of LAP-plus-DOX-induced cardiac dysfunction. Importantly, in a murine SK-BR-3 breast cancer xenograft model, we observed that co-administration of L-NIL with LAP-plus-DOX resulted in reduced cardiac injury and dysfunction without compromising the anti-cancer potency of the combination treatment. Therefore, although previous studies have already validated the cardioprotective effect of iNOS inhibition in the context of DOX alone-mediated cardiotoxicity [22], this study expands the potential scope of this strategy to include cardiotoxicity induced by LAP-plus-DOX combined treatment.
The use of trastuzumab over LAP, in combination with ANTs, has been shown to result in higher risk of heart failure. Relatively low rates of cardiotoxicity have been reported with LAP, regardless of whether used alone or in conjunction with ANTs as a combination therapy. However, our study clearly shows that combining LAP-plus-DOX results in synergistic induction of apoptosis in hPSC-CMs. Based on previous studies showing that LAP treatment significantly increases DOX accumulation in cancer cells and hepatocytes by inhibiting transporter drug efflux [25,26], it may also be possible that LAP facilitates the uptake and retention of DOX in cardiomyocytes. Therefore, LAP-plus-DOX synergistic cardiotoxicity may arise from simultaneous LAP-mediated inhibition of both DOX efflux and HER2 survival signaling, thereby exacerbating the severity of DOX-induced nitrosative stress and cardiac injury.
Moreover, LAP-plus-DOX-induced upregulation of iNOS expression was evident not only in vitro in hPSC-CMs (Figure 2A), but also in vivo in wild type mice (Figure 3F-G) and a breast cancer xenograft model (Figure 5A). The expression of myocardial iNOS has been associated with increased myocardial peroxynitrite, myocardial fibrosis, heart failure and cardiac sudden death [27]. This stress may induce the compensatory activation of multiple intracellular signaling pathways such as PI3K/Akt as shown by our data. Consistently, iNOS expression has been shown to be associated with increased oxidative stress [28] and subsequent activation of PI3K-dependent signaling via inactivation of tumor suppressor phosphatase and tensin homolog PTEN [29]. PI3K/Akt signaling has also been demonstrated to trigger NF-κB activation [24] and indeed, enhanced expression levels of phosphorylated PI3K/Akt and increased nuclear NF-κB were observed in our study (Figure 2H). Collectively, LAP-plus-DOX may activate PI3K/Akt/NF-κB signal transduction via iNOS, possibly leading to augmented inflammatory cell infiltration in the myocardium (Figure 3F) and subsequent cardiotoxicity.
Our findings that hPSC-CM can reproducibly model synergistic LAP-plus-DOX-induced cardiotoxicity show that there is considerable promise in using this in vitro human model to screen for new cardioprotectants. However, it is important to note that there are major differences between the cardiomyocyte microenvironment in our experimental set-up compared to that in the normal physiological context of the heart. In particular, treating a pure population of cardiomyocytes in vitro may not replicate the repertoire of processes occurring in vivo, given the lack of other cardiac cell types such as fibroblasts and endothelial cells. Regardless, our findings emphasize the importance of more studies and long-term follow-ups to fully elucidate the risk-benefit of combining LAP with ANT-based chemotherapies. Further insights into the molecular mechanisms behind drug-induced cardiotoxicity may be gained from using hiPSC-CMs generated from heart failure patients who have been treated with ANT and HER2-targeted drugs. Furthermore, gene editing may be utilized alongside iPSC reprogramming to explore the impact of genetic risk factors, such as the position of HER2 655 Val/Ile polymorphisms [30], in the pathology of HER2 inhibitor/ANT combination therapy-induced cardiotoxicity. These approaches will extend our findings to realize the goal of precision medicine for cardio-oncology and make these treatments safer for patients.
Abbreviations
7-AAD: 7-aminoactinomycin D; ANP: atrial natriuretic peptide; ANT: anthracycline; BNP: brain natriuretic peptide; CM: cardiomyocyte; cTnI: cardiac troponin I; DOX: doxorubicin; HER2: human epidermal growth factor receptor 2; hESC: human embryonic stem cell; hiPSC: human induced pluripotent stem cell; hPSC: human pluripotent stem cell; iNOS: inducible nitric oxide synthase; IVIS: In Vivo Imaging System; LAP: lapatinib; L-NIL: N6- (1-iminoethyl)-L-lysine; NF-κB: nuclear factor kappa B; PI3K: phosphoinositide 3-kinase; RNS: reactive nitrogen species; ROS: reactive oxygen species; TraZ: trastuzumab.
Supplementary Material
Supplementary methods and figures.
Acknowledgements
This work was supported by the Ministry of Science and Technology, Taiwan (MOST 106-3114-B-001-001 and MOST 106-2319-B-001-003), the National Health Research Institutes grant EX106-10512SI and the Academia Sinica Program for Technology Supporting Platform Axis (TSPA) Scheme. We thank the Pathology Core Laboratory, IBMS, Academia Sinica, for assistance in tissue embedding.
Ethics committee approval
All protocols were approved by the Institutional Review Board of Biomedical Science Research at Academia Sinica (approval number AS-IRB01-104027) and written informed consent was obtained from research subjects.
Competing Interests
Patrick Hsieh receives research grants from AstraZeneca, Gilead and Takeda, all of which did not participate in funding this work. The other authors report no conflicts.
References
1. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177-82
2. Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J. et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525-6
3. Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr, Davidson NE. et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353:1673-84
4. Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T. et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733-43
5. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A. et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783-92
6. Perez EA, Koehler M, Byrne J, Preston AJ, Rappold E, Ewer MS. Cardiac safety of lapatinib: pooled analysis of 3689 patients enrolled in clinical trials. Mayo Clin Proc. 2008;83:679-86
7. Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M. et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66:1630-9
8. Scaltriti M, Chandarlapaty S, Prudkin L, Aura C, Jimenez J, Angelini PD. et al. Clinical benefit of lapatinib-based therapy in patients with human epidermal growth factor receptor 2-positive breast tumors coexpressing the truncated p95HER2 receptor. Clin Cancer Res. 2010;16:2688-95
9. Zhao Y, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM. et al. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res. 2010;70:9287-97
10. Burridge PW, Li YF, Matsa E, Wu H, Ong SG, Sharma A. et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med. 2016;22:547-56
11. Nediani C, Raimondi L, Borchi E, Cerbai E. Nitric oxide/reactive oxygen species generation and nitroso/redox imbalance in heart failure: from molecular mechanisms to therapeutic implications. Antioxid Redox Signal. 2011;14:289-331
12. Song W, Lu X, Feng Q. Tumor necrosis factor-alpha induces apoptosis via inducible nitric oxide synthase in neonatal mouse cardiomyocytes. Cardiovasc Res. 2000;45:595-602
13. Simeone AM, Broemeling LD, Rosenblum J, Tari AM. HER2/neu reduces the apoptotic effects of N-(4-hydroxyphenyl)retinamide (4-HPR) in breast cancer cells by decreasing nitric oxide production. Oncogene. 2003;22:6739-47
14. Gordon LI, Burke MA, Singh AT, Prachand S, Lieberman ED, Sun L. et al. Blockade of the erbB2 receptor induces cardiomyocyte death through mitochondrial and reactive oxygen species-dependent pathways. J Biol Chem. 2009;284:2080-7
15. Gabrielson K, Bedja D, Pin S, Tsao A, Gama L, Yuan B. et al. Heat shock protein 90 and ErbB2 in the cardiac response to doxorubicin injury. Cancer Res. 2007;67:1436-41
16. Jay SM, Murthy AC, Hawkins JF, Wortzel JR, Steinhauser ML, Alvarez LM. et al. An engineered bivalent neuregulin protects against doxorubicin-induced cardiotoxicity with reduced proneoplastic potential. Circulation. 2013;128:152-61
17. Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y. et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation. 2013;127:1677-91
18. Maillet A, Tan K, Chai X, Sadananda SN, Mehta A, Ooi J. et al. Modeling Doxorubicin-induced cardiotoxicity in human pluripotent stem cell derived-cardiomyocytes. Sci Rep. 2016;6:25333
19. Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N. et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med. 2017;9:eaaf2584
20. Feaster TK, Cadar AG, Wang L, Williams CH, Chun YW, Hempel JE. et al. Matrigel Mattress: A method for the generation of single contracting human-induced pluripotent stem cell-derived cardiomyocytes. Circ Res. 2015;117:995-1000
21. Burris HA 3rd, Taylor CW, Jones SF, Koch KM, Versola MJ, Arya N. et al. A phase I and pharmacokinetic study of oral lapatinib administered once or twice daily in patients with solid malignancies. Clin Cancer Res. 2009;15:6702-8
22. Mukhopadhyay P, Rajesh M, Batkai S, Kashiwaya Y, Hasko G, Liaudet L. et al. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am J Physiol Heart Circ Physiol. 2009;296:H1466-83
23. Brown GC. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995;369:136-9
24. Cheng JC, Chou CH, Kuo ML, Hsieh CY. Radiation-enhanced hepatocellular carcinoma cell invasion with MMP-9 expression through PI3K/Akt/NF-kappaB signal transduction pathway. Oncogene. 2006;25:7009-18
25. Dai C, Ma S, Wang F, Zhao H, Wu X, Huang Z. et al. Lapatinib promotes the incidence of hepatotoxicity by increasing chemotherapeutic agent accumulation in hepatocytes. Oncotarget. 2015;6:17738-52
26. Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG. et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905-14
27. Mungrue IN, Gros R, You X, Pirani A, Azad A, Csont T. et al. Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J Clin Invest. 2002;109:735-43
28. Zhang P, Xu X, Hu X, van Deel ED, Zhu G, Chen Y. Inducible nitric oxide synthase deficiency protects the heart from systolic overload-induced ventricular hypertrophy and congestive heart failure. Circ Res. 2007;100:1089-98
29. Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003;22:5501-10
30. Fleishman SJ, Schlessinger J, Ben-Tal N. A putative molecular-activation switch in the transmembrane domain of erbB2. Proc Natl Acad Sci U S A. 2002;99:15937-40
Author contact
![]() Corresponding author: Patrick C.H. Hsieh, M.D., Ph.D. Institute of Biomedical Sciences, Academia Sinica, 128 Academia Road, Section 2, Nankang District, Taipei 115, Taiwan. Phone: +886-2-2789-9170; E-mail phsiehsinica.edu.tw
Corresponding author: Patrick C.H. Hsieh, M.D., Ph.D. Institute of Biomedical Sciences, Academia Sinica, 128 Academia Road, Section 2, Nankang District, Taipei 115, Taiwan. Phone: +886-2-2789-9170; E-mail phsiehsinica.edu.tw