Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(10):2862-2883. doi:10.7150/thno.23547 This issue Cite
Research Paper
MicroRNA-193b-3p regulates chondrogenesis and chondrocyte metabolism by targeting HDAC3
Fangang Meng#, Zhiwen Li#, Zhiqi Zhang, Zibo Yang, Yan Kang, Xiaoyi Zhao, Dianbo Long, Shu Hu, Minghui Gu, Suiwen He, Peihui Wu, Zongkun Chang, Aishan He ![]() , Weiming Liao
, Weiming Liao ![]()
Department of Joint Surgery, First Affiliated Hospital of SunYat-sen University, Guangzhou, Guangdong 510080, China
#Co-first authors: These authors contributed equally to this work.
Received 2017-10-20; Accepted 2018-3-12; Published 2018-4-15
Abstract

Histone deacetylase 3 (HDAC3) plays a pivotal role in the repression of cartilage-specific gene expression in human chondrocytes. The aim of this study was to determine whether microRNA-193b-3p (miR-193b-3p) regulates the expression of HDAC3 during chondrogenesis and chondrocyte metabolism.
Methods: miR-193b-3p expression was assessed in a human mesenchymal stem cell (hMSC) model of chondrogenesis, in interleukin-1β (IL-1β)-treated primary human chondrocytes (PHCs), and in non-degraded and degraded cartilage. hMSCs and PHCs were transfected with miR-193b-3p or its antisense inhibitor. A direct interaction between miR-193b-3p and its putative binding site in the 3′-untranslated region (3′-UTR) of HDAC3 mRNA was confirmed by performing luciferase reporter assays. Chondrocytes were transfected with miR-193b-3p before performing a chromatin immunoprecipitation assay with an anti-acetylated histone H3 antibody. To investigate miR-193b-3p-transfected PHCs in vivo, they were seeded in tricalcium phosphate-collagen-hyaluronate (TCP-COL-HA) scaffolds, which were then implanted in nude mice. In addition, plasma exosomal miR-193b-3p in samples from normal controls and patients with osteoarthritis (OA) were measured.
Results: miR-193b-3p expression was elevated in chondrogenic and hypertrophic hMSCs, while expression was significantly reduced in degraded cartilage compared to non-degraded cartilage. In addition, miR-193b-3p suppressed the activity of reporter constructs containing the 3′-UTR of HDAC3, inhibited HDAC3 expression, and promoted histone H3 acetylation in the COL2A1, AGGRECAN, COMP, and SOX9 promoters. Treatment with the HDAC inhibitor trichostatin A (TSA) increased cartilage-specific gene expression and enhanced hMSCs chondrogenesis. TSA also increased AGGRECAN expression and decreased MMP13 expression in IL-1β-treated PHCs. Further, 8 weeks after implanting PHC-seeded TCP-COL-HA scaffolds subcutaneously in nude mice, we found that miR-193b overexpression strongly enhanced in vivo cartilage formation compared to that found under control conditions. We also found that patients with OA had lower plasma exosomal miR-193b levels than control subjects.
Conclusions: These findings indicate that miR-193b-3p directly targets HDAC3, promotes H3 acetylation, and regulates hMSC chondrogenesis and metabolism in PHCs.
Keywords: microRNA-193b-3p, HDAC3, histone acetylation, chondrogenesis, cartilage
Introduction
Osteoarthritis (OA) is the most prevalent joint disease, characterized by the degeneration of joint cartilage, thickening of subchondral bone, and osteophyte formation [1]. Histone deacetylases (HDACs) constitute a major enzyme family that modulates histone acetylation, a process shown to play an important role in chondrogenesis and OA progression [2, 3]. For example, HDAC4 (a member of the class-IIa HDAC family) prevents chondrocyte hypertrophy by inhibiting runt-related transcription factor-2 expression [4], and HDAC4 overexpression decreases the levels of the matrix-degrading enzymes matrix metallopeptidase-1 (MMP-1), MMP-3, and MMP-13. In contrast, HDAC1, HDAC2, and HDAC3, which are members of the class-I HDAC family, inhibit chondrogenesis by repressing cartilage-specific genes, such as COL2A1, SOX9, and AGGRECAN [5], while their overexpression increases interleukin-1β (IL-1β)-induced MMP expression and catabolic activity in chondrocytes [2, 6].
Chondrogenesis and cartilage degeneration are both regulated by many different factors, including retinoic acid receptor signaling, microRNAs (miRNAs), and short endogenous oligonucleotides (22-24 nucleotides), all of which play a profound role in the regulation of post-transcriptional gene expression by binding to the 3′ untranslated region (UTR) of target genes [7]. Previously, we reported a 2.751-fold up-regulation of miR-193b-3p when human adipose-derived stem cells differentiated into chondrocytes [8] and provided evidence that miR-193b-3p may regulate early chondrogenesis and suppress the expression of TNF-alpha in IL-1β-induced mouse chondrocytes [9]. Recent findings have shown that miR-193b-3p is an important regulator of chondrogenesis and cartilage degeneration [9-11]; however, the mechanism by which miR-193b-3p regulates chondrogenesis and cartilage degradation remains unclear. Using miRNA target-prediction algorithms, we found that miR-193b-3p has the potential to regulate HDAC3 expression. Thus, we hypothesized that miR-193b-3p plays an important role in modulating histone acetylation by regulating HDAC3 expression. In this study, we investigated the role of miR-193b-3p in chondrogenesis and cartilage degradation by characterizing the molecular mechanism by which miR-193b-3p influences histone acetylation to regulate the expression of cartilage-specific genes.
Methods
This study adhered to the standards of the Ethics Committee on Human Experimentation at The First Affiliated Hospital at Sun Yat-sen University, China (IRB: 2011011) and the Helsinki Declaration (2000). All participants provided informed consent.
Cell isolation and culture
Bone marrow samples were obtained by iliac crest aspiration from 4 normal donors (2 males and 2 females; age range: 29-38 years). Human mesenchymal stem cells (hMSCs) were isolated using density-gradient centrifugation methods, as described previously [12]. hMSCs were cultured in Alpha-modified Eagle's medium (α-MEM) (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 IU/mL penicillin, and 100 μg/mL streptomycin. For chondrocyte isolation, cartilage samples were obtained from five participants (3 males and 2 females; age range: 20-29 years) with no previous history of OA or rheumatoid arthritis, who underwent lower limb amputation surgery because of sarcomas not involving the knee joint. For OA chondrocytes, cartilage samples were obtained from 3 participants (all males; age range: 62-68 years) undergoing total knee arthroplasty (TKA) because of knee joint OA. Primary human chondrocytes (PHCs) were isolated from cartilage as described previously [13]. PHC and SW1353 human chondrosarcoma cells (American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco's Modified Eagle Medium/Ham's F-12 Nutrient Mixture (DMEM/F12) (Gibco) containing 10% FBS, 100 IU/mL penicillin, and 100 μg/mL streptomycin. All cells were cultured at 37 °C in a 5% CO2 atmosphere. The culture media were changed every 3 days. When the cultures neared 80% confluence, the cells were detached by treatment with 0.05% trypsin/ethylenediaminetetraacetic acid (EDTA) and passaged in culture. All PHCs were used at passage 1 and all hMSCs were used at passage 3.
Inducing hMSC chondrogenesis
hMSCs at passage 3 were collected and cultured by micromass culture, as previously described [8]. Briefly, hMSCs were resuspended at 2 × 107 cells/mL in incomplete chondrogenic medium (97 mL human mesenchymal stem cell chondrogenic differentiation basal medium, 10 μL dexamethasone, 300 μL ascorbate, 1 mL of ITS [insulin, transferrin, selenium] supplement, 100 μL sodium pyruvate, and 100 μL proline; Cyagen, Guangzhou, China). Droplets of resuspended cells (12.5 µL) were carefully dropped in each well of a 24-well plate and hMSCs were incubated at 37 °C for 90 min to stimulate adherence of the cells to the plate. Droplets were divided into two groups: the first group was cultured in 500 µL incomplete chondrogenic medium in each well, whereas the second group was cultured in 500 µL complete chondrogenic induction medium, which was prepared by the addition of 10 µL TGF-β3 to 1 mL incomplete chondrogenic medium (Cyagen). Samples were collected for experiments at different selected time points.
Collection of human cartilage
Human articular cartilage samples were obtained during TKA procedures for OA. After collecting patient information regarding age, gender, comorbidities, and medications, samples were de-identified and coded to ensure patient privacy. Subjects scheduled for arthroplasty for rheumatoid or other inflammatory arthritides were excluded, as were subjects who were taking medications that may influence cartilage metabolism (e.g., corticosteroids). Cartilage was obtained from 30 eligible subjects undergoing TKA (16 males and 14 females; age range: 59-78 years). Regions of all articular surfaces of tissue from the patella, femur, and tibia discarded during TKA were partitioned with a #10 blade and graded clinically by the surgeons (ZbY and SwH), according to the Outerbridge classification scale [14]: grade 0, normal appearance; grade 2, minor fragmentation and fissuring involving an area of less than half an inch; and grade 3, fragmentation and fissuring in an area greater than half an inch.
Cartilage with a grade of 0 was defined as non-degraded cartilage, while grade 2 and 3 cartilages were collected and pooled as degraded cartilage. Each sample was dissected into two portions: one was immediately snap-frozen in liquid nitrogen for quantitative real-time polymerase chain reaction (qRT-PCR), while the other was fixed in formaldehyde solution for 24 h for histological analyses.
Isolation and identification of exosomes from human plasma
Nineteen control subjects (10 males and 9 females; age range: 40-64 years) were recruited from patients admitted for cosmetic surgery in our hospital. Individuals without potential risk factors of OA were enrolled. None of the control subjects had evidence of OA as assessed by clinical examination, questionnaire, and X-ray autoradiography. Nineteen patients with knee OA (8 males and 11 females; age range: 40-64 years; plasma and cartilage samples were not from the same patients) included in this study were recruited from patients admitted to our hospital for TKA and arthroscopic surgery for at least one knee with a Kellgren Lawrence grade of II to IV (graded clinically by ZbY and SwH) [15]. Subjects scheduled for arthroplasty for rheumatoid or other inflammatory arthritides were excluded, as were subjects who were taking medications that may influence cartilage metabolism. Following a 12-h overnight fast, venous blood samples were collected from all subjects into EDTA-containing Vacutainer tubes. Samples were maintained at 4 °C until processed within 1 h of collection. After plasma separation by centrifugation (1000 × g for 15 min), plasma was divided into aliquots and stored at -80 °C until analyzed. All plasma samples were obtained prior to any treatment and were analyzed within 3 months.
Exosomes were isolated from 4 mL human plasma. Nanoparticle-tracking analysis (NTA) and transmission electron microscopy (TEM) were used to identify exosomes. Exosomal RNA was extracted using an miRNeasy Serum/Plasma Kit (Qiagen), and miR-39 was used as a reference gene according to the manufacturer's instructions. Proteins were extracted from exosomes using a Total Exosome Protein Isolation Kit (Invitrogen, Carlsbad, USA) for further analysis. The experimental details are described in Supplementary Material.
RNA extraction, reverse transcription, and qRT-PCR
Cartilage and cell-seeded scaffolds were ground in liquid nitrogen prior to RNA isolation. Total RNA from cells, cartilage samples, and cell-seeded scaffolds was extracted using a miRNA Mini Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions.
Next, cDNA was synthesized from miRNA and mRNA using a Mir-X™ miRNA First-Strand Synthesis Kit (Clontech Laboratories, Inc., Mountain View, CA, USA) and a PrimeScript™ RT Master Mix (Takara, Shiga, Japan), respectively. qRT-PCR of target genes was performed using SYBR® Premix Ex Taq™ II (Takara) and a CFX96 real-time qPCR instrument (Bio-Rad, Hercules, CA, USA), according to the manufacturer's instructions. Transcript levels were normalized to that of the reference gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) for mRNA, the small U6 RNA for miRNA, or miR-39 for exosomal miRNA. The specific primers used for these analyses are shown in Supplementary Material. The mRQ 3′ Primer (Clontech) was used as the reverse primer for miRNA-193b-3p, and the miR-39 primer was supplied in the miRNeasy Serum/Plasma Kit. Gene expression was calculated using the 2-ΔΔCt method, and each experiment was performed in triplicate.
Western blot analysis
Western blotting of was performed as described previously [17]. Total protein was isolated from hMSCs and PHCs. Thirty micrograms of protein from each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). Membranes were incubated with primary antibodies specific for HDAC3, SOX9 (1:1,000 dilution, Cell Signaling Technology, Boston, MA, USA), COL2A1, AGGRECAN, MMP-13 (1:2,000 dilution, Abcam, Cambridge, MA, USA) acetylated histone H3 (ac-H3), total histone H3 (H3) (1:1,500 dilution, Millipore, Darmstadt, Germany), CD63, CD9 (1:1,000 dilution, System Biosciences, Palo Alto, CA, USA), and GAPDH (1:3,000 dilution, Cell Signaling Technology). The blots were then incubated with appropriate secondary antibodies conjugated with horseradish peroxidase (1:3,000 dilution, Cell Signaling Technology) at room temperature (22-26 ºC) for 1 h, after which they were developed with an ECL Chemiluminescence Kit (Santa Cruz Biotech, Santa Cruz, CA, USA). Quantitative data were expressed by normalizing the densitometric units to the reference gene using Image J (http://imagej.nih.gov/ij/).
Transfection of small-interfering RNA (siRNA) molecules, and miR-193b-3p mimics and inhibitors
hMSCs and PHCs were transfected with an agomiR (50 nM) or an antagomiR (100 nM) (RiboBio, Guangzhou, China) of miR-193b-3p. PHCs were also transfected with siHDAC3 (100 nM) or siNC (RiboBio). Lipofectamine® 2000 Transfection Reagent (Life Technologies, Carlsbad, CA, USA) was used to transfect cells, according to the manufacturer's instructions. Non-specific microRNA (miR-Control and anti-miR-Control; RiboBio) was used as a control. RNA-free nuclease water was used as a blank. For chondrogenic differentiation of hMSCs by micromass culture, hMSC monolayers were transfected twice, first on the day after plating and again after 3 days.
In situ hybridization, immunohistochemistry, and histology staining
Samples were fixed in 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA), decalcified (in the case of samples from human cartilage), embedded in paraffin, and cut into 5-μm sections that were deparaffinized, rehydrated, and stained with Alcian blue and Safranin O to visualize the glycosaminoglycan (GAG) distribution. For human cartilage samples, following Safranin O staining, cartilage destruction was blindly scored by two surgeons using the OARSI grading system (ZbY and SwH) [16].
COL2A1, AGGRECAN, SOX9, and HDAC3 expression were analyzed by immunohistochemistry, as described previously [17]. Briefly, the sections were deparaffinized, rehydrated, and pretreated with pepsin solution for 15 min at 37 °C followed by processing with the EnVision™ Detection Kit (GeneTech, Shanghai, China), according to the manufacturer's instructions. Sections were incubated for 10 min with 50 mL of 3% H2O2, then blocked in phosphate-buffered saline (PBS) plus 0.025% Tween 20 with 10% FBS, followed by overnight incubation with primary antibodies specific to HDAC3 (1:50 dilution, Abcam) and COL2A1 (1:100 dilution, Abcam) at 4 °C. Negative controls were prepared by substituting PBS for the primary antibody. The sections were then incubated for 30 min with a secondary antibody supplied with the kit. Sections were stained with 3, 3′-diaminobenzidine tetrahydrochloride and counterstained with hematoxylin.
For in situ hybridization of microRNA expression, we used the method previously reported by Akhtar et al. [18] and a probe for human miR-193b-3p (Exiqon, Copenhagen, Denmark). Immunoreactivity was analyzed by determining the integrated optical density (IOD) per stained area in μm2, using Image-Pro Plus software, version 6.0 (Media Cybernetics, Rockville, MD, USA).
Luciferase constructs and reporter assay
The DNA sequence of the HDAC3 3′-UTR was amplified by PCR using the following primers: forward 5′-CCGCTCGAGGAGTGGCTTGGGATGCTGTGTCC-3′ and reverse 5′-ATAAGAATGCGGCCGCTAAAGGTACAAAAAATACCGAC-3′. The seed sequences were mutated using standard PCR techniques with the following primers: forward 5′-GACATTATTGGCAGTGGGCCCTGGAAATTCAGCCCTAGCCCCCCTTGCCCCTTATTT-3′ and reverse 5′-AAATAAGGGGCAAGGGGGGCTAGGGCTGAATTTCCAGGGCCCACTGCCAATAATGTC-3′. The amplified DNA sequences (593bp) were inserted into the pmiR-RB-REPORT™ Vector (Landbiology, Guangzhou, China) to generate the HDAC3 3′-UTR and mutated (MUT) HDAC3 3′-UTR luciferase vectors. For the reporter assay, 1.2 × 104 SW1353 cells were transfected in a 96-well plate with 50 nM miR-193b-3p, 50 nM miR-Control, 100 nM anti-miR-193b-3p, or 100 nM anti-miR-Control (RiboBio). The cells were then co-transfected with 2 mg/mL of vector with the wild-type or mutant 3′-UTR of HDAC3. Luciferase activity was measured 48 h post-transfection using the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI, USA). Firefly luciferase activity was normalized to the corresponding Renilla luciferase activity. Luciferase assays were performed in quadruplicate and repeated in three independent experiments.
Chromatin immunoprecipitation (ChIP) assays
PHCs were grown to confluency in 100-mm dishes, treated for 24 h with 50 nM miR-193b-3p or miR-Control, and used for ChIP assay analysis. The Simple ChIP Enzymatic Chromatin IP Kit (Millipore) was used following the manufacturer's recommended protocol. An anti-ac-H3 antibody (Millipore) was used to pull down DNA-protein complexes, and mouse IgG was used as a negative control. Purified DNA was quantified by qPCR. The sequences of primers used to amplify the SOX9, COL2A1, AGGRECAN, and COMP promoters are provided in Supplementary Material. The percent input method was used to identify enrichment.
Biochemical assay for detecting GAGs
Assays were performed to measure the reaction between GAGs and dimethyl-methylene blue (Sigma-Aldrich). Samples were collected at different time points, washed with PBS, and digested with papain solution for 16 h at 60 °C. A 15-μL aliquot of supernatant was transferred to a new tube and reacted with 1, 9-dimethylmethylene (DMMB) solution, as previously described [12, 17]. The results of GAG quantification were normalized to double-stranded DNA (dsDNA) content, as previously described [12, 17].
Enzyme-linked immunosorbent assays (ELISAs)
ELISAs were performed using 100 μL plasma, and IL-6 and IL-8 immunoassay kits (CusaBio, Wuhan, China), according to the manufacturer's instructions. Plates were read at 450 nM using an HT Microplate Reader (BioTek, Winooski, VT, USA), and the plasma concentrations of IL-6 and IL-8 were calculated using a standard curve.
PHC scaffold implantation in nude mice
Procedures were carried out according to the guidelines of the Ethics Committee of the First Affiliated Hospital of Sun Yat-sen University (IRB: 2014C-028). Porous tricalcium phosphate-collagen-hyaluronate (TCP-COL-HA) (height × diameter: 3.5 × 1.75 mm) cartilage scaffolds were generated by freeze-drying followed by chemical cross-linking, as previously described [12]. PHCs were divided into four groups: miR-193b-3p, miR-Control, anti-miR-193b-3p, and anti-miR-Control. PHC monolayers PHCs were transfected twice, first on the day after plating and again after 3 days. PHCs were transfected with 50 nM agomiR-193b-3p (miR-193b-3p group) or 100 nM antagomiR-193b-3p (anti-miR-193b-3p group), while nonspecific microRNAs (50 nM miR-Control and 100 nM anti-miR-Control groups; RiboBio) were used as respective controls as described above. Next, the TCP-COL-HA scaffolds were soaked in DMEM/F12, blotted dry, seeded with transfected PHCs (2 × 107 cells/mL) in 40 μL DMEM/F12, and placed in a 24-well plate for 1 day in vitro to allow the cells to firmly attach onto the scaffolds.
BALB/c-nude mice (6-8-week-old; Sun Yat-sen University Laboratory Animal Center, Guangdong, China) were anesthetized with 1% sodium pentobarbital in water (1 mL/0.1 kg). A cell-seeded scaffold was subcutaneously implanted into the back of each animal (n = 6 mice in each group). An empty scaffold was subcutaneously implanted as an empty control sample. The incisions were closed with 6-0 sterile sutures, and animals were returned to the housing facility where they could recover with ad libitum access to food and water, as previously described [19]. Mice were sacrificed 8 weeks after implantation, and the scaffolds were collected. Fibrous capsules and other host tissues were carefully removed using fine, straight, toothless forceps and #15 scalpel blades in #3 handles under a stereoscope (10× magnification), as described previously [19]. Scaffolds were rinsed with PBS and used for biochemical, qRT-PCR, histological, and immunohistochemical analyses.
Statistical analysis
All experiments were performed with at least three biological replicates. The data were evaluated as the mean ± standard deviation (SD). Both parametric and non-parametric inferential statistics were utilized in this study, depending on whether the data were normally distributed. Student's t-test and Mann-Whitney U test were used to identify differences between groups as appropriate. One-way analysis of variance and Kruskal-Wallis tests were performed for multiple-group comparisons. Gaussian distribution of the data was confirmed using the Shapiro-Wilk test, and the equality of variance was confirmed using Levene's test. P values less than 0.05 were considered statistically significant. All analyses were performed using SPSS software, version 13.0 (IBM Corporation, Armonk, NY, USA).
Results
Expression patterns of miR-193b-3p and HDAC3 during hMSCs chondrogenesis
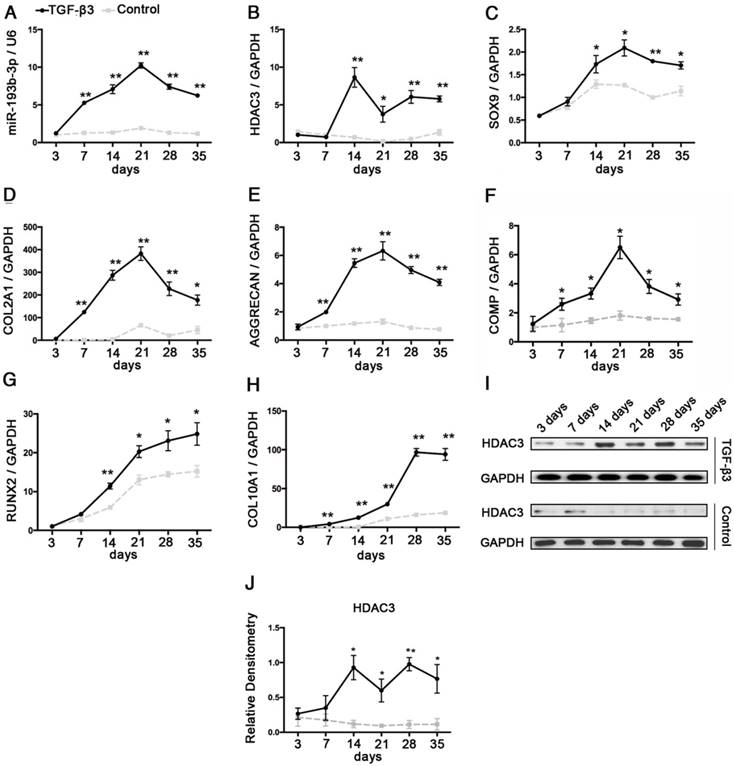
hMSCs were induced to differentiate to chondrocytes in vitro. We observed a rapid up-regulation of miR-193b-3p in chondrogenic hMSCs beginning at 3 days, which peaked at 21 days, followed by a marked decrease in expression at 28 days and 35 days (Figure 1A). Further, opposing expression patterns between miR-193b-3p and HDAC3 were observed during the chondrogenic differentiation of hMSCs at 14-28 days, a finding that suggests miR-193b-3p may affect HDAC3 expression (Figure 1A-B, I-J). Chondrocyte differentiation was verified by screening for increased expression of the chondrogenic markers SOX9, COL2A1, COMP and AGGRECAN during early chondrogenesis (21 days) and the markers of degeneration RUNX2 and COL10A1 during late-stage chondrogenesis (28-35 days) (Figure 1C-H).
Analysis of the relative expression levels of miR-193b and HDAC3 during the chondrogenesis of hMSCs. The relative expression levels of (A) miR-193b-3p and (B) HDAC3. (C-E) The relative expression levels of the chondrogenic markers SOX9, COL2A1, AGGRECAN, and COMP. (G-H) The relative expression levels of the hypertrophic markers COL10A1 and RUNX2. (I) The protein-expression levels of HDAC3. (J) Western blots data from three experiments were quantified by densitometric analysis. RNU6B and GAPDH were detected as endogenous controls. The data shown represent the mean ± standard error (SE) of three independent experiments in samples from three different donors. *P < 0.05; ** P < 0.001
miR-193b-3p regulated the expression of HDAC3 in hMSCs during chondrogenesis
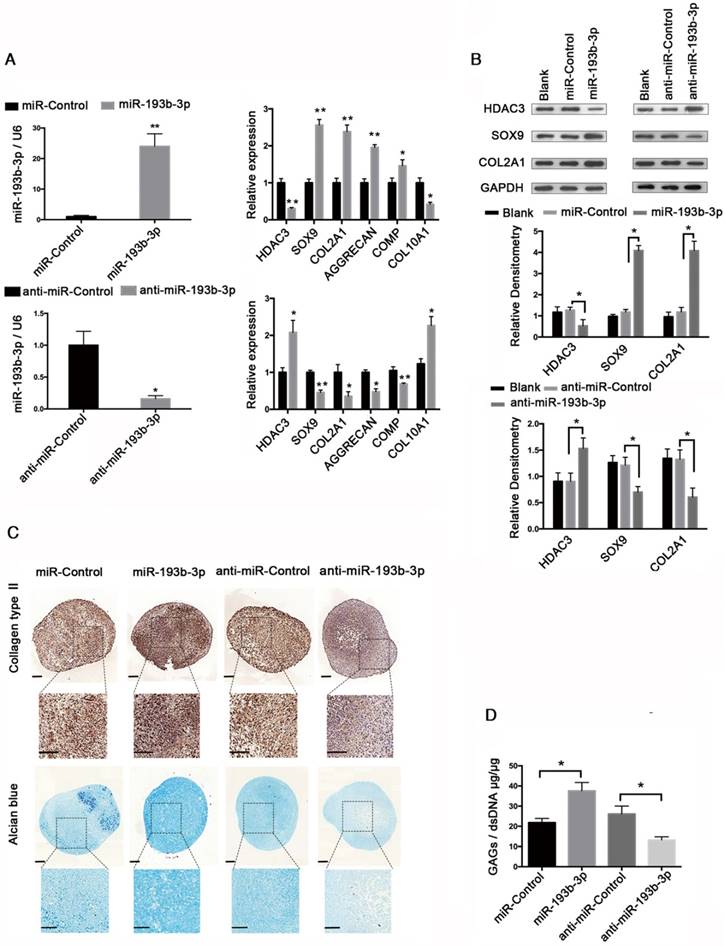
To further investigate whether miR-193b-3p regulates HDAC3 expression during chondrogenesis, we inhibited or overexpressed miR-193b-3p in hMSCs. hMSCs were transfected with either miR-193b-3p or anti-miR-193b-3p and then induced to differentiate into chondrocytes for 21 days (Figure 2). The relative expression levels of HDAC3, SOX9, COL2A1, AGGRECAN, and COMP mRNAs were assessed by qRT-PCR (Figure 2A), and HDAC3, SOX9, and COL2A1 protein-expression levels were assessed by western blotting (Figure 2B). The relative expression levels of miR-193b-3p on days 7 and 14 are shown in Figure S1. Overexpression of miR-193b-3p induced a significant decrease in the expression of HDAC3 and an evident increase in the expression of SOX9, COL2A1, AGGRECAN, and COMP. In contrast, the miR-193b-3p inhibitor induced a significant increase in HDAC3 expression and a significant decrease in SOX9, COL2A1, AGGRECAN, and COMP expression (Figure 2A). Further, as determined by western blotting, miR-193b-3p overexpression significantly decreased HDAC3 protein expression and increased SOX9 and COL2A1 protein expression, while transfection with anti-miR-193b-3p significantly increased HDAC3 protein expression and reduced SOX9 and COL2A1 protein expression in hMSCs (Figure 2B). Collagen type II immunohistochemical staining, Alcian blue staining, and a biochemical assay for GAGs were performed to assess the chondrogenic differentiation of hMSCs at day 21. We found that miR-193b-3p overexpression enhanced the chondrogenic differentiation of hMSCs, while the miR-193b-3p inhibitor prevented chondrogenic differentiation (Figure 2C-D). No significant difference in the size of pellets from the different groups was found.
miR-193b regulates HDAC3 expression during hMSCs chondrogenesis. hMSCs were transfected with anti-miR-193b-3p, anti-nonspecific control miRNA (anti-miR-Control), and miR-193b-3p or miR-Control, and then induced to differentiate into chondrocytes for 21 days. (A) The relative expression levels of miR-193b-3p, HDAC3, SOX9, COL2A1, AGGRECAN, COMP, and COL10A1 were estimated by qRT-PCR. (B) The protein-expression levels of HDAC3, SOX9, and COL2A1 were estimated by western blotting. Western blots data from three experiments were quantified by densitometric analysis. (C) Alcian blue staining and immunohistochemical staining of collagen type II at 21 days following chondrogenic induction. Upper panels: 8× magnification; lower panel: magnified view of the areas enclosed by black boxes, 16× magnification. Scale bar = 100 μm. (D) Quantification of the GAG/dsDNA ratio after 21 days of chondrogenic culture. RNU6B and GAPDH were detected as endogenous controls. The data shown represent the mean ± standard error (SE) of three independent experiments performed with samples from three different donors. *P < 0.05; ** P < 0.001.
Inverse correlation between IL-1β-regulated HDAC3 and miR-193b-3p expression in human chondrocytes
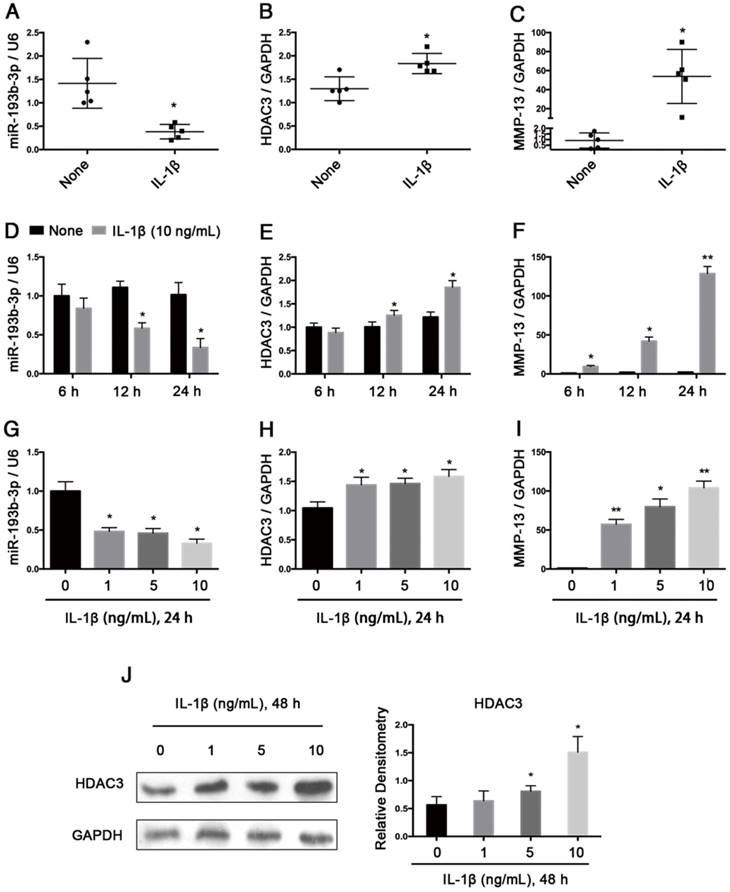
Next, we verified the regulation of miR-193b-3p expression by IL-1β, which is a potent proinflammatory cytokine in OA cartilage degradation. In normal chondrocytes from 5 participants (3 males and 2 females; age range: 20-29 years), stimulating these chondrocytes for 24 h with IL-1β (1 ng/mL) resulted in decreased miR-193b-3p expression and increased HDAC3 and MMP-13 expression (Figure 3A-C). Further study showed that in PHCs from a 20-year-old male, IL-1β stimulation resulted in decreased miR-193b-3p expression and increased the levels of HDAC3 and MMP-13 mRNA in time- and dose-dependent manners (Figure 3D-I). IL-1β stimulation of chondrocytes also resulted in increased levels of the HDAC3 protein in a dose-dependent manner (Figure 3J). These results indicate that the reduced miR-193b-3p expression in response to IL-1β correlated with increased HDAC3 expression.
Relative expression levels of miR-193b-3p and HDAC3 in IL-1β-treated PHCs from 5 subjects and an inverse correlation between IL-1β-regulated HDAC3 and miR-193b-3p expression in PHCs. (A-C) The relative expression levels of miR-193b-3p, HDAC3, and MMP-13 in PHCs from 5 subjects (3 males and 2 females; age range: 20-29 years) at 24 h post-treatment with IL-1β (1 ng/mL) or in untreated PHCs. (D-F) The relative expression levels of miR-193b-3p, HDAC3, and MMP-13 at 6 h, 12 h, or 24 h post-treatment with IL-1β (10 ng/mL) or in untreated PHCs from a 20-year-old male subject. (G-I) The relative expression levels of miR-193b-3p, HDAC3, and MMP-13 in PHCs (a 20-year-old male subject) treated with various concentrations (0, 1, 5, and 10 ng/mL) of IL-1β after 24 h. The experiments were performed in triplicate with independent cultures. (J) The protein levels of HDAC3 in PHCs (a 20-year-old male subject) treated with different concentrations (0, 1, 5, and 10 ng/mL) of IL-1β after 48 h. Western blots data from three experiments were quantified by densitometric analysis. RNU6B and GAPDH were detected as endogenous controls. The data shown represent the mean ± standard error (SE) of at least three independent experiments. *P < 0.05; ** P < 0.001
miR-193b-3p regulated histone H3 acetylation and SOX9, COL2A1, and AGGRECAN expression by targeting HDAC3
To assess whether miR-193b-3p regulates HDAC3 expression in chondrocytes, both PHCs and OA chondrocytes were transfected with miR-193b-3p, miR-Control, anti-miR-193b-3p, or anti-miR-Control for 24 h. In PHCs and OA chondrocytes, overexpression of miR-193b decreased the expression of HDAC3, COL10A1 (in PHCs), and MMP-13 (in OA chondrocytes) and increased the expression of SOX9, COL2A1, COMP, and AGGRECAN. In contrast, suppression of miR-193b-3p expression with anti-miR-193b-3p led to a significant increase in HDAC3, COL10A1 (in PHCs), and MMP-13 (in OA chondrocytes) expression and a corresponding decrease in SOX9, COL2A1, COMP, and AGGRECAN expression (Figure 4A-D). Additionally, western blot analysis showed that, in normal PHCs, miR-193b-3p overexpression significantly decreased HDAC3 protein expression and increased SOX9, COL2A1, AGGRECAN, and histone H3 acetylation protein expression. Conversely, transfection with anti-miR-193b-3p significantly increased HDAC3 protein expression and decreased SOX9, COL2A1, AGGRECAN, and histone H3 acetylation protein expression (Figure 4E). These results suggest that HDAC3 expression was down-regulated by miR-193b-3p in PHCs.
miR-193b-3p regulated histone H3 acetylation and SOX9, COL2A1, and AGGRECAN expression by targeting HDAC3. (A-B) The relative expression levels of miR-193b-3p, HDAC3, SOX9, COL2A1, AGGRECAN, COMP, and COL10A1 in PHCs 24 h after transfection with miR-Control, miR-193b-3p, anti-miR-Control, or anti-miR-193b-3p. (C-D) The relative expression levels of miR-193b-3p, HDAC3, SOX9, COL2A1, AGGRECAN, COMP, and MMP-13 in OA chondrocytes 24 h after transfection with miR-Control, miR-193b-3p, anti-miR-Control, or anti-miR-193b-3p. (E) Acetylation of histone H3 and protein levels of HDAC3, SOX9, COL2A1, and AGGRECAN 48 h after transfection with miR-Control, miR-193b-3p, anti-miR-Control, or anti-miR-193b-3p. Western blots data from three experiment were quantified by densitometric analysis. (F) Sequence of the putative or mutated miR-193b-3p binding site within the 3′-UTR of HDAC3 mRNA. (G) The HDAC3 3′-UTR (Luc-HDAC3-UTR) reporter plasmid or a mutant HDAC3 3′-UTR (Luc-HDAC3-UTR-Mut) reporter plasmid was co-transfected with miR-320-3p, miR-Control, anti-miR-Control, or anti-miR-193b-3p into SW1353 cells. Cells transfected with Luc-HDAC3-UTR or Luc-HDAC3-UTR-Mut were used as controls. RNU6B and GAPDH were detected as endogenous controls. Total histone H3 was detected as an internal control for ac-H3. The data shown represent the mean ± standard error (SE) of three independent experiments in samples from three different donors (for chondrocytes) or the mean ± standard error (SE) of three independent experiments (for SW1353 cells). *P < 0.05; ** P < 0.001
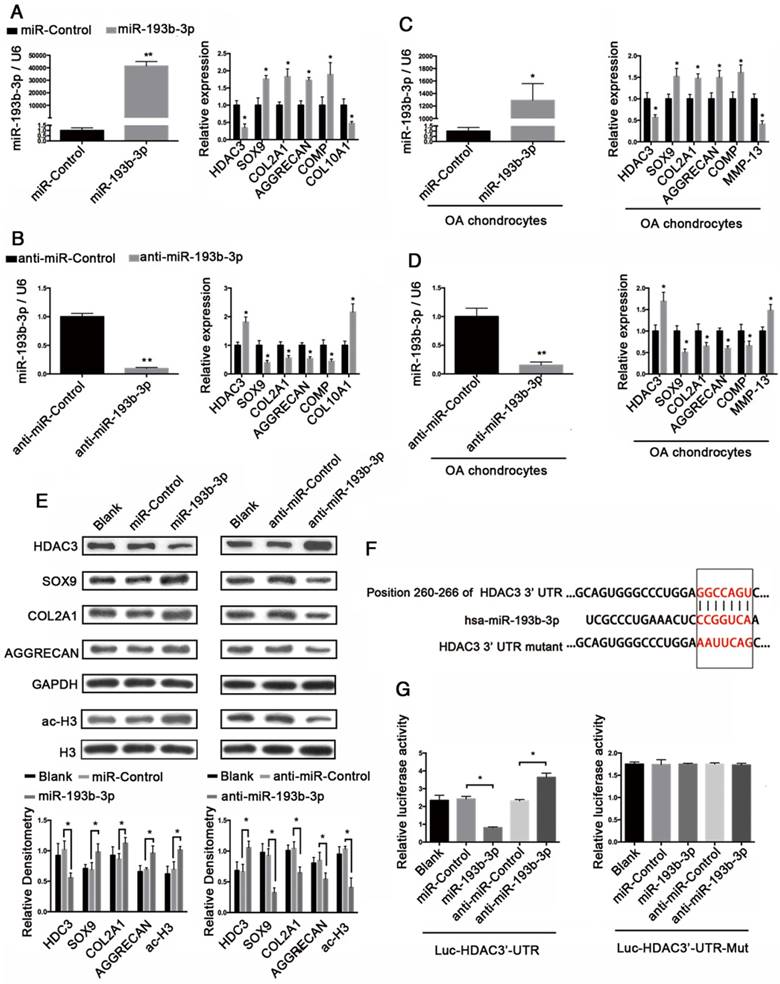
To further clarify the molecular mechanisms that underlie the regulation of HDAC3 expression by miR-193b-3p, we analyzed the sequence of the 3′-UTR of human HDAC3 mRNA. Using predictive bioinformatics programs, such as TargetScan (http://www.targetscan.org), miRanda (http://www.microrna.org), and miRWalk (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/), revealed that the 3′-UTR of human HDAC3 contains a potential miR-193b-binding site (Figure 4F). Luciferase reporter assays with 3′-UTR HDAC3 and mutated 3′-UTR HDAC3 were performed in the presence or absence of miR-193b-3p overexpression and inhibition. We found that co-transfection of miR-193b-3p with HDAC3 3′-UTR luciferase reporter plasmids significantly reduced luciferase activity, while co-transfection of anti-miR-193b-3p with HDAC3 3′-UTR luciferase reporter plasmids significantly increased luciferase activity (Figure 4G). Further, the mutated 3′-UTR sequence prevented the binding of miR-193b-3p to HDAC3 mRNA. Mutation of the miR-193b-3p-binding sequence abrogated both miR-193b-3p-mediated repression and the anti-miR-193b-3p-mediated increase of HDAC3 3′-UTR reporter activity (Figure 4G). These results demonstrated that miR-193b-3p reduced luciferase activity by binding to the 3′-UTR of HDAC3 and that HDAC3 is a target for miR-193b-3p-mediated repression.
Trichostatin A induced histone acetylation in hMSCs and enhanced chondrogenic differentiation of hMSCs
To evaluate the effects of TSA on chondrogenic differentiation of hMSCs, we induced hMSC chondrogenesis in complete chondrogenic medium in the presence or absence of 100 nM TSA (Chondrogenic culture group and Chondrogenic culture + TSA group) for 21 days. hMSCs cultured in incomplete chondrogenic medium for 21 days were used as negative controls (NC group). The expression levels of SOX9, COL2A1, AGGRECAN, COMP, COL10A1, and RUNX2 at day 21were examined. The highest levels of gene expression were observed in the Chondrogenic culture + TSA group, followed by the Chondrogenic culture and NC groups (Figure 5A-F).
TSA induced histone acetylation in hMSCs and enhanced chondrogenic differentiation of hMSCs. (A-F) The relative expression levels of SOX9, COL2A1, AGGRECAN, COMP, COL10A1, and RUNX2 in hMSCs after 21 days of chondrogenesis in the NC, Chondrogenic culture, or Chondrogenic culture + TSA (100 nM) groups. (G) Histone H3 acetylation and COL2A1 protein expression in hMSCs after 21 days of chondrogenesis in the NC, Chondrogenic culture, and Chondrogenic culture + TSA (100 nM) groups. Western blots data from three experiments were quantified by densitometric analysis. (H) Immunohistochemistry of collagen type II and Alcian blue staining after 21 days of chondrogenesis in the NC, Chondrogenic culture, and Chondrogenic culture + TSA (100 nM) groups. Upper panels: 8× magnification; lower panel: magnified view of the areas enclosed by black boxes, 16× magnification. Scale bar = 100 μm. (I) Quantification of the GAG/dsDNA ratio 21 days following chondrogenesis in the NC, Chondrogenic culture, and Chondrogenic culture + TSA (100 nM) groups. GAPDH was detected as an internal control for SOX9, COL2A1, AGGRECAN, and COMP, whereas total histone H3 was used as an internal control for ac-H3. The data shown represent the mean ± standard error (SE) of three independent experiments performed with samples from three different donors. *P < 0.05; ** P < 0.001
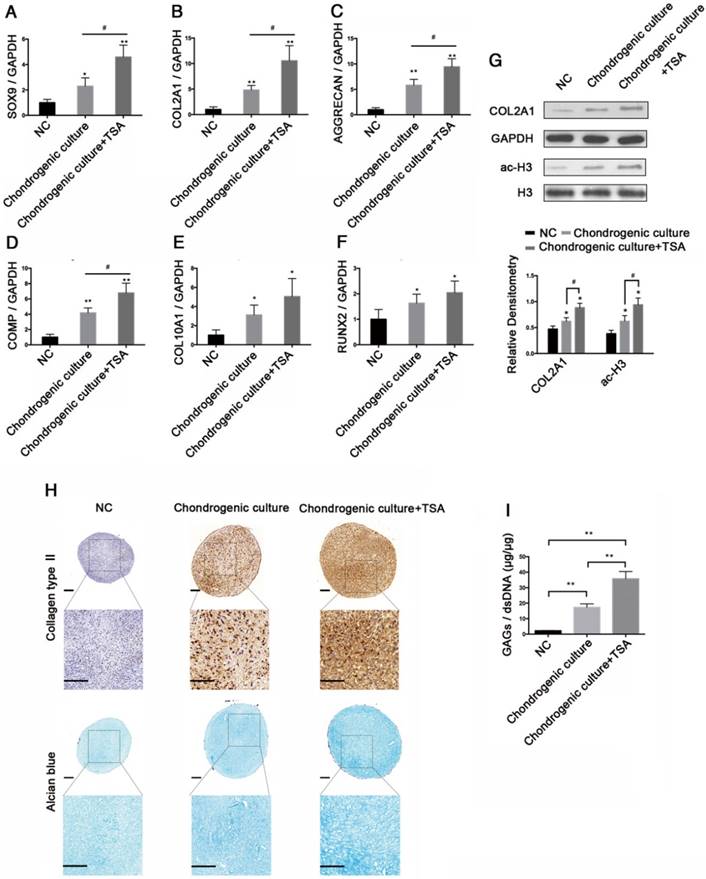
COL2A1 protein expression and histone H3 hyperacetylation were highest in the Chondrogenic culture + TSA group, followed by the Chondrogenic culture and NC groups (Figure 5G). We fixed and stained the pellets via Alcian blue and collagen type II immunohistochemistry to detect GAGs and collagen type II deposition after 21 days of chondrogenesis. We found that chondrogenic hMSCs in the Chondrogenic culture + TSA group exhibited stronger Alcian blue and collagen type II immunohistochemical staining than chondrogenic hMSCs in the Chondrogenic culture group, in which staining was much stronger for the Chondrogenic culture group than for the NC group (Figure 5H). Additionally, pellets specimens in the NC group were approximately 686±76.17 μm in diameter, which was significantly smaller than the pellets in the Chondrogenic culture group (~822.17±32.91 μm in diameter) (p = 0.0468) and in the Chondrogenic culture+TSA group (~828.4±37.02 μm in diameter) (p = 0.0436). However, no significant differences in diameters were found between pellets in the Chondrogenic culture group and those in the Chondrogenic culture + TSA group (p = 0.8381). We also found that chondrogenic hMSCs in the Chondrogenic culture + TSA group had the highest GAG/dsDNA ratio and that the GAG/dsDNA ratio was higher in chondrogenic hMSCs in the Chondrogenic culture group than in the NC group (Figure 5I).
The effects of TSA on PHCs and the protective effect of TSA on cytokine-induced degradation
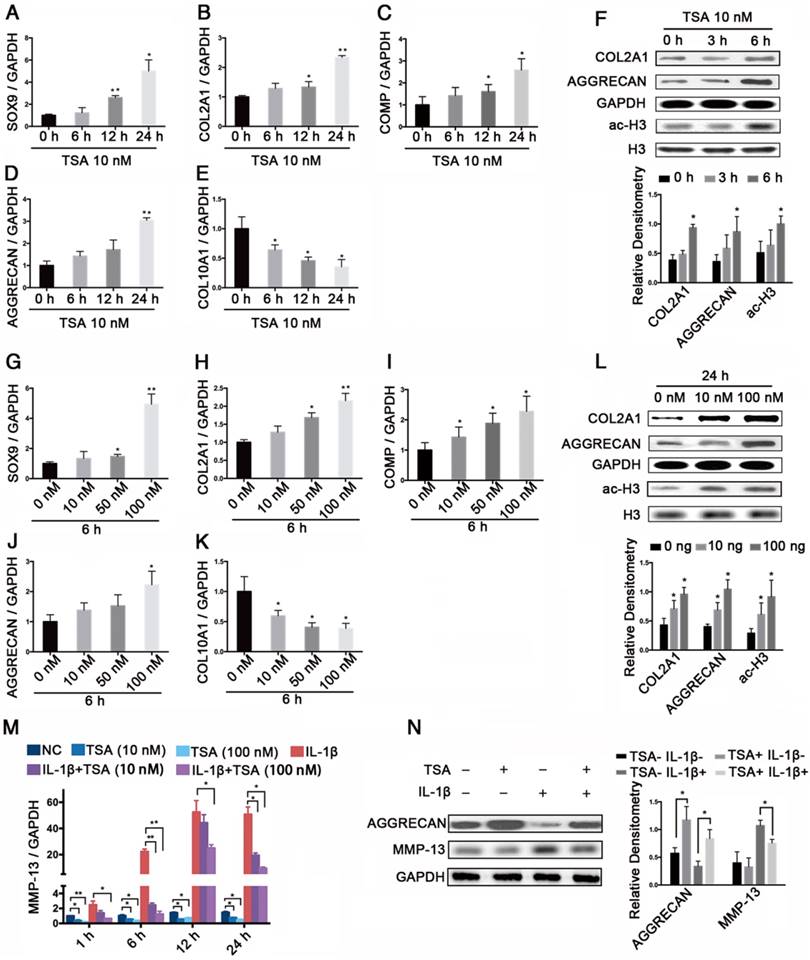
We next assessed the effects of TSA on histone acetylation in PHCs and found that TSA increased the levels of SOX9, COL2A1, COMP, and AGGRECAN mRNA and decreased the level of COL10A1 mRNA in time- and dose-dependent manners (Figure 6A-E, G-K). Notably, TSA also promoted COL2A1 and AGGRECAN protein expression, and induced histone H3 acetylation in time- and dose-dependent manners (Figure 6F, L). Moreover, we found that the significant increase in MMP-13 expression observed at 1 h, 6 h, 12 h, and 24 h in PHCs treated with IL-1β (1 ng/mL) were dose-dependently suppressed by TSA treatment (Figure 6M). Finally, TSA (100 nM) promoted AGGRECAN expression in both IL-1β-treated (1 ng/mL) and non-IL-1-treated PHCs, and inhibited the expression of MMP-13 in IL-1β-treated PHCs (Figure 6N).
The effects of TSA on PHCs and the protective effect of TSA on cytokine-induced degradation. (A-E) The relative expression levels of SOX9, COL2A1, COMP, AGGRECAN, and COL10A1 treated with 10 nM TSA for the indicated times (0 h, 6 h, 12 h, 24 h) in PHCs. (F) Histone H3 acetylation and the protein levels of COL2A1 and AGGRECAN in PHCs treated with 10 nM TSA for 0 h, 3 h, or 6 h. Western blots data from three experiments were quantified by densitometric analysis. (G-K) The relative expression levels of SOX9, COL2A1, COMP, AGGRECAN, and COL10A1 in PHCs at 6 h after treatment with various concentrations of TSA (0 nM, 10 nM, 50 nM, or 100 nM). (L) Histone H3 acetylation and the protein levels of COL2A1 and AGGRECAN in PHCs at 24 h after treatment with different concentrations of TSA (0 nM, 10 nM, 100 nM). Western blots data from three experiments were quantified by densitometric analysis. (M) The relative expression levels of MMP-13 in PHCs treated with IL-1β (1 ng/mL) and/or TSA (10 nM, 100 nM) for the indicated times (1 h, 6 h, 12 h, or 24 h). (N) Protein levels of AGGRECAN and MMP-13 in PHCs treated with IL-1β (1 ng/mL) and/or TSA (100 nM) for 24 h. GAPDH was detected as an internal control for SOX9, COL2A1, AGGRECAN, COMP, COL10A1, and MMP-13, whereas total histone H3 was detected as an internal control for ac-H3. The data shown represent the mean ± standard error (SE) of three independent experiments performed with samples from three different donors. *P < 0.05; ** P < 0.001
HDAC3 knockdown regulated cartilage-specific gene expression and blocked the effect of the miR-193b-3p inhibitor in PHCs
To evaluate the regulatory effects of HDAC3 expression on cartilage-specific gene expression, siRNA-mediated knockdown of HDAC3 expression was performed in PHCs. PHCs were transfected with an HDAC3 siRNA (siHDAC3-1, siHDAC3-2), which resulted in decreased HDAC3 protein expression, and increases COL2A1, SOX9, and acetylated histone H3 protein expression, as shown by western blotting (Figure 7B). This reduction in HDAC3 protein levels correlated well with increased cartilage-specific gene expression (Figure 7A), in which the relative expression levels of COMP, COL2A1, SOX9, and AGGRECAN were up-regulated, whereas the expression of COL10A1 was down-regulated. These findings indicate that by lowering HDAC3 expression, the expression of cartilage-specific genes was elevated and the expression of a hypertrophy-related gene was down-regulated.
HDAC3 knockdown regulated cartilage-specific gene expression and blocked the effect of an miR-193b-3p inhibitor in PHCs. (A) The relative expression levels of HDAC3, COMP, COL2A1, AGGRECAN, SOX9, and COL10A1 in PHCs transfected with siNC, siHDAC3-1, or siHDAC3-2. (B) Histone H3 acetylation and HDAC3, SOX9, and COL2A1 expression in PHCs transfected with siNC, siHDAC3-1, or siHDAC3-2. Western blots data from three experiments were quantified by densitometric analysis. (C) The relative expression levels of HDAC3, COMP, COL2A1, AGGRECAN, SOX9, and COL10A1 in PHCs transfected with anti-miR-Control, anti-miR-193b-3p, anti-miR-193b-3p + siNC or anti-miR-193b-3p + siHDAC3. (D) Histone H3 acetylation and HDAC3, SOX9, and COL2A1 expression in PHCs transfected with anti-miR-Control, anti-miR-193b-3p, anti-miR-193b-3p + siNC or anti-miR-193b-3p + siHDAC3. Western blots data from three experiments were quantified by densitometric analysis. GAPDH was detected as an internal control for HDAC3, SOX9, COL2A1, COMP, AGGRECAN, and COL10A1, whereas total histone H3 was detected as an internal control for ac-H3. The data shown represent the mean ± standard error (SE) of three independent experiments performed with samples from three different donors. *P < 0.05; ** P < 0.001
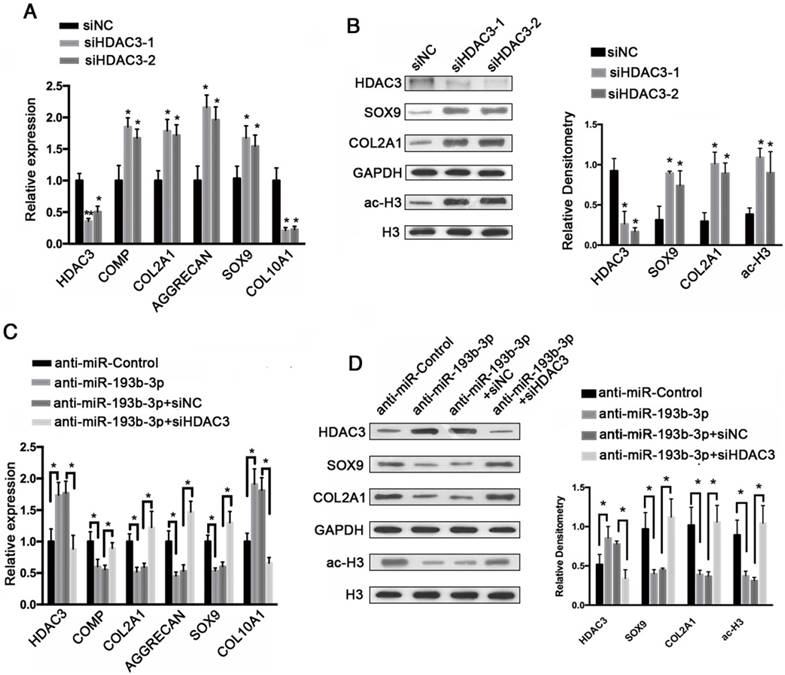
To functionally confirm that miR-193b-3p regulates chondrocytes metabolism by targeting HDAC3, an HDAC3 siRNA (siHDAC3-2) was used to examine whether the effect of the miR-193b-3p inhibitor could be blocked by HDAC3 knockdown. As shown in Figure 7D, the miR-193b-3p inhibitor significantly increased the protein expression of HDAC3 and reduced the protein levels of SOX9, COL2A1, and acetylated histone H3. More importantly, the HDAC3 siRNA significantly blocked the miR-193b-3p inhibitor-mediated up-regulation of HDAC3 and down-regulation of SOX9, COL2A1, and histone H3 acetylation. Similar results were observed by qRT-PCR (Figure 7C). These data demonstrate that the effect of the miR-193b-3p inhibitor could be blocked by HDAC3 knockdown in PHCs.
miR-193b-3p enhanced acetylation of histone H3 in the SOX9, COL2A1, AGGRECAN, and COMP promoters
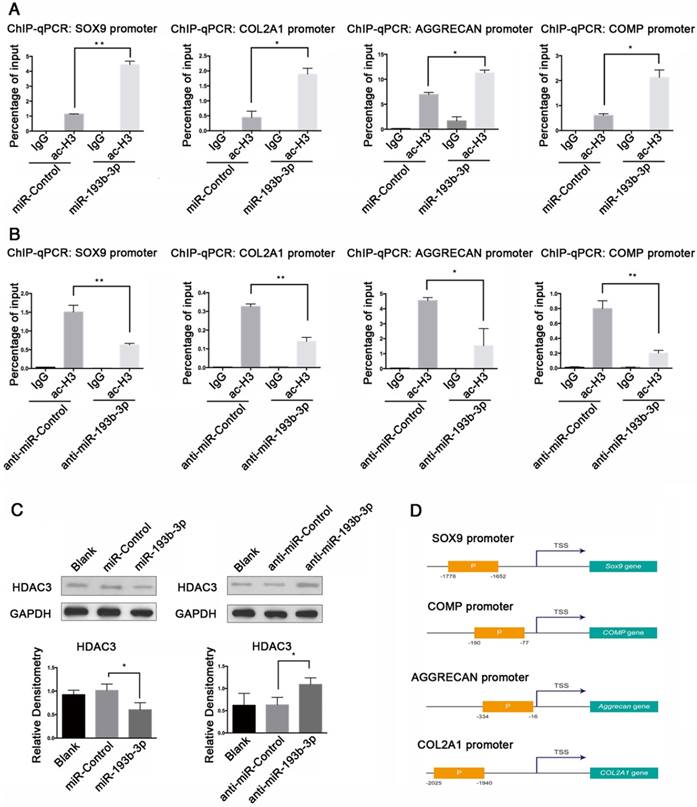
We performed ChIP assays to determine whether histone proteins associated with the promoter regions of SOX9, COL2A1, AGGRECAN, and COMP were hyperacetylated after transfection with miR-193b-3p. Histone proteins show various patterns of acetylation with differential effects on transcription. Among them, ac-H3 is mostly associated with the enhancement of gene expression. Therefore, for our ChIP assays, we treated PHCs with 50 nM miR-193b-3p or the miR-Control, and 100 nM anti-miR-193b-3p or the anti-miR-Control, and used an anti-ac-H3 antibody. By qPCR, we found that overexpression of miR-193b-3p decreased HDAC3 protein expression (Figure 8C), and increased acetylation of histones associated with the SOX9, COL2A1, AGGRECAN, and COMP promoters (Figure 8A), whereas the suppression of miR-193b-3p increased HDAC3 protein expression (Figure 8C), and decreased acetylation of histones associated with the promoters of these genes (Figure 8B). Therefore, our findings indicate that miR-193b-3p promoted the transcriptional activation of SOX9, COL2A1, AGGRECAN, and COMP by directly inhibiting the expression of HDAC3, thereby increasing the level of histone H3 acetylation of the SOX9, COL2A1, AGGRECAN, and COMP promoter regions (Figure 8D).
miR-193b-3p enhanced acetylation of histone H3 in the SOX9, COL2A1, AGGRECAN, and COMP promoters. (A-B) PHCs were transfected with miR-193b-3p or miR-Control (NC), and anti-miR-193b-3p or anti-miR-Control for 24 h before a ChIP assay was performed, using an anti-ac-H3 antibody. ChIP followed by qRT-PCR analysis detected acetylated H3 at the SOX9, COL2A1, AGGRECAN, and COMP promoters. (C) HDAC3 protein expression at 48 h after transfection with miR-Control, miR-193b-3p, anta-miR-Control, or anta-miR-193b-3p. Western blots data from three experiments were quantified by densitometric analysis. GAPDH was detected as an internal control for HDAC3. (D) Upstream promoter regions of SOX9, COL2A1, AGGRECAN, and COMP. The promoter region amplified for ChIP-qPCR (performed with the ac-H3 antibody) and primers labeled “P” are shown in orange box. P: the promoter region amplified for ChIP-qPCR, with the ac-H3 antibody and primers; TSS: transcription start site. The data shown are normalized to input DNA and represent the mean ± standard error (SE) of three independent experiments performed with samples from three different donors. *P < 0.05; ** P < 0.001
miR-193b-3p enhanced ectopic cartilage formation in nude mice
An ectopic cartilage-formation model in nude mice was used to test the effect of miR-193b-3p on cartilage regeneration in vivo. We evaluated cartilage formation efficiency using nude mice and an ectopic cartilage-formation model at 8 weeks after implantation, finding that Safranin O and Alcian blue staining and collagen type II, AGGRECAN, and SOX9 immunoreactivity were stronger in the miR-193b-3p group than in the miR-Control group. In contrast, we found that Safranin O and Alcian blue staining and collagen type II, AGGRECAN, and SOX9 immunoreactivity were less intense in the anti-miR-193b-3p group compared to the anti-miR-Control group (Figure 9A-B). In addition, HDAC3 immunoreactivity was less intense in the miR-193b-3p group compared to the miR-Control group, whereas HDAC3 immunoreactivity was greater in the anti-miR-193b group compared to that in the anti-miR-Control group (Figure 9A). Few cells were seen in the center of the empty control scaffold after carefully removing fibrous capsules and other host tissues from the sample (Figure S3). We also examined the expression of miR-193b-3p, HDAC3, AGGRECAN, COL2A1, and SOX9 at 8 weeks after scaffold implantation. We found that the expression level of miR-193b-3p was significantly higher in the miR-193b-3p group compared to the miR-Control group, whereas miR-193b-3p expression was significantly lower in the anti-miR-193b-3p group compared to the anti-miR-Control group. Further, overexpression of miR-193b-3p decreased HDAC3 expression and increased the expression of SOX9, COL2A1, and AGGRECAN, whereas suppression of miR-193b-3p using anti-miR-193b-3p significantly increased HDAC3 expression and significantly decreased SOX9, COL2A1, and AGGRECAN expression (Figure 9C).
miR-193b-3p enhanced ectopic cartilage formation in nude mice. (A) TCP-COL-HA scaffolds seeded with PHCs were stained with Safranin O and Alcian blue, and subjected to HDAC3 immunohistochemical staining 8 weeks after implantation. (B) TCP-COL-HA scaffolds seeded with PHCs were subjected to collagen type II, AGGRECAN, and SOX9 immunohistochemical staining 8 weeks after implantation. Upper panels: 20× magnification; lower panel: magnified view of areas enclosed by the black boxes, 50× magnification. (C) The relative expression levels of miR-193b-3p, HDAC3, SOX9, COL2A1, and AGGRECAN in implanted PHCs seeded with TCP-COL-HA. (D) Quantification of the GAG/dsDNA ratio in implanted PHCs seeded with TCP-COL-HA. Top left panel: overall view of the sample. Lower panel: magnified view. Scale bar = 100 μm. RNU6B and GAPDH were detected as endogenous controls. The data shown represent the mean ± standard error (SE) of at least three independent experiments. *P < 0.05; ** P < 0.001
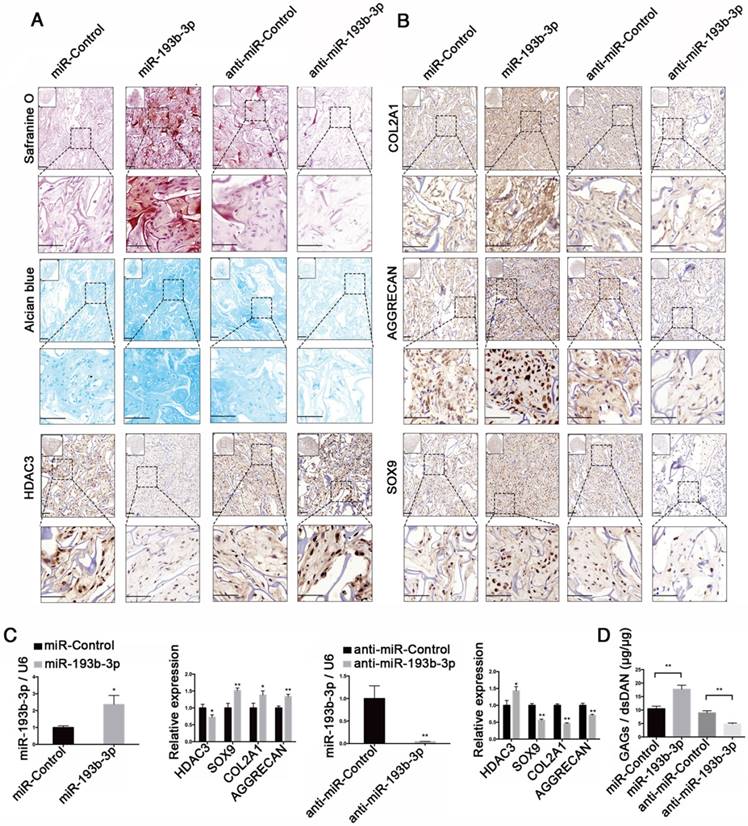
Moreover, using a biochemical assay for GAGs, we found that overexpression of miR-193b-3p increased the GAG/dsDNA ratio, whereas inhibiting miR-193b-3p expression decreased the GAG/dsDNA ratio (Figure 9D). These subcutaneous-implantation results indicated that miR-193b-3p suppressed HDAC3 expression and enhanced cartilage formation, whereas the miR-193b-3p inhibitor increased HDAC3 expression and inhibited cartilage formation in vivo.
Expression levels of miR-193b-3p in clinical cartilage and plasma samples
To determine whether miR-193b-3p expression changes during OA progression, we compared its expression level in samples from 30 pairs of non-degraded cartilage and corresponding degraded cartilage. By qRT-PCR analysis and in situ hybridization, we found that miR-193b-3p expression was significantly lower in degraded cartilage compared to non-degraded cartilage (Figure 10A, qRT-PCR analysis; Figure 10G-J, in situ hybridization), whereas degraded cartilage exhibited higher levels of HDAC3 mRNA (Figure 10B) and protein (Figure 10E-F) compared to non-degraded cartilage. In addition, we found that Safranin O staining in degraded cartilage was significantly lower than that in non-degraded cartilage (Figure 10C-D), and degraded cartilage samples showed higher OARSI grade scores compared to scores from non-degraded samples (Figure 10K). Histomorphometric analyses confirmed that the expression level of miR-193b-3p was higher in non-degraded cartilage compared to degraded cartilage, whereas HDAC3 expression was higher in degraded cartilage compared to that found in non-degraded cartilage (Figure 10L-M).
The relative expression levels of miR-193b-3p in clinical cartilage and plasma samples. (A-B) The relative expression levels of miR-193b-3p and HDAC3 in 30 pairs of non-degraded and degraded cartilages. (C-D) Safranin O staining of non-degraded and degraded cartilages at 20× magnification. (E-F) HDAC3 immunohistochemistry of non-degraded and degraded cartilages at 20× magnification. (G-H) miR-193b immunohistochemistry of non-degraded and degraded cartilages at 20× magnification. Note that miR-193b expression is indicated by red fluorescence, and the cell nuclei are indicated by blue fluorescence. (I-J) Magnified view of areas enclosed by white boxes in panels G and H at 50× magnification. Representative results from three paired non-degraded and degraded cartilages are shown. (K) OARSI grade scores of three paired representative non-degraded and degraded cartilages samples. (L) Average IOD/area for miR-193b-3p. (M) Average IOD/area for HDAC3. (N) Particle-size distribution of exosomes measured by NTA. (O) Morphology of exosomes observed by TEM at 1.0 × 104 magnification. Exosomes are indicated by the white arrows. (P) Exosome surface markers (CD63 and CD9) measured by western blotting. The supernatant was used as a negative control. (Q) Relative expression levels of exosomal miR-193b-3p in 19 normal and OA plasma samples. (R-S) The concentrations of IL-6 and IL-8 in 19 normal and OA plasma samples were measured by ELISA. RNU6B and GAPDH were detected as endogenous controls. miR-39 was detected as a reference gene for plasma exosomal miR-193b-3p expression. Scale bar = 100 μm. The data shown represent the mean ± standard error (SE) of at least three independent experiments. *P < 0.05; ** P < 0.001
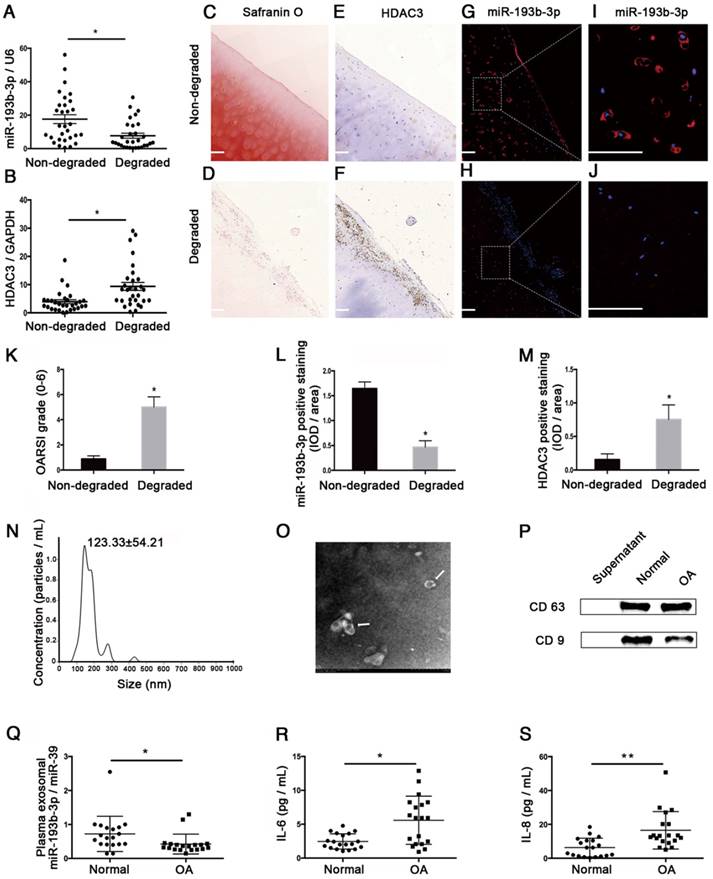
We also compared exosomal miR-193b in plasma from normal subjects and patients with OA. Using NTA analysis, TEM, and western blotting to identify exosomes isolated from human plasma, we found that most of these vesicles ranged from 50 to 200 nm in size (Figure 10N) and that hollow spherical microvesicles (Figure 10O) were seen by TEM. In addition, the expression of the exosome markers CD63 and CD9 were significantly enriched in our isolated exosomes (Figure 10P). Thus, these findings indicated that exosomes were successfully isolated. Furthermore, we found that plasma exosomal miR-193b expression was significantly lower in the OA group compared to that in the normal group (Figure 10Q). In addition, the results of ELISA assays indicated that the expression levels of common proinflammatory indicators, such as IL-6 and IL-8, were significantly higher in the OA group compared to those in the normal group (Figure 10R-S).
Discussion
miR-193b-3p has gained increasing interest because of its important regulatory functions in a wide range of organs and tissues, including cartilage [9, 10, 17-19, 20-22]. We previously demonstrated that miR-193b regulated early chondrogenesis in ATDC5 cells and suppressed TNF-alpha expression in IL-1β-induced chondrocytes [9]. However, the role of miR-193b in cartilage development and degeneration in humans, and the mechanism by which this RNA regulates gene expression, remain unclear. In this study, we demonstrated that miR-193b-3p promoted chondrogenesis of hMSCs and regulated chondrocyte metabolism by enhancing histone H3 acetylation via targeting and suppressing HDAC3 expression.
Studies have demonstrated that the class I HDACs (HDAC1, HDAC2, HDAC3, and HDAC8) play important roles in the regulation of chondrocyte development and hypertrophy response [3, 5]. Class I HDACs tend to inhibit chondrogenesis by repressing cartilage-specific genes, such as COL2A1 and AGGRECAN [5, 6], and depletion of a class I HDAC dramatically increased expression of a wide range of extracellular matrix-related genes [23]. HDAC3 is highly expressed by chondrocytes and osteoblasts, and controls chondrocyte hypertrophy and endochondral bone formation [24-25]. HDAC3 is also required for mesenchymal progenitor cell differentiation and long bone development [26]. siRNA-mediated gene knockdown represents a crucial method for understanding the effects of individual isoforms of class I HDACs on the expression of cartilage-specific genes [27]. In mouse chondrocytes, the knockdown of HDAC3 significantly increased SOX9 expression [6], whereas in this study using human chondrocytes, we found that knockdown of HDAC3 resulted in up-regulated COMP, COL2A1, SOX9, and AGGRECAN expression and down-regulated COL10 expression. Similar results were seen in miR-193b overexpression studies in PHCs. Both microRNA overexpression and siRNA treatment are attractive options for cell-based cartilage repair [27, 31]. It is unclear whether modulation of cartilage-specific gene expression is more affected by siRNA-mediated HDAC3 silencing or miR-193b overexpression in PHCs. However, it has been shown that, in contrast with overexpression treatment, siRNA techniques require a limited utilization of cellular resources and do not overload the cells or the extracellular environment with transgene products [27]. Therefore, cells may be less stressed by siRNAs than by transgene overexpression. Further studies could be performed to clarify this issue.
In this study, opposing expression patterns between miR-193b-3p and HDAC3 were observed during the chondrogenic differentiation of hMSCs on days 14-28 (middle and late stage of chondrogenesis); however, on day 3-14 (early stage of chondrogenesis), both miR-193b-3p and HDAC3 expression increased. Similar phenomena were observed in previous studies [17, 28]. miR-320 regulated MMP-13 expression in chondrogenesis and opposing expression patterns between miR-320 and MMP-13 were observed on days 14-28, but not on days 3-14, during ATDC5 cell chondrogenesis [17]. miR-92a regulated HDAC2 in chondrogenesis of hMSCs and opposing expression patterns between miR-92a and HDAC2 were seen during MSC chondrogenesis on days 14-28 [28]. A microRNA could regulate a specific phase of chondrocyte differentiation [29], and miR-193b-3p may mainly regulate the chondrogenesis of hMSCs through HDAC3 on days 14-28 (middle and late stage of chondrogenesis). Further studies are needed to elucidate the underlying mechanisms.
We previously demonstrated that miR-193b-3p could regulate early chondrogenesis in ATDC5 chondrocyte-like cells in mice [9]. In this study, we found that miR-193b-3p could enhance the chondrogenic differentiation of hMSCs. A miRNA may have distinct targets in different cell types and in different species [30-31]. Predictive bioinformatics analyses using TargetScan, miRanda, and miRWalk revealed that miR-193b-3p can potentially bind the specific seed sequence of the 3'-UTR of HDAC3 in humans, but not in mice. Thus, the mechanism by which miR-193b-3p regulates gene expression could differ between human and mouse cells. Links between miRNA and members of the HDAC family during cartilage development and degradation have been previously reported [32-34]. For example, miR-365, miR-29b, miR-222, and miR-381 have been reported to directly target HDAC4 and to participate in chondrocyte differentiation and OA pathogenesis [32-34]. In this study, miR-193b-3p was found to negatively regulate HDAC3 expression during hMSC chondrogenesis. Subsequent analyses using dual luciferase reporting, qRT-PCR, and western blotting confirmed that miR-193b targeted and inhibited HDAC3 expression. Together, these results indicate that miR-193b promoted chondrogenic differentiation of hMSCs by suppressing HDAC3 expression, thereby maintaining cartilage-specific gene expression.
Inflammation plays an important role in OA progression [35]. Cartilage destruction begins when chondrocytes are stimulated by proinflammatory cytokines such as IL-1β, IL-6, IL-8, and TNF-alpha [36], and anti-inflammatory therapies have been considered as innovative osteoarthritis approaches [27, 37]. miR-193b-3p was shown to inhibit MMP-19 and iNOS expression in IL-1β-induced human chondrocytes by direct targeting MMP-19 [38]. In this study, we found that miR-193b-3p was down-regulated in time (6 h, 12 h, 24 h)- and dose (0, 1, 5, and 10 ng/mL)-dependent manners in chondrocytes stimulated with IL-1β. Furthermore, miR-193b-3p could inhibit MMP-13 expression in OA chondrocytes and increase expression of cartilage-specific genes, like COL2, AGGRECAN, and SOX9, by targeting HDAC3 in PHCs. In vivo, miR-193b-3p expression was significantly lower in degraded cartilage than in non-degraded cartilage. Based on these results, miR-193b may help in negatively regulating the local inflammatory environment and in limiting the strength of inflammation. Similar results have been reported in previous studies. For example, Ikeda et al. found that activation of the mitogen-activated protein kinase pathway (a key regulator of proinflammatory cytokines) induced significant down-regulation of miR-193b [39], whereas Aner et al. found that miR-193b inhibited expression of the proinflammatory cytokines CCL2 and IL-6 [20]. Our previous study also showed that miR-193b could suppress the expression of TNF-alpha in IL-1β-induced chondrocytes [9]. However, conflicting results were reported in a study by Ukai et al. [10], who found that miR-193b down-regulated expression of cartilage-specific genes, such as COL2, AGGRECAN, and SOX9, in chondrocytes. The discrepancy between their findings and those of the present study may be attributable to the different harvesting times following transfection of miR-193b into chondrocytes. Ukai et al. harvested cells 6 h after miR-193b transfection into chondrocytes, whereas in our study, we harvested cells 24 h after transfecting miR-193b-3p into chondrocytes (for qRT-PCR) or 48 h after transfection (for western blotting analysis). In our study, we found no significant changes of HDAC3 6 h after transfection (data not shown). Consistent with our qRT-PCR results, we also found that protein levels of COL2, AGGRECAN, and SOX9 were increased by miR-193b in chondrocytes at 48 h after transfection. Using an in vivo ectopic cartilage-formation assay in nude mice further confirmed that miR-193b could promote COL2, AGGRECAN, and SOX9 expression in chondrocytes. Overall, these results strongly indicate that miR-193b can increase cartilage-specific gene expression by suppressing HDAC3 in chondrocytes, thereby promoting ectopic cartilage formation in nude mice.
In our previous study, we found miR-193b-3p was up-regulated in PHCs at 4 h after treatment with IL-1β. In this study, miR-193b expression showed a tendency to increase, but no significant change was found after 4 h of IL-1β treatment compared with the non-treated group (Figure S2). The discrepancy between these results could be attributable to the different donor ages and different passage numbers of PHCs used in the two studies. In our previous study, we used PHCs at passage 4, and the cells were isolated from the femoral head of a 62-year old male. In this study, we used PHCs at passage 1, and the cells were isolated from the knee joint of a 20-year old male. Considering that miR-193b expression in chondrocytes changed with donor ages [10], the expression of miR-193b in response to IL-1β treatment could differ with donor ages in PHCs. In a follow-up study, we will use PHCs from different ages to further study the expression and function of miR-193b-3p in both IL-1β-induced chondrocytes responses and chondrogenic differentiation.
HDAC3 was previously shown to be up-regulated during inflammation and to play a central role in this process [40-41]. In this study, we found that the expression of HDAC3 increased when human PHCs were stimulated with IL-1β, suggesting a biological role that HDAC3 could play in IL-1β-induced PHC responses. The cause of increased HDAC3 expression in IL-1β-stimulated PHCs is unclear, but may be attributable to the down-regulation of miR-193b in IL-1β-stimulated PHCs. Previous data showed a positive role for HDAC3 in the transcription of most IL-1-induced human genes. The effect could be mediated by the HDAC3-mediated deacetylation of NF-Kb p65 at lysines 122, 123, 314, and 315 [42]. Further studies are needed to clarify the exact function of HDAC3 in IL-1β-induced PHC responses.
The HDAC inhibitor TSA has previously shown efficacy in treating arthritis in several models of inflammatory arthritis [2, 43-45]. TSA was found to suppress the expression of MMP13 in IL-1β-induced chondrocytes [2]. In this study, we found that TSA decreased MMP13 and increased AGGRECAN expression in IL-1β-treated PHCs and enhanced the chondrogenic differentiation of hMSCs. Similar results have been reported in previous studies. For example, EI-Serafi et al. found that TSA enhanced hMSCs chondrogenesis in pellets cultured in chondrogenic media [46]. Kim et al. found TSA treatment increased type II collagen expression over control levels seen during hMSC chondrogenesis [47]. The mechanism by which TSA affects cartilage development and degradation may be attributable to histone modification by HDACs. TSA increases the level of histone H3 acetylation near the COL2A1 enhancer region [48, 2]. In this study, we found that miR-193b-3p functions, at least to some extent, in a manner similar to an HDAC inhibitor. We found that miR-193b inhibited HDAC3 expression and enhanced histone H3 hyperacetylation near the AGGRECAN, SOX9, COL2, and COMP promoter regions in chondrocytes.
It has been reported that most serum miRNAs are enriched in exosomes [49]. Serum/plasma exosomal microRNAs have received considerable attention as potential biomarkers for diagnosing diseases [49-52]. Serum exosomal expression of miR-193b, but not that of total miR-193b, was lower in patients with dementia of Alzheimer's type and mild cognitive impairment compared to that found in controls, a finding that indicates its potential as a novel blood-based biomarker [22]. Similarly, plasma exosomal miR-21 has been demonstrated to be a useful biomarker for predicting recurrence and poor prognosis in colorectal cancer [50]. In this study, we found that plasma exosomal miR-193b expression was significantly lower in patients with OA compared to that found in normal controls. Using a qRT-PCR array, Murata et al. found that miR-193b expression in plasma was lower in patients with rheumatoid arthritis compared to healthy controls [51], whereas Wang et al. found that serum miR-193b* expression was negatively correlated with inflammation and sepsis [52]. Therefore, miR-193b has the potential to serve as a novel non-invasive blood biomarker of OA.
The present study has some limitations. First, in this study, the effects of HDAC3 overexpression on reversing the effect of miR-193b-3p overexpression were not clarified in PHCs. However, we demonstrated an opposite effect of HDAC3 knockdown versus the miR-193b inhibitor, which confirmed that miR-193b regulates chondrocyte metabolism by targeting HDAC3. In hMSCs, the effects of HDAC3 overexpression on countering the effect of miR-193b-3p overexpression (and likewise the effect of HDAC3 knockdown on reversing the effect of miR-193b-3p inhibition) in chondrogenesis were not clarified. We plan to generate stable HDAC3-overexpressing, HDAC3-knockdown hMSC cell lines and miR-193b-overexpressing, miR-193b-knockdown hMSCs cell lines in a future study, to further elaborate the role of HDAC3 and miR-193b-3p in hMSC chondrogenesis. Second, we used TSA, but not an HDAC3-specific inhibitor, to study the role of HDAC3 in PHCs and hMSCs. Although an HDAC3-specific inhibitor would be more appropriate, TSA has also been widely used in HDAC3 studies [41-43], and we used an HDAC3 siRNA to further demonstrate the role of HDAC3 in PHCs in this study. Lastly, the results from this study have shown that miR-193b-3p could play some roles in IL-1β-induced chondrocyte responses and OA pathogenesis; however, the mechanistic insights into the role of miR-193b-3p in IL-1β-mediated chondrocyte responses were not clarified. We plan to study inflammatory cytokine (like IL-1β and TNF-alpha)-induced chondrocyte responses and cell-signaling pathways to further clarify the specific functions of miR-193b in inflammatory responses in chondrocytes. Further research will be conducted to study the effects of inflammatory cytokines on cartilage generation using human chondrocytes seeded on scaffolds in nude mice and to further study the role of miR-193b in inflammation-induced chondrocyte responses in vivo. Previously, we found that miR-193b-3p could affect the TGF-beta2-signaling pathway in mice [9]; however, the functional relationship between miR-193b-3p and TGF-beta2 signaling (which might also affect chondrocyte metabolism and chondrogenesis, while miR-193b-3p, TGF-beta2, and HDAC3 might have some synergistic effect on hMSC chondrogenesis) is unclear in humans. Further studies are needed to clarify this issue.
In summary, we demonstrated that the novel and crucial microRNA miR-193b is expressed during hMSC chondrogenesis and is differentially expressed in degraded and non-degraded cartilage. We found that miR-193b-3p functions as a negative regulator by down-regulating HDAC3 expression in chondrogenesis and by increasing histone H3 acetylation near the promoter regions of AGGRECAN, SOX9, COL2A1, and SOX9 in chondrocytes. Furthermore, miR-193b enhanced tissue engineering of cartilage formation in vivo and may serve as a novel non-invasive biomarker of OA. These findings provide new insights into chondrogenic regulation by class I HDACs and miRNAs and raise the possibility of using miR-193b-3p to influence chondrogenesis and chondrocyte metabolism.
Abbreviations
α-MEM: alpha-modified Eagle's medium; ac-H3: acetylated histone H3; ChIP: chromatin immunoprecipitation; DMEM: Dulbecco's Modified Eagle Medium; dsDNA: double-stranded DNA; EDTA: ethylenediaminetetraacetic acid; ELISA: enzyme-linked immunosorbent assay; F12: Ham's F-12 Nutrient Mixture; FBS: fetal bovine serum; GAG: glycosaminoglycan; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; HDAC3: histone deacetylase 3; hMSC: human mesenchymal stem cell; IL-1β: interleukin-1β; IOD: integrated optical density; miR-193b-3p: microRNA-193b-3p; miRNA: microRNA; MMP1: matrix metallopeptidase 1; NTA: nanoparticle-tracking analysis; OA: osteoarthritis; PBS: phosphate-buffered saline; PHC: primary human chondrocyte; qRT-PCR: quantitative real-time polymerase chain reaction; SD: standard deviation; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SE: standard error; siRNA: small-interfering RNA; TCP-COL-HA: tricalcium phosphate-collagen-hyaluronate; TEM: transmission electron microscopy; TKA: total knee arthroplasty; TSA: trichostatin A; UTR: untranslated region.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Gang Wu (South China University of Technology, Guangzhou, China) for providing scaffold materials and technical assistance. We thank Xuerong Li and Mei Shang (Department of Parasitology at the Zhongshan School of Medicine, Sun Yat-sen University, People's Republic of China) for their technical assistance. These studies were funded by the National Natural Science Foundation of China (grant numbers 81702142 and 81672145), the China Postdoctoral Science Foundation (grant number 2016M602590), and the Doctoral Start-up Fund of the Natural Science Foundation of Guangdong Province (grant number 2015A030310204).
Author contributions
Weiming Liao and Aishan He conceived and designed the study. Fangang Meng, Zhiwen Li, Zhiqi Zhang, Zibo Yang, and Dianbo Long performed the experiments. Aishan He, Yan Kang, Shu Hu, Minghui Gu, and Suiwen He collected the cartilage and plasma samples. Zibo Yang and Suiwen He clinically graded the cartilage samples and scored the Safranin O staining of cartilage samples. Fangang Meng and Zhiwen Li wrote the paper. Peihui Wu, Zongku Chang, Dianbo Long, Zhiqi Zhang, Aishan He, and Weiming Liao reviewed and edited the manuscript. All authors read and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213:626-34
2. Culley KL, Hui W, Barter MJ, Davidson RK, Swingler TE, Destrument AP. et al. Class I histone deacetylase inhibition modulates metalloproteinase expression and blocks cytokine-induced cartilage degradation. Arthritis Rheum. 2013;65:1822-30
3. Carpio LR, Westendorf JJ. Histone deacetylases in cartilage homeostasis and osteoarthritis. Curr Rheumatol Rep. 2016;18:52
4. Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E. et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555-66
5. Hong S, Derfoul A, Pereira-Mouries L, Hall DJ. A novel domain in histone deacetylase 1 and 2 mediates repression of cartilage-specific genes in human chondrocytes. FASEB J. 2009;23:3539-52
6. Chen W, Chen L, Zhang Z, Meng F, Huang G, Sheng P. et al. MicroRNA-455-3p modulates cartilage development and degeneration through modification of histone H3 acetylation. Biochim Biophys Acta. 2016;1863:2881-91
7. Engels BM, Hutvagner G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene. 2006;25:6163-9
8. Zhang Z, Kang Y, Zhang Z, Zhang H, Duan X, Liu J. et al. Expression of microRNAs during chondrogenesis of human adipose-derived stem cells. Osteoarthritis Cartilage. 2012;20:1638-46
9. Hou C, Yang Z, Kang Y, Zhang Z, Fu M, He A. et al. MiR-193b regulates early chondrogenesis by inhibiting the TGF-beta2 signaling pathway. FEBS Lett. 2015;589:1040-7
10. Ukai T, Sato M, Akutsu H, Umezawa A, Mochida J. MicroRNA-199a-3p, microRNA-193b, and microRNA-320c are correlated to aging and regulate human cartilage metabolism. J Orthop Res. 2012;30:1915-22
11. Li BF, Zhang Y, Xiao J, Wang F, Li M, Guo XZ. et al. Hsa_circ_0045714 regulates chondrocyte proliferation, apoptosis and extracellular matrix synthesis by promoting the expression of miR-193b target gene IGF1R. Hum Cell. 2017;30:311-8
12. Meng F, He A, Zhang Z, Zhang Z, Lin Z, Yang Z. et al. Chondrogenic differentiation of ATDC5 and hMSCs could be induced by a novel scaffold-tricalcium phosphate-collagen-hyaluronan without any exogenous growth factors in vitro. J Biomed Mater Res A. 2014;102:2725-35
13. Hou C, Meng F, Zhang Z, Kang Y, Chen W, Huang G. et al. The role of microRNA-381 in chondrogenesis and interleukin-1-β induced chondrocyte responses. Cell Physiol Biochem. 2015;36:1753-66
14. Outerbridge RE. The etiology of chondromalacia patellae. 1961. Clin Orthop Relat Res. 2001Aug;389:5-8
15. Altman RD, Gold GE. Atlas of individual radiographic features in osteoarthritis, revised. Osteoarthritis Cartilage. 2007;15:A1-A56
16. Pritzker KPH, Gay S, Jimenez SA. et al. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage. 2006;14:13-29
17. Meng F, Zhang Z, Chen W, Huang G, He A, Hou C. et al. MicroRNA-320 regulates matrix metalloproteinase-13 expression in chondrogenesis and interleukin-1β-induced chondrocyte responses. Osteoarthritis Cartilage. 2016;24:932-41
18. Akhtar N, Rasheed Z, Ramamurthy S, Anbazhagan AN, Voss FR, Haqqi TM. MicroRNA-27b regulates the expression of matrix metalloproteinase 13 in human osteoarthritis chondrocytes. Arthritis Rheum. 2010;62:1361-71
19. Meng FG, Zhang ZQ, Huang GX, Chen WS, Zhang ZJ, He AS. et al. Chondrogenesis of mesenchymal stem cells in a novel hyaluronate-collagen-tricalcium phosphate scaffolds for knee repair. Eur Cell Mater. 2016;30:79-94
20. Arner E, Mejhert N, Kulyté A, Balwierz PJ, Pachkov M, Cormont M. et al. Adipose tissue microRNAs as regulators of CCL2 production in human obesity. Diabetes. 2012;61:1986-93
21. Haetscher N, Feuermann Y, Wingert S, Rehage M, Thalheimer FB, Weiser C. et al. STAT5-regulated microRNA-193b controls haematopoietic stem and progenitor cell expansion by modulating cytokine receptor signalling. Nat Commun. 2015;6:8928
22. Liu CG, Song J, Zhang YQ, Wang PC. MicroRNA-193b is a regulator of amyloid precursor protein in the blood and cerebrospinal fluid derived exosomal microRNA-193b is a biomarker of Alzheimer's disease. Mol Med Rep. 2014;10:2395-400
23. Whetstine JR, Ceron J, Ladd B, Dufourcq P, Reinke V, Shi Y. Regulation of tissue-specific and extracellular matrix-related genes by a class I histone deacetylase. Mol Cell. 2005;18:483-90
24. Carpio LR, Bradley EW, McGee-Lawrence ME, Weivoda MM, Poston DD, Dudakovic A. et al. Histone deacetylase 3 supports endochondral bone formation by controlling cytokine signaling and matrix remodeling. Sci Signal. 2016;9:ra79
25. Bradley EW, Carpio LR, Westendorf JJ. Histone deacetylase 3 suppression increases PH domain and leucine-rich repeat phosphatase (Phlpp)1 expression in chondrocytes to suppress Akt signaling and matrix secretion. J Biol Chem. 2013;288:9572-82
26. Feigenson M, Shull LC, Taylor EL, Camilleri ET, Riester SM, van Wijnen AJ. Histone deacetylase 3 deletion in mesenchymal progenitor cells hinders long bone development. J Bone Miner Res. 2017;32:2453-65
27. Lolli A, Penolazzi L, Narcisi R, Osch GJVM, Piva R. Emerging potential of gene silencing approaches targeting anti-chondrogenic factors for cell-based cartilage repair. Cell Mol Life Sci. 2017;74:3451-65
28. Mao G, Zhang Z, Huang Z, Chen W, Huang G, Meng F. et al. MicroRNA-92a-3p regulates the expression of cartilage-specific genes by directly targeting histone deacetylase 2 in chondrogenesis and degradation. Osteoarthritis Cartilage. 2016;25:521-32
29. McAlinden A, Varghese N, Wirthlin L, Chang L-W. Differentially expressed microRNAs in chondrocytes from distinct regions of developing human cartilage. PloS One. 2013;8:e75012
30. Maatouk D, Harfe BD. MicroRNAs in development. Sci World J. 2006;6:1828-40
31. Mirzamohammadi F, Papaioannou G, Kobayashi T. microRNAs in cartilage development, homeostasis, and disease. Curr Osteoporosis Rep. 2014;12:410-19
32. Guan Y-J, Yang X, Wei L, Chen Q. MiR-365: a mechanosensitive microRNA stimulates chondrocyte differentiation through targeting histone deacetylase 4. FASEB J. 2011;25:4457-66
33. Song J, Jin EH, Kim D, Kim KY, Chun CH, Jin EJ. MicroRNA-222 regulates MMP-13 via targeting HDAC-4 during osteoarthritis pathogenesis. BBA Clin. 2014;3:79-89
34. Chen W, Sheng P, Huang Z, Meng F, Kang Y, Huang G. et al. MicroRNA-381 regulates chondrocyte hypertrophy by inhibiting histone deacetylase 4 expression. Int J Mol Sci. 2016;17:1377
35. Goldring MB, Otero M. Inflammation in osteoarthritis. Curr Opin Rheumatol. 2011;23:471-8
36. Bian Q, Wang YJ, Liu SF, Li YP. Osteoarthritis: genetic factors, animal models, mechanisms, and therapies. Front Biosci. 2012;4:74-100
37. Calich AL, Domiciano DS, Fuller R. Osteoarthritis: can anti-cytokine therapy play a role in treatment? Clin Rheumatol. 2010;29:451-5
38. Chang ZK, Meng FG, Zhang ZQ, Mao GP, Huang ZY, Liao WM. et al. MicroRNA-193b-3p regulates matrix metalloproteinase 19 expression in interleukin-1β-induced human chondrocytes. J Cell Biochem. 2018 [Epub ahead of print]
39. Ikeda Y, Tanji E, Makino N, Kawata S, Furukawa T. MicroRNAs associated with mitogen-activated protein kinase in human pancreatic cancer. Mol Cancer Res. 2012;10:259-69
40. Kim Y, Eom S, Park D, Kim H, Jeoung D. The hyaluronic acid-HDAC3-miRNA network in allergic inflammation. Front Immunol. 2015;6:210
41. Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci USA. 2012;109:E2865-74
42. Ziesché E, Kettner-Buhrow D, Weber A, Wittwer T, Jurida L, Soelch J. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-κB. Nucleic Acids Res. 2012;41:90-109
43. Young DA, Lakey RL, Pennington CJ, Jones D, Kevorkian L, Edwards DR. et al. Histone deacetylase inhibitors modulate metalloproteinase gene expression in chondrocytes and block cartilage resorption. Arthritis Res Ther. 2005;7:R503-12
44. Chabane N, Zayed N, Afif H, Mfuna-Endam L, Benderdour M, Boileau C. et al. Histone deacetylase inhibitors suppress interleukin-1beta-induced nitric oxide and prostaglandin E2 production in human chondrocytes. Osteoarthritis Cartilage. 2008;16:1267-74
45. Saito T, Nishida K, Furumatsu T, Yoshida A, Ozawa M, Ozaki T. Histone deacetylase inhibitors suppress mechanical stress-induced expression of RUNX-2 and ADAMTS-5 through the inhibition of the MAPK signaling pathway in cultured human chondrocytes. Osteoarthritis Cartilage. 2013;21:165-74
46. El-Serafi AT, Oreffo RO, Roach HI. Epigenetic modifiers influence lineage commitment of human bone marrow stromal cells: differential effects of 5-aza-deoxycytidine and trichostatin A. Differentiation. 2011;81:35-41
47. Kim H J, Kwon Y-R, Bae Y-J, Kim Y-J. Enhancement of human mesenchymal stem cell differentiation by combination treatment with 5-azacytidine and trichostatin A. Biotechnol Lett. 2015;38:167-74
48. T Furumatsu, M Tsuda, K Yoshida, N Taniguchi, T Ito. et al. Sox9 and p300 cooperatively regulate chromatin-mediated transcription. J Biol Chem. 2005;28:35203-8
49. Li Y, Zhang L, Liu F, Xiang G, Jiang D, Pu X. Identification of endogenous controls for analyzing serum exosomal miRNA in patients with hepatitis B or hepatocellular carcinoma. Dis Markers. 2015;2015:893594
50. Tsukamoto M, Iinuma H, Yagi T, Matsuda K, Hashiguchi Y. Circulating exosomal microRNA-21 as a biomarker in each tumor stage of colorectal cancer. Oncology. 2017;92:360-70
51. Murata K, Furu M, Yoshitomi H, Ishikawa M, Shibuya H, Hashimoto M. et al. Comprehensive microRNA analysis identifies miR-24 and miR-125a-5p as plasma biomarkers for rheumatoid arthritis. PLoS One. 2013;8:e69118
52. Wang HJ, Zhang PJ, Chen WJ, Feng D, Jia YH, Xie LX. Four serum microRNAs identified as diagnostic biomarkers of sepsis. J Trauma Acute Care Surg. 2012;73:850-4
Author contact
![]() Corresponding author: Weiming Liao, Department of Joint Surgery, First Affiliated Hospital of SunYat-sen University, Guangzhou, Guangdong 510080, China. E-mail: liaowmsysucom; Aishan He, Department of Sports Medicine, First Affiliated Hospital of SunYat-sen University, Guangzhou, Guangdong 510080, China. E-mail: heaishan2006com.
Corresponding author: Weiming Liao, Department of Joint Surgery, First Affiliated Hospital of SunYat-sen University, Guangzhou, Guangdong 510080, China. E-mail: liaowmsysucom; Aishan He, Department of Sports Medicine, First Affiliated Hospital of SunYat-sen University, Guangzhou, Guangdong 510080, China. E-mail: heaishan2006com.