Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(10):2884-2895. doi:10.7150/thno.23824 This issue Cite
Research Paper
G9a stimulates CRC growth by inducing p53 Lys373 dimethylation-dependent activation of Plk1
Jie Zhang1,2#, Yafang Wang1#, Yanyan Shen1, Pengxing He3, Jian Ding1 ![]() , Yi Chen1
, Yi Chen1 ![]()
1. Division of Anti-Tumor Pharmacology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;
2. School of life Science and Technology, ShanghaiTech University, Shanghai 201210, China;
3. School of Pharmaceutical Sciences, Zhengzhou University, Zhengzhou 450001, China.
#These authors contributed equally to this work.
Received 2017-11-3; Accepted 2018-3-1; Published 2018-4-15
Abstract

Rationale: G9a is genetically deregulated in various tumor types and is important for cell proliferation; however, the mechanism underlying G9a-induced carcinogenesis, especially in colorectal cancer (CRC), is unclear. Here, we investigated if G9a exerts oncogenic effects in CRC by increasing polo-like kinase 1 (Plk1) expression. Thus, we further characterized the detailed molecular mechanisms.
Methods: The role of Plk1 in G9a aberrant CRC was determined by performing different in vitro and in vivo assays, including assessment of cell growth by performing cell viability assay and assessment of signaling transduction profiles by performing immunoblotting, in the cases of pharmacological inhibition or short RNA interference-mediated suppression of G9a. Detailed molecular mechanisms underlying the effect of G9a on Plk1 expression were determined by performing point mutation analysis, chromatin immunoprecipitation analysis, and luciferase reporter assay. Correlation between G9a and Plk1 expression was determined by analyzing clinical samples of patients with CRC by performing immunohistochemistry.
Results: Our study is the first to report a significant positive correlation between G9a and Plk1 levels in 89 clinical samples of patients with CRC. Moreover, G9a depletion decreased Plk1 expression and suppressed CRC cell growth both in vitro and in vivo, thus confirming the significant correlation between G9a and Plk1 levels. Further, we observed that G9a-induced Plk1 regulation depended on p53 inhibition. G9a dimethylated p53 at lysine 373, which in turn increased Plk1 expression and promoted CRC cell growth.
Conclusions: These results indicate that G9a-induced and p53-dependent epigenetic programing stimulates the growth of colon cancer, which also suggests that G9a inhibitors that restore p53 activity are promising therapeutic agents for treating colon cancer, especially for CRC expressing wild-type p53.
Keywords: G9a, polo-like kinase 1 (Plk1), p53, colorectal cancer, proliferation
Introduction
G9a, also known as lysine methyltransferase 1C (KMT1C) or euchromatic histone methyltransferase 2 (EHMT2), is the second identified histone methyltransferase (HMTase) that mainly catalyzes the mono- and dimethylation of histone H3K9 (H3K9me1/H3K9me2) [1]. Recent studies have reported that G9a also methylates some non-histone proteins such as CDYL1, WIZ, ACINUS, p53, and G9a itself [2, 3]. G9a is involved in embryonic development, genomic imprinting, transcriptional regulation, proliferation, apoptosis, senescence, autophagy, etc. [1, 4, 5]. Increasing evidence suggests that G9a is required for promoting cancer cell proliferation and maintaining a malignant phenotype. G9a dysregulation has been found in different human cancers, including lung cancer, breast cancer, and hepatocellular carcinoma [6, 7]. G9a promotes cell growth, invasion and metastasis of lung cancer and triple-negative breast cancer [8-10]. Previously, we found that G9a is highly expressed in colorectal cancer (CRC) and is important for stimulating CRC growth [11]. G9a depletion increases the rate of chromosome aberration, induces DNA double strand breaks, and promotes CRC cell senescence [11]. However, the tumorigenic role of G9a in CRC is still unclear.
Polo-like kinases (Plks) are highly conserved serine/threonine protein kinases that play a key role in cell division and checkpoint regulation during mitosis. Approximately 80% of human tumors of different origins express high levels of Plk transcripts [12]. Moreover, Plk overexpression is associated with poor prognosis and decreased overall survival rate in patients with different cancers [13-15]. Plk1 could be used as a progression marker in patients with CRC, and Plk1 depletion inhibits the migration and invasion of CRC cells [16, 17]. Therefore, therapeutic strategies targeting Plk1 are a promising new therapeutic approach for treating CRC.
In the present study, we examined the correlation between the level of Plk1, a crucial driver of cancer cell proliferation, and expression of G9a in clinical samples obtained from patients with CRC. We found that G9a increased Plk1 expression in a p53-dependent manner and stimulated CRC cell proliferation. Moreover, we attempted to elucidate mechanisms underlying G9a-induced upregulation of Plk1 transcription.
Methods
Chemicals and antibodies
UNC0638 and BIX01294 were purchased from Sigma (St. Louis, MO, USA), BI2536 was purchased from Selleck (Shanghai, China), BRD9539 and A-366 were purchased from MedChem Express (Shanghai, China). RIPA were purchased from Beyotime (Nantong, China). The study included the following primary antibodies: anti-G9a antibody (Cell Signaling Technology, Danvers, MA, USA), anti-Plk1 antibody (Cell Signaling Technology), rabbit anti-p53 antibody (Abcam, Cambridge, MA, USA), mouse anti-p53 antibody (Santa Cruz Biotechnology, Dallas, USA), anti-pan-methyl lysine antibody (Abcam), anti-histone H3 antibody (Santa Cruz Biotechnology), anti-histone H3 dimethyl (K9) antibody (Cell Signaling Technology), anti-β-actin antibody (Cell Signaling Technology), anti-GAPDH antibody (Cell Signaling Technology), and anti-GFP antibody (Santa Cruz Biotechnology). Horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit secondary antibodies were purchased from KangChen Bio-Tech (Shanghai, China), and HRP-conjugated donkey anti-goat IgG was purchased from Santa Cruz Biotechnology. Protein A/G agarose beads were purchased from Santa Cruz Biotechnology, and protease inhibitor cocktail was purchased from Roche Applied Science (Basel, Switzerland).
Immunohistochemistry
CRC tissues were provided by Shanghai Biochip Company Ltd. CRC tissue samples included 89 pairs of tumor and matched peritumoral tissues. IHC was performed as described previously [11]. Briefly, tissue samples were fixed overnight in 10% neutral-buffered formalin, processed, embedded in paraffin, sectioned, and incubated overnight with the primary antibodies at 4 °C in a humidified chamber, followed by incubation with the HRP-conjugated secondary antibodies for 2 h. Staining was completed by incubating the samples with 3.30-diaminobenzidine for 5-10 min, which produced a brown precipitate at the antigen site. The stained tissue sections were reviewed and scored separately by two pathologists who were blinded to clinical parameters. Any disagreements were arbitrated by a third pathologist.
Results of immunostaining were scored to 0, 0% positive cells; 1, 1%-25% positive cells; 2, 26%-50% positive cells; 3, 51%-75% positive cells; and 4, >75% positive cells according to the positive staining rate and divided into 0, 1+, 2+, 3+ according to the staining intensity. The final score was determined by combining the intensity and quantity scores, which yielded scores in the range from 0 to 12. Final staining scores of 0-5 indicated low relative protein expression and of 6-12 indicated high relative protein expression.
Cell culture
Human colon adenocarcinoma LoVo, HCT116, and HT29 cells and human embryonic kidney 293FT and 293T cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in a humidified atmosphere of 5% CO2 at 37 °C, according to the instructions of ATCC. HCT116 p53+/+ and HCT116 p53-/- cells were grown in Dulbecco's modified Eagle medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum.
Plasmids
Plasmid pLKO.1-shG9a was generously gifted by Dr. Jin Jian (University of North Carolina, USA). Plasmids pEGFP-N, pEGFP-hG9a, pEGFP-ΔSET-hG9a, and pcDNA3.1-Flag-p53 were obtained from Addgene [18]. Human G9a and p53 mutants were generated using Muta-direct Site-directed Mutagenesis Kit (Beijing SBS Genetech Co, Ltd, Beijing, China).
siRNA and plasmid transfection
siRNAs specifically targeting human genes encoding G9a, Plk1, and p53 were synthesized by GenePharma (Shanghai, China). These siRNAs were transfected using Lipofectamine RNAiMAX (Invitrogen). The plasmids were transfected using Lipofectamine 2000, according to the manufacturer's instructions, and were harvested at 24 or 48 h after transfection.
RT-PCR
Total cellular RNA was isolated using TRIzol reagent (Invitrogen, Oregon, USA) and was used for synthesizing first-strand cDNA by using a Reverse Transcription System (Roche). The mRNA expression of all target genes was quantified by performing qPCR with Power SYBR Green PCR Master Mix and ABI PRISM 7500 sequence detection system (Applied Biosystems, Foster City, CA, USA), with GAPDH as an internal control. Sequences of primers used for performing qPCR are provided in Table S1.
Colony formation assay
HCT116 p53+/+ and HCT116 p53-/- cells were seeded in six-well plates at a density of 500 cells/well. On the next day, UNC0638 or BIX01294 were added to the wells at different concentrations, and the cells were cultured for 10-15 days until colonies were visible. The colonies were fixed in 10% formaldehyde and 10% acetic acid for 10 min at room temperature and were stained with 1% crystal violet.
Sulforhodamine B assay
Cells were seeded in 96-well plates at the same density as that described in the previous assay and were transfected with siG9a or siPlk1. After incubation for 48 h, the cells were fixed with 10% trichloroacetic acid, washed with distilled water, and stained with sulforhodamine B (SRB; Sigma) in 1% acetic acid. SRB present in the cells was dissolved using 10 mM Tris-HCl, and optical density was measured at 560 nm by using spectraMAX190 (Molecular Devices, Sunnyvale, CA, USA).
Immunoprecipitation assay
To investigate the interaction between G9a and p53 in 293T cells, the cells were transfected with the indicated expression constructs for 48 h and were lysed in NP40 buffer (50 mM Tris-HCL (pH 8.0), 150 mM NaCl, and 0.5% NP40) containing complete TM protease inhibitors (Roche). Whole-cell extracts were immunoprecipitated by incubating overnight with mouse anti-GFP (G9a) or anti-Flag (p53) monoclonal antibody at 4 °C. Next, protein A/G beads were added to the cells, and the cells were incubated for another 2 h. Next, the beads were washed, and proteins were eluted using 0.2% SDS loading buffer, separated by performing SDS-PAGE, and processed for performing immunoblotting analysis. Anti-p53 antibody used for immunoblotting was a rabbit monoclonal antibody.
Western blotting analysis
Immunoprecipitates or cell lysates prepared using RIPA supplemented with a protease inhibitor cocktail were resolved by performing SDS-PAGE on 10% gels and were transferred onto nitrocellulose membranes. For performing western blotting analysis, the nitrocellulose membranes were incubated overnight with the appropriate antibodies at 4 °C, followed by incubation with a secondary antibody.
Generation of subcellular fraction
Cells were harvested in PBS buffer and were resuspended in 500 μL CLB buffer (10 mM Hepes, 10 mM NaCl, 1 mM KH2PO4, 5 mM NaHCO3, 5 mM EDTA, 1 mM CaCl2, and 0.5 mM MgCl2) on ice for 10 min. Cell lysates were homogenized by performing sonication on ice. Next, 50 μL 2.5 M sucrose was added to restore isotonic conditions. The first round of centrifugation was performed at 6300 ×g and 4 °C for 5 min. Pellet obtained was washed with TSE buffer (10 mM Tris, 300 mM sucrose, 1 mM EDTA, and 0.1% NP40 [pH 7.5]) and was centrifuged at 4000 ×g and 4 °C for 5 min until the supernatant was clear. Next, the supernatant was discarded, and pellets containing the nuclei were retained. The supernatant from the first round of differential centrifugation was precipitated for 15 min at 14000 ×g. The resulting pellets contained cell membranes, and the supernatant contained cytoplasm.
Chromatin immunoprecipitation
ChIP analysis was performed as described previously. Briefly, cells were crosslinked with 1% formaldehyde for 15 min at room temperature, and the reaction was stopped using 125 mM glycine. Nuclei were isolated by resuspending the cells in a swelling buffer containing 5 mM PIPES (pH 8.0), 85 mM KCl, 1% NP-40, and complete protease inhibitors for 20 min at 4° C. The isolated nuclei were resuspended in a nuclei lysis buffer (50 mM Tris (pH 8.0), 10 mM EDTA, and 1% SDS) and were sonicated using Bioruptor Sonicator (Diagenode). Samples were immunoprecipitated by incubating overnight with the appropriate antibodies at 4 °C. Immunoprecipitates obtained were washed three times with ChIP wash low-salt buffer (100 mM Tris (pH 8.0), 500 mM LiCl, 1% NP-40, and 1% deoxycholic acid sodium salt) and once with high-salt buffer. After reverse crosslinking, DNA was eluted and purified using a PCR purification kit (Qiagen). Primer sequences used for performing ChIP analysis are listed in Table S1.
Luciferase reporter assay
For this, 293T cells were co-transfected with 250 ng pGL3-plk1-promoter-luc, 1 μg pEGFP-hG9a (WT, SET domain deleted or Y1154F mutant), or siNC or siG9a and 25 ng pRL-TK-Renilla luciferase plasmids. After 24 h, the cells were lysed and luciferase activity was measured using Dual-Luciferase Reporter Assay System (Promega), according to the manufacturer's instructions.
Animal studies
Athymic BALB/c nude mice (age, 4-6 weeks; Beijing HFK Bioscience Co., Ltd, Beijing, China) were housed in a specific pathogen-free room maintained at a temperature of 25 °C ± 1 °C and 12 h/12 h light/dark cycle and were fed autoclaved chow diet and water ad libitum. All animal experiments were performed according to the institutional ethical guidelines on animal care and were approved by the Institute Animal Care and Use Committee at Shanghai Institute of Materia Medica. HCT116 and LoVo cells were subcutaneously injected into the right flank of the nude mice at a density of 5 × 106 cells/mouse (six mice per group). Tumor diameters were measured two times per week, and tumor volumes (V) were calculated using the formula ½ × length × width2.
Statistical analyses
Statistical analyses, including the determination of mean, SD, and SEM, were performed using GraphPad Prism. Statistical differences between the groups were determined using two-tailed Student's t-test, two-way ANOVA, or one-way ANOVA with Dunnett's multiple comparisons test. P < 0.05 was considered statistically significant.
Results
Positive correlation between Plk1 and G9a levels in patients with CRC
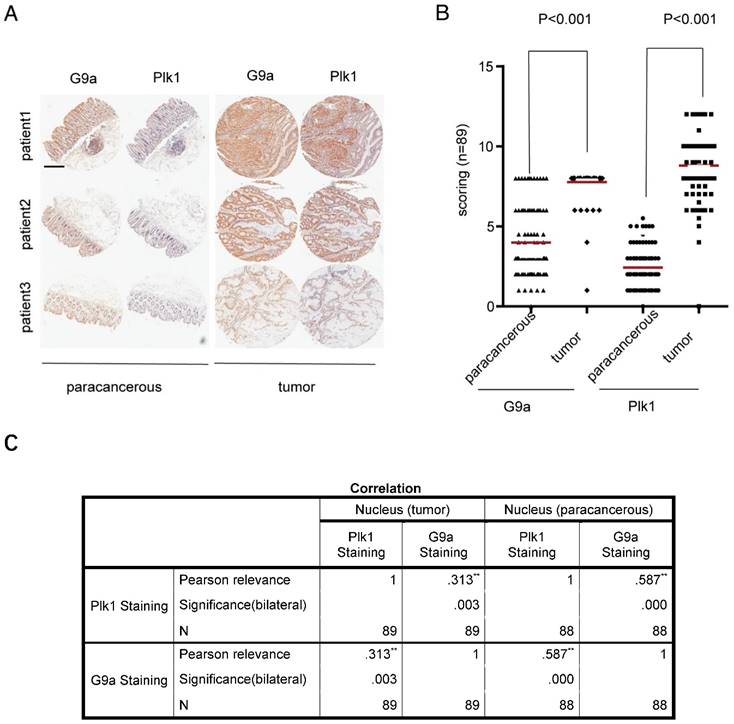
Previously, we found that G9a depletion induced DNA damage in CRC cells and arrested some kinds of CRC cells in G2/M phase [11]. Because Plk1 is an important regulator of several events during mitosis and DNA damage, especially mitotic entry and exit, we examined the correlation between G9a and Plk1 levels by performing immunohistochemistry (IHC) with a tissue microarray containing 89 CRC tissue samples (linked to detailed clinic-pathological and long term clinical outcome data with matched adjacent normal tissues). Staining score was classified as high or low based on the sum of proportion and intensity scores. In all the tested samples, G9a and Plk1 staining were mainly observed in the nucleus. Examination of paired CRC specimens showed significantly higher G9a and Plk1 levels in tumor tissues than in peritumoral tissues (P < 0.001; Figure 1A-B). Interestingly, we observed a significant positive correlation between nuclear G9a and Plk1 expression in tumor as well as peritumoral tissue of patients with CRC (P < 0.01; Figure 1C).
High G9a level is correlated with high Plk1 level in CRC. (A) Representative photomicrographs of IHC staining of G9a and Plk1 in CRC tissues and corresponding peritumoral tissues (scale bar = 14 mm). (B) IHC scores of G9a and Plk1 were plotted for each specimen. (C) Pearson correlation between G9a and Plk1 expression in clinical samples of patients with CRC (n = 89).
Depletion of G9a represses Plk1 expression in vitro and in vivo
To further clarify the correlation between G9a and Plk1 expression, we transfected CRC cell lines HCT116 and LoVo with small interfering RNA (siRNA) or shRNA against the gene encoding G9a to establish transiently silenced or stable G9a knockdown cells. As expected, G9a depletion suppressed Plk1 expression in HCT116 and LoVo cells (Figure 2A-B). Moreover, G9a overexpression increased Plk1 expression in these cells (Figure 2C). Next, G9a-knockdown cells were subcutaneously inoculated into nude mice. We observed that G9a silencing profoundly impaired the growth of xenograft tumors derived from HCT116 and LoVo CRC cells in nude mice (Figure 2D), which was consistent with that reported previously [11]. Moreover, as expected, Plk1 and Ki67 expression was downregulated in tumor xenografts derived from stable G9a-knockdown HCT116 cells (Figure 2E-F). However, G9a depletion had no obvious effect on the expression of other Plk proteins such as Plk2, Plk3, and Plk4 (Figure S1A).
G9a positively regulates Plk1 expression. Treatment with specific (A) siRNA or (B) shRNA reduced G9a expression and decreased Plk1 protein level in HCT116 and LoVo cells. (C) Plk1 level increased in G9a-overexpressing HCT116 cells. (D) Growth of HCT116 and LoVo cells after G9a knockdown and inoculation in nude mice. (E) Representative images showing IHC staining of G9a and Plk1 in xenografts derived from stable G9a-knockdown HCT116 cells (scale bar = 25 μM). (F) Immunoblotting of G9a, Plk1, and GAPDH (loading control) in xenografts derived from HCT116 cells. (G) Relative Plk1 mRNA and protein levels in G9a-knockdown, WT G9a-expressing, and G9a SET domain-deleted rescued HCT116 cells. All data are representative of at least three independent experiments and are expressed as mean ± SD; *P < 0.05, **P < 0.01, and ***P < 0.001.
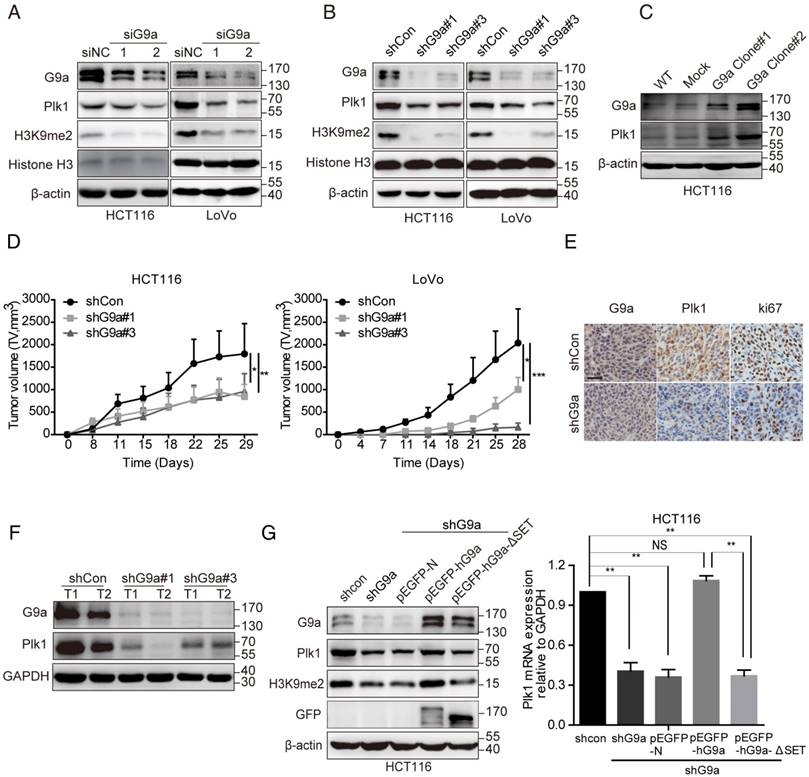
Next, we investigated whether Plk1 regulated G9a level. We found that Plk1 knockdown by using a specific siRNA (siPlk1) or treatment with Plk1 enzyme inhibitor BI2536 [19] for 72 h did not affect G9a expression, suggesting that Plk1 is a downstream signaling protein of G9a (Figure S1B).
G9a regulates Plk1 transcription in a p53-dependent manner
It has been reported that G9a induces transcriptional activation in an HMTase-independent manner or by methylating non-histone targets [2, 3]. To evaluate whether the HMTase activity of G9a regulated Plk1 expression, we transfected G9a-depleted HCT116 (HCT116 shG9a) cells with plasmids expressing wild-type (WT) G9a (pEGFP-hG9a WT) or non-activated G9a (lacking SET domain, pEGFP-hG9a-ΔSET, and enzyme-dead point mutation, pEGFP-hG9a-Y1154F [21]) and evaluated Plk1 mRNA and protein levels. We found that decreased Plk1 expression was restored in pEGFP-hG9a WT-transfected HCT116 shG9a cells and that Plk1 expression remained suppressed in pEGFP-N (mock)-, pEGFP-hG9a-ΔSET- or pEGFP-hG9a-Y1154F- transfected HCT116 shG9a cells (Figure 2G and Figure S1C). Similar results were obtained for LoVo shG9a cells (Figure S1D).
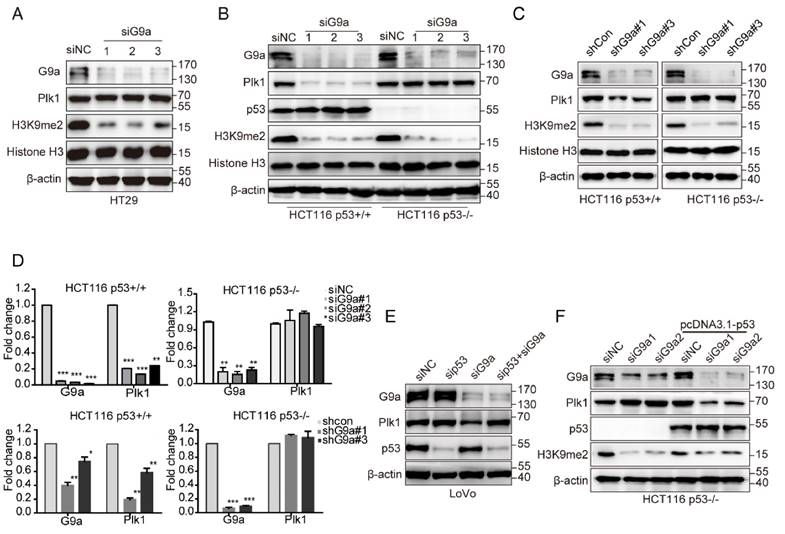
Plk1 is directly repressed by p53 [22], and p53 is an important non-histone substrate of G9a [3]. Therefore, we hypothesized that p53 is necessary for G9a-induced regulation of Plk1 expression. As expected, G9a depletion did not affect Plk1 expression in HT-29 cells, a p53 mutant cell line (Figure 3A). Next, we transfected HCT116 p53+/+ and HCT116 p53-/- cells with siG9a and observed a significant decrease in Plk1 mRNA and protein levels in p53-competent cells, with no obvious change of Plk1 mRNA and protein levels in p53-null cells (Figure 3B, D). To further assess the role of p53 in G9a-Plk1 pathway, we generated stable HCT116 p53+/+ and HCT116 p53-/- cells showing limited G9a expression. We observed that Plk1 mRNA and protein expression reduced only in stable G9a-knockdown HCT116 p53+/+ cells (Figure 3C, D). These data strongly indicate that G9a-induced Plk1 expression depends on WT p53. Similar results were obtained using WT p53-expressing LoVo cells. G9a silencing did not exert any obvious effect on Plk1 expression in LoVo p53 depletion cells (Figure 3E). To further verify these observations, HCT116 p53-/- cells were re-transfected with the p53 gene. G9a silencing remarkably decreased Plk1 level in p53-re-transfected cells (Figure 3F). Together, these data indicate that G9a upregulates Plk1 expression in a p53-dependent manner.
G9a regulates Plk1 expression in a p53-dependent manner. (A) Plk1 expression was analyzed in G9a-depleted HT29 cells. G9a, Plk1, p53, H3K9me2, histone H3, and GAPDH (loading control) protein levels in HCT116 p53+/+ and HCT116 p53-/- cells after (B) siRNA- or (C) shRNA-mediated G9a knockdown. (D) Plk1 mRNA level in HCT116 p53+/+ and HCT116 p53-/- cells after siRNA- or shRNA-mediated G9a knockdown. (E) G9a, Plk1, p53, and β-actin (loading control) protein levels in LoVo cells after G9a knockdown or G9a and p53 double knockdown. (F) HCT116 p53-/- cells were transfected with a p53-overexpressing plasmid to restore G9a-mediated regulation of Plk1 expression. All data are representative of at least three independent experiments and are expressed as mean ± SD; *P < 0.05, **P < 0.01, and ***P < 0.001.
G9a inactivates p53 through a methylation-dependent mechanism
Next, we attempted to determine the detailed mechanism underlying G9a-induced p53-dependent regulation of Plk1 expression. To determine the physical interaction between p53 and G9a, we performed co-immunoprecipitation (co-IP) analysis by using HEK293T cells co-transfected with pcDNA3.1-Flag-p53 and pEGFP-hG9a. G9a and p53 were detected in immunoprecipitates obtained using Flag-specific (p53) or GFP-specific (G9a) antibody but not in immunoprecipitates obtained using control IgG (Figure 4A). Moreover, stable G9a-overexpressing 293T cells showed increased Plk1 expression (Figure 4B). Results of co-IP assay by using the anti-GFP antibody showed that G9a interacted with p53 in 293T-hG9a cells (Figure 4C), suggesting a definite interaction between G9a and p53.
G9a interacts with and methylates p53. (A) Identification of the exogenous interaction between G9a and p53 in 293T cells. HEK293T cells were transfected with pEGFP-G9a or Flag-p53, and immunoprecipitates obtained using anti-GFP (G9a) or anti-Flag (p53) antibody were analyzed by performing western blotting analysis. (B) Plk1 expression increased in stable G9a-overexpressing 293T cells. (C) G9a co-precipitated with endogenous p53 in 293T cells. (D) Methylated p53 level increased in stable G9a-overexpressing 293T cells.
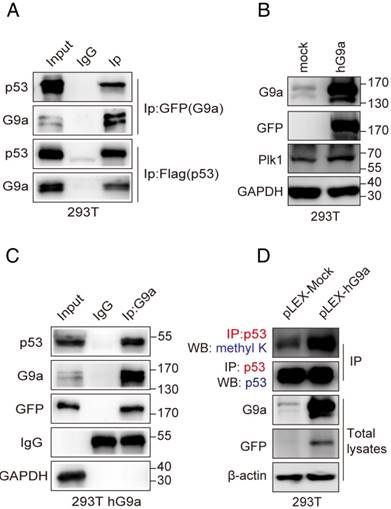
Since p53 is a non-histone substrate of G9a, we hypothesized that G9a regulated Plk1 expression by methylating p53. Results of immunoprecipitation with an anti-p53 antibody, followed by immunoblotting with a pan-methyl lysine antibody, showed increased levels of methylated p53 in G9a-overexpressing 293T cells (Figure 4D). It has also been revealed that human G9a promotes p53 activity by inducing its acetylation [23]. Therefore, we investigated whether G9a affected p53 acetylation in CRC. G9a depletion or the HMTase-specific inhibitor did not affect p53 pan-acetylation levels (Figure S2A-B), suggesting that G9a affected p53 activity in a methylation-dependent manner to facilitate Plk1 expression.
G9a-mediated p53 Lys373me2 is important for Plk1 transactivation
Modification of p53 by methylation mainly occurs at specific C-terminal lysines such as Lys370, Lys372, Lys373 and Lys381 (K370, K372, K373 and K381) [24]. Therefore, we substituted these lysine residues with arginine (K370R, K372R, K373R and K381R) to identify the lysine residue in p53 that was methylated by G9a for regulating p53 activity and Plk1 expression. HCT116 p53-/- cells were transfected with Flag-tagged p53 wildtype (WT), p53K370R, p53K372R, p53K373R or p53K381R plasmids for 24 h, followed by transfection with siG9a for another 48 h. Western blotting analysis detected reduced Plk1 levels in whole-cell extracts of cells transfected with plasmids expressing p53 WT, p53K370R, p53K372R or p53K381R together with siG9a but no change in Plk1 levels in the extracts of cells transfected with p53K373R and siG9a (Figure 5A). Thus, these results suggest that G9a regulates Plk1 expression by methylating p53K373.
p53 K373me2 is important for Plk1 transactivation. (A) Western blotting analysis detected reduced Plk1 levels in the whole-cell extracts of cells transfected with plasmids expressing WT p53, p53K370R, p53K372R, p53K373R or p53K381R after G9a knockdown. (B) Results of western blotting analysis showing Plk1 level in separated nuclear and cytoplasmic fractions. (C) Immunoprecipitates obtained from the extracts of G9a-knockdown HCT116 cells by using anti-p53 antibody were analyzed by performing western blotting analysis with anti-p53 K373me2 and anti-p53 antibodies. (D) Identification of p53K373me2 in stable G9a-overexpressing 293T cells. (E) Interaction between p53 and G9a was determined after transfecting 293T cells with p53K373R mutant plasmid. Immunoprecipitates obtained from whole-cell extracts of 293T cells transfected with Flag-p53, p53K372R, or p53K373R plasmid by using anti-p53 antibody were analyzed by performing western blotting analysis with anti-p53 K373me2 and anti-G9a antibodies. (F) Binding levels of p53 and abundance of p53 K373me2 in the p53RE2 region (-816 to -785) of the Plk1 promoter in G9a-knockdown HCT116 cells were determined by performing ChIP-qPCR analysis. Data obtained using the GAPDH coding region was used as positive control. Dashed line indicates IgG control. Error bars represent SD of triplicate experiments and are representative of two independent experiments. (G) HEK293T cells were co-transfected with pGL3-Plk1-luc (0.25 µg) and pEGFP-G9a (1 µg), pEGFP-G9a-ΔSET (1 µg), or pEGFP-N (1 µg; control) along with the pRL-TK-Renilla luciferase expression plasmid (0.05 µg). Cell extracts were assayed for luciferase activity. Results are expressed as mean ± SD of three independent experiments; *P < 0.05, **P < 0.01, and ***P < 0.001.
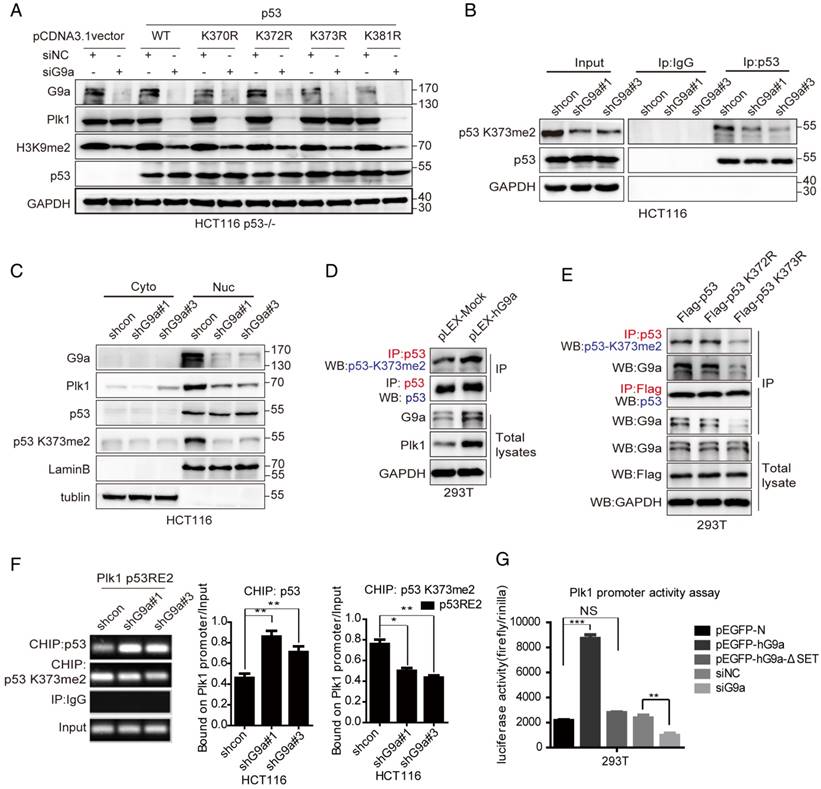
We developed an antibody against p53K373me2 that specifically recognized p53 peptides dimethylated at K373 and did not cross-react with unmodified p53 or other methylated p53 residues, as determined by performing ELISA, dot assay and western-blot (Figure S3A-C). Immunoprecipitation of p53 followed by immunoblotting with anti-p53K373me2 antibody showed reduced p53K373me2 level in G9a-depleted HCT116 cells (Figure 5B). Moreover, nuclear Plk1 expression decreased in G9a-depleted HCT116 cells, which was consistent with the decrease in nuclear p53K373me2 level (Figure 5C). Similar results were obtained by treatment with G9a inhibitor BRD9539[25], which competes for SAM. Plk1 levels were decreased companied with p53K373me2 decline in HCT116 cells treated with BRD9539 (Figure S3D). These results suggest that inhibition of p53K373 methylation by G9a depletion or its enzymatic activity activates p53 and represses Plk1 expression. We also observed that G9a interacted with p53K373me2 in G9a-overexpressing 293T cells and that p53 showed increased K373-dimethylation level in 293T-hG9a cells (Figure 5D). In addition, G9a easily interacted with p53K372R compared with p53K373R (Figure 5E). Next, we performed chromatin immunoprecipitation (ChIP) analysis to investigate whether G9a regulated Plk1 transcription in a p53-dependent manner. p53 binds to the Plk1 promoter in the region from -816bp to -785bp, which is termed p53-responsive element 2 (p53RE2). G9a depletion profoundly increased p53 activity and its recruitment to the p53RE2 in the Plk1 promoter. Conversely, p53K373me2 recruitment to the p53RE2 in the Plk1 promoter was significantly decreased in HCT116 shG9a cells (Figure 5F).
To further understand mechanisms underlying the transcriptional regulation of Plk1 by the G9a-p53 pathway, we performed a luciferase reporter assay by using pGL3-Plk1-luc reporter system. Plk1 transcription was repressed after G9a depletion and was stimulated after G9a overexpression in 293T cells transfected with the pGL3-Plk1-luc system (Figure 5G), which was consistent with the results of real-time PCR. Moreover, results obtained using the SET domain-deleted G9a mutant showed that G9a depletion-induced transcriptional repression of Plk1 was dependent on its methyltransferase activity (Figure 5G). To further verify that G9a increases Plk1 expression by regulating p53 K373me2, we also tested effects of p53 WT and K373R mutant on Plk1 promoter activity. As shown in Figure S3E, p53 WT significantly suppressed Plk1 transcription while p53 K373R had no effect. These data clearly indicate that G9a positively regulates Plk1 expression by interfering with the binding of p53 to the Plk1 promoter (-816bp to -785bp region), which was dependent on p53K373 dimethylation.
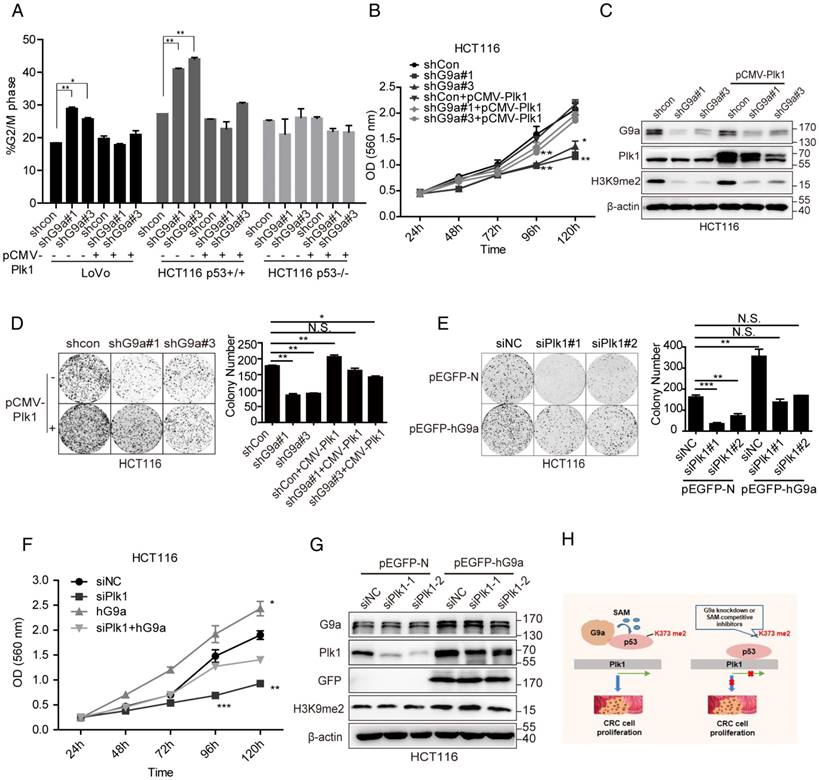
Plk1 plays an important role in G9a-induced cell proliferation
Next, we determined whether Plk1 promoted G9a-induced phenotypes. If the stimulative effect of G9a on Plk1 expression is important for the growth-promoting function of G9a, we would expect restoration of Plk1 expression in G9a-depleted CRC cells may facilitate their survival. G9a knockdown drastically arrested WT p53-expressing CRC cells (LoVo and HCT116 p53+/+ cells) in the G2/M phase of the cell cycle (Figure 6A) and reduced cell growth in vivo (Figure 2D). However, no obvious G2/M phase cell cycle arrest was observed in HCT116 p53-/- cells and p53 mutant-expressing HT29 cells lacking G9a (Figure 6A and Figure S4A). As expected, re-introduction of Plk1 partially restored cell cycle arrest and cell growth of G9a-knockout HC116 p53+/+ cells (Figure 6A-D and Figure S4B-D). In contrast, G9a overexpression accelerated clonogenic activity, and Plk1 knockdown by using siPlk1 partially restricted the growth of G9a-overexpressing cells (Figure 6E-G). Together, these data suggest that decreased Plk1 expression induced by G9a depletion in WT p53-expressing CRC cells decreases their proliferation. Another interesting finding of this study was that G9a and Plk1 inhibitors exerted synergistic effects in CRC cells. The effect of combination therapy with G9a and Plk1 inhibitors was determined by calculating the combination index (CI) with the CalcuSyn program, which is based on the Chou-Talalay method [27]. A CI < 0.8 indicates a synergistic effect. We observed that Plk1 inhibitor BI2536 and G9a inhibitors UNC0638 or BIX01294, which compete for histone, exerted synergistic effects in HCT116 cells irrespective of their p53 expression status, especially in HCT116 p53+/+ cells (Figure S5A-B). Together, the results of the present study suggest that G9a overexpression contributes to the accelerative proliferation of WT p53-expressing CRC by inducing Plk1 expression, while G9a knockdown decreased p53K373me2 level and accompanying Plk1 level, which led to cell cycle arrest and growth inhibition.
G9a regulates CRC growth by regulating Plk1. (A) Cell cycle arrest induced by G9a inhibition in WT p53-expressing cells was reduced after transfecting a Plk1-overexpressing plasmid pCMV-Plk1. (B) Growth curves of HCT116 shG9a cells with or without Plk1 restoration. (C) Western blotting analysis of Plk1 restoration in HCT116 shG9a cells. (D) Plk1 restoration promoted colony formation by HCT116 cells. (E) Colony formation ability and (F) cell proliferation reduced after siPlk1-induced Plk1 depletion in G9a-overexpressing cells. (G) Western blotting analysis of G9a and Plk1 expression levels in G9a-overexpressing HCT116 cells after Plk1 knockdown. (H) A schematic model of G9a function in CRC cell proliferation. All data are expressed as a mean of three independent experiments. Error bars indicate ±SD; *P < 0.05, **P < 0.01, and ***P < 0.001.
Discussion
Although G9a promotes cancer cell proliferation, the detailed molecular mechanisms underlying this have not been elucidated. Previously, we highlighted the importance of G9a in colon cancer progression and provided insights on the possible benefits associated with G9a downregulation in CRC [11]. The present study is the first to show a significant positive correlation between G9a and Plk1 levels in 89 clinical samples obtained from patients with CRC. This was further validated by the finding that G9a depletion decreased the expression of Plk1 and suppressed the proliferation of WT p53-expressing CRC cells (Figure 6H). Further, our data showed that G9a-induced Plk1 regulation was mediated by p53 inhibition. G9a dimethylated p53 at lysine 373, which in turn increased Plk1 expression and promoted colon cancer cells growth.
Plk1 is the most well-known protein belonging to the Plk family, and its role in mitosis has been characterized in detail. Plk1 overexpression contributes to oncogenesis by inducing chromosome instability and other mitotic defects associated with the dysfunction of the checkpoint system, a hallmark of cancer. Plk1 is involved in tumorigenesis and progression of CRC and is a predictive marker of poor prognosis [28]. Our present study highlighted the key role of G9a in Plk1 regulation in CRC expressing WT p53. Several studies have shown that Plk1 is a key molecule of a regulatory network that controls entry into mitosis at the G2/M transition. In the present study, we found that G9a silencing induced Plk1 downregulation and G2/M phase cell cycle arrest. We proved that G9a played an upstream regulatory role in p53-mediated Plk1 expression and stimulated the progression of CRC expressing WT p53. Thus, our findings suggest that G9a is a promising new therapeutic target for treating CRC, especially CRC expressing WT p53.
The tumor suppressor protein p53 plays a critical role in preventing human cancers and in tumor response to chemotherapy. p53 is a transcription factor that activates and/or represses its target genes, indicating the presence of a highly complex regulatory network for fine-tuning the responses of p53 to different stress signals [29]. Many mechanisms regulate the transcriptional activation of p53, including nuclear localization, DNA binding, post-translational modification, and co-factor recruitment, which are interdependent of each other. A recent study showed that lysine methylation regulates p53 activity [1]. Multiple lysines in p53, including Lys370, Lys372, and Lys282, are methylated by histone methylases such as Smyd2, Set7/9, and Pe-Set7, respectively [30, 31]. Similar to that observed with histones, variable levels of p53 methylation induce different biological outcomes. For example, Lys370me1 represses p53 function, whereas Lys370me2 activates p53. Lys382me1 antagonizes Lys382 acetylation, whereas Lys382me2 is associated with p53 response to DNA damage [32, 33]. Moreover, p53 methylation is mediated by G9a [3]. Rada et al. showed that human G9a augmented the activity of p53 toward its target genes irrespective of its methylation status, which relied on the recruitment of at least one co-activator, including HAT and CBP, by G9a [23]. The latter event increased the acetylation of p53 and activation of p53 target genes such as Puma. In contrast, Huang et al. found that G9a inhibited p53 activity by methylating it at K373 [3], which is inconsistent with the result of Rada et al. [23]. However, target genes regulated by p53K373 were not determined in that study. In the present study, we found that G9a inhibited p53 by dimethylating it at K373 and positively regulated Plk1 expression at both the transcriptional and translational levels. Results of the Plk1-Luc reporter and ChIP assays showed that G9a increased the promoter activity and expression of Plk1 in CRC cells expressing WT p53. G9a knockdown decreased the Plk1 promoter activity in a SET domain-dependent manner. And as expected, G9a inhibitor BRD9539 declined the expression of Plk1 accompanied with significantly decreased p53K373me2. All results shown in the manuscript strongly suggest that G9a modulates Plk1 expression by dimethylating p53 at lysine 373.
Unbiased screening approaches have identified essential G9a in tumors, leading to the development of potent and selective inhibitors. However, compounds targeting G9a have not shown sufficient in vivo efficacy in various cancer models to date despite showing promising selectivity and potency in vitro. These findings strongly suggest that G9a, which negatively regulates p53 activity, acts as an oncogene when expressed at aberrantly high levels. These results also suggest a new strategy involving G9a inhibitors that restore p53 activity. This concept is particularly intriguing for treating cancers expressing the WT p53 gene. In summary, the results of the present study strongly indicate that G9a inhibition may be a promising therapeutic target for treating CRC, especially CRC expressing WT p53.
Abbreviations
G9a: lysine methyltransferase 1C or euchromatic histone methyltransferase 2; Plk1: polo-like kinase 1; CRC: colorectal cancer; CDYL1: Chromodomain Y Like; WIZ: widely interspaced zinc finger motifs; GAPDH: Glyceraldehyde-3-Phosphate Dehydrogenase; GFP: Green fluorescent protein; HAT: histone acetyltransferase; siRNA: small interfering RNA; WT: wildtype; IHC: Immunohistochemistry; SRB: sulforhodamine B; CI: combination index; ChIP: chromatin immunoprecipitation; IP: immunoprecipitation; co-IP: co-immunoprecipitation.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Professor Jian Jin (University of North Carolina at Chapel Hill) for providing the plasmids pLKO.1-shControl and pLKO.1-shG9a and Martin J. Walsh for providing the plasmids pEGFP-hG9a and pEGFP-ΔSET-hG9a. We are grateful to Thomas Roberts for providing the plasmid pcDNA3.1-Flag-p53 and Dr B. Vogelstein (Johns Hopkins) for providing HCT116 p53+/+ and HCT116 p53-/- cells. This work was supported by grants from the National Natural Science Foundation of China (81773763 and 81321092).
Authors' contributions
Jie Zhang, Yafang Wang and Pengxing He conceived and performed the experiments. Yanyan Shen and Yi Chen performed the animal experiments. Jie Zhang, Yafang Wang and Yi Chen analyzed the data and wrote the manuscript. Yi Chen and Jian Ding conceived the study, reviewed and edited the manuscript, acquired funding, provide resources, and supervised and administered the project.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tachibana M, Sugimoto K, Fukushima T, Shinkai Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem. 2001;276:25309-17
2. Rathert P, Dhayalan A, Murakami M, Zhang X, Tamas R, Jurkowska R. et al. Protein lysine methyltransferase G9a acts on non-histone targets. Nat Chem Biol. 2008;4:344-6
3. Huang J, Dorsey J, Chuikov S, Perez-Burgos L, Zhang X, Jenuwein T. et al. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J Biol Chem. 2010;285:9636-41
4. Shankar SR, Bahirvani AG, Rao VK, Bharathy N, Ow JR, Taneja R. G9a, a multipotent regulator of gene expression. Epigenetics. 2013;8:16-22
5. De Narvajas AAM, Gomez TS, Zhang J, Mann AO, Taoda Y, Gorman JA. et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol Cell Biol. 2013;33:3983-93
6. Watanabe H, Soejima K, Yasuda H, Kawada I, Nakachi I, Yoda S. et al. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 2008;8:15
7. Kondo Y, Shen L, Suzuki S, Kurokawa T, Masuko K, Tanaka Y. et al. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol Res. 2007;37:974-83
8. Chen MW, Hua KT, Kao HJ, Chi CC, Wei LH, Johansson G. et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010;70:7830-40
9. Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG. et al. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. 2012;122:1469-86
10. Wang YF, Zhang J, Su Y, Shen YY, Jiang DX, Hou YY. et al. G9a regulates breast cancer growth by modulating iron homeostasis through the repression of ferroxidase hephaestin. Nat Commun. 2017;8:274
11. Zhang J, He P, Xi Y, Geng M, Chen Y, Ding J. Down-regulation of G9a triggers DNA damage response and inhibits colorectal cancer cells proliferation. Oncotarget. 2015;6:2917-27
12. Li L, Wang X, Chen J, Ding H, Zhang Y, Hu T. et al. The natural product Aristolactam AIIIa as a new ligand targeting the polo-box domain of polo-like kinase 1 potently inhibits cancer cell proliferation. Acta Pharmacol Sin. 2009;30:1443-53
13. Lowery DM, Lim D, Yaffe MB. Structure and function of Polo-like kinases. Oncogene. 2005;24:248-59
14. De Carcer G, Manning G, Malumbres M. From Plk1 to Plk5: functional evolution of polo-like kinases. Cell Cycle. 2011;10:2255-62
15. Ng WTW, Shin J, Roberts TL, Wang B, Lee CS. Molecular interactions of polo-like kinase 1 in human cancers. J Clin Pathol. 2016;69:557-62
16. Han D, Zhu Q, Cui J, Wang P, Qu S, Cao Q. et al. Polo-like kinase 1 is overexpressed in colorectal cancer and participates in the migration and invasion of colorectal cancer cells. Med Sci Monit. 2012;18:237-46
17. Weichert W, Kristiansen G, Schmidt M. et al. Polo-like kinase 1 expression is a prognostic factor in human colon cancer. World J Gastroenterol. 2005;11:5644-50
18. Gjoerup O, Zaveri D, Roberts TM. Induction of p53-independent apoptosis by simian virus 40 small t antigen. J Virol. 2001;75:9142-55
19. Gleixner KV, Ferenc V, Peter B, Gruze A, Meyer RA, Hadzijusufovic E. et al. Polo-like kinase 1 (Plk1) as a novel drug target in chronic myeloid leukemia: overriding imatinib resistance with the Plk1 inhibitor BI 2536. Cancer Res. 2010;70:1513-23
20. Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V. et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7:566-74
21. Ling BMT, Bharathy N, Chung TK, Kok WK, Li SD, Tan YH. et al. Lysine methyltransferase G9a methylates the transcription factor MyoD and regulates skeletal muscle differentiation. Proc Natl Acad Sci. USA. 2012;109:841-6
22. Mckenzie L, King S, Marcar L, Nicol S, Dias S, Schumm K. et al. p53-dependent repression of polo-like kinase-1 (PLK1). Cell Cycle. 2010;9:4200-12
23. Rada M, Vasileva E, Lezina L, Marouco D, Antonov AV, Macip S. et al. Human EHMT2/G9a activates p53 through methylation-independent mechanism. Oncogene. 2017;36:922-32
24. Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA. et al. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629-32
25. Yuan Y, Wang Q, Paulk J, Kubicek S, Kemp MM, Adams DJ. et al. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem Biol. 2012;7:1152-7
26. Pappano WN, Guo J, He Y, Ferguson D, Jagadeeswaran S, Osterling DJ. et al. The Histone Methyltransferase Inhibitor A-366 Uncovers a Role for G9a/GLP in the Epigenetics of Leukemia. PloS one. 2015;10:e0131716
27. Chou TC, Talalay P. Analysis of combined drug effects: a new look at a very old problem. Trends Pharmacol Sci. 1983;4:450-54
28. Degenhardt Y, Lampkin TA. Targeting Polo-like Kinase in Cancer Therapy. Clin Cancer Res. 2010;16:384-89
29. Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609-22
30. Chuikov S, Kurash J, Wilson JR, Xiao B, Justin N, Ivanov GS. et al. Regulation of p53 activity through lysine methylation. Nature. 2004;432:353-60
31. Huang J, Perezburgos L, Placek BJ, Sengupta R, Richter M, Dorsey J. et al. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629-32
32. Shi X, Kachirskaia I, Yamaguchi H, West LE, Wen H, Wang EW. et al. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol Cell. 2007;27:636-46
33. Huang J, Sengupta R, Espejo A, Lee MG, Dorsey J, Richter M. et al. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105-08
Author contact
![]() Corresponding author: Yi Chen, Email: ychenac.cn; Jian Ding, Email: jdingac.cn
Corresponding author: Yi Chen, Email: ychenac.cn; Jian Ding, Email: jdingac.cn