Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(9):2549-2564. doi:10.7150/thno.22523 This issue Cite
Research Paper
CAPG enhances breast cancer metastasis by competing with PRMT5 to modulate STC-1 transcription
Sheng Huang1,2#, Yayun Chi1,#, Yi Qin3,#, Ziliang Wang4, Bingqiu Xiu1, Yonghui Su1, Rong Guo1, Liang Guo1, Hefen Sun1, Chujia Zeng1, Shuling Zhou5, Xin Hu1, Sheng Liu6, Zhimin Shao1, Zhaohui Wu7, Wei Jin1, ![]() , Jiong Wu1,
, Jiong Wu1, ![]()
1. Department of Breast Surgery, Breast Cancer Institute, Shanghai Cancer Center, Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, People's Republic of China
2. The Second Department of Breast Surgery, The Third Affiliated Hospital of Kunming Medical University (Tumor Hospital of Yunnan Province), Kunming, Yunnan, People's Republic of China
3. Department of Pancreas & Hepatobiliary Surgery, Pancreas & Hepatobiliary Cancer Institute, Shanghai Cancer Center, Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, People's Republic of China
4. Cancer Institute, Shanghai Cancer Center, Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, People's Republic of China
5. Department of Pathology, Shanghai Cancer Center, Fudan University, Shanghai, People's Republic of China
6. Department of Breast Surgery and Pharmacology Laboratory of Traditional Chinese Medicine, Long Hua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, People's Republic of China
7. Department of Pathology and Laboratory Medicine; Center for Cancer Research, University of Tennessee Health Science Center, Memphis, Tennessee
# These authors contributed equally to this work.
Received 2017-8-24; Accepted 2018-2-22; Published 2018-4-3
Abstract

Macrophage-capping protein (CAPG) has been shown to promote cancer cell metastasis, although the mechanism remains poorly understood.
Methods: Breast cancer (BC) tissue microarray was used to test the role of CAPG in the prognosis of BC patients. Xenograft mice model was used to validate the metastasis promotion role of CAPG in vivo. Gene expression array, chromatin immunoprecipitation and luciferase report assay were performed to search for the target genes of CAPG. Protein immunoprecipitation, MS/MS analysis, tissue microarray and histone methyltransferase assay were used to explore the mechanism of CAPG regulating stanniocalcin 1 (STC-1) transcription.
Results: We demonstrate a novel mechanism by which CAPG enhances BC metastasis via promoting the transcription of the pro-metastatic gene STC-1, contributing to increased metastasis in BC. Mechanistically, CAPG competes with the transcriptional repressor arginine methyltransferase 5 (PRMT5) for binding to the STC-1 promoter, leading to reduced histone H4R3 methylation and enhanced STC-1 transcription. Our study also indicates that both CAPG and PRMT5 are independent prognostic factors for BC patient survival. High CAPG level is associated with poor survival, while high PRMT5 expression favors a better prognosis in BC patients.
Conclusion: Our findings identify a novel role of CAPG in the promotion of BC metastasis by epigenetically enhancing STC-1 transcription.
Keywords: breast cancer, metastasis, CAPG, PRMT5, STC-1
Introduction
Breast cancer (BC) is the most frequently diagnosed cancer and the leading global cause of cancer deaths in women, accounting for 23% of cancer diagnoses (1.38 million women) and 14% of cancer deaths (458,000 women) each year [1]. Metastatic disease is responsible for the vast majority of cancer patient deaths and represents the main clinical challenge of solid tumors. The importance of understanding the mechanisms underlying the metastatic process and the complex interactions between the tumor and the host during disease progression is widely recognized, and these topics have attracted considerable research interest [2].
Macrophage-capping protein (CAPG, also known as gCap39 or MCP) is a member of the gelsolin family of actin assembly regulators [3]. CAPG localizes to both the cytoplasm and the nucleus due to the lack of a nuclear export sequence, whereas other members of the gelsolin family are present only in the cytoplasm [4]. In the cytoplasm, CAPG interacts with the actin cytoskeleton to ensure actin filaments are correctly polymerized, arranged and localized for cells to move or change shape [3, 5-7]. Intriguingly, nuclear CAPG plays a much stronger role in eliciting invasion than the cytoplasmic CAPG fraction [8]. The CAPG gene shares sequence homology with genes encoding basic helix-loop-helix-family DNA-binding proteins (bHLH), including the c-Myc oncogene. Therefore, the CAPG protein was also termed myc basic motif homolog-1 (mbh-1) [9]. Nuclear CAPG has been suggested to modulate gene transcription, potentially by controlling actin nucleation and assembly, which is involved in regulating gene transcription [10-12]. However, whether and how the nuclear CAPG-regulated gene transcription enhances BC aggressiveness is still largely unknown.
STC is a glycoprotein hormone that was first identified in bony fish, wherein it inhibits hypercalcemia by decreasing calcium uptake from the gills [13]. The human homolog STC-1 shares 73% homology with the fish STC gene and encodes a 247-amino-acid protein. Owing to a signal peptide sequence, dimerized STC-1 can be secreted into the extracellular space, where it regulates calcium and phosphate homeostasis [14-16]. Previous studies have indicated that STC-1 may promote cancer metastasis [17, 18] and elevated STC-1 levels correlate with a poor prognosis in cancer patients [19-22]. In BC, STC-1 was shown to enhance cancer cell invasion by activating the phosphoinositide 3-kinase (PI3K) pathway [18]. However, the regulatory factors involved in STC-1 transcription are not clear.
In this study, we found that CAPG expression was significantly increased in the highly metastatic MDA-MB-231 HM cells compared with that in its parental MDA-MB-231 cells, and CAPG up-regulation in tumors was associated with a worse prognosis in BC patients, suggesting a pathological link between CAPG expression and BC aggressiveness. We further showed that CAPG was involved in epigenetic regulation of STC-1 gene transcription in BC cells by binding to the arginine methyltransferase PRMT5. PRMT5 serves as an epigenetic repressor by methylating histones H2A, H3R2, H3R8, and H4R3 [23-26], which may in turn suppress STC-1 transcription. Our data indicated that CAPG association with PRMT5 decreased PRMT5 binding at the STC-1 promoter, resulting in increased STC-1 transcription and enhanced BC metastasis. Therefore, CAPG may promote breast cancer metastasis by epigenetically enhancing STC-1 transcription.
Results
CAPG expression is increased in BC tissues and is associated with aggressiveness
To identify genes and their parallel protein responsible for increased metastasis, RNA expression array analysis and iTRAQ-nano-HPLC-mass spectrometer (MS)/MS analysis were performed using MDA-MB-231 HM and its parental cell line MDA-MB-231. The highly metastatic MDA-MB-231(MDA-MB-231 HM) cell line was developed from parental MDA-MB-231 cells by in vivo selection in mice, which was reported previously [27]. Another highly bone-metastatic cell line MDA-MB-231BO we also used in this study was obtained from Dr. Toshiyuki Yoneda (The University of Texas, Houston, TX), which was isolated via in vivo selection and also had stronger lung metastasis capacity compared with MDA-MB-231 cells [28, 29]. Both analyses found that CAPG expression was significantly higher in MDA-MB-231 HM cells than in MDA-MB-231 cells (Figure 1A-C and Figure S1), which was further confirmed by qPCR (Figure 1D) and western blot assays (Figure 1E). To explore the role of CAPG in human BC metastasis, we first examined a panel of 7 human BC cell lines. As shown in Figure 1E, CAPG levels in most of the non-invasive cell lines (except ZR75-1) were hardly detectable, whereas CAPG expression was substantially higher in more aggressive and metastatic cell lines, such as MDA-MB-231, MDA-MB-231 HM and MBA-MD-231 BO. We then analyzed primary BC tissues from 250 BC patients using immunohistochemistry (IHC) to determine CAPG expression (Figure 1F). The relationship between CAPG and clinical-pathological characteristics was assessed and the data are shown in Table S1. CAPG was negatively correlated with estrogen receptor (ER) (p=0.002) and positively correlated with human epidermal growth factor receptor 2 (HER2) (p=0.005), while it displayed no correlationship with other statuses. Upon Kaplan-Meier analysis, patients with high CAPG expression had a significantly worse disease-free survival (DFS; p=0.028; Figure 1G).
Increased CAPG correlates with poor prognosis in BC. (A) The human gene expression array showed that CAPG is higher in MDA-MB-231 HM cells than in MDA-MB-231 cells. Fold change represents the fold change of CAPG expression in MDA-MB231 HM cells, which was normalized by CAPG expression in MDA-MB231. (B) CAPG protein detection data from iTRAQ-nano-HPLC-MS/MS. (C) iTRAQ-nano-HPLC-MS/MS analysis of the CAPG fold change ratio in MDA-MB-231 and MDA-MB-231 HM cells. (D) qPCR analysis of CAPG mRNA in MDA-MB231 and MDA-MB231 HM cells to validate the human gene expression array result. (E) CAPG expression in seven BC cell lines, with their classification and metastatic ability (Bo: bone only; Bo*: predominantly bone; L*: predominantly lung; BaB: basal B; Lu: luminal). (F) Tissue array from 250 BC patients using immunohistochemistry to determine CAPG expression. The scale bars is 50μm. (G-I) Kaplan-Meier analysis of DFS (p=0.028), DDFS (p=0.064) and OS (p=0.170), respectively, in BC patients with differential CAPG expression levels (CAPG-: n=48, blue line; CAPG+: n=196, green line) according to the tissue microarray. *** p<0.001.
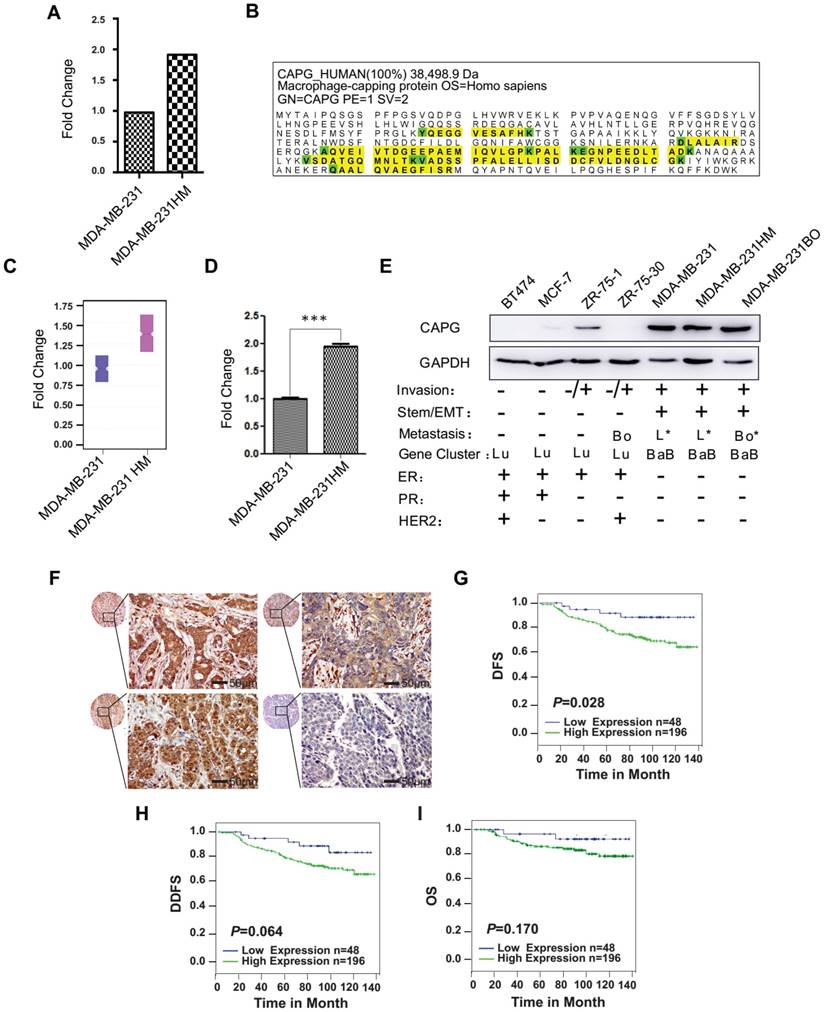
High CAPG expression also correlated with poor distant disease-free survival (DDFS; p=0.064; Figure 1H) and overall survival (OS; p=0.170; Figure 1I). These two parameters were correlated although they were not statistically significant. A univariate analysis demonstrated that high CAPG expression in cancer tissue (HR=2.963; 95% CI: 1.069-8.207; p=0.037), along with a primary BC diagnosis at 35 years of age or younger (HR=2.494; 95% CI: 1.032-6.027; p=0.042) and axillary lymph node metastasis (HR=1.410; 95% CI: 1.032-1.927; p=0.031) were significant unfavorable prognostic factors for DFS in BC patients (Table 1). Multivariate analysis also demonstrated that high CAPG expression (HR=3.514; 95% CI: 1.052-11.743; p=0.041) and primary BC diagnosis at 35 years of age or younger (HR=3.500; 95% CI: 1.152-10.624; p=0.027) were independent unfavorable prognostic factors for DFS in BC patients (Table 1). These data suggest that increased CAPG expression is associated with BC aggressiveness.
Univariate and multivariate analysis for disease-free survival in 250 cases
| Univariate analysis | Multivariate analysis | ||||
|---|---|---|---|---|---|
| HR (95% CI) | pa value | HR (95% CI) | pa value | ||
| Age | 2.494 (1.032-6.027) | 0.042 | 3.500 (1.153-10.624) | 0.027 | |
| Menopausal status | 1.084 (0.552-2.126) | 0.815 | 1.120 (0.437-2.872) | 0.814 | |
| Tumor grade | 0.893 (0.459-1.738) | 0.740 | 0.480 (0.222-1.036) | 0.061 | |
| Tumor size | 1.517 (0.984-2.337) | 0.059 | 1.282 (0.513-3.202) | 0.595 | |
| Lymph node status | 1.410 (1.032-1.927) | 0.031 | 1.212 (0.635-2.314) | 0.559 | |
| ER status | 0.665 (0.335-1.316) | 0.241 | 0.435 (0.179-1.061) | 0.067 | |
| HER-2/neu status | 0.923 (0.459-1.856) | 0.822 | 1.106 (0.476-2.572) | 0.814 | |
| CAPG | 2.963 (1.069-8.207) | 0.037 | 3.514 (1.052-11.743) | 0.041 | |
| PRMT5 | 0.302 (0.116-0.787) | 0.014 | 0.199 (0.058-0.676) | 0.010 | |
CAPG: macrophage-capping protein; PRMT5: protein arginine methyltransferase 5; ER: estrogen receptor; HER-2: human epidermal growth factor receptor 2; HR: hazard ratio; CI: confidence interval; a. p is based on the Cox regression test.
CAPG promotes BC metastasis in vitro and in vivo
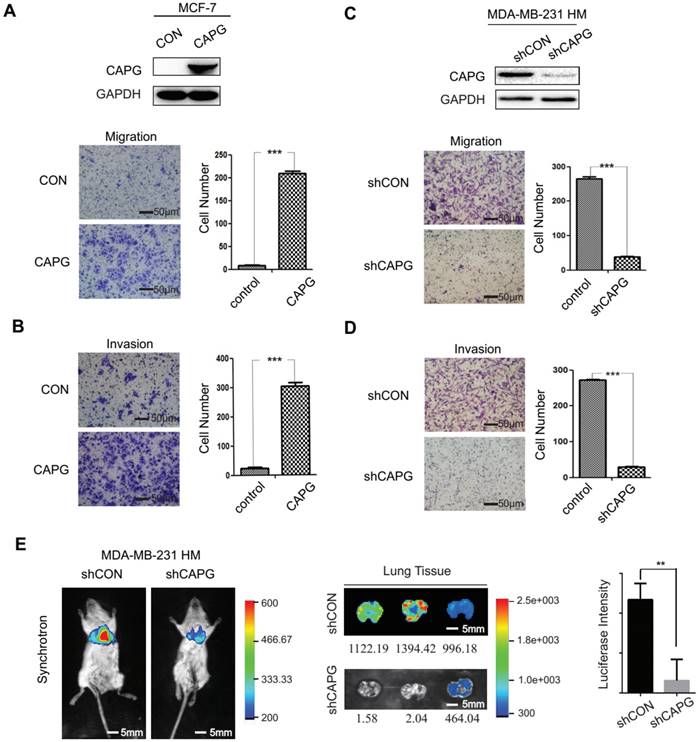
To investigate the role of CAPG in BC metastasis, we established a CAPG-overexpressing stable MCF-7 cell line (MCF-7-CAPG). CAPG level was examined by Western Blot (Figure 2A, upper). Compared with the control group, MCF-7-CAPG cells exhibited increased migration and invasion (Figure 2A-B). In parallel, CAPG-knockdown stable cell lines were established in MDA-MB-231 HM cells. CAPG was significantly knocked down in CAPG-knockdown stable cell lines (Figure 2C, upper). Migration and invasion were significantly decreased in MDA-MB-231 HM-shCAPG (Figure 2C-D) and MDA-MB-231 BO-shCAPG (Figure S2A-B) cells compared with those in their respective control cells. To further validate the potential significance of CAPG in breast cancer metastasis, we generated an orthotopic breast cancer model using MDA-MB-231 HM-shCAPG cells. After six weeks, we observed significantly reduced lung metastasis in mice transplanted with HM-shCAPG cells relative to HM-CON-transplanted mice. Quantified bioluminescent signals are shown as mean on the right of Figure 2E (p=0.0063). These data suggested that CAPG played a pro-metastatic role in BC.
CAPG promotes BC metastasis. (A) Migration and (B) invasion assays comparing MCF-7-control and MCF-7-CAPG cells after 22 h or 36 h. The scale bars is 50μm. (C) Migration and (D) invasion assays comparing MDA-BM-231 HM-control and MDA-MB-231 HM-shCAPG cells. The scale bars is 50μm. (E) Bioluminescence imaging was performed at the end point of the experiment to visualize lung metastasis. The scale bars is 5mm. Mean quantified bioluminescent signals are shown on the right. **P< 0.01.
Screening for a CAPG-modulating gene
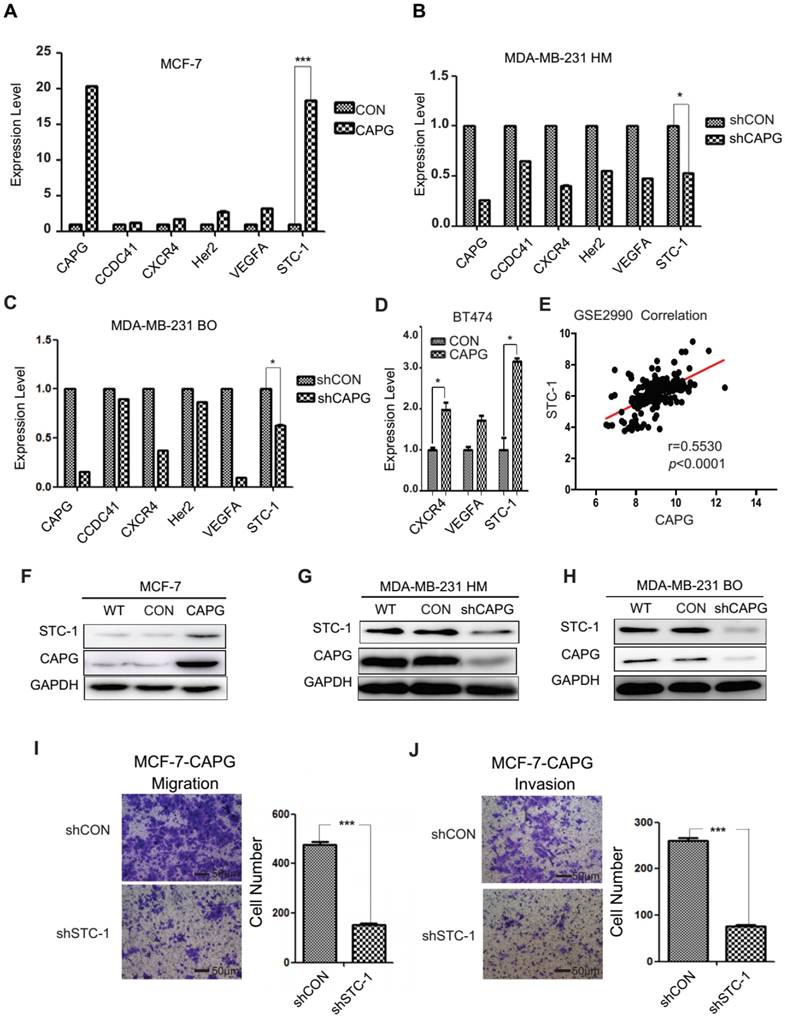
A gene expression array analysis was performed to identify genes with altered expression induced by CAPG. A total of 397 genes displayed a great change in MCF-7-CAPG stable cells compared with control cells (Figure S3A). Gene ontology (GO) enrichment analysis indicated that these genes were involved in 50 cellular processes (Figure S3B). Five metastasis-related genes were chosen to validate the array results. Expression of centrosomal protein 83 kDa (CCDC41), chemokine C-X-C motif receptor 4 (CXCR4), HER2, vascular endothelial growth factor A (VEGFA), and STC-1 were quantified by qPCR in MCF-7-CAPG, MDA-MB-231 HM-shCAPG, MDA-MB-231 BO-shCAPG cells and their respective control cells (Figure 3A-C). STC-1 mRNA levels were remarkably increased in MCF-7-CAPG cells (Figure 3A) and decreased in the two shCAPG cell lines compared with those in their parental cells (Figure 3B-C). Then, we tried the BT474 cells, the other luminal breast cancer cell line: VEGFA was not upregulated by CAPG transfection; but, CXCR4 was found to be upregulated after CAPG transfection (Figure 3D). To further determine the exact genes regulated by CAPG, the GSE database was used for the correlation analysis. Consistently, CAPG mRNA level positively correlated with STC-1 gene (p<0.0001, r=0.5530) in breast cancer samples from the GSE2990 database (Figure 3E). No positive correlation was found between CAPG and the other two genes (data not shown). In support of these observations, we believed that STC-1 was the main factor regulated by CAPG. Consistent STC-1 protein level changes were also observed in these paired cell lines (Figure 3F-H). Additionally, knockdown of STC-1 led to decreased migration and invasion in MCF-7-CAPG cells (Figure 3I-J). These results indicate that STC-1 may be the main downstream factor of CAPG.
Screening for CAPG target genes. (A-C) PCR analysis of 5 genes from the gene expression array in MCF-7-CAPG, MDA-MB-231 HM-shCAPG and MDA-MB-231 BO-shCAPG cells with control cells. (D) PCR analysis of CXCR4, VEGFA and STC-1 in control and CAPG transiently transfected BT474 cells. (E) Pearson analysis of gene expression data from breast cancer patients (GSE2990) was used for depicting the correlation between CAPG and STC-1. (F) MCF-7 cells were transfected with control or CAPG, as shown. STC-1 expression was determined by Western blot. GAPDH was used as loading control. (G-H) MDA-MB231 HM and BO cells were transfected with control or shCAPG, as shown. STC-1 expression was determined by Western blot. GAPDH was used as loading control. (I) Migration and (J) invasion assays for the analysis of control and STC-1 knockdown treatments in MCF-7 CAPG cells. The scale bars is 50μm. * p<0.05, *** p<0.001.
CAPG modulates STC-1 transcription
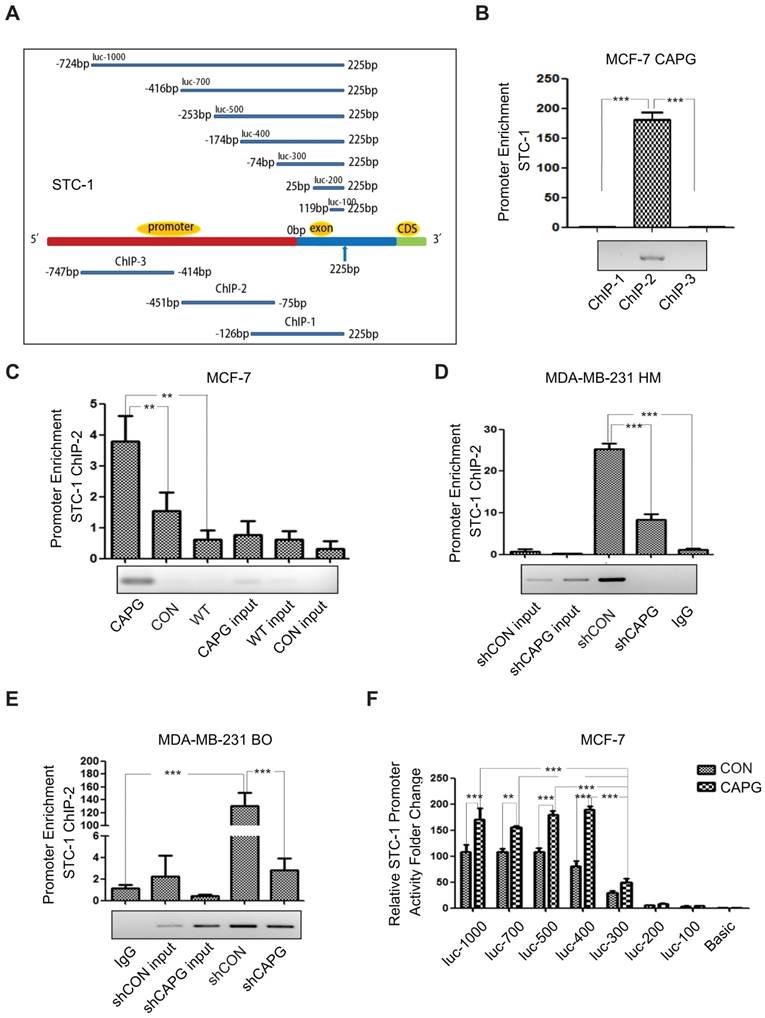
As our data suggested that STC-1 mRNA expression was enhanced by CAPG (Figure 3A), and a domain within CAPG shares homology with members of bHLH transcription regulators [9], we examined whether CAPG was involved in STC-1 transcriptional regulation. To this end, a chromatin immunoprecipitation (ChIP) assay was conducted to determine whether CAPG was able to bind to the STC-1 promoter. Three pairs of ChIP PCR primers corresponding to the STC-1 promoter were designed, each spanning an approximately 350-bp fragment within the 1000-bp STC-1 promoter (Figure 4A, lower). We found that a fragment spanning the positions -451 bp to -75 bp (ChIP-2) of the STC-1 promoter was enriched in CAPG-precipitated chromosomal DNA in MCF-7-CAPG cells (Figure 4B). Compared with MCF-7 cells or MCF-7 control cells, CAPG recruitment to the STC-1 promoter region (-451 bp to -75 bp) was substantially increased when CAPG was transiently transfected (Figure 4C). In contrast, decreased CAPG recruitment to the region (-451 bp to -75 bp) was detected in MDA-MB-231 HM-shCAPG cells (Figure 4D) and MDA-MB-231 BO-shCAPG cells (Figure 4E). In order to examine the exact main region for CAPG binding, STC-1 promoter fragment mutants with different lengths were constructed. They are shown schematically in Figure 4A, upper. The dual-luciferase reporter assay was employed to identify the exact promoter region for CAPG binding. We found that the -174 bp to -74 bp locus was the main binding region of CAPG, as the activity of luc-300 mutant was dramatically decreased in MCF-7 cells (Figure 4F) and human embryonic kidney (HEK) 293T cells (Figure S4). Together, our data indicate that CAPG may enhance STC-1 transcription by binding to its promoter region, and the main region responsible for CAPG binding was the -174 bp to -74 bp locus in the STC-1 promoter.
CAPG is involved in the activation of STC-1 transcription. (A) The primer design scheme for the STC-1 promoter and its fragment mutant in ChIP assay and luciferase assay. (B) qPCR-ChIP analysis of CAPG binding to different-length fragments of the STC-1 promoter. (C) qPCR-ChIP analysis of CAPG binding to the STC-1 ChIP-2 promoter in MCF-7-CAPG cells (column 1, transfected pCMV-CAPG), MCF-7-CON cells (column 2, transfected pCMV-FLAG control) and wild-type (WT) MCF-7 cells (column 3, wild-type cells without transfection). (D) qPCR-ChIP analysis of CAPG binding to the STC-1 ChIP-2 promoter in MDA-MB-231 HM-control (column 3) and MDA-MB-231 HM-shCAPG (column 4) cells and control IgG (column 5). (E) qPCR-ChIP analysis of CAPG binding to the STC-1 ChIP-2 promoter in MDA-MB-231 BO-control (column 4) and MDA-MB-231 BO-shCAPG (column 5) cells and control IgG (column 1). (F) Dual-luciferase reporter assay analyzing luciferase activity in MCF-7 cells transiently co-transfected with FLAG-CAPG or FLAG along with Renilla luciferase and various STC-1 promoter fragments expressed from the pGL3-basic reporter vector, as indicated. * p<0.05, ** p<0.01, *** p<0.001.
CAPG directly interacts with PRMT5
To explore how CAPG regulates STC-1 transcription, we performed a proteomic screen of CAPG-associated immune complexes. We ectopically expressed HA-CAPG in HEK 293T cells and then used HA beads to capture CAPG-binding proteins. The immunoprecipitated protein complexes were visualized by silver staining (Figure 5A) and analyzed by mass spectrometry to determine CAPG-associated proteins. Interestingly, PRMT5 was identified as a CAPG-associated protein in this complex (the exact band was marked by an arrow). PRMT5 belongs to the arginine methyltransferase type II family of PRMTs that catalyze the formation of symmetrical dimethylarginine (ù-NG,N'G-dimethylarginine or SDMA) [30, 31], which has been reported to regulate gene transcription [32].
PRMT5 interacts with CAPG and predicts a favorable prognosis in BC. (A) Silver staining for total protein extracted by HA agarose beads in HEK 293T cells expressing HA (lane 2) or HA-CAPG (lane 3). The distinct band is marked by an arrow. (B) HA-CAPG or HA were co-expressed with FLAG-PRMT5 or FLAG in HEK 293T cells. An anti-FLAG antibody was used to precipitate HA-associated proteins (rows 3, 4). An anti-HA antibody was used to precipitate FLAG-associated proteins (rows 5, 6). (C) HA-CAPG and FLAG-PRMT5 were expressed separately in MDA-MB-231 HM cells. An anti-HA antibody was used to precipitate CAPG-associated proteins, and an anti-FLAG antibody was used to precipitate PRMT5-associated proteins. Antibodies against CAPG and PRMT5 were used to probe for endogenous CAPG (row 3) and PRMT5 (row 4) in MDA-MB-231 HM cell extracts. (D) The endogenous binding proteins of CAPG and PRMT5. An anti-CAPG antibody was used to precipitate endogenous CAPG-associated proteins (lane 4), and an anti-PRMT5 antibody was used to precipitate endogenous PRMT5-associated proteins (lane 5). (E) The purified full-length GST-PRMT5 was used to bind full-length His-CAPG. An anti-His antibody was used to probe for His-tagged proteins by Western blotting. (F) Migration and invasion assays in MDA-BM-231 HM-control and MDA-MB-231 HM-shPRMT5 cells after 10 h or 18 h. The scale bars is 50μm. (G) Migration and invasion assays in MDA-BM-231 HM-control and MDA-MB-231 HM-PRMT5 cells after 18 h or 30 h. (H-I) Kaplan-Meier analysis of DFS (p=0.009) and DDFS (p=0.013) associated with different PRMT5 expression levels (PRMT5-: n=92, blue line; PRMT5+: n=152, green line) based on the BC tissue microarray. ** p<0.01, *** p<0.001.
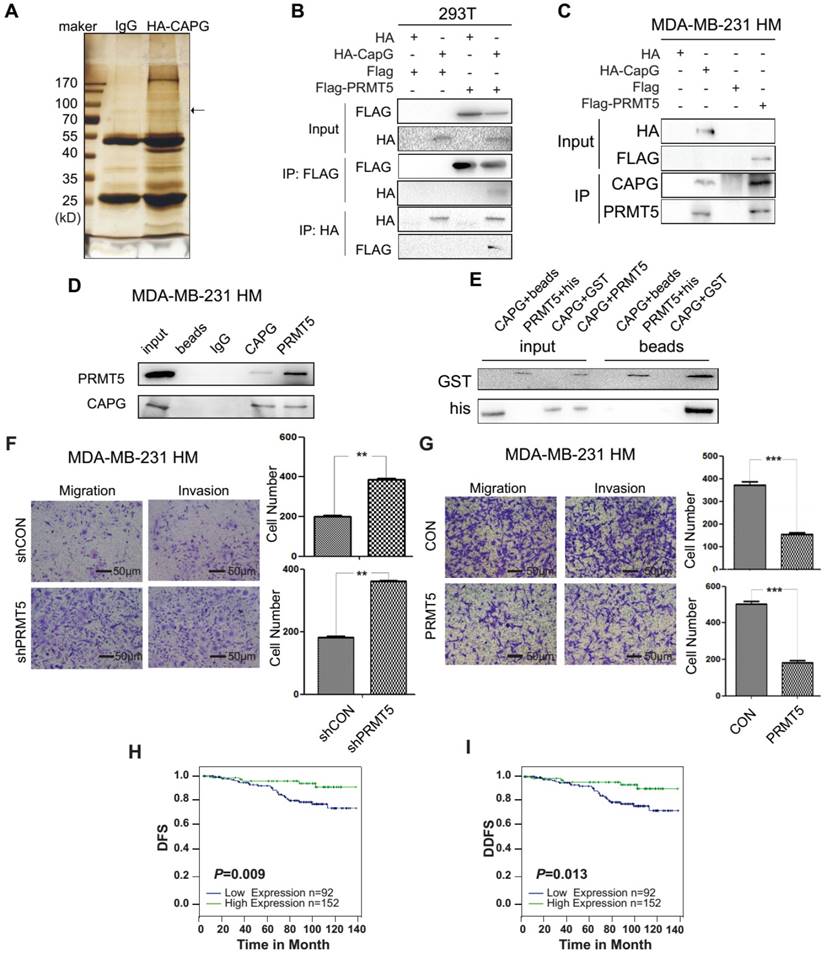
To validate the interaction between CAPG and PRMT5, HEK 293T cells were co-transfected with HA-CAPG and FLAG-PRMT5. CAPG and PRMT5 were found to interact with each other in reciprocal Co-IP experiments (Figure 5B). Similar results were also observed in MDA-MB-231 HM cells transfected with either HA-CAPG or FLAG-PRMT5 (Figure 5C). This interaction was further confirmed between endogenous CAPG and PRMT5 in MDA-MB-231 HM cells (Figure 5D). To determine whether CAPG could directly interact with PRMT5, we generated recombinant His-CAPG and GST-PRMT5 proteins in bacteria (Figure S5A). Our in vitro pull-down assay indicated that CAPG directly bound with PRMT5 (Figure 5E).
PRMT5 is a marker for favorable prognosis in BC and decreases BC metastasis
To evaluate the role of PRMT5 in BC metastasis, we depleted PRMT5 in MDA-MB-231 HM cells using shRNA. We found that depletion of PRMT5 increased the migration and invasion of MDA-MB-231 HM cells (Figure 5F). In contrast, cancer cell migration and invasion were decreased by PRMT5 overexpression in MDA-MB-231 HM cells (Figure 5G). These data indicated that PRMT5 played a metastasis-suppressive role in BC.
We then determined PRMT5 expression in the same tissue microarray used for analyzing CAPG expression. We found that BC patients with high PRMT5 expression displayed improved DFS (p=0.009; Figure 5H) and DDFS (p=0.013; Figure 5I), although OS was not significantly altered (p=0.117; Figure S6). Both the univariate and multivariate analyses revealed that in addition to CAPG expression, high PRMT5 expression (univariate HR=0.302, 95% CI: 0.116-0.787, p=0.014; multivariate HR=0.199, 95% CI: 0.058-0.676, p=0.010) was an independent prognostic factor for DFS in BC patients (Table 1).
PRMT5 represses CAPG-dependent STC-1 induction through interfering with PRMT5-induced methylation of histone H4
Due to the complex formation of CAPG and PRMT5, we further investigated whether PRMT5 could regulate STC-1 expression. In MDA-MB-231 HM cells, PRMT5 depletion by shRNA led to increased STC-1 protein and mRNA levels (Figure 6A-B). Furthermore, PRMT5 inhibited STC-1 protein level in MCF-7-CAPG cells (Figure 6C). Also, increased PRMT5 mitigated the increases of STC-1 mRNA level increased by CAPG in MCF-7 cells (Figure 6D). To validate the functional association between CAPG and PRMT5 in regulating STC-1 transcription, we measured STC-1 promoter-driven trans-activation with a reporter assay. In MCF-7 cells, CAPG-enhanced STC-1 promoter transcription activity was repressed by PRMT5 (Figure 6E).
PRMT5 inhibits STC-1 transcription. (A) Total cell extracts of the shPRMT5 stable cells, wild type cells (WT) and its corresponding control cells (shCON) were analyzed by immunoblotting with the indicated antibodies. (B) qPCR analysis of STC-1 mRNA in the shPRMT5-stable cells, WT and shCON groups *P<0.05. (C-D) STC-1 protein and mRNA levels in MCF-7 cells CPAG co-transfected with pWPI or with PRMT5. (E) Dual-luciferase reporter assay analysis of luciferase activity in MCF-7 cells, which were transiently co-transfected with pCMV-C-FLAG-CAPG or pCMV-C-FLAG and pcDNA3.0-HA-PRMT5 or pcDNA3.0-HA, Renilla luciferase and the pGL3-basic STC-1 -174 bp to 225 bp promoter vector. (F) qPCR-ChIP analysis of PRMT5 binding to the STC-1 ChIP-2 promoter in MDA-MB-231 HM cells. (G) Re-ChIP analysis of CAPG-PRMT5 corecruitment to the STC-1 promoter region was carried out in MDA-MB-231 BO cells. Re-ChIP analysis of CPRMT5 (column 1) and rabbit IgG (column 2) recruitment in the cross-linking protein-DNA complex eluted from CAPG. Re-ChIP analysis of CAPG (column 3) and mouse IgG (column 4) recruitment in the cross-linking protein-DNA complex eluted from PRMT5. * p<0.05, ** p<0.01, *** p<0.001.
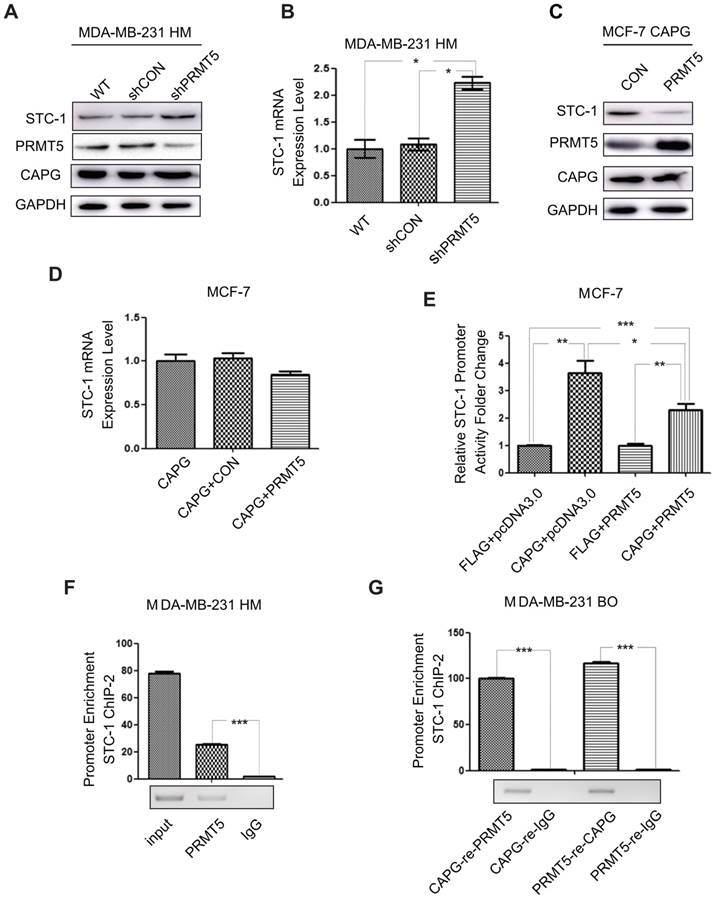
To assess whether PRMT5 regulates STC-1 transcription by binding to the same promoter region as CAPG, we performed ChIP and re-ChIP assays. In MDA-MB-231 HM cells, we found that PRMT5 was recruited to the same STC-1 promoter region (-451 bp to -75 bp) where CAPG bound (Figure 6F). Furthermore, ChIP-reChIP assay using sequential IP with CAPG and PRMT5 antibodies confirmed the co-recruitment of CAPG and PRMT5 to the same region of the STC-1 promoter (-451 bp to -75 bp) in MDA-MB-231 BO cells (Figure 6G).
Interestingly, PRMT5 overexpression in MCF-7-CAPG cells was able to decrease CAPG binding to the STC-1 promoter region (Figure 7A), which was also confirmed in MDA-MB-231 HM and MDA-MB-231 BO cells (Figure S7A and Figure 7B). Reciprocally, CAPG overexpression decreased PRMT5 binding to the STC-1 promoter in MCF-7-CAPG cells (Figure 7C). Moreover, the CAPG-induced decrease in PRMT5 binding to the STC-1 promoter was not through changing PRMT5 protein levels. The overexpression or down-regulation of CAPG did not affect PRMT5 protein levels (Figure S5B). Furthermore, we also detected the recruitment of PRMT5 in the STC1 promoter when CAPG was knocked down and the recruitment of CAPG in the STC1 promoter when PRMT5 was knocked down. The data showed enhanced protein recruitment by PRMT5 to the STC1 promoter along with decreased levels of CAPG, and CAPG (Figure 7D-E). These results suggest that PRMT5 and CAPG may mutually antagonize each other for binding at the STC-1 promoter, thereby modulating STC-1 expression to reach a dynamic balance.
CAPG competes with PRMT5 to bind the exact location of the STC-1 promoter. (A-B) qPCR-ChIP analysis of CAPG binding to the STC-1 ChIP-2 promoter in MCF-7-CAPG cells and MDA-BM-231 BO transfected with pWPI or with pWPI-PRMT5 separately. (C) qPCR-ChIP analysis of PRMT5 binding to the STC-1 ChIP-2 promoter in MCF-7 cells transfected with pCDH (column 3) or with pCDH-CAPG (column 4). (D) ChIP analyses of PRMT5 recruitment to the STC-1 promoter region was carried out in CAPG knocked down MDA-MB-231 BO cells. (E) ChIP analysis of CAPG recruitment to the STC-1 promoter region was carried out in PRMT5 knocked down MDA-MB-231 BO cells. (F) Purified GST and full-length GST-PRMT5 were incubated with core histone and His or His-CAPG for 2 h. Proteins were confirmed by Coomassie staining (input). An anti-histone H4 antibody was used to probe PRMT5-associated histone H4 via Western blotting (IP). (G) In vitro histone methylation (HMT) assays for histone H4. GST (lane 1), GST-PRMT5 (lane 2), GST-PRMT5 and His (lane 3), or GST-PRMT5 and His-CAPG (lane 4) were incubated with AdoMet SAM in HMT buffer and detected by Western blot analysis. (H-I) Kaplan-Meier analysis of DFS (p=0.034) and DDFS (p=0.026) associated with different CAPG plus PRMT5 statuses in 241 patients. CAPG-/PRMT5- patients: n=33; CAPG-/PRMT5+ patients: n=15; CAPG+/PRMT5- patients: n=117; CAPG+/PRMT5+ patients: n=76. (J) The schematic diagram shows the mechanism by which CAPG and PRMT5 regulate STC-1 transcription. * p<0.05, *** p<0.001.
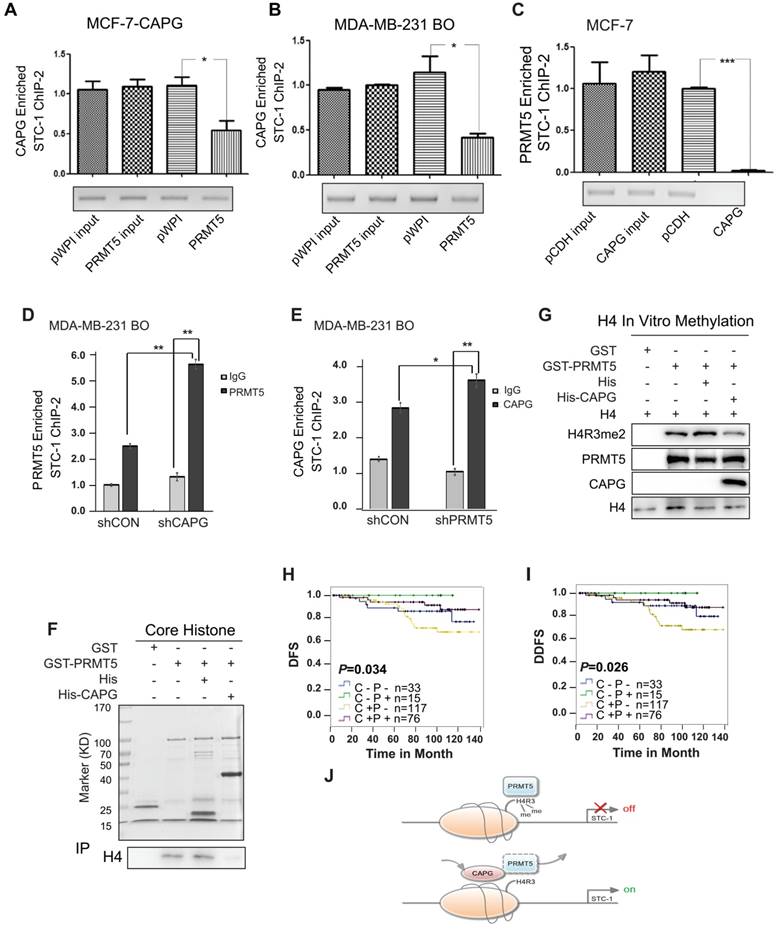
PRMT5 was previously shown to mediate the symmetric dimethylation of arginine residues within histones (e.g., H2AR3me2s, H3R2me2s, H3R8me2s, and H4R3me2s) [24-26]. Using re-ChIP assays, we found that depletion of CAPG increased the co-enrichment between PRMT5 with H4R3me2 but not H3R8me2 at the STC-1 promoter in MDA-MB-231 BO cells (Figure S7B). GST pull-down experiments further demonstrated that CAPG directly interfered with the binding between PRMT5 and histone H4 (Figure 7F). Consistently, we found that CAPG repressed PRMT5-dependent methylation of histone H4R3 using an in vitro histone methyltransferase (HMT) assay (Figure 7G).
For prognosis, both CAPG and PRMT5 are independent prognostic factors for the DFS of BC patients. However, CAPG predicts an unfavorable prognosis, whereas PRMT5 predicts a favorable one. We then analyzed whether CAPG was associated with PRMT5 expression in 241 patients. As expected, CAPG-positive and PRMT5-negative patients had the worst DFS and DDFS (n=117), whereas CAPG-positive and PRMT5-positive patients had a better DFS and DDFS (n=76). Patients who were CAPG negative and PRMT5 positive (n=15) showed the best DFS (p=0.034) and DDFS (p=0.026; Figure 7H-I).
Discussion
In previous studies, CAPG has been implicated in promoting tumor metastasis [4, 8, 33-37]. However, the underlying mechanisms are not completely understood. It was proposed that CAPG may increase metastasis via regulating the cytoskeleton in the cytoplasm. For cells to move or change shape, actin filaments must be correctly assembled, arranged and localized [7], which depend on CAPG-mediated 5' capping of actin. This capping activity is activated by Ca2+ and inhibited by phosphatidylinositol 4,5-bisphosphate [3]. A previous study also found that a CAPG nanobody blocks the interaction between CAPG and actin monomers or actin filaments to prevent BC cell metastasis [36]. We also found that actin was among the CAPG-interacting proteins identified by MS (data not shown). Another pro-metastasis mechanism may involve CAPG-participated signaling pathways. The reduction in CAPG expression in human pulmonary artery smooth muscle cells (hPAMSCs) was accompanied by significantly impaired migration in vitro, particularly under hypoxia [35]. However, this finding needs validation.
In this study, we discovered a novel mechanism by which CAPG enhances metastasis by regulating transcription of STC-1, a metastasis-related gene. We found that STC-1 enhanced BC metastasis, which is consistent with other studies [18, 38]. STC-1 may promote metastasis by activating the PI3K pathway rather than by increasing intracellular calcium levels [18]. In BC, STC-1 is a potential molecular marker for the detection of BC metastasis in the blood and bone marrow [20]. Additionally, with the other two biomarkers N-acetylgalactosaminyltransferase (GalNacT) and melanoma antigen gene family-A3 (MAGE-A3), the determination of STC-1 levels may be an efficient method to assess the presence of circulating tumor cells that induce tumor metastasis to the axillary lymph nodes [21]. Previous studies found that STC-1 transcription was elicited by p53 and NF-κB, whereas expression was repressed by histone deacetylase (HDAC) [39, 40], specificity protein 1 (Sp1) and retinoblastoma protein (Rb) [41]. Here, we demonstrated that CAPG could bind to STC-1 promoter at -451 bp to -75 bp. This binding activated STC-1 transcription. Moreover, we found PRMT5 also bound to the same STC-1 promoter sequence, but elicited the opposite effect. PRMT5 repressed STC-1 promoter activation.
PRMT5 methylates arginines in both histones and other cellular proteins, such as p53, SPT5, and epidermal growth factor receptor (EGFR) [42-44], indicating that it is involved in multiple cellular processes such as signal transduction, histone modification, chromatin remodeling, transcriptional silencing and activation, and protein synthesis [31, 45-47]. Many proteins, such as RIO kinase 1 (RioK1) [48], chloride nucleotide-sensitive channel 1A (pICLn) [48], methyl-CpG binding domain protein (MBD) [49], WD repeat domain 77 (MEP50) [50], DNA methyltransferase 3a (DNMT3A) [30], cyclin D1 [51, 52] have been shown to function as partners of PRMT5 to regulate these cellular processes. PRMT5 depletion accelerates the growth of some melanoma cell lines (including A375 and MeWo) [53]. Moreover, PRMT5 also can attenuate GATA4 transcription [54]. GATA4 closely correlates with tumor recurrence and acts as an independent factor that predicts unfavorable prognosis in BC patients [23]. PRMT5 also methylates EGFR on Arg 1175. Abolishment of EGFR Arg 1175 methylation may enhance EGF-stimulated extracellular signal-regulated kinases (ERK) activation by reducing SHP1 recruitment to EGFR, resulting in augmented cell proliferation, migration and invasion of EGFR-expressing cells [44]. In our study, we found that PRMT5 repressed the invasiveness of BC cells and acted as an independent favorable clinical prognostic factor in BC patients. This effect is likely achieved through the PRMT5-mediated transcriptional repression of the oncogene STC-1. Our data demonstrated that both CAPG and PRMT5 bound to the same chromatin region in the STC-1 promoter and that CAPG repressed the binding of PRMT5 to the STC-1 promoter. Consistently, CAPG interrupted the binding of PRMT5 to histone H4 in the core histone protein complex and decreased PRMT5-dependent symmetrical methylation of H4R3. This dimethylation may induce transcriptional repression of STC-1, as H4R3me2s serve as a direct binding site for DNMT3A, leading to the subsequent DNA methylation of CpG dinucleotides [30].
In conclusion, we here show that CAPG promotes tumor invasion and correlates with a poor prognosis in BC. The underlying mechanism reveals CAPG-dependent transcript regulation of STC-1. CAPG suppressed PRMT5 binding to the STC-1 gene promoter, which decreased PRMT5-dependent histone H4R3 methylation and subsequent decrease of CpG DNA methylation, resulting in enhanced STC-1 transcription (Figure 7I). Therefore, CAPG and PRMT5 expression may be a promising biomarker to determine prognosis in BC. Furthermore, silencing the oncogene CAPG and enhancing PRMT5-mediated histone H4R3me2 and CpG DNA methylation, thereby inhibiting STC-1 expression, may serve as a promising strategy to mitigate BC progression.
Methods
Cell lines, culture, plasmids and transfection
MCF-10A, MCF-7, ZR-75-30, ZR-75-1, 293T and MDA-MB-231 cells were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA) and cultured according to ATCC instructions. The high-metastasis (HM) MDA-MB-231 HM cell line was derived from the parental MDA-MB-231 cell line, as established by our institute [55]. The high-osteolytic-metastasis (BO) MDA-MB-231 BO cell line was obtained as a generous gift from Dr. Toshiyuki Yoneda (The University of Texas, USA) [28, 29]. MDA-MB-231 HM and MDA-MB-231 BO cells were cultured similarly to MDA-MB-231 cells [27].
pCMV6-entry-CAPG-MYC-FLAG and pCMV6-entry were purchased from OriGene Technologies, Inc. pcDNA3-HA and pCMV-FLAG were purchased from Beyotime Biotechnology, Inc. pCDH-puro, pET28a, pGEX-4T-1 and pWPI-puro were purchased from Addgene, Inc. pGL3-Basic was purchased from Promega. The pLKO.1-TRC cloning vector was a gift from David Root [56]. The shRNA sequences for shCAPG, shPRMT5 and shSTC-1 are provide in Table S2. All other vectors were constructed by our lab, and the strategies are provided in Table S3. Transfection and lentiviral particles generation were performed as previously described [57, 58].
RNA, gene array and real-time PCR
Total RNA was extracted from cells or tissues using the TRIzol reagent (Invitrogen Life Technologies, Inc., Canada). RT- and qPCR were performed as previously described [59]. Primers for qPCR are provided in Table S4. Gene expression in MCF-7 CAPG-stable cells and its control cells was analyzed using an Agilent PrimeView human gene expression array containing 36,000 gene transcripts and variants via UniGene annotation (Affymetrix, Canada).
Protein purification, immunoprecipitation and Western blotting
The recombinant plasmids pET28a-His-CAPG and pGEX-4T-1-GST-PRMT5 were individually transformed into BL21(DE3) cells and incubated for 16 h on LB agar plates at 37 °C. Selected colonies were extracted and cultured in LB medium, followed by induction with 0.1 mM IPTG upon reaching an OD at 600 nm of 0.4. The transformed E. coli were lysed via sonication (VCX130/VCX130PB, Sonics & Materials, Inc., USA). The supernatant was collected after centrifugation at 10,000 ×g at 4 °C for 20 min. The supernatant was also passed through a Ni-NTA His-tag high-performance nickel-Sepharose column (GE Healthcare, Sweden). The GST and GST-tagged proteins were purified from bacterial lysates using glutathione-agarose beads (Amersham Biosciences). Purified core histone protein was purchased from Millipore (Millipore, Merck KGaA, Darmstadt, Germany).
Protein immunoprecipitation, GST pull-downs, and Western blotting were performed as described previously [57, 60, 61]. The following antibodies were used: anti-CAPG, anti-STC-1 (Santa Cruz Biotechnology, Inc., USA); mouse anti-FLAG (Sigma-Aldrich, USA); anti-PRMT5, rabbit anti-HA, mouse anti-HA, anti-His, rabbit anti-FLAG, anti-H3R8me2s, and anti-H4R3me2s (Abcam Inc., USA); anti-H4 (Millipore, Germany); and anti-GAPDH (ProteinTech Group, USA).
Migration, invasion assay and luciferase assay
Migration and invasion assays were performed as previously described [57]. The cells were incubated at 37 °C for different times according to their invasive ability. Luciferase reporter assays were performed as previously described [58].
Animals
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of Fudan University. The protocol was approved by the Committee on the Ethics of Animal Experiments of Fudan University (Permit Number, SYXK 2012-0001). Female NOD scid gamma mice (6 weeks old) were maintained in Shanghai Medical College of Fudan University and housed in standard animal cages under specific pathogen-free conditions in the college's animal facility. 2×106 MDA-MB-231 HM parental or shCAPG stable cells were injected into the mammary fat pads (1 per mouse, 5 mice per group) with a total of two groups. Six weeks later, after excising the orthotopic tumors, bioluminescence imaging of mice and the lung tissues was performed to visualize the lung metastasis by Bruker In-Vivo FX PRO System (Bruker company, Germany) and analyzed by Bruker MI software.
Statistical analysis
All in vitro experiments were performed at least three times.
Comparisons between two groups were performed using unpaired two-sided t-tests. The results are reported as the mean ± standard deviation (SD) or ± standard error of the mean (SEM) unless otherwise noted.
The follow-up period was defined as the time from surgery to the last observation for the censored cases or relapse/death for the complete observations. The categories analyzed for DFS were the first recurrence of disease at a local, regional, or distant site; the diagnosis of contralateral BC; and breast-cancer-specific death. All of the categories that are listed above were considered DFS events. Those patients with a study end date and a loss of follow-up were considered censored. Correlations between the clinicopathological parameters and markers of interest were evaluated using contingency tables and Pearson's χ2 or Fisher's exact tests. The DFS probability was derived from the Kaplan-Meier estimate and compared using the log-rank test. Univariate and multivariate analyses were performed using the Cox risk proportion model. Statistical analyses were performed using SPSS (version 13.0; SPSS Co., USA). All of the p values are two-sided, and p values of less than 0.05 were considered significant. All of the analyses were based on the observed data with the assumption that missing data were random.
iTRAQ-nano-HPLC-MS/MS analysis
The cell lysates were quantified and labeled with iTRAQ labeling reagents (Applied Biosystems). The trypsin-digested peptides were fractionated and separated using nanoscale high-performance liquid chromatography (nano-HPLC) (Eksigent Technologies) on a secondary reverse-phase (RP) analytical column. A Triple TOF 4600 MS was operated for information-dependent data acquisition mode to switch automatically between MS and tandem MS (MS/MS) acquisition. MS/MS spectra were extracted and charged state deconvoluted using an MS Data Converter from AB Sciex.
Chromatin immunoprecipitation
The procedure and principle for the ChIP assay were described previously [58]. For the ChIP-re-ChIP assay, briefly, sonicated chromatin was incubated overnight with 4 μg of anti-CAPG or anit-PRMT5. The beads were washed four times and then incubated with 50 μL of buffer containing 0.5% SDS and 0.1 M NaHCO3 for 2 h at 10 °C. The supernatant was collected after centrifugation, diluted with buffer (1 mM EDTA, 150 mM NaCl, 50 mM HEPES (pH 7.5), 0.1% SDS, 1% Triton X-100, and 0.1% sodium deoxycholate), and incubated overnight with 4 μg of anti-PRMT5, anti-CAPG, anti-H3R8, anti-H4R3, or IgG as a negative control (Millipore Corp., MA). After the samples were washed and digested by proteinase K, they were quantified by qPCR. The primers used for ChIP qPCR are provided in Table S4.
In vitro histone methyltransferase (HMT) assay
Histone H4 protein was produced by TNT®T7Quick Coupled Transcription Translation (Promega, USA) with pcDNA3-HA-H4 according to the manufacturer's protocol. For the HMT assay, with or without 4 µg of His-CAPG, 1 µg of GST-PRMT5 and 10 μL of H4 mixture were added to 600 μM AdoMet SAM incubated in 30 μL of HMT buffer (20 mM Tris (pH 8.8), 0.4 mM EDTA, 200 mM NaCl) at 30 °C for 2 h. The reaction was stopped by adding SDS loading buffer at 70 °C for 10 min, and the proteins were resolved on SDS-PAGE gels.
Patient tissue and microarray construction
Informed consent forms were signed by each participant, and appropriate ethical committee approval was obtained. For the tissue microarray, 250 primary BC tissue samples from female invasive ductal carcinoma patients (no co-morbidities reported) were randomly collected at the Department of Breast Surgery of the Fudan University Shanghai Cancer Center (FDUSCC, Shanghai, P.R. China) between 2002 and 2006. The median follow-up time was 96 months (84-141 months). All patients were female and had a median age of 53 years at the time of diagnosis. The clinicopathological characteristics are summarized in Table S1.
Immunohistochemical staining of CAPG and PRMT5
The primary antibodies used were anti-CAPG (Santa Cruz Biotechnology, Inc.; Cat#: sc-166428; 1:100 dilution) and anti-PRMT5 (Abcam, Inc.; Cat#: ab109451; 1:100 dilution). The procedure and principle for the staining ratio and intensity were described previously [59].
Abbreviations
CAPG: macrophage-capping protein; BC: breast cancer; STC-1: stanniocalcin 1; PRMT5: arginine methyltransferase 5; bHLH: basic helix-loop-helix-family DNA-binding proteins; mbh-1: myc basic motif homolog-1; PI3K: phosphoinositide 3-kinase; IHC: immunohistochemistry; ER: estrogen receptor; HER2: human epidermal growth factor receptor 2; DFS: disease-free survival; DDFS: distant disease-free survival; OS: overall survival; HR: hazard ratio; CI: confidence interval; CCDC41: centrosomal protein 83 kDa; CXCR4: chemokine C-X-C motif receptor 4; VEGFA: vascular endothelial growth factor A; ChIP: chromatin immunoprecipitation; HEK: human embryonic kidney; HMT: histone methyltransferase; hPAMSCs: human pulmonary artery smooth muscle cells; GalNacT: N-acetylgalactosaminyltransferase; MAGE-A3: melanoma antigen gene family-A3; HDAC: histone deacetylase; Sp1: specificity protein 1; Rb: retinoblastoma protein; RioK1: RIO kinase 1; pICLn: chloride nucleotide-sensitive channel 1A; MBD: methyl-CpG binding domain protein; MEP50: WD repeat domain 77; DNMT3A: DNA methyltransferase 3a; EGFR: epidermal growth factor receptor; ERK: extracellular signal-regulated kinases; nano-HPLC: nanoscale high-performance liquid chromatography.
Acknowledgements
We thank Dr. Toshiyuki Yoneda (The University of Texas) for the high-osteolytic-metastasis (BO) MDA-MB-231 BO cells. Financial Support: Supported by the National Basic Research Program of China (2010CB834305), the National Natural Scientific Foundation of China (81472456, 81528016) and the Shanghai International Fund for scientific and technological cooperation (15410724100).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
2. Redig AJ, McAllister SS. Breast cancer as a systemic disease: a view of metastasis. J Intern Med. 2013;274:113-26
3. Johnston PA, Yu FX, Reynolds GA, Yin HL, Moomaw CR, Slaughter CA. et al. Purification and expression of gCap39. An intracellular and secreted Ca2(+)-dependent actin-binding protein enriched in mononuclear phagocytes. J Biol Chem. 1990;265:17946-52
4. Watari A, Takaki K, Higashiyama S, Li Y, Satomi Y, Takao T. et al. Suppression of tumorigenicity, but not anchorage independence, of human cancer cells by new candidate tumor suppressor gene CapG. Oncogene. 2006;25:7373-80
5. Witke W, Li W, Kwiatkowski DJ, Southwick FS. Comparisons of CapG and gelsolin-null macrophages: demonstration of a unique role for CapG in receptor-mediated ruffling, phagocytosis, and vesicle rocketing. J Cell Biol. 2001;154:775-84
6. Parikh SS, Litherland SA, Clare-Salzler MJ, Li W, Gulig PA, Southwick FS. CapG(-/-) mice have specific host defense defects that render them more susceptible than CapG(+/+) mice to Listeria monocytogenes infection but not to Salmonella enterica serovar Typhimurium infection. Infect Immun. 2003;71:6582-90
7. Pollard TD, Blanchoin L, Mullins RD. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct. 2000;29:545-76
8. De Corte V, Van Impe K, Bruyneel E, Boucherie C, Mareel M, Vandekerckhove J. et al. Increased importin-beta-dependent nuclear import of the actin modulating protein CapG promotes cell invasion. J Cell Sci. 2004;117:5283-92
9. Prendergast GC, Ziff EB. Mbh 1: a novel gelsolin/severin-related protein which binds actin in vitro and exhibits nuclear localization in vivo. EMBO J. 1991;10:757-66
10. Oma Y, Harata M. Actin-related proteins localized in the nucleus: from discovery to novel roles in nuclear organization. Nucleus. 2011;2:38-46
11. Nurnberg A, Kitzing T, Grosse R. Nucleating actin for invasion. Nat Rev Cancer. 2011;11:177-87
12. de Lanerolle P, Serebryannyy L. Nuclear actin and myosins: life without filaments. Nat Cell Biol. 2011;13:1282-8
13. Wagner GF, Hampong M, Park CM, Copp DH. Purification, characterization, and bioassay of teleocalcin, a glycoprotein from salmon corpuscles of Stannius. Gen Comp Endocrinol. 1986;63:481-91
14. Chang AC, Janosi J, Hulsbeek M, de Jong D, Jeffrey KJ, Noble JR. et al. A novel human cDNA highly homologous to the fish hormone stanniocalcin. Mol Cell Endocrinol. 1995;112:241-7
15. Olsen HS, Cepeda MA, Zhang QQ, Rosen CA, Vozzolo BL, Wagner GF. Human stanniocalcin: a possible hormonal regulator of mineral metabolism. Proc Natl Acad Sci U S A. 1996;93:1792-6
16. Jellinek DA, Chang AC, Larsen MR, Wang X, Robinson PJ, Reddel RR. Stanniocalcin 1 and 2 are secreted as phosphoproteins from human fibrosarcoma cells. Biochem J. 2000:350 Pt 2: 453-61
17. Pena C, Cespedes MV, Lindh MB, Kiflemariam S, Mezheyeuski A, Edqvist PH. et al. STC1 expression by cancer-associated fibroblasts drives metastasis of colorectal cancer. Cancer Res. 2013;73:1287-97
18. Murai R, Tanaka M, Takahashi Y, Kuribayashi K, Kobayashi D, Watanabe N. Stanniocalcin-1 promotes metastasis in a human breast cancer cell line through activation of PI3K. Clin Exp Metastasis. 2014;31:787-94
19. Song H, Xu B, Yi J. Clinical significance of stanniocalcin-1 detected in peripheral blood and bone marrow of esophageal squamous cell carcinoma patients. J Exp Clin Cancer Res. 2012;31:35
20. Wascher RA, Huynh KT, Giuliano AE, Hansen NM, Singer FR, Elashoff D. et al. Stanniocalcin-1: a novel molecular blood and bone marrow marker for human breast cancer. Clin Cancer Res. 2003;9:1427-35
21. Nakagawa T, Martinez SR, Goto Y, Koyanagi K, Kitago M, Shingai T. et al. Detection of circulating tumor cells in early-stage breast cancer metastasis to axillary lymph nodes. Clin Cancer Res. 2007;13:4105-10
22. Tamura S, Oshima T, Yoshihara K, Kanazawa A, Yamada T, Inagaki D. et al. Clinical significance of STC1 gene expression in patients with colorectal cancer. Anticancer Res. 2011;31:325-9
23. Takagi K, Moriguchi T, Miki Y, Nakamura Y, Watanabe M, Ishida T. et al. GATA4 immunolocalization in breast carcinoma as a potent prognostic predictor. Cancer Sci. 2014;105:600-7
24. Eckert D, Biermann K, Nettersheim D, Gillis AJ, Steger K, Jack HM. et al. Expression of BLIMP1/PRMT5 and concurrent histone H2A/H4 arginine 3 dimethylation in fetal germ cells, CIS/IGCNU and germ cell tumors. BMC Dev Biol. 2008;8:106
25. Migliori V, Muller J, Phalke S, Low D, Bezzi M, Mok WC. et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat Struct Mol Biol. 2012;19:136-44
26. Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol. 2004;24:9630-45
27. Jiang H, Sun HF, Gao SP, Li LD, Huang S, Hu X. et al. SSBP1 Suppresses TGF-beta-Driven Epithelial-to-Mesenchymal Transition and Metastasis in Triple-Negative Breast Cancer by Regulating Mitochondrial Retrograde Signaling. Cancer Res. 2015
28. Hiraga T, Kizaka-Kondoh S, Hirota K, Hiraoka M, Yoneda T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007;67:4157-63
29. Stark AM, Anuszkiewicz B, Mentlein R, Yoneda T, Mehdorn HM, Held-Feindt J. Differential expression of matrix metalloproteinases in brain- and bone-seeking clones of metastatic MDA-MB-231 breast cancer cells. J Neurooncol. 2007;81:39-48
30. Zhao Q, Rank G, Tan YT, Li H, Moritz RL, Simpson RJ. et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol. 2009;16:304-11
31. Karkhanis V, Hu YJ, Baiocchi RA, Imbalzano AN, Sif S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem Sci. 2011;36:633-41
32. Fabbrizio E, El Messaoudi S, Polanowska J, Paul C, Cook JR, Lee JH. et al. Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 2002;3:641-5
33. Van den Abbeele A, De Corte V, Van Impe K, Bruyneel E, Boucherie C, Bracke M. et al. Downregulation of gelsolin family proteins counteracts cancer cell invasion in vitro. Cancer Lett. 2007;255:57-70
34. Thompson CC, Ashcroft FJ, Patel S, Saraga G, Vimalachandran D, Prime W. et al. Pancreatic cancer cells overexpress gelsolin family-capping proteins, which contribute to their cell motility. Gut. 2007;56:95-106
35. Zhang R, Zhou L, Li Q, Liu J, Yao W, Wan H. Up-regulation of two actin-associated proteins prompts pulmonary artery smooth muscle cell migration under hypoxia. Am J Respir Cell Mol Biol. 2009;41:467-75
36. Van Impe K, Bethuyne J, Cool S, Impens F, Ruano-Gallego D, De Wever O. et al. A nanobody targeting the F-actin capping protein CapG restrains breast cancer metastasis. Breast Cancer Res. 2013;15:R116
37. Glaser J, Neumann MH, Mei Q, Betz B, Seier N, Beyer I. et al. Macrophage capping protein CapG is a putative oncogene involved in migration and invasiveness in ovarian carcinoma. Biomed Res Int. 2014;2014:379847
38. Chang AC, Doherty J, Huschtscha LI, Redvers R, Restall C, Reddel RR. et al. STC1 expression is associated with tumor growth and metastasis in breast cancer. Clin Exp Metastasis. 2015;32:15-27
39. Ching LY, Yeung BH, Wong CK. Synergistic effect of p53 on TSA-induced stanniocalcin 1 expression in human nasopharyngeal carcinoma cells, CNE2. J Mol Endocrinol. 2012;48:241-50
40. Law AY, Lai KP, Lui WC, Wan HT, Wong CK. Histone deacetylase inhibitor-induced cellular apoptosis involves stanniocalcin-1 activation. Exp Cell Res. 2008;314:2975-84
41. Law AY, Yeung BH, Ching LY, Wong CK. Sp1 is a transcription repressor to stanniocalcin-1 expression in TSA-treated human colon cancer cells, HT29. J Cell Biochem. 2011;112:2089-96
42. Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B. et al. Arginine methylation regulates the p53 response. Nat Cell Biol. 2008;10:1431-9
43. Kwak YT, Guo J, Prajapati S, Park KJ, Surabhi RM, Miller B. et al. Methylation of SPT5 regulates its interaction with RNA polymerase II and transcriptional elongation properties. Mol Cell. 2003;11:1055-66
44. Hsu JM, Chen CT, Chou CK, Kuo HP, Li LY, Lin CY. et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat Cell Biol. 2011;13:174-81
45. Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37-50
46. Pal S, Baiocchi RA, Byrd JC, Grever MR, Jacob ST, Sif S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007;26:3558-69
47. Wang L, Pal S, Sif S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol Cell Biol. 2008;28:6262-77
48. Guderian G, Peter C, Wiesner J, Sickmann A, Schulze-Osthoff K, Fischer U. et al. RioK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J Biol Chem. 2011;286:1976-86
49. Le Guezennec X, Vermeulen M, Brinkman AB, Hoeijmakers WA, Cohen A, Lasonder E. et al. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol Cell Biol. 2006;26:843-51
50. Friesen WJ, Wyce A, Paushkin S, Abel L, Rappsilber J, Mann M. et al. A novel WD repeat protein component of the methylosome binds Sm proteins. J Biol Chem. 2002;277:8243-7
51. Aggarwal P, Vaites LP, Kim JK, Mellert H, Gurung B, Nakagawa H. et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell. 2010;18:329-40
52. Li Y, Chitnis N, Nakagawa H, Kita Y, Natsugoe S, Yang Y. et al. PRMT5 is required for lymphomagenesis triggered by multiple oncogenic drivers. Cancer Discov. 2015;5:288-303
53. Nicholas C, Yang J, Peters SB, Bill MA, Baiocchi RA, Yan F. et al. PRMT5 is upregulated in malignant and metastatic melanoma and regulates expression of MITF and p27(Kip1.). PLoS One. 2013;8:e74710
54. Chen M, Yi B, Sun J. Inhibition of cardiomyocyte hypertrophy by protein arginine methyltransferase 5. J Biol Chem. 2014;289:24325-35
55. Xu SG, Yan PJ, Shao ZM. Differential proteomic analysis of a highly metastatic variant of human breast cancer cells using two-dimensional differential gel electrophoresis. J Cancer Res Clin Oncol. 2010;136:1545-56
56. Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G. et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283-98
57. Jin W, Chen BB, Li JY, Zhu H, Huang M, Gu SM. et al. TIEG1 inhibits breast cancer invasion and metastasis by inhibition of epidermal growth factor receptor (EGFR) transcription and the EGFR signaling pathway. Mol Cell Biol. 2012;32:50-63
58. Jiang HL, Sun HF, Gao SP, Li LD, Huang S, Hu X. et al. SSBP1 Suppresses TGFbeta-Driven Epithelial-to-Mesenchymal Transition and Metastasis in Triple-Negative Breast Cancer by Regulating Mitochondrial Retrograde Signaling. Cancer Res. 2016;76:952-64
59. Chi Y, Huang S, Wang L, Zhou R, Xiao X, Li D. et al. CDK11p58 inhibits ERalpha-positive breast cancer invasion by targeting integrin beta3 via the repression of ERalpha signaling. BMC Cancer. 2014;14:577
60. Chi Y, Hong Y, Zong H, Wang Y, Zou W, Yang J. et al. CDK11p58 represses vitamin D receptor-mediated transcriptional activation through promoting its ubiquitin-proteasome degradation. Biochem Biophys Res Commun. 2009;386:493-8
61. Chi Y, Zhang C, Zong H, Hong Y, Kong X, Liu H. et al. Thr-370 is responsible for CDK11(p58) autophosphorylation, dimerization, and kinase activity. J Biol Chem. 2011;286:1748-57
Author contact
![]() Corresponding author: Jiong Wu, Department of Breast Surgery, Building 2, No. 270 Dong An Road, Shanghai, 200032 Email: wujiong1122sina.com; Tel: 86-21-64175590-3423; Fax: 86-21-64172585 or Wei Jin, Breast Cancer Institute, Building 7, No. 270 Dong An Road, Shanghai, 200032 Email: jinwei7207com; Tel: 86-21-64175590-3423; Fax: 86-21-64172585;
Corresponding author: Jiong Wu, Department of Breast Surgery, Building 2, No. 270 Dong An Road, Shanghai, 200032 Email: wujiong1122sina.com; Tel: 86-21-64175590-3423; Fax: 86-21-64172585 or Wei Jin, Breast Cancer Institute, Building 7, No. 270 Dong An Road, Shanghai, 200032 Email: jinwei7207com; Tel: 86-21-64175590-3423; Fax: 86-21-64172585;