Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(11):3164-3175. doi:10.7150/thno.22164 This issue Cite
Research Paper
Pre-existing anti-polyethylene glycol antibody reduces the therapeutic efficacy and pharmacokinetics of PEGylated liposomes
Yuan-Chin Hsieh1,4*, Hsin-Ell Wang2*, Wen-Wei Lin1,4,12, Steve R. Roffler5, Ta-Chun Cheng4, Yu-Cheng Su5, Jia-Je Li2, Chao-Cheng Chen2, Chun-Han Huang2, Bing-Mae Chen5, Jaw-Yuan Wang4,7, Tian-Lu Cheng1,3,4,6,8 ![]() , Fang-Ming Chen4,9,10,11
, Fang-Ming Chen4,9,10,11 ![]()
1. Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, 100 Shih-Chuan First Road, Kaohsiung 80708, Taiwan
2. Department of Biomedical Imaging and Radiological Sciences and Institute of Microbiology and Immunology, National Yang-Ming University, Taipei, Taiwan
3. Institute of Biomedical Sciences, National Sun Yat-sen University, Kaohsiung, Taiwan
4. Center for Biomarkers and Biotech Drugs, Kaohsiung Medical University, 100 Shih-Chuan First Road, Kaohsiung 80708, Taiwan
5. Institute of Biomedical Sciences, Academia Sinica, 128 Academia Road, Section 2, Taipei 11529, Taiwan
6. Department of Biomedical Science and Environmental Biology, Kaohsiung Medical University, 100 Shih-Chuan First Road, Kaohsiung 80708, Taiwan
7. Department of Surgery, Faculty of Medicine, College of Medicine, Kaohsiung Medical University, 100 Shih-Chuan First Road, Kaohsiung 80708, Taiwan
8. Department of Medical Research, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan
9. Department of Surgery, Kaohsiung Municipal Ta-Tung Hospital, 68 Jhonghua third Road, Kaohsiung 80145, Taiwan
10. Cancer Center, Kaohsiung Municipal Ta-Tung Hospital, 68 Jhonghua third Road, Kaohsiung 80145, Taiwan
11. Division of Breast Surgery, Department of Surgery, Kaohsiung Medical University Hospital, Kaohsiung Medical University Hospital, 100 Tzyou first Road, Kaohsiung 80708, Taiwan
12. Department of Laboratory Medicine, School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
* Yuan-Chin Hsieh and Hsin-Ell Wang contributed equally to this work.
Received 2017-7-31; Accepted 2018-4-2; Published 2018-5-9
Abstract

Rationale: Increasing frequency of human exposure to PEG-related products means that healthy people are likely to have pre-existing anti-PEG antibodies (pre-αPEG Ab). However, the influence of pre-αPEG Abs on the pharmacokinetics (PK) and therapeutic efficacy of LipoDox is unknown.
Methods: We generated two pre-αPEG Ab mouse models. First, naïve mice were immunized with PEGylated protein to generate an endogenous αPEG Ab titer (endo αPEG). Second, monoclonal αPEG Abs were passively transferred (αPEG-PT) into naïve mice to establish a αPEG titer. The naïve, endo αPEG and αPEG-PT mice were intravenously injected with 111in-labeled LipoDox to evaluate its PK. Tumor-bearing naïve, endo αPEG and αPEG-PT mice were intravenously injected with 111in-labeled LipoDox to evaluate its biodistribution. The therapeutic efficacy of LipoDox was estimated in the tumor-bearing mice.
Results: The areas under the curve (AUC)last of LipoDox in endo αPEG and αPEG-PT mice were 11.5- and 15.6- fold less, respectively, than that of the naïve group. The biodistribution results suggested that pre-αPEG Ab can significantly reduce tumor accumulation and accelerate blood clearance of 111In-labeled LipoDox from the spleen. The tumor volumes of the tumor-bearing endo αPEG and αPEG-PT mice after treatment with LipoDox were significantly increased as compared with that of the tumor-bearing naïve mice.
Conclusions: Pre-αPEG Abs were found to dramatically alter the PK and reduce the tumor accumulation and therapeutic efficacy of LipoDox. Pre-αPEG may have potential as a marker to aid development of personalized therapy using LipoDox and achieve optimal therapeutic efficacy.
Keywords: PEG, Polyethylene glycol, anti-PEG antibodies, liposome, LipoDox, ELISA
Introduction
PEGylated agents are widely used in clinical therapy because PEGylation of drugs improves therapeutic efficacy, reduces cytotoxicity, and enhances half-life. For example, modification of nano drug carriers (liposomes or micelles) with PEG provides a steric barrier preventing recognition by cells of the mononuclear phagocyte system [1, 2]. PEGylation also enhances the enhanced permeability and retention (EPR) effect of liposomal doxorubicin (Doxil/Caelyx/LipoDox) that is used to treat ovarian and breast carcinomas and Kaposi's sarcoma [3-5], and a PEG-micelle drug (Genexol-PM) is currently undergoing phase I/II clinical trials for treatment of lung cancer and solid tumors [6, 7]. The long half-life of PEGylated epoetin beta (Mircera) was used to help erythropoiesis in the human body [8]. In addition, modification of interferon with PEG (PEG-Intron, Pegasys) increased its serum half-life, therapeutic efficacy and the quality of life of hepatitis C patients [9, 10]. PEG is currently the gold standard polymer for development of long-circulating drugs and is regarded as the third generation of therapeutic agents.
Alongside the increasing use of PEGylated agents, generation of αPEG antibodies (αPEG Abs) has resulted in reduction of the therapeutic efficacy and alteration of the PK of PEGylated agents. For example, Ganson et al. found that 38% of hyperuricemia patients have anti-PEG IgM and IgG Abs, which resulted in a reduced half-life and therapeutic efficacy of PEG-uricase (pegloticase). [11]. Ganson and colleagues reported that a patient with acute coronary syndrome receiving Pegnivacogin (a PEGylated coagulation factor IXa inhibitor) treatment developed serious life-threatening allergic reactions due to formation of a pre-existing αPEG Ab (pre-αPEG Ab)-pegnivacogin immune complex, a finding that led to early termination of the phase 2b clinical trial [12]. In addition, Zhao et al. found the αPEG Ab decreased the serum half-life of PEGylated solid lipid nanoparticle (SLN) carriers by 50% in mice and beagles [13]. Wang et al. repeatedly administered PEGylated liposomal carriers to induce αPEG Abs, which dramatically increased the hepatic clearance of PEG-liposome from 0.0045 to 28.5 mL/h in vivo [14]. These studies all suggested that the αPEG Ab can affect the PK and therapeutic efficacy of PEGylated pharmaceuticals or nano-carriers. However, the effect of pre-αPEG Abs on the therapeutic efficacy and pharmacokinetics of PEGylated liposomal doxorubicin (LipoDox) remains unexplored.
In this study, we generated two αPEG animal models with pre-existing αPEG titer to evaluate the PK, tumor accumulation and therapeutic efficacy of LipoDox. In the first model, naïve BALB/c mice were immunized with PEGylated molecules to induce an endogenous αPEG Ab titer (endo αPEG). In the second model, naïve mice were intravenously (IV) passively transferred with αPEG Ab to generate a pre-existing αPEG Ab titer (αPEG-PT). The concentration and specificity of αPEG Ab in endo αPEG mice were confirmed by αPEG Ab quantitative ELISA and competition ELISA. For PK study, the naïve, endo αPEG, and αPEG-PT mice were IV injected with 111In-labeled LipoDox and the blood radioactivity was measured. CT26 tumor-bearing naïve, endo αPEG and αPEG-PT mice were IV injected with DIR-labeled liposome to estimate its accumulation efficacy in tumors. These CT26 tumor-bearing mice were IV injected with 111In-labeled LipoDox to evaluate its biodistribution in different organs. Finally, these tumor-bearing mice were injected with LipoDox and tumor volumes were measured to evaluate the therapeutic efficacy. These results suggest that αPEG Ab affects the therapeutic efficacy of LipoDox and αPEG Ab could be used as a marker in the development of personal therapies using PEGylated nanomedicines.
Methods
Drug and reagents
LipoDox (pegylated liposomal doxorubicin HCl, 2 mg/mL) was purchased from Taiwan Tung Yang Biopharm Company Ltd. Lipo-DIR (pegylated liposomal, 1'-Dioctadecyl-3,3,3',3'-Tetramethylindotricarbocyanine Iodide), ABTS solution (0.4 g/mL), ABTS [2-azinobis (3-ethylbenzthiazoline-6-sulfonic acid)] was purchased from Sigma-Aldrich. Anti-PEG IgG antibody 6.3 [15] and anti-IgM AGP4 [16] monoclonal Ab were from Academia Sinica. Complete adjuvant (F5581 SIGMA Freund's Adjuvant, Complete) and incomplete Freund's Adjuvant (F5506 SIGMA Freund's Adjuvant, Incomplete) was purchased from Sigma-Aldrich.
Cells and animals
CT26 colon cancer cells (American Type Culture Collection) were cultured in DMEM (Sigma-Aldrich) supplemented with 10% bovine calf serum, 100 units/mL penicillin, and 100 μg/mL streptomycin at 37 °C in an atmosphere of 5% CO2. Specific pathogen-free female BALB/c mice were obtained from the National Laboratory Animal Center, Taipei, Taiwan. All animal experiments were conducted in accordance with institutional guidelines and approved by the Animal Care and Use Committee of the Kaohsiung Medical University.
The preparation of PEGylated eβG and HSA
β-glucuronidase from Escherichia coli (eβG) and human serum albumin (HSA) were passed through a Sephadex G-25 column equilibrated with 0.1 M NaHCO3, pH 8.0, and then concentrated by ultrafiltration to 2 mg/mL. CH3-PEG5000-NHS (molar ratio protein: CH3-PEG5000-NHS=1:200) was added for 2 h at room temperature. One-tenth volume of a saturated solution of 1 M Tris-buffer (pH 8.0) was added to stop the reaction. Unreacted PEG was removed by dialysis. Protein concentrations were determined by the bicinchoninic acid assay (Pierce, Rockford, IL) with bovine serum albumin used as the reference protein. The molecular weights of PEGylated eβG and HSA were confirmed by SDS-polyacrylamide gel electrophoresis (data not shown).
Preparation of 111In-labeled PEGylated liposomal doxorubicin
The labeling protocol was according to our previous reports [17, 18]. An adequate amount of 111InCl (in 0.05 M HCl), 10 μL 8-hydroxyquinline (68 mM oxine in ethanol; Sigma-Aldrich Corp., St. Louis, MO, USA), and 500 μL acetate buffer (0.1 M, pH 5.5; J.T. Baker Inc., Phillipsburg, NJ, USA) were added into a sample vial and then incubated at 50 °C for 30 min. After cooling to ambient temperature, 111In-oxine was extracted by chloroform and dried by rotary evaporator. 111In-oxine residue was dissolved in 20 μL of ethanol and added to 80 μL of distilled water and then incubated with 1 mL of PEGylated liposomal doxorubicin at 45 ºC for 30 min and cooled down to 4 ºC for another 30 min. The crude mixture was purified by a Sepharose 4B gel size-exclusion column to get the final product 111In-labeled PEGylated liposomal doxorubicin (111In-Lipodox). The radiochemical yield and radiochemical purity of 111In-LipoDox were approximately 20% and >90%, respectively.
Generation of mice with the αPEG Ab in vivo
We developed two pre-existing anti-PEG antibody mice, endogenous αPEG (endo αPEG), and passive transfer anti-PEG antibody (αPEG-PT). The endo αPEG mouse model (N=36) was generated by a traditional protocol developed by Leenaars et al. [19]. We choose four-week-old female BALB/c mice and immunized them once a month for two months. At the 1st boost, the mice were subcutaneously injected with 60 μg PEG5000-eβG containing 50% complete adjuvant to a total volume (antigen volume: adjuvant volume = 1:1) of 100 μL. At the 2nd boost, the mice were subcutaneously injected with 60 μg PEG5000-HSA containing 50% incomplete adjuvant to a total volume (antigen volume: adjuvant volume = 1:1) of 100 μL. The anti-PEG titer of the mice was analyzed by antibody ELISA. The anti-PEG PT was developed by choosing eight-week-old female BALB/c mice and IV passively transferring 4 mg/kg anti-PEG monoclonal antibody (IgG1, Clone 6.3).
Assessment of the pre-existing αPEG Ab by αPEG quantitative ELISA
96-well plates (Nalge Nunc International, Roskilde, Denmark) were coated with 20 μg/mL of CH3-PEG2000-NH2 in 0.1 M NaHCO3 for 1 h at 37 °C. After blocking the plates with 5% skim milk in PBS overnight at 4 °C, the plates were washed with PBS twice. Mice serum samples at dilutions of 25- and 50-fold and serially-diluted IgG-6.3 and IgM-AGP4 antibody standards (in duplicate) were added to separate plates (50 μL/well) at 37 °C for 1 h. Unbound antibodies were removed by washing the plates three times with PBS. Horseradish peroxidase-conjugated isotype-specific goat anti-mouse IgG or IgM secondary antibodies were added to the well to further detect the isotype of pre-existing αPEG in mice. The plates were washed with PBS three times, and bound antibody was measured by adding 150 μL/well ABTS solution (0.003% H2O2 and 100 mM phosphate-citrate, pH=4.0) for 30 min at room temperature. The absorbance was measured at 405 nm with blank subtraction. All incubations were conducted in triplicate and data is presented as mean ± SD. The relative concentrations of anti-PEG IgG or IgM in mice serum samples were calculated by comparison with IgG-6.3 and IgM-AGP4 standard curves, respectively.
Assessment of the specificity of pre-existing αPEG Ab by αPEG competition ELISA
The specific ability of monoclonal antibody IgG, IgM αPEG antibody and αPEG mice serum to bind LipoDox was evaluated using a competition-based ELISA. The αPEG-positive mouse serum was collected to confirm the binding specificity to LipoDox. Each pre-existing αPEG-positive mouse was labeled by an ear-punching system; for example, the number '3-2' represents animal number 2 in cage 3. CH3-PEG2000-NH2 with a similar molecular weight as the PEG on LipoDox was coated on 96-well plates (Nalge Nunc International, Roskilde, Denmark). The 96-well plates were coated with 20 μg/mL of CH3-PEG2000-NH2 in 0.1 M NaHCO3 for 1 h at 37 ºC. After blocking the plates with 5% skim milk in PBS overnight at 4 ºC, the wells were washed 3 times with 200 µL of PBS-T (PBS, 0.05% Tween-20) and once with 200 µL of PBS. 5 μg/mL of IgG, IgM or 50-fold diluted mouse serum were co-incubated with competitor (200 μg/mL LipoDox) and added to the wells coated with PEG for 1 h at room temperature. After washing, horseradish peroxidase-conjugated goat anti-mouse IgG+M body was added to the wells (50 μL/well) at room temperature for 1 h. The plates were washed with PBST three times and PBS once and bound antibody was measured by adding 150 μL/well ABTS solution, 0.003% H2O2, and 100 mM phosphate-citrate (pH = 4.0) for 30 min at room temperature. The absorbance was measured at 405 nm with blank subtraction. All incubations were conducted in triplicate and data is presented as mean ± SD.
Pharmacokinetics of 111In-labeled LipoDox in mice
The mice (N=4) were intravenously injected with 111In-labeled LipoDox. The mouse blood was harvested at different times using a 1 μL capillary tube. The radioactivity of the serum was counted on a Wallac 1470 Wizard gamma counter (Perkin-Elmer) and expressed as percentage of injected dose per milliliter (%ID/mL). A sample of the 111In-labeled LipoDox injection was used as the decay-corrected standard. The serum half-life of 111In-labeled LipoDox was estimated by fitting the data to a non-compartmental model using WinNonlin 6.3 software (Pharsight Corp., Mountain View, California, USA).
In vivo optical imaging of fluorescently labeled PEG-liposome (Lipo-DIR)
BALB/c mice (N=3) bearing CT26 tumors (~100 mm3) on their right hind leg, were IV injected with anti-PEG antibody, anti-myc antibody, or saline buffer and then injected with Lipo-DIR820 (0.625 mg/kg body weight) 6 h later. The mice were anesthetized with isoflurane (Abbott Laboratories) using a vaporizer system (A.M. 272 Bickford). The distribution and accumulation of the fluorescent probes were measured by an IVIS Spectrum imaging system (Caliper Life Sciences) at 24, 48 and 72 h after injection. The regions-of-interest in tumor areas were drawn and analyzed with Living Image software version 4.2 (Caliper Life Sciences).
The Biodistribution of 111In-labeled LipoDox
BALB/c mice bearing CT26 tumors (~50 mm3) on their right flanks were used for biodistribution studies. Each group (N=4) of mice was IV injected with 100 μCi 111In-labeled LipoDox. For biodistribution, mice were sacrificed by CO2 asphyxiation at 4, 24 and 48 h (4 mice per group) after administration of 111In-labeled LipoDox. At each time point, the organs of interest were dissected and the blood was sampled via cardiac puncture. The tissue samples were rinsed in water, blotted dry, weighed, and then counted using a Wallac 1470 Wizard gamma counter. A sample of the 111In-labeled LipoDox injection was used as the decay-corrected standard. Data are expressed as the percentage of the injected dose per gram of tissue (%ID/g).
In vivo treatment of CT26 tumor-bearing mice with LipoDox
Naïve, endo αPEG and αPEG-PT (αPEG IgG-6.3 Abs passively transferred) BALB/c mice bearing CT26 tumors (~100 mm3; N=4) in their right flanks were IV injected with LipoDox (3 mg/kg body weight) every 4 days for a total of 3 doses. The therapeutic efficacy was monitored by measuring tumor volume (length × width × height × 0.5) on a weekly basis. We independently repeated the experiment twice and combined the results (N=8) for statistical analysis.
Statistical analysis
The detection signal of competition ELISA in Figure 2 was defined as the anti-PEG antibody against CH3-PEG2000-NH2 molecule that produced a statistically higher signal than the signal produced from the blank. Statistical significance was calculated using GraphPad Prism 6.0 with Student's t-test. In Figures 1, 3-6, the statistical difference between the αPEG group and naïve group was analyzed by comparing multiple t-tests. Data were considered statistically different at *, P < 0.05 and **, P < 0.0001.
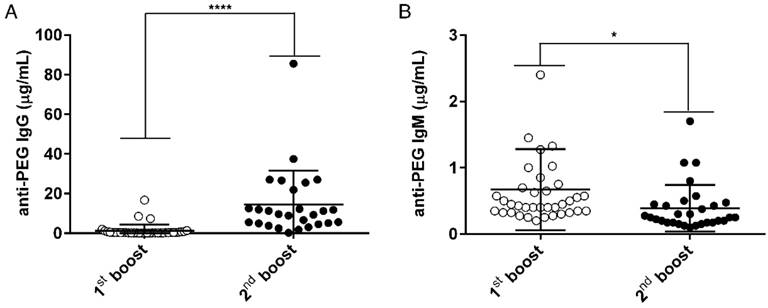
Characterization of the αPEG Ab concentration in the endo αPEG mice. BALB/c mice (N=36, one symbol represents one animal) were immunized with PEGylated protein (PEGylated-eβG and -HSA). The serum of each mouse was collected after the 1st (○) and 2nd (●) boost. The serum from each mouse was diluted with 2% skim milk buffer, and then added to a CH3-PEG2000-NH2-coated plate. (A) Pre-existing IgG and (B) pre-existing IgM were incubated with HRP-conjugated goat anti-IgG and IgM specific antibodies. The relative concentrations of anti-PEG IgG or IgM in mice serum samples were calculated by comparison with IgG-6.3 and IgM-AGP4 standard curves, respectively. Data are represented as mean ± SD. Statistical analysis was performed by comparing multiple t tests, *, P < 0.05 and, ****, P < 0.0001.
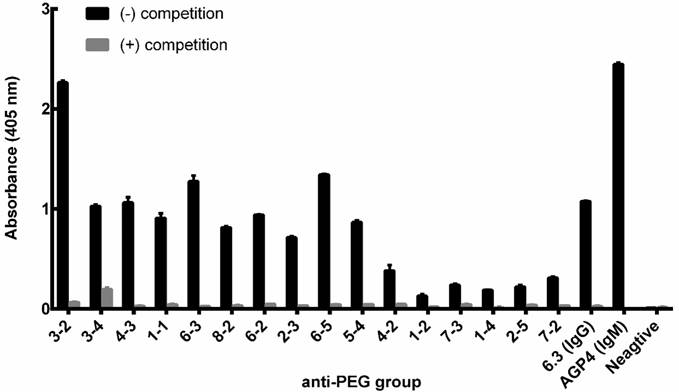
Competition assay of αPEG Ab from endogenous mouse serum. The αPEG monoclonal Abs AGP4 (IgM; 5 μg/mL), 6.3 (IgG; 5 μg/mL) and 50-fold diluted αPEG mouse serum sample were added to wells pre-coated with CH3-PEG2000-NH2. LipoDox (200 μg/mL) was added as a competitor to compete for binding of the αPEG Ab to the CH3-PEG2000-NH2 well coating. Each pre-existing αPEG-positive mouse was labeled using an ear-punching system; for example, the number '3-2' represents animal number 2 in cage 3. The mean absorbance values (405 nm) of duplicate determinations are shown. (Bars, SD).
Results
Characterization of αPEG Ab in mice
In order to generate an animal model (endo αPEG) to mimic humans who have pre-existing αPEG Abs and to evaluate whether αPEG Abs affect the PK, bio-distribution or therapeutic efficacy of PEG-liposomes in vivo, we generated two mouse models with pre-αPEG Abs (endo αPEG, αPEG-PT). Naïve BALB/c mice were immunized with PEGylated molecules to generate an endogenous αPEG Ab titer (endo αPEG). Additionally, naïve BALB/c mice were passively transferred with αPEG Ab (6.3) to generate an αPEG Ab titer (αPEG-PT). In order to measure the concentration of αPEG Ab in endo αPEG mice, the individual mice sera were collected at the times of the 1st (○) and 2nd (●) boosts. The concentrations of αPEG IgG and IgM Abs in endo αPEG were measured by mouse αPEG Ab quantitative ELISA. The diluted standard αPEG Ab and the endo αPEG mice serum were added to a CH3-PEG2000-NH2 (mPEG2000)-coated plate. The isotype-specific IgG and IgM secondary antibodies were added to the well to further confirm the isotype of pre-existing αPEG in mice. Figure 1A shows that the pre-existing Ab concentration after the PEGylated protein immunization of anti-PEG IgG was initially 1.29±0.53 μg/mL and then 14.57±3.29 μg/mL, and that of anti-IgM was initially 0.67±0.10 μg/mL and then 0.39±0.06 μg/mL, indicating the pre-αPEG Ab isotype switched to IgG-dominant in endo αPEG mice.
In order to confirm whether the αPEG Ab of endo αPEG or αPEG-PT mice could specifically bind to PEG molecules, the endo αPEG mouse serum, αPEG IgM Ab (AGP4) and αPEG IgG Ab (6.3) were incubated with or without PEGylated-liposomal doxorubicin (LipoDox) to compete for binding with the αPEG Abs, then added to a 96-well plate coated with CH3-PEG2000-NH2 antigen to measure the specificity of αPEG Ab. Figure 2 shows that the binding ability of αPEG and the pre-αPEG Ab to mPEG2000-NH2 was reduced by incubation with LipoDox, demonstrating that the pre- αPEG Ab in endo αPEG mice specifically bound to LipoDox. Together, we have successfully established a pre-αPEG mouse model that can mimic the human condition to investigate the influence on the PK, biodistribution and therapeutic efficacy of the PEG-liposomal drug in vivo.
Influence of αPEG Abs on the PK of 111In-labeled LipoDox
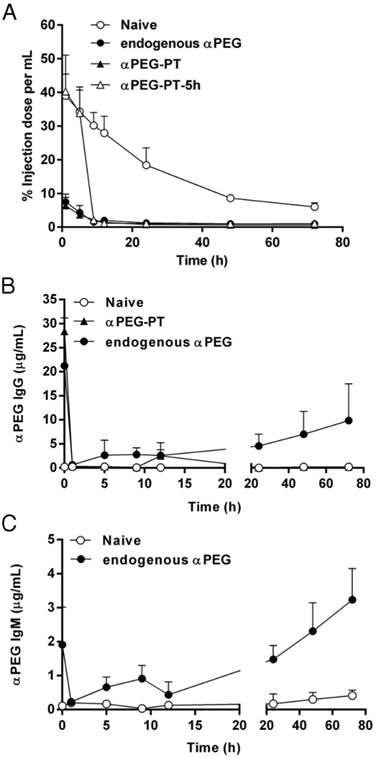
To understand whether pre-αPEG Abs alter the PK of PEG-liposomal drugs (PEGylated liposomal doxorubicin; LipoDox), the naïve, endo αPEG and αPEG-PT mice were injected with 111In-labeled LipoDox. The naïve mice were further injected with αPEG Ab at 5 h post-inoculation (αPEG-PT-5h) of the 111In-labeled LipoDox to confirm whether the αPEG Ab is the key factor for the alteration of the PK of LipoDox. The radioactivity of blood samples was counted by a gamma counter at different time points and the AUC parameter was used to determine the alteration of pharmacokinetics as calculated by WinNonlin program (Table 1). Figure 3A shows that, compared with the naïve group, the radioactivity of the 111In-labeled LipoDox was significantly reduced in endo αPEG and αPEG-PT. When αPEG Abs were passively injected at 5 h post-inoculation of the 111In-labeled LipoDox, the radioactivity of the blood was dramatically decreased in naïve mice. The AUClast values in naïve, endo αPEG, αPEG-PT, and αPEG-PT-5 h were 1388.07, 120.35, 88.79 and 256.51 h·%ID/mL, respectively. At the end of the observed point, the AUClast of LipoDox in endo αPEG, αPEG-PT and PEG-PT-5 h were 11.5-, 15.6- and 5.41-fold less than that of the naïve group, respectively (Table 1). These results suggest that the pre-existing and post inoculated αPEG Abs dramatically reduced the pharmacokinetics of 111In-labeled LipoDox in vivo. In order to understand the blood clearance level of αPEG Ab at different time points after injecting αPEG Ab (in anti-PEG PT model), we collected the blood samples from naïve, endo αPEG and αPEG-PT BALB/c mice and measured the concentration of αPEG Ab by quantitative αPEG ELISA. The results show that the pre-existing αPEG Ab levels in the αPEG-PT and endo αPEG group (Figure 3B-C) decreased after injecting 111In-labeled LipoDox. In addition, the pre-existing αPEG Ab level in the endo αPEG group was recovered, accompanied by a decrease in 111In-labeled LipoDox, indicating the pre-existing αPEG Abs could specifically accelerate the clearance of LipoDox in vivo.
Pharmacokinetics of 111In-labeled LipoDox in vivo. The serum half-life of 111In-labeled LipoDox was estimated by fitting the data to a non-compartmental model using WinNonlin 6.3 software. (N = 4 for each group)
| Naïve | Endo αPEG | αPEG-PT | αPEG-PT-5h | |
|---|---|---|---|---|
| AUClast (h·%ID/mL) | 1388.07 | 120.35 | 88.79 | 256.51 |
| AUC0→∞ (h·%ID/mL) | 1628.25 | 311.84 | 186.64 | 286.47 |
| CL (mL/h) | 0.06 | 0.32 | 0.54 | 0.35 |
| MRTlast (h) | 24.96 | 24.35 | 20.93 | 12.69 |
AUClast: area under curve from time 0 to the observed time point; AUC0→∞: area under curve calculated from time 0 to infinity; CL: clearance rate; MRTlast: mean residence time from time 0 to the observed time point
Influence of αPEG Ab on the pharmacokinetics of 111In-labeled LipoDox. (A) Naïve (○), endo αPEG (●) and αPEG-PT (▲) BALB/c mice were IV injected with 111In-labeled LipoDox. The αPEG-PT-5 h (△) mice were injected with αPEG Ab and injected with 111In-labeled LipoDox 5 h later. The blood was harvested at different times. The pharmacokinetics of 111In-labeled LipoDox was analyzed using a γ-counter. (B) The sera of mice with pre-existing αPEG IgG and (C) pre-existing αPEG IgM were also measured by quantitative αPEG Ab ELISA. Data are represented as mean ± SD. Statistical analysis was performed by comparing multiple t-tests. **, P < 0.0001 as compared to the naïve group.
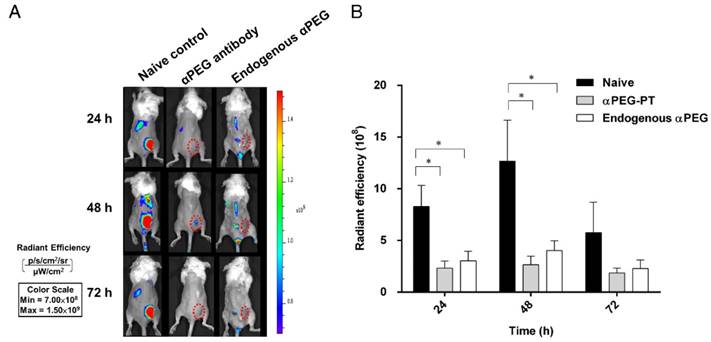
Influence of αPEG Abs on the accumulation of fluorescently labeled PEG-liposome in tumors
In order to assess whether the pre-αPEG Ab alters the tumor accumulation of PEG-liposome, the CT26 tumor xenografted naïve, endo αPEG, αPEG-PT mice were IV injected with Lipo-DIR probe. The fluorescence intensity in the tumor region was acquired by an IVIS spectrum image system at 24, 48, and 72 h. Figure 4A shows that the fluorescence intensity of tumors in naïve mice was significant higher than that in endo αPEG or αPEG-PT mice. The fluorescence intensity of Lipo-DIR in the tumor region in αPEG-PT mice was 3.55-, 4.76-, and 3.09-fold less and in endo αPEG mice was 2.74-, 3.15-, and 2.52-fold less than that in the naïve mice at 24, 48 and 72 h, respectively (Figure 4B). These results indicate that pre-αPEG Abs could significantly reduce (P < 0.05) the tumor accumulation of PEG-liposome.
Influence of αPEG Ab on the accumulation of fluorescently labeled PEG-liposome in tumors. (A) Naïve, αPEG-PT and endo αPEG BALB/c mice bearing CT26 tumors were IV injected with fluorescently labeled PEG-liposome (Lipo-DIR). Mice were sequentially imaged at 24, 48 and 72 h with an IVIS spectrum optical imaging system. (B) The fluorescence intensity of Lipo-DIR in the tumor was analyzed with Living Image software version 4.2. Statistical analysis was performed by compare multiple t-tests. *, P<0.05 as compared to the naïve group.
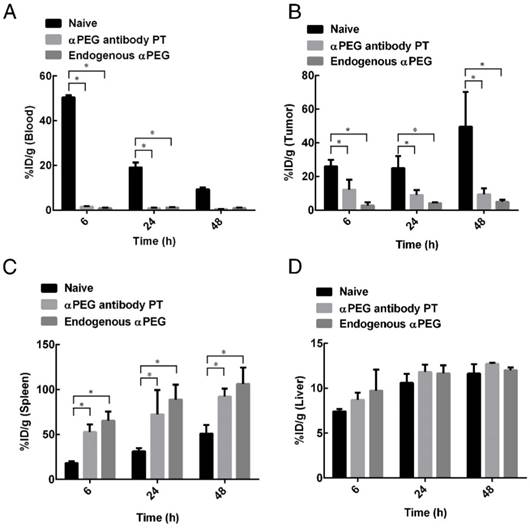
Biodistribution of 111In-labeled LipoDox in αPEG Ab mice
In order to assess whether pre-αPEG Abs alter the biodistribution of LipoDox in mice, 111In-labeled LipoDox (100 μCi) was IV injected into CT26 tumor-bearing naïve, endo αPEG and αPEG-PT mice. The radioactivity of 111In-labeled LipoDox was measured in blood samples, tumors and various organs collected at different times (Tables 2-4) and is summarized as follows: (a) The radioactivity of 111In-labeled LipoDox in the blood of αPEG-PT mice was 32.94-, 22.78-, and 16.73-fold less, and in endo αPEG was 54.20-, 15.68-, and 9.86-fold less than in naïve mice at 6, 24 and 48 h, respectively (Figure 5A), indicating that pre-αPEG Ab accelerates clearance of 111In-labeled LipoDox in blood. (b) Figure 5B also shows that the radioactivity of 111In-labeled LipoDox in tumors of αPEG-PT mice was 2.12-, 2.78-, and 5.30-fold less, and in endo αPEG was 9.30-, 6.13-, and 10.27-fold less than in naïve mice at 6, 24 and 48 h, respectively, indicating that pre-αPEG reduced the accumulation of 111In-labeled LipoDox in tumors. (c) The radioactivity of 111In-labeled LipoDox in the spleen and liver of αPEG-PT mice was 2.89-, 2.30-, and 1.80-fold for spleen and 1.17-, 1.11-, and 1.09-fold for liver and that of endo αPEG was 3.58-, 2.81-, and 2.08-fold for spleen and 1.31-, 1.10-, and 1.03-fold for liver in comparison with naïve mice at 6, 24 and 48 h, respectively (Figure 5C-D). The bio-distribution level of 111In-labeled LipoDox seems to be slightly increased in the liver and spleen of the pre-existing αPEG Ab mice as compared with that of the naïve group, but only spleen showed a significant difference in the statistical analysis, indicating that 111In-labeled LipoDox was mainly cleared from the spleen.
Biodistribution of 111In-LipoDox 6 h after intravenous injection in CT26 tumor-bearing mice. Values are presented as percentage of injected dose per gram (%ID/g; mean ± standard error of the mean; N= 3 at each time point).
| Organ | Naïve | αPEG antibody PT | Endogenous αPEG |
|---|---|---|---|
| Blood | 50.41 ± 1.07 | 1.53 ± 0.32 | 0.93 ± 0.15 |
| Tumor | 25.96 ± 3.90 | 12.23 ± 5.78 | 2.79 ± 1.82 |
| Spleen | 18.24 ± 2.04 | 52.83 ± 8.47 | 65.33 ± 10.25 |
| Liver | 7.43 ± 0.26 | 8.71 ± 0.81 | 9.74 ± 2.33 |
| SI | 14.24 ± 6.06 | 21.08 ± 9.58 | 2.11 ± 0.95 |
| LI | 1.27 ± 0.64 | 2.39 ± 0.68 | 0.31 ± 0.04 |
| Muscle | 0.38 ± 0.05 | 3.08 ± 2.22 | 0.39 ± 0.27 |
| Bone | 0.70 ± 0.20 | 0.99 ± 0.60 | 0.47 ± 0.25 |
| Marrow | 7.11 ± 3.33 | 9.69 ± 1.10 | 18.15 ± 1.87 |
| Bladder | 1.49 ± 0.17 | 2.39 ± 0.87 | 0.22 ± 0.25 |
| Kidney | 7.08 ± 0.42 | 3.86 ± 0.47 | 1.43 ± 0.14 |
| Urine | 0.27 ± 0.23 | 1.01 ± 1.19 | 0.80 ± 0.30 |
| Feces | 0.52 ± 0.11 | 0.47 ± 0.07 | 0.31 ± 0.11 |
SI: small intestine; LI: large intestine
Biodistribution of 111In-LipoDox 24 h after intravenous injection in CT26 tumor-bearing mice. Values are presented as percentage of injected dose per gram (%ID/g; mean ± standard error of the mean; N= 3 at each time point).
| Organ | Naïve | αPEG antibody PT | Endogenous αPEG |
|---|---|---|---|
| Blood | 19.14 ± 2.11 | 0.84 ± 0.25 | 1.22 ± 0.16 |
| Tumor | 25.02 ± 7.08 | 8.97 ± 2.96 | 4.08 ± 0.55 |
| Spleen | 31.49 ± 3.37 | 72.45 ± 27.16 | 88.75 ± 16.51 |
| Liver | 10.60 ± 1.00 | 11.82 ± 0.80 | 11.67 ± 0.87 |
| SI | 28.16 ± 5.74 | 6.91 ± 1.52 | 2.03 ± 0.66 |
| LI | 3.44 ± 0.97 | 1.20 ± 0.43 | 0.93 ± 0.31 |
| Muscle | 0.68 ± 0.27 | 1.16 ± 1.28 | 0.52 ± 0.07 |
| Bone | 0.76 ± 0.49 | 0.73 ± 0.31 | 0.33 ± 0.14 |
| Marrow | 18.76 ± 24.22 | 5.60 ± 5.57 | 32.07 ± 20.21 |
| Bladder | 1.78 ± 0.67 | 1.47 ± 0.85 | 1.52 ± 0.72 |
| Kidney | 8.47 ± 1.54 | 5.08 ± 0.56 | 4.10 ± 0.33 |
| Urine | 1.12 ± 0.32 | 2.32 ± 0.13 | 1.56 ± 1.37 |
| Feces | 0.34 ± 0.15 | 1.03 ± 0.37 | 1.08 ± 0.72 |
SI: small intestine; LI: large intestine
Biodistribution of 111In-LipoDox 48 h after intravenous injection in CT26 tumor-bearing mice. Values are presented as percentage of injected dose per gram (%ID/g; mean ± standard error of the mean; N= 3 at each time point).
| Organ | Naïve | αPEG antibody PT | Endogenous αPEG |
|---|---|---|---|
| Blood | 9.37 ± 0.73 | 0.56 ± 0.03 | 0.95 ± 0.26 |
| Tumor | 49.65 ± 20.59 | 9.36 ± 3.53 | 4.83 ± 1.38 |
| Spleen | 51.08 ± 9.56 | 92.12 ± 8.89 | 106.30 ± 18.00 |
| Liver | 11.63 ± 1.07 | 12.71 ± 0.14 | 12.01 ± 0.32 |
| SI | 20.42 ± 4.24 | 9.07 ± 2.55 | 1.89 ± 0.34 |
| LI | 3.90 ± 0.33 | 1.66 ± 0.33 | 0.65 ± 0.05 |
| Muscle | 0.88 ± 0.41 | 1.41 ± 0.70 | 0.31 ± 0.15 |
| Bone | 0.91 ± 0.29 | 1.17 ± 0.57 | 0.92 ± 0.68 |
| Marrow | 8.64 ± 3.07 | 18.58 ± 1.07 | 40.33 ± 12.27 |
| Bladder | 1.30 ± 0.02 | 1.46 ± 0.08 | 1.03 ± 0.22 |
| Kidney | 10.24 ± 0.98 | 6.86 ± 0.29 | 6.33 ± 0.88 |
| Urine | 1.39 ± 0.12 | 0.94 ± 0.83 | 1.07 ± 0.44 |
| Feces | 0.71 ± 0.09 | 2.64 ± 1.75 | 1.68 ± 1.37 |
SI: small intestine; LI: large intestine
Biodistribution of 111In-labeled LipoDox in αPEG Ab mice. Naïve, endo αPEG, and αPEG-PT BALB/c mice bearing CT26 tumors were IV injected with 111In-labeled LipoDox. The mice were sacrificed at different times and the organs and blood were collected and weighed. The radioactivities of (A) blood, (B) tumor, (C) spleen and (D) liver were analyzed using a γ-counter. Statistical analysis was performed by comparing multiple t-tests. *, P < 0.05 as compared to the naïve group.
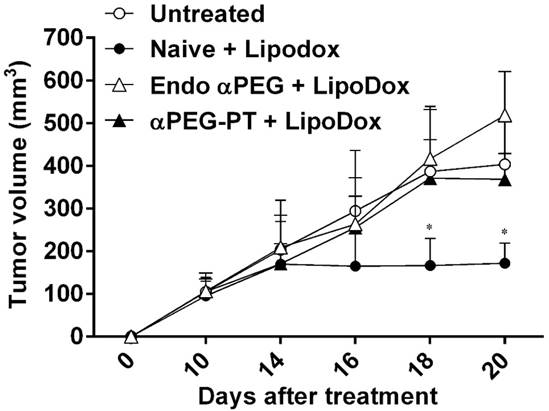
Effect of a PEG Ab on the therapeutic efficacy of LipoDox in vivo
In order to assess whether the pre-αPEG Abs reduced the therapeutic efficacy of LipoDox, the CT26 tumor-bearing naïve, endo αPEG, and αPEG-PT mice were IV injected with PBS or LipoDox (3 mg/kg/dose every 4 days, for three doses). The tumor volume was measured every two days. Figure 6 shows that the tumor sizes of the two LipoDox-treated groups (endo αPEG and αPEG-PT mice) were similar to that of the untreated controls but was larger than that of treated naïve mice. These results indicate that the pre-αPEG Ab might significantly (P < 0.05) reduce the therapeutic efficacy of LipoDox.
Effect of αPEG Ab on the therapeutic efficacy of LipoDox in vivo. CT26 tumor-bearing untreated (○), naïve (●), endo αPEG (△) and αPEG-PT (▲) BALB/c mice were IV injected with LipoDox (3 mg/kg body weight) every 4 days, 3 times. The tumor volume (length × width × height × 0.5) was measured using calipers every 2 days. The results are shown as mean tumor size (n = 8) ± SD. Statistical analysis was performed by comparing multiple t-tests. *, P < 0.05 as compared to the untreated group.
In addition, in order to investigate whether repeated injection of LipoDox would induce the production of αPEG Ab, the concentration of αPEG IgG and IgM Ab in naïve, endo αPEG and αPEG-PT BALB/c mice treated with LipoDox was measured by quantitative αPEG Ab ELISA. The result showed that repeated injection of LipoDox did not induce the production of αPEG IgG and IgM Abs in the naïve and passive transfer (PT) groups (Figure S1).
Discussion
In this study, we demonstrated that the presence of αPEG Ab or pre-αPEG Ab in mice alters the PK and reduces the tumor accumulation efficacy of LipoDox. In addition, pre-αPEG Ab accelerated the clearance of 111In-labeled LipoDox in the blood and tumor region and enhanced spleen elimination, resulting in decreased therapeutic efficacy. These results indicate that αPEG Ab may have potential use as a marker in the development of personalized therapy using LipoDox and help to achieve optimal therapeutic efficacy.
An understanding of the clearance of PEGylated agents by αPEG Abs is important as αPEG Abs have been reported to reduce serum half-life and therapeutic efficacy in the clinic. Understanding the possible mechanism of elimination of PEGylated agents may help to use these therapies effectively. For example, Ishida et al. proposed repeated injection of PEGylated liposome induced the formation of anti-PEG IgM and activated the complement system. Activated C3 fragments (iC3b) led to opsonization and enhanced uptake of PEGylated liposomes by Kupffer cells in the liver [20]. In our study, we found the pre-αPEG Abs reduced the tumor accumulation of Lipo-DIR in endo αPEG mice. The biodistribution of 111In-labeled LipoDox in endo αPEG mice was 9.86-fold less in the blood, 10.2-fold less in the tumor region, 2.08-fold more in the spleen and 1.03-fold more in the liver in comparison with the naive mice at 48 h (Figure 5). We found that most of the 111In-labeled LipoDox was significantly eliminated from the spleen. This may be because the αPEG IgG-LipoDox complex could be recognized by the IgG receptor on red pulp macrophages [21]. Thus, preventing the presence of αPEG Abs would help to maintain the serum half-life and therapeutic efficacy during administration of PEGylated drugs.
The occurrence of pre-αPEG Abs has been found to be increased in naïve healthy blood donors (pre-αPEG Ab) who have never been administrated PEGylated drugs or nanocarriers [22]. For instance, Ricardo et al. first reported that occurrence of pre- αPEG Ab in healthy blood donors has increased from 0.2% to 25% over the last two decades [23]. And, a recent study found healthy people have 18% IgG, 25% IgM, and 30% both IgG and IgM pre-αPEG Abs by quantitative competitive ELISA assays [24]. In our previous study, we also found that 1504 healthy donors had 25.7% IgG and 27% IgM pre-αPEG Ab in serum by long exposure with PEG derivatives [25]. pre-αPEG Abs were found in an acute coronary syndrome patient treated with PEGylated coagulation factor IX inhibitor that led to serious life-threatening allergic reactions [12]. PEGylated-uricase [11], -SLN, and -liposomes [13, 14] have also been reported to have reduced serum half-lives or reduced therapeutic efficacy due to induction of αPEG Abs. In this study, we generated an animal model (endo αPEG) to mimic humans who have pre-existing αPEG Abs. We found that the pre-αPEG Abs reduced the therapeutic efficacy and dramatically accelerated the clearance of LipoDox from the blood in vivo, resulting in decreased therapeutic efficacy of LipoDox.
Development of a convenient and specific approach to measure the presence of pre-αPEG Abs may ensure the precise use of PEGylated medicine in the clinic. Richter et al. utilized the hemagglutination assay to measure αPEG Abs in human or rabbit serum. Serum containing αPEG Abs was incubated with mPEG6000-modified fresh red blood cells (RBC) to cause agglutination of the PEG-coated RBCs. They found that rabbit serum treated with PEG-modified enzymes [26] or serum from 0.2% of healthy donors caused agglutination of the PEG-coated RBCs [27]. However, it may be inconvenient to freshly purify RBCs and modify them with PEG. Liu et al. developed a double antigen bridging immunogenicity ELISA to measure the concentration of αPEG Abs from human serum samples. Human serum was incubated with hapten-labeled PEG 40 kDa and then added to the pre-coated PEG 40 kDa plate to form a bridge complex. HRP-labeled anti-hapten antibody was used to detect the αPEG-positive signals. The ELISA identified 15 pre-αPEG Ab-positive human serum samples with 100% accuracy in a total population of 350 naïve donors, indicating that the antigen bridging immunogenicity ELISA assay could rapidly and sensitively detect pre-αPEG Abs in serum [28]. However, the bridging ELISA-based detection system may have the disadvantage of failing to detect low affinity or monovalent antibodies [29]. Zhang et al. developed a sensitive methoxy-PEG5000-coated surface plasmon resonance sensor to measure anti-PEG IgG and IgM antibodies in human serum [30, 31]; however, the high cost of the instrument may limit its wide application. In our study, we developed an αPEG quantitative ELISA system as a simple and sensitive method to measure the concentrations of pre-existing αPEG Abs in serum. The convenient approach can screen and monitor pre-αPEG Abs in serum before administration of LipoDox and may help attain optimal therapeutic efficacy of LipoDox in the clinic.
The specificity and affinity of pre-existing αPEG Abs may influence the therapeutic efficacy or PK of PEGylated drug. For example, Sabine and colleagues immunized mice with carrier-free PEG molecules (40 kDa PEG-Diol) to develop PEG-specific IgM Ab, and the pre-existing αPEG IgM antibodies did not affect the PK of PEG-rASNase MC0609 [32]. In their study, they mention that probably the titer or affinity of these anti-PEG IgM antibodies were too low to influence the pharmacokinetics of PEG-rASNase. In contrast, we immunized mice with PEGylated protein (PEGylated-eβG and -HSA), which lead to pre-existing αPEG Ab isotype switching from low affinity IgM to higher affinity IgG (Figure 1). The specificity of pre-existing αPEG Abs was analyzed using a LipoDox-based competition ELISA. The result indicated that the therapeutic efficacy of LipoDox was decreased in αPEG-positive CT26 tumor-bearing mice (Figure 6). Taken together, it is important to confirm the isotypes, affinity and specificity of pre-existing αPEG Abs before using PEGylated medicines for effective therapy.
The administration of PEGylated medicines should be personalized in the clinic. Choosing the appropriate PEGylated or non-PEGylated medicine to avoid binding with αPEG Abs could maintain the serum half-life and achieve optimal therapeutic efficacy in disease treatment [22]. Armstrong et al. found that in acute lymphoblastic leukemia patients individually treated with PEGylated or non-PEGylated asparaginase, formation of αPEG Abs was induced in 32% of patients treated with PEGylated asparaginase, resulting in complete loss of enzyme activity. No αPEG Abs were found to reduce the serum ASNase activity in patients treated with unmodified ASNase [33]. Our study found that pre-αPEG Abs reduce the therapeutic efficacy of LipoDox, and tumor volume was significantly increased compared with the naïve group. These results suggest that patients who screen αPEG Ab positive should use a non-PEGylated drug. PEG alternatives such as chitosan, XTEN peptide, poly(carboxybetaine), poly(glycerol) and poly(2-oxazaline) molecules have been used in drug modification, which enhanced serum half-life and had good biocompatibility [34-37]. Development of various kinds of long serum half-life treatments may be warranted for clinical use. The αPEG Ab may act as a marker to help patients choose PEGylated therapies or alternative therapies to achieve optimal therapeutic efficacy.
PEGylation technology is widely used in pharmaceuticals, processed foods and cosmetics in everyday life. These have significantly increased the occurrence of pre-αPEG Ab in naïve donors. A previous study has already proved that pre-αPEG Abs can cause a serious life-threating allergic reaction in the treatment of acute coronary syndrome by a coagulation factor IX inhibitor, Pegnivacogin [12]. In the current study, we proved that the pre-αPEG Ab could alter the PK and reduce the accumulation and therapeutic efficacy of LipoDox in vivo. These results reveal that determining the pre-αPEG Ab and avoiding drug contact with pre-αPEG Abs is important before using LipoDox; this point should be carefully considered to prevent the influence of pre-αPEG Abs in the development of novel liposomal-based drug carriers. Further, using drugs modified with PEG alternatives will prevent recognition by pre-αPEG Abs. Accurate measurement of the existence of αPEG Abs may aid the development of personal therapy using LipoDox or other PEGylated agents in the clinic in the future.
Abbreviations
pre-αPEG Ab: pre-existing anti-PEG antibodies; endo αPEG: endogenous αPEG Ab titer; αPEG-PT: αPEG Abs were passively transferred; AUC: under the curve; PK: pharmacokinetics; PEG: polyethylene glycol; EPR: permeability and retention; eβG: β-glucuronidase from Escherichia coli; HSA: human serum albumin; αPEG-PT-5h: αPEG Ab at 5 h post-inoculation; RBC: red blood cells.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by grants from the Ministry of Science and Technology, Taiwan (MOST 106-2632-B-037-003, MOST 106-2320-B-041-001, MOST106-2314-B-037-019, MOST106-2311-B-037-001-MY2 and MOST 107-2321-B-037-003); the National Health Research Institutes, Taiwan (NHRI-EX107-10729EI); Academia Sinica, Taiwan (AS-107-TP-B11); the Program for Translational Innovation of Biopharmaceutical Development - Technology Supporting Platform Axis (Grant No. 107-0210-01-19-04), Academia Sinica, Taiwan; the Grant of Biosignature in Colorectal Cancers, Academia Sinica, Taiwan; the KMU-KMUH Co-Project of Key Research (KMU-DK107001) from Kaohsiung Medical University, Taiwan; and the Center for Biomarkers and Biotech Drugs, Kaohsiung Medical University, Taiwan. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors declare no competing financial interest.
Author contributions
YC Hsieh, BM Chen, SR Roffler and TL Cheng design the ELISA system (Figure 1-2). YC Hsieh, YC Su, TC Cheng, and WW Lin prepared Figure 4. YC Hsieh, HE Wang, JJ Li, CH Huang and FM Chen prepared Figure 3 and Figure 5. YC Hsieh, JY Wang and JJ Li prepared Figure 6. YC Hsieh, CC Chen and HE Wang helped in data analysis. YC Hsieh, FM Cheng, WW Lin and TL Cheng writing, review, and/or revision of the manuscript. YC Hsieh and HE Wang contributed equally to this work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Koning GA, Morselt HW, Kamps JA, Scherphof GL. Uptake and intracellular processing of PEG-liposomes and PEG-immunoliposomes by kupffer cells in vitro 1 *. J Liposome Res. 2001;11:195-209
2. Hume DA, Ross IL, Himes SR, Sasmono RT, Wells CA, Ravasi T. The mononuclear phagocyte system revisited. J Leukoc Biol. 2002;72:621-7
3. Gabizon AA, Patil Y, La-Beck NM. New insights and evolving role of pegylated liposomal doxorubicin in cancer therapy. Drug Resist Updat. 2016;29:90-106
4. Lao J, Madani J, Puertolas T, Alvarez M, Hernandez A, Pazo-Cid R. et al. Liposomal Doxorubicin in the treatment of breast cancer patients: a review. J Drug Deliv. 2013;2013:456409
5. Poizot-Martin I, Giovannini M, Rosello R, Viallat JR, Sauniere JF, Dalmas AM. et al. Liposomal-doxorubicin in human immunodeficiency virus-associated Kaposi's sarcoma. J Clin Oncol. 1994;12:645
6. Ahn HK, Jung M, Sym SJ, Shin DB, Kang SM, Kyung SY. et al. A phase II trial of Cremorphor EL-free paclitaxel (Genexol-PM) and gemcitabine in patients with advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2014;74:277-82
7. Lee KS, Chung HC, Im SA, Park YH, Kim CS, Kim SB. et al. Multicenter phase II trial of Genexol-PM, a Cremophor-free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer. Breast Cancer Res Treat. 2008;108:241-50
8. Sanchez-Fructuoso A, Guirado L, Ruiz JC, Torregrosa V, Gonzalez E, Suarez ML. et al. Anemia control in kidney transplant patients treated with methoxy polyethylene glycol-epoetin beta (mircera): the Anemiatrans Group. Transplant Proc. 2010;42:2931-4
9. Zeuzem S, Diago M, Gane E, Reddy KR, Pockros P, Prati D. et al. Peginterferon alfa-2a (40 kilodaltons) and ribavirin in patients with chronic hepatitis C and normal aminotransferase levels. Gastroenterology. 2004;127:1724-32
10. Glue P, Fang JW, Rouzier-Panis R, Raffanel C, Sabo R, Gupta SK. et al. Pegylated interferon-alpha2b: pharmacokinetics, pharmacodynamics, safety, and preliminary efficacy data. Hepatitis C Intervention Therapy Group. Clin Pharmacol Ther. 2000;68:556-67
11. Ganson NJ, Kelly SJ, Scarlett E, Sundy JS, Hershfield MS. Control of hyperuricemia in subjects with refractory gout, and induction of antibody against poly(ethylene glycol) (PEG), in a phase I trial of subcutaneous PEGylated urate oxidase. Arthritis Res Ther. 2006;8:R12
12. Ganson NJ, Povsic TJ, Sullenger BA, Alexander JH, Zelenkofske SL, Sailstad JM. et al. Pre-existing anti-polyethylene glycol antibody linked to first-exposure allergic reactions to pegnivacogin, a PEGylated RNA aptamer. J Allergy Clin Immunol. 2016;137(e7):1610-3
13. Zhao Y, Wang L, Yan M, Ma Y, Zang G, She Z. et al. Repeated injection of PEGylated solid lipid nanoparticles induces accelerated blood clearance in mice and beagles. Int J Nanomedicine. 2012;7:2891-900
14. Wang X, Ishida T, Kiwada H. Anti-PEG IgM elicited by injection of liposomes is involved in the enhanced blood clearance of a subsequent dose of PEGylated liposomes. J Control Release. 2007;119:236-44
15. Lin WW, Hsieh YC, Cheng YA, Chuang KH, Huang CC, Chuang CH. et al. Optimization of an anti-poly(ethylene glycol) (anti-PEG) cell-based capture system to quantify PEG and PEGylated molecules. Anal Chem. 2016;88:12371-9
16. Su YC, Chen BM, Chuang KH, Cheng TL, Roffler SR. Sensitive quantification of PEGylated compounds by second-generation anti-poly(ethylene glycol) monoclonal antibodies. Bioconjug Chem. 2010;21:1264-70
17. Lin YY, Kao HW, Li JJ, Hwang JJ, Tseng YL, Lin WJ. et al. Tumor burden talks in cancer treatment with PEGylated liposomal drugs. PLoS One. 2013;8:e63078
18. Lin YY, Li JJ, Chang CH, Lu YC, Hwang JJ, Tseng YL. et al. Evaluation of pharmacokinetics of 111In-labeled VNB-PEGylated liposomes after intraperitoneal and intravenous administration in a tumor/ascites mouse model. Cancer Biother Radiopharm. 2009;24:453-60
19. Leenaars M, Hendriksen CF. Critical steps in the production of polyclonal and monoclonal antibodies: evaluation and recommendations. ILAR J. 2005;46:269-79
20. Ishida T, Kashima S, Kiwada H. The contribution of phagocytic activity of liver macrophages to the accelerated blood clearance (ABC) phenomenon of PEGylated liposomes in rats. J Control Release. 2008;126:162-5
21. Nusrat AR, Wright SD, Aderem AA, Steinman RM, Cohn ZA. Properties of isolated red pulp macrophages from mouse spleen. J Exp Med. 1988;168:1505-10
22. Zhang P, Sun F, Liu S, Jiang S. Anti-PEG antibodies in the clinic: Current issues and beyond PEGylation. J Control Release. 2016;244:184-93
23. Garay RP, El-Gewely R, Armstrong JK, Garratty G, Richette P. Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert Opin Drug Deliv. 2012;9:1319-23
24. Yang Q, Jacobs TM, McCallen JD, Moore DT, Huckaby JT, Edelstein JN. et al. Analysis of pre-existing IgG and IgM antibodies against polyethylene glycol (PEG) in the general population. Anal Chem. 2016;88:11804-12
25. Chen BM, Su YC, Chang CJ, Burnouf PA, Chuang KH, Chen CH. et al. Measurement of pre-existing IgG and IgM antibodies against polyethylene glycol in healthy individuals. Anal Chem. 2016;88:10661-6
26. Richter AW, Akerblom E. Antibodies against polyethylene glycol produced in animals by immunization with monomethoxy polyethylene glycol modified proteins. Int Arch Allergy Appl Immunol. 1983;70:124-31
27. Richter AW, Akerblom E. Polyethylene glycol reactive antibodies in man: titer distribution in allergic patients treated with monomethoxy polyethylene glycol modified allergens or placebo, and in healthy blood donors. Int Arch Allergy Appl Immunol. 1984;74:36-9
28. Liu Y, Reidler H, Pan J, Milunic D, Qin D, Chen D. et al. A double antigen bridging immunogenicity ELISA for the detection of antibodies to polyethylene glycol polymers. J Pharmacol Toxicol Methods. 2011;64:238-45
29. Wadhwa M, Knezevic I, Kang HN, Thorpe R. Immunogenicity assessment of biotherapeutic products: An overview of assays and their utility. Biologicals. 2015;43:298-306
30. Zhang P, Sun F, Hung HC, Jain P, Leger KJ, Jiang S. Sensitive and quantitative detection of anti-poly(ethylene glycol) (PEG) antibodies by methoxy-PEG-coated surface plasmon resonance sensors. Anal Chem. 2017;89:8217-22
31. Zhang P, Sun F, Tsao C, Liu S, Jain P, Sinclair A. et al. Zwitterionic gel encapsulation promotes protein stability, enhances pharmacokinetics, and reduces immunogenicity. Proc Natl Acad Sci USA. 2015;112:12046-51
32. Poppenborg SM, Wittmann J, Walther W, Brandenburg G, Krahmer R, Baumgart J. et al. Impact of anti-PEG IgM antibodies on the pharmacokinetics of pegylated asparaginase preparations in mice. Eur J Pharm Sci. 2016;91:122-30
33. Armstrong JK, Hempel G, Koling S, Chan LS, Fisher T, Meiselman HJ. et al. Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer. 2007;110:103-11
34. Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed Engl. 2010;49:6288-308
35. Cavadas M, Gonzalez-Fernandez A, Franco R. Pathogen-mimetic stealth nanocarriers for drug delivery: a future possibility. Nanomedicine. 2011;7:730-43
36. Abu Lila AS, Nawata K, Shimizu T, Ishida T, Kiwada H. Use of polyglycerol (PG), instead of polyethylene glycol (PEG), prevents induction of the accelerated blood clearance phenomenon against long-circulating liposomes upon repeated administration. Int J Pharm. 2013;456:235-42
37. Ishihara T, Maeda T, Sakamoto H, Takasaki N, Shigyo M, Ishida T. et al. Evasion of the accelerated blood clearance phenomenon by coating of nanoparticles with various hydrophilic polymers. Biomacromolecules. 2010;11:2700-6
Author contact
![]() Corresponding authors: fchenkmu.edu.tw, tlchengedu.tw
Corresponding authors: fchenkmu.edu.tw, tlchengedu.tw