Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(9):2477-2487. doi:10.7150/thno.24173 This issue Cite
Research Paper
Graphene oxide sensitizes cancer cells to chemotherapeutics by inducing early autophagy events, promoting nuclear trafficking and necrosis
Kuan-Chen Lin1,*, Mei-Wei Lin1,2 *, Mu-Nung Hsu1, Guan Yu-Chen3,4, Yu-Chan Chao5, Hsing-Yu Tuan1, Chi-Shiun Chiang6, Yu-Chen Hu1,7 ![]()
1. Department of Chemical Engineering, National Tsing Hua University, Hsinchu, Taiwan 30013
2. Biomedical Technology and Device Research Laboratories, Industrial Technology Research Institute, Hsinchu, Taiwan 31057
3. Institute of Biomedical Engineering, National Chiao Tung University, Hsinchu 30010, Taiwan
4. Department of Biological Science and Technology, National Chiao Tung University, Hsinchu 30010, Taiwan
5. Institute of Molecular Biology, Academia Sinica, Taipei, Taiwan, 11529
6. Department of Biomedical Engineering and Environmental Science, National Tsing Hua University, Hsinchu, Taiwan 30013
7. Frontier Research Center on Fundamental and Applied Sciences of Matters, National Tsing Hua University, Hsinchu, Taiwan 30013
*These two authors contributed equally to this manuscript
Received 2017-12-4; Accepted 2018-2-20; Published 2018-3-28
Abstract

Rationale: Cisplatin (CDDP) is a broad-spectrum anticancer drug but chemoresistance to CDDP impedes its wide use for cancer therapy. Autophagy is an event occurring in the cytoplasm and cytoplasmic LC3 puncta formation is a hallmark of autophagy. Graphene oxide (GO) is a nanomaterial that provokes autophagy in CT26 colon cancer cells and confers antitumor effects. Here we aimed to evaluate whether combined use of GO with CDDP (GO/CDDP) overcomes chemoresistance in different cancer cells and uncover the underlying mechanism.
Methods: We treated different cancer cells with GO/CDDP and evaluated the cytotoxicity, death mechanism, autophagy induction and nuclear entry of CDDP. We further knocked down genes essential for autophagic flux and deciphered which step is critical to nuclear import and cell death. Finally, we performed immunoprecipitation, mass spectrometry and immunofluorescence labeling to evaluate the association of LC3 and CDDP.
Results: We uncovered that combination of GO and CDDP (GO/CDDP) promoted the killing of not only CT26 cells, but also ovarian, cervical and prostate cancer cells. In the highly chemosensitized Skov-3 cells, GO/CDDP significantly enhanced concurrent nuclear import of CDDP and autophagy marker LC3 and elevated cell necrosis, which required autophagy initiation and progression but did not necessitate late autophagy events (e.g., autophagosome completion and autolysosome formation). The GO/CDDP-elicited nuclear trafficking and cell death also required importin α/β, and LC3 also co-migrated with CDDP and histone H1/H4 into the nucleus. In particular, GO/CDDP triggered histone H4 acetylation in the nucleus, which could decondense the chromosome and enable CDDP to more effectively access chromosomal DNA to trigger cell death.
Conclusion: These findings shed light on the mechanisms of GO/CDDP-induced chemosensitization and implicate the potential applications of GO/CDDP to treat multiple cancers.
Keywords: graphene oxide, autophagy, cisplatin, chemoresistance, nuclear import, LC3
Introduction
Autophagy is a multi-step cellular process by which cytoplasmic materials are delivered to lysosomes for degradation and is linked to a number of diseases including cancer [1]. Autophagy induction requires the repression of mTOR kinase, which subsequently stimulates ULK1 activity and initiates phagophore formation. The phagophore continues to elongate to form the autophagosome and enclose cytosolic proteins and organelles. Autophagosomes undergo maturation by fusion with lysosomes to create autolysosomes, in which the inner membrane and the cargo are degraded by lysosomal enzymes [1]. The complete autophagic flux involves multiple proteins. For instance, microtubule-associated light chain 3 (LC3) plays roles in cargo selection, phagophore elongation and autophagosome formation, and typically forms puncta in the cytoplasm upon autophagy induction. ATG7 is important in phagophore elongation and autophagosome completion. The adaptor protein p62 interacts with polyubiquitinated target proteins and the p62-cargo complex is selectively tethered to autophagosomes by the interaction of p62 and LC3 [1]. At the late stage, receptor proteins such as lysosome-associated membrane protein 2 (Lamp-2) and SNAP-29 [2, 3] mediate the fusion of autophagosomes with lysosomes to form autolysosomes.
Graphene oxide (GO) is a nanomaterial that has captured immense attention for many biological and medical applications including gene/drug delivery [4-6], imaging [7], cellular probing [8], cellular differentiation [9] and photothermal therapy [10, 11]. GO can be phagocytosed by macrophages and colon cancer cell CT26, hence simultaneously activating autophagy and toll-like receptor (TLR)-4 and -9 responses [12-15]. More recently, we uncovered that both GO and the chemotherapy drug cisplatin (CDDP) hardly kill CT26 cells due to the emergence of chemoresistance. Intriguingly, co-treatment with GO and CDDP (GO/CDDP) induces significant CT26 cell death via the necrosis pathway [16]. Intratumoral injection of GO/CDDP into mice bearing CT26 colon tumors augments immune cell infiltration, cell death and autophagy, thereby synergistically potentiating the antitumor effects. These studies collectively unveil the potential of GO as a chemosensitizer to overcome chemoresistance [16].
In this study, we further explored whether GO/CDDP chemosensitized other cancer cells and examined the underlying mechanisms. We unraveled that GO sensitized not only CT26 cells, but also ovarian (Skov-3), cervical (HeLa) and prostate (Tramp-C1) cancer cells to the CDDP-mediated killing. The autophagy marker LC3 is normally reported to be located in the cytoplasm. In the highly chemosensitized Skov-3 cells, GO/CDDP, but not GO or CDDP, induced autophagic flux that is non-canonical to conventional autophagy, and enhanced concurrent nuclear import of LC3 and CDDP in an importin α/β-dependent manner. Moreover, we uncovered that GO/CDDP treatment stimulated the co-delivery of LC3 with histones H1 and H4 into the nucleus. Specifically, GO/CDDP enhanced the acetylation of histone H4 (H4K16ac) in the nucleus, which could decondense the chromatin, thus enabling CDDP trafficking into the nucleus to more easily access chromosomal DNA and trigger cell death. Altogether, this study demonstrates the potentials of GO as a chemosensitizer not only for colon cancer, but also for ovarian cancer and other cancers, and unveils a novel mechanism that overcomes the chemoresistance.
Methods
Cell culture
Human cervical cancer (HeLa), lung cancer (A549) and prostate cancer (Tramp-C1) cells were cultured in high-glucose DMEM medium (Sigma) containing 10% fetal bovine serum (FBS, Biological Industries). Human ovarian cancer cell Skov-3 was cultured in McCoy's 5A (Sigma) medium containing 15% FBS. Mouse colon cancer cell CT26 was cultured in RPMI-1640 medium (Gibco) supplemented with 10% FBS and 1% antibiotics (PEN-STREP-AMPHO SOL, Biological Industries). All cancer cells were cultured in T75 flasks at 37 ºC and 5% CO2 and passaged when confluence exceeded 70%.
Preparation of GO nanosheets and CDDP stock solutions
The GO nanosheets (thickness < 2 nm, lateral size ~100-800 nm, mean diameter ~450 nm) were prepared and characterized as described [12, 13] and dispersed in water at 250 μg/mL as the stock solution. The details for GO nanosheets preparation and characterization are described in Supplementary Material. The chemotherapy drug CDDP (Millipore, cat. no. 2321-20) was dissolved in dimethyl sulfoxide (DMSO) at 25 mg/mL, aliquoted and stored at 4 ºC.
Treatment of cells with GO and CDDP
The cancer cells (CT26, Skov-3, HeLa, A549 and Tramp-C1) were seeded in 6-well plates (2×105 cells/well) or 24-well plates (7.5×104 cells/well), cultured for 12 h and the medium was removed. For GO treatment, the GO stock solution (250 μg/mL) was mixed with an equal volume of concentrated complete medium (2X), and further diluted to 50 μg/mL by mixing with complete medium (1X). After PBS washes, the cells were treated with GO (50 μg/mL) by culturing in the GO-containing medium for 12 or 24 h. For CDDP treatment, the drug stock solution was added to the complete medium (1X) to 200 μg/mL and the cells were cultured in the drug-containing medium for 12 or 24 h. For co-treatment with GO and CDDP (GO/CDDP group), the CDDP stock solution was added to the GO-containing medium to 200 μg/mL and the cells were cultured for 12 or 24 h. For the untreated control, the cells were simply cultured using the complete medium (1X).
For siRNA pre-treatment, 6 μL (60 pmol/mL) siRNA (Santa Cruz Biotechnology, specific for ULK1, ATG7, SNAP29, karyopherin alpha 2 or karyopherin beta 2) was added to 94 μL Opti-MEM medium (Invitrogen), while 6 μL Lipofectamine® 3000 (Invitrogen) was added to 94 μL Opti-MEM. Both solutions were mixed for 30 min and added to 800 μL Opti-MEM. In parallel, Skov-3 cells were seeded in 6-well plates (2×105 cells/well) and cultured overnight. After washing with Opti-MEM (1 mL/well), the siRNA-containing medium (1 mL/ well) was added to the wells. After incubation at 37 ºC for 5-6 h, the cells were treated with GO/CDDP as described above for 24 h.
Cell death analyses
The cells were seeded into 24-well plates (7.5×104 cells/well), cultured for 12 h and treated with GO, CDDP or GO/CDDP. After 24 h incubation, the cell viability was evaluated by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, Sigma) assays. The data were normalized against those of untreated cells to yield relative cell viability. Alternatively, the cells were seeded into 6-well plates, treated with GO, CDDP or GO/CDDP. Apoptosis and necrosis were quantified using the PE Annexin V/7 amino-actinomycin (7-AAD) Apoptosis Detection Kit I (BD Biosciences) coupled with flow cytometry analysis [12].
Immunofluorescence microscopy and quantitative analysis
Cells were seeded to coverslips in the 6-well plates (3.0×105 cells/well) and treated with GO, CDDP or GO/CDDP for 24 h, with or without siRNA pretreatment. After PBS washes, the cells were fixed with 4% formalin and permeabilized as described [13], washed with PBS and incubated with the primary antibody (1:200 dilution) for 1 h at room temperature. The primary antibody was specific for LC3 (LC3B isoform, Cell Signaling, 3868), p62 (Abnova, H00008878), Lamp-2 (GeneTex, GTX13524), karyopherin alpha 2 (importin α) (BD Biosciences, cat no. 610485), karyopherin beta 2 (importin β) (BD Biosciences, cat no. 610559), acetyl-α-tubulin (Lys40) (Cell Signaling, cat no. 5335), histone H1 (Merck, cat no. 05-457), phosphorylated histone H1 (Merck, cat no. 06-597), acetylated histone H4 (H4K16ac, Genetex GTX632097) or histone H4 (D2X4V) (Cell Signaling, 13919). For double labeling, two primary antibodies were added. After PBS washes, the cells were incubated with the secondary antibody (1:200 dilution) for 1 h at room temperature in the dark. The secondary antibodies were all purchased from Jackson ImmunoResearch for green (Alexa Fluor® 488 goat anti-rabbit IgG (H+L)) or red (Alexa Fluor® 594 goat anti-mouse or anti-rat IgG (H+L)) fluorescence. After washing, the cells were counterstained with 4,6-diamidino-2-phenylindole (DAPI, Vector Labs) and visualized with a confocal microscope. The images were captured and analyzed using Image-Pro Plus 6.0 (Media Cybernetics).
For quantitative analyses, ~100-150 cells from 3 independent culture experiments were counted for each group. The number of cells with LC3/p62 or LC3/Lamp-2 co-localizing in the cytosol was counted and divided by the number of DAPI+ cells. The cells containing 3 or more LC3 puncta in the nucleus were scored as LC3+. Dividing the number of LC3+ cells by the number of DAPI+ cells yielded the percentage of cells with LC3 puncta in the nucleus. Likewise, the percentage of cells with acetyl-tubulin, importin α or β, histone H1, phosphorylated histone H1, histone H4 and acetylated histone H4 were quantified.
Nuclear fractionation
The cells were harvested to microfuge tubes and washed with PBS twice. After resuspension with 500 μL RIPA lysis buffer (Millipore), the cells were passed through a 27G needle 50 times to disrupt plasma membranes. The cell slurry was centrifuged (500 ×g for 5 min) and the supernatant (containing the cytoplasm) was transferred to new microfuge tubes. The pellets (containing the nuclei) were resuspended with PBS or RIPA buffer. The nuclear fractions were stored at -20 ºC until analysis.
CDDP concentration measurement
The amount of CDDP imported into the nucleus was quantified by measuring the platinum (Pt) concentration in the nuclear fraction by an inductively coupled plasma mass spectrometer (ICP-MS, Agilent 7500ce).
Immunoprecipitation, SDS-PAGE and Western blot
The nuclear fractions and lysates from the untreated Skov-3 and GO/CDDP-treated Skov-3 cells were resuspended into 500 μL RIPA buffer and the protein concentration was quantified using the Bradford protein assay (Pierce). Same amount of proteins was subjected to immunoprecipitation using LC3B MAb (Cell Signaling, cat. no. 3868) and Dynabeads® Protein A (Thermo Fisher, cat. 10001D). The immunocomplex associated with LC3 was separated by 12% SDS-PAGE and the bands that were apparently upregulated after GO/CDDP treatment were cut for Liquid Chromatography/Mass Spectrometry (LC/MS/MS) analysis. The LC3-immunocomplex was also subjected to SDS-PAGE and subsequent Western blot using the following primary antibodies: rabbit MAb (1:500 dilution) specific for histone H4 (D2X4V, Cell Signaling) or acetyl-histone H4(Lys16) (Cell Signaling), mouse MAb specific for histone H1 (Merck) or rabbit polyclonal Ab specific for phosphorylated histone H1 (Merck). The secondary antibodies were HRP-conjugated goat anti-rabbit (Genetex) or anti-mouse (SeraCare) IgG. The images were developed using the chemiluminescence reagent (Western Lightning® ECL Pro, PerkinElmer) and captured using the GeneGnome system (Syngene). The band intensities were quantified using Image Pro Plus 6.0.
Statistical analysis
The quantitative in vitro data were statistically analyzed by student's t-test and represent the mean ± standard deviation (s.d.) of at least 3 independent culture experiments. p<0.05 was considered significant.
Results
Effects of GO/CDDP on cancer cell killing
To explore whether combined use of GO and CDDP (GO/CDDP) possessed the potential to overcome chemoresistance for different cancers, we selected cells derived from colon (CT26), ovarian (Skov-3), cervical (HeLa), prostate (Tramp-C1) and lung (A549) cancers. The cells were separately treated for 24 h with GO (50 μg/mL) or CDDP (200 μg/mL), or co-treated with GO (50 μg/mL) and CDDP (200 μg/mL) at concentrations that could exert synergistic killing effects to CT26 cells [16]. When compared with the untreated control, GO/CDDP triggered tremensdous detachment of CT26 and Skov-3 cells, but relatively less detachment of HeLa, Tramp-C1 and A549 cells (Figure 1A).
Effects of GO and CDDP, alone or in combination, on the cancer cell viability. (A) Microscopic observations. (B) Relative cell viability. Cancer cells were seeded in 6-well plates (2×105 cells/mL) and cultured overnight, followed by treatment with GO (50 μg/mL), CDDP (200 μg/mL) or GO/CDDP. The viability was quantified by MTT assay and the data were normalized to that of the untreated cell. The data represent mean ± s.d. of 3 independent culture experiments.
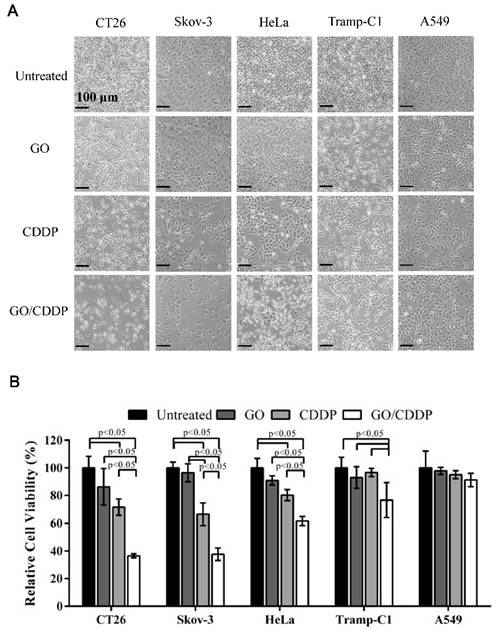
The MTT assay (Figure 1B) showed that the viability of CT26 and Skov-3 cells remained at ~71.5% and ~66.4% after CDDP treatment, indicating that both types of cells evolved chemoresistance to CDDP. Nonetheless, GO/CDDP resulted in a precipitous drop in the viability of CT26 and Skov-3 cells to ~36.5% and ~37.7%, respectively, demonstrating that GO chemosensitized CT26 and Skov-3 cells to CDDP. Likewise, CDDP alone did not effectively kill HeLa and Tramp-C1 but GO/CDDP enhanced the killing effects (Figure 1B). Only A549 cells were resistant to the treatment of GO, CDDP and GO/CDDP and maintained high viability.
Effects of GO/CDDP on autophagic flux, nuclear import and cell death
Autophagy is commonly regarded as an event occurring in the cytoplasm and cytoplasmic LC3 puncta formation is a major hallmark of autophagy [17]. To evaluate whether GO/CDDP induced autophagy, nuclear import and death in different cancer cells, we chose CT26 and Skov-3 that were most pronouncedly chemosensitized by GO, and A549 that was resistant to the GO-induced chemosensitization. The cells were treated with GO, CDDP or GO/CDDP for 24 h and subjected to immunofluorescence double labeling for LC3/p62 or LC3/Lamp-2 because co-localization of LC3/p62 and LC3/Lamp-2 are indicators of early (autophagosome formation) and late (autolysosome formation) stages of autophagy [18].
In CT26 and Skov-3 cells, GO/CDDP triggered evident co-localization of LC3/p62 (Figure 2A) and LC3/Lamp-2 (Figure 2B) in the cytosol (yellow dots), indicating the induction of autophagy. Notably, GO/ CDDP also gave rise to LC3 accumulation in the nucleus, yet the nuclear LC3 did not co-localize with p62 or Lamp-2 (sky blue dots, Figure 2A-B). Quantitative analysis depicted that GO/CDDP provoked significantly (p<0.05) higher degrees of cytosolic co-localization of LC3/p62 (Figure 2C) and LC3/Lamp-2 (Figure 2D) than the untreated control in CT26 and Skov-3 cells. Meanwhile, GO/CDDP triggered nuclear accumulation of LC3 puncta in significantly more CT26 and Skov-3 cells than GO and CDDP (Figure 2E). Furthermore, GO/CDDP triggered the nuclear entry of significantly more CDDP than CDDP only (Figure S1). However, GO and CDDP, either alone or in combination, neither provoked pronounced autophagic flux nor triggered nuclear entry of LC3 in A549 cells.
Effects of GO, CDDP and GO/CDDP on autophagic flux, nuclear import and cell death in different cancer cells. (A-B) Immunofluorescence microscopy for co-localization of LC3/p62 and LC3/Lamp-2. (C-D) Quantitative analysis of LC3/p62 and LC3/Lamp-2 in the cytosol. (E) Quantitative analysis of LC3 puncta in the nucleus. (F) Necrosis and apoptosis. CT26, Skov-3 and A549 cells were treated with GO, CDDP or GO/CDDP and subjected to immunofluorescence double labeling for LC3/p62 or LC3/Lamp-2. The images were analyzed using Image-Pro Plus 6.0. Approximately 100-150 cells from 3 independent culture experiments were counted for each group. The cells containing ≥ 3 yellow dots were considered to have LC3/p62 (LC3/Lamp-2) co-localization. The number of cells with LC3/p62 or LC3/Lamp-2 co-localizing in the cytosol was counted and divided by the number of DAPI+ cells. The cells containing 3 or more LC3 puncta in the nucleus were scored as LC3+. Dividing the number of LC3+ cells by the number of DAPI+ cells yielded the percentage of cells with LC3 puncta in the nucleus. The cells were also detached for Annexin V/7-AAD labeling and flow cytometry analysis to measure necrosis and apoptosis. The data represent mean ± s.d of at least 3 independent culture experiments.
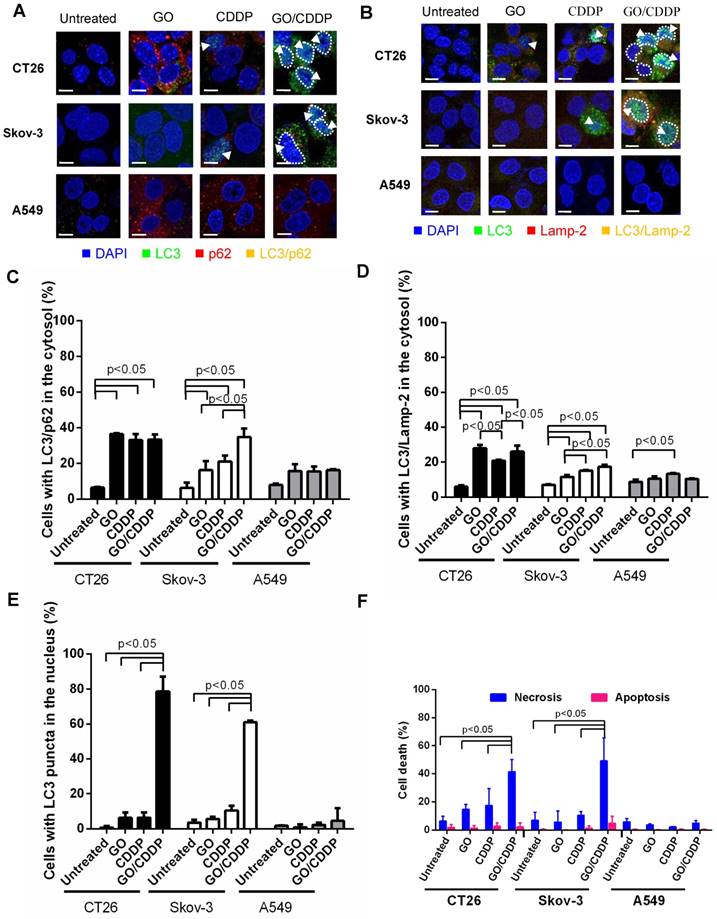
To analyze necrosis and apoptosis, we also performed Annexin V/7-AAD double staining followed by flow cytometry (Figure 2F), which unraveled that in CT26 and Skov-3 cells GO/CDDP provoked significantly higher degrees (>40% of cells) of necrosis than GO and CDDP. The necrosis induction in the GO/CDDP-treated CT26 cells was also confirmed by other necrosis hallmarks such as HMGB1 release, RIP1 decrease and RIP3 increase [16]. However, GO/CDDP barely elicited necrosis in A549, and GO/CDDP did not trigger appreciable apoptosis in the 3 cell types. Figure 2 collectively proved that, compared with GO and CDDP alone, GO/CDDP elicited more evident autophagic flux, triggered concurrent nuclear entry of LC3 and provoked more pronounced necrosis in CT26 and Skov-3 cells, but not in A549 cells. Since CT26 cell has been investigated previously [16], Skov-3 cell was selected for ensuing experiments.
Roles of autophagy and importin α/β on nuclear import and chemosensitization
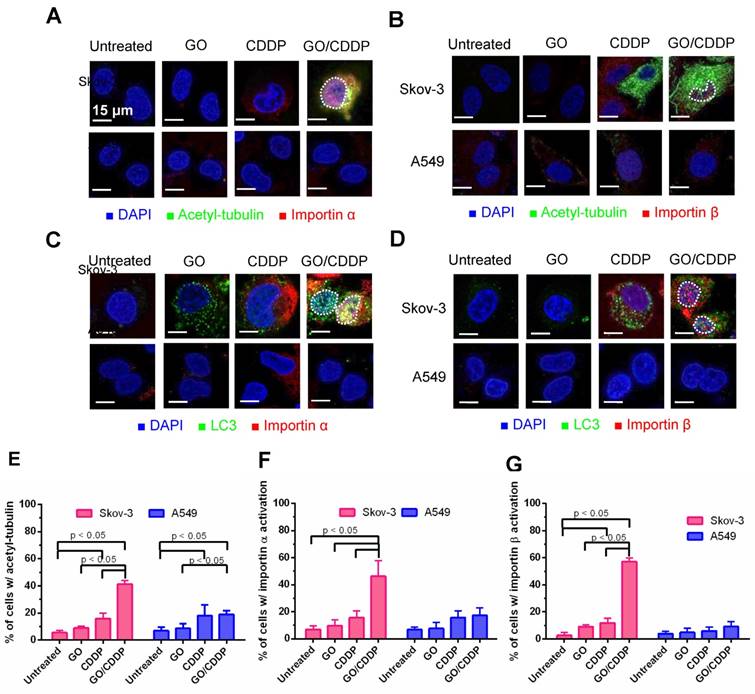
Importin α and β are proteins that play important roles in nuclear transport [19]. Meanwhile, acetylation of tubulin increases the stability of microtubules, and acetyl tubulin orchestrates with importin α/β to boost nuclear trafficking [20]. To assess the roles of tubulin acetylation and importin α/β on LC3 nuclear import, we performed immunofluorescence double labeling.
Figure 3A-B illustrate that, in Skov-3 cells, GO/CDDP triggered tubulin acetylation in the cytoplasm and abundant importin α/β (pink dots) accumulation in the nucleus, indicating the nuclear trafficking of importin α/β. The acetyl-tubulin co-localized with importin α in the cytoplasm (yellow/orange dots, Figure 3A) but did not co-localize with importin β (Figure 3B). Immunofluorescence double labeling for LC3 and importin α (Figure 3C) or β (Figure 3D) also confirmed that GO/CDDP augmented the nuclear entry of LC3 and importin α/β (Figure 3C-D). Notably, LC3 co-localized with importin α (yellow dots in Figure 3C) in the nucleus of Skov-3 cells, yet LC3 did not co-localize with importin β in the nucleus (Figure 3D). The quantitative data (Figure 3E-G) confirmed that, in Skov-3 cells, GO/CDDP induced significantly higher degrees of tubulin acetylation and importin α/β accumulation than GO and CDDP. In A549 cells, GO and CDDP, either alone or in combination, induced low levels of tubulin acetylation and importin α/β accumulation.
Roles of tubulin acetylation and importin α/β on nuclear import. (A-B) Immunofluorescence microscopy for acetyl-tubulin/importin α or acetyl-tubulin/importin β. (C-D) Immunofluorescence microscopy for co-localization of LC3/importin α or LC3/importin β. (E-G) Quantitative analysis of percentage of cells with acetyl-tubulin, importin α and importin β accumulation. The data represent mean ± s.d of at least 3 independent culture experiments.
To further assess the correlation between autophagy, nuclear entry and cell death, we treated Skov-3 cells with scramble siRNA as the control, or siRNAs to knockdown genes associated with autophagy initiation (ULK1), phagophore elongation (ATG7) or autolysosome formation (SNAP-29) [18, 21]. Meanwhile, we treated Skov-3 cells with siRNAs specific for genes (KPNA/KPNB) that encode importin α/β. The siRNA-transfected cells were treated with GO/CDDP for 24 h and subjected to immunofluorescence microscopy (Figure 4A) and necrosis/apoptosis (Figure 4B) analyses.
Verification of roles of autophagy and importin α/β on nuclear import and cell death. (A) Immunofluorescence microscopy for LC3/importin α and LC3/importin β co-localization. (B) Necrosis and apoptosis. The data represent mean ± s.d of at least 3 independent culture experiments. Skov-3 cells were treated with scramble siRNA as the control or siRNAs to knockdown ULK1, ATG7, SNAP-29, KPNA (encoding importin α) or KPNB (encoding importin β). The siRNA-transfected cells were treated with GO/CDDP for 24 h and subjected to immunofluorescence microscopy. The cells were also detached for Annexin V/7-AAD labeling and flow cytometry analysis to measure necrosis and apoptosis. The data represent mean ± s.d of at least 3 independent culture experiments.
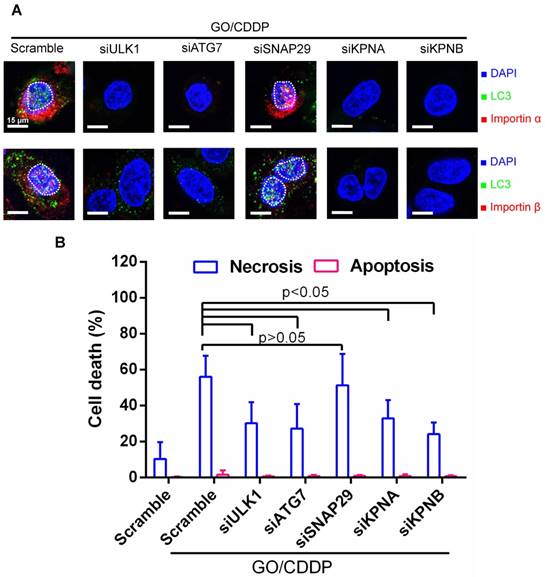
When compared with scramble siRNA treatment, siRNA knockdown of ULK1, ATG7, KPNA and KPNB effectively suppressed the nuclear entry of LC3 puncta and importin α/β (Figure 4A). Figure 4B depicts that attenuating these genes reduced the GO/CDDP-induced necrosis with statistical significance. In contrast, suppressing SNAP-29 neither abolished the nuclear entry of LC3 puncta and importin α/β (Figure 4A), nor abrogated the GO/CDDP-induced necrosis (Figure 4B). Figure 4 collectively proves that autophagy initiation, phagophore elongation and importin α/β are critical for the enhanced nuclear import of LC3 after GO/CDDP treatment, which potentiate the chemosensitization effects to kill Skov-3 cells. However, the late stage of autophagic flux (autolysosome formation) is dispensable for the nuclear entry of LC3 as well as the chemosensitization.
Histone and CDDP co-migrates with LC3 into the nucleus
Figure 3-4 indicate that importin α/β and LC3 puncta were concurrently transported into the nucleus of Skov-3 cells. To dissect what other proteins were associated with LC3 and concomitantly imported into the nucleus, we untreated or treated Skov-3 cells with GO/CDDP, collected the cell lysate, isolated the nuclear fraction and performed immunoprecipitation using the anti-LC3 antibody.
SDS-PAGE analysis of the LC3-immunocomplex (Figure 5A) showed that GO/CDDP treatment led to the upregulation and accumulation of some proteins associated with LC3 in the lysate and nuclear fraction when compared with the untreated (UN group) Skov-3 cells. Among these, two proteins (~11 kDa and ~35 kDa) were particularly prominent. LC/MS/MS analysis of these LC3-associated proteins (Table S1-2) unraveled that the proteins were presumably histone H1 (~35 kDa) and histone H4 (~11 kDa).
GO/CDDP triggered direct association of histones H1/H4 and CDDP with LC3. (A) SDS-PAGE analysis of LC3-immunocomplex from the lysate (Lys) and nuclear (Nuc) fractions of cells with or without GO/CDDP treatment. (B) Western blot analysis of proteins (~11 kDa and ~35 kDa) derived from the nuclear LC3-immunocomplex. (C) Immunofluorescence microscopy of cells with or without GO/CDDP treatment. The cells were immunolabeled with primary antibody for histone H1 or H4 and counter stained with DAPI. (D) Percentages of cells positive with total histone H1, phosphorylated histone H1 (p-histone H1), histone H4 and acetylated histone H4 (H4K16ac). (E) Amount of CDDP associated with nuclear LC3 immunocomplex. The nuclear LC3 immunocomplex was analyzed by ICP-MS to quantify the amount of platinum (Pt) in CDDP. The data represent mean ± s.d of at least 3 independent culture experiments.
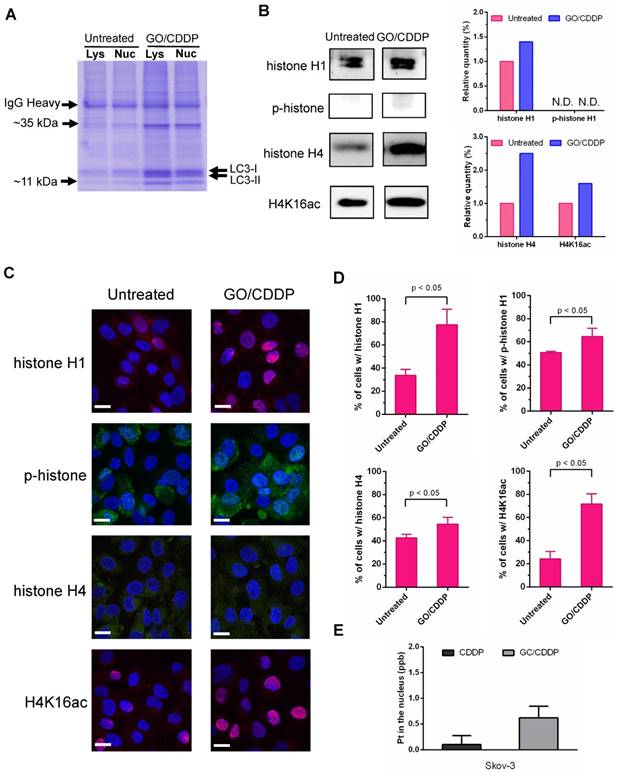
Histone H1 phosphorylation (p-histone H1) and acetylation of histone H4 at lysine 16 (H4K16ac) are crucial for modulating the higher-order chromatin structure. To evaluate the effects of GO/CDDP on histone H1/H4 modification, we further analyzed the LC3-immunocomplex in the nuclear fraction by Western blot. Figure 5B shows that GO/CDDP, when compared with the untreated control, increased the amount of total histone H1, total histone H4 and acetylated histone H4 that were co-immunoprecipitated with LC-3. Immunofluorescence microscopy (Figure 5C) and subsequent quantitative (Figure 5D) data indicated that GO/CDDP elevated the amount of histone H1 and H4, either unmodified or modified, in Skov-3 cells. In particular, the acetylated histone H4 triggered by GO/CDDP was mainly localized in the nucleus.
Moreover, we treated Skov-3 cells with CDDP only or GO/CDDP and performed nuclear fractionation and immunoprecipitation, followed by ICP-MS analysis to quantify the platinum (Pt) in CDDP (a Pt-containing compound) in the nucleus. ICP-MS analysis of the nuclear LC3 complex confirmed that GO/CDDP significantly promoted the direct association of LC3 and CDDP and co-migration into the nucleus (Figure 5E).
Discussion
CDDP is one of the most effective broad-spectrum anticancer drugs, but chemoresistance to CDDP impedes its wide use for cancer therapy [22]. We have previously shown that GO and CDDP alone are able to induce autophagy in CT26 cells, but do not elicit significant cell death [16]. Nonetheless, GO chemosensitizes CT26 cells to CDDP and triggers substantial CT26 cell death via necrosis [16].
In this study, we further uncovered that GO/CDDP gave rise to similar chemosensitization effects to ovarian (Skov-3), cervical (HeLa) and prostate (Tramp-C1) cancer cells, albeit to different degrees (Figure 1). In the highly chemosensitized Skov-3 cells, GO/CDDP, but not GO or CDDP, triggered the initiation and progression of autophagic flux as well as enhanced nuclear accumulation of LC3 and CDDP, which concurred with elevated necrosis (Figure 2 and Figure S1). In contrast, GO/CDDP did not induce autophagy, nuclear entry or cell death in A549 cells (Figure 1-3). These data suggested that autophagic flux progression and enhanced nuclear entry of CDDP contributed to the killing of Skov-3 cells. siRNA knockdown of ULK1 and ATG7 abolished the autophagic flux, nuclear import of LC3 and necrosis (Figure 4), indicating that autophagy initiation and phagophore formation are necessary for the nuclear import and necrosis. However, knockdown of SNAP-29 neither abrogated the nuclear import nor ablated GO/CDDP-induced necrosis (Figure 4), and the nuclear LC3 did not co-localize with p62 (Figure 2). Since SNAP-29 is crucial for the late stage of autophagy (i.e., fusion of autophagosome and lysosome), while p62 is the adaptor protein that carries the polyubiquitinated cargo to autophagosomes by the interaction of p62 and LC3, these data suggested that tethering of p62 to LC3 on the elongating phagophore was dispensable. Therefore, GO/ CDDP induced nuclear import of LC3 and CDDP as well as necrosis in Skov-3 cells, which require early events of autophagic flux (initiation and phagophore elongation) but do not necessitate late autophagy events (e.g., autophagosome completion and autolysosome formation). Such diversion of autophagic flux and nuclear entry of LC3 and CDDP in Skov-3 cells is consistent with the findings observed in CT26 cells [16], suggesting that this might be a mechanism common for certain cancer cells.
Intriguingly, CDDP generally kills cells by inducing DNA lesions and subsequent apoptosis, but we unraveled that GO/CDDP elicited necrosis rather than apoptosis in both CT26 and Skov-3 cells (Figure 2). Such findings appeared contradictory to typical notions, but mounting evidence has demonstrated the correlation between autophagy and necrosis [23], and has shown that CDDP triggers not only apoptosis but also necrosis [24], especially in cancer cells resistant to chemotherapy drugs [25, 26]. Moreover, GO may also induce necrosis [14]. As such, the induction of autophagy flux and nuclear import of CDDP and LC3 might have altered the distribution of CDDP and autophagy-related proteins in the cytosol and nucleus, leading to the switch of death mechanisms. Such necrosis induction is beneficial to the chemotherapy efficacy because necrosis triggers the release of a number of danger signals (e.g., HMGB1) that potentiate the inflammation and host's antitumor immune responses [27].
In addition, autophagy is commonly regarded as an event occurring in the cytoplasm, and cytoplasmic LC3 puncta formation is a major hallmark of autophagy [17]. Although recent evidence implicates that LC3 shuttles between cytoplasm and the nucleus [21, 28, 29], how LC3 is transported in and out of the nucleus remains elusive. Conventional nuclear import involves importin β, which binds the cargo protein directly, or indirectly via an adaptor protein importin α [30]. The importin-cargo complex is actively transported into the nucleus, after which the cargo is released from importin β [31]. Such nuclear import may play critical roles in cancer by transporting key oncogenesis mediators across the nuclear membrane in cancer cells [31]. Here we unraveled that the GO/CDDP-induced nuclear trafficking of LC3 and CDDP was accompanied by abundant accumulation of importin α/β within the nucleus (Figure 3) and was abolished by knocking down importin α/β expression (Figure 4). Furthermore, importin α/β inhibitor ivermectin abrogated the nuclear import of LC3 and CDDP in CT26 cells [16]. These data confirmed that LC3 and CDDP were actively transported into the nucleus by importin α/β. Since LC3 co-localized with importin α but not importin β inside the nucleus (Figure 3), it was possible that LC3 was incorporated into the growing phagophore and the complex engaged with importin α/β for active transport into the nucleus. The LC3-cargo was released from importin β after being transported across the nuclear pore complex while maintaining the association with importin α.
It is also noteworthy that LC3 is suggested to interact with high-molecular weight protein complexes [32, 33]. Recent studies have identified a number of proteins in direct association with LC3, including ATG-related proteins (e.g., ATG16L [34]), SOS1 [35], TP53INP2 [36], extracellular signal regulated kinase [37], DOR [28], lamin B1 [38], MAP1B, tubulin and several 40S ribosomal proteins [32]). Some of these are transcription factors or nuclear proteins that translocate to the nucleus upon stimulation. By immunoprecipitation and LC/MS/MS, we uncovered that GO/CDDP treatment stimulated the association of LC3 with histone H1 and H4 (Figure 5A-B and Table S1-2) and promoted the acetylation of nuclear histone H4 (H4K16ac) (Figure 5C-D). Importantly, LC3 was directly associated with CDDP (Figure 5E). It is known that after translation in the cytoplasm, histone is bound by histone chaperones, subjected to post-translational modifications such as acetylation and is actively transported into the nucleus by karyopherins such as importin α/β [39]. Since LC3 was imported into the nucleus via importin α/β, one may reason that LC3 engaged with histones H1/H4 and CDDP in the cytosol, which were co-imported into the nucleus with the aid of importin α/β. Inside the nucleus, histones are assembled into nucleosomes and modulate the chromatin structure [40]. In particular, H4K16ac is sufficient to induce chromatin structure reorganization [41] and cancer cell resistance to chemotherapeutics [42]. GO/CDDP treatment might have enhanced H4K16ac in the nucleus and resulted in chromatin relaxation, hence enabling the co-imported CDDP to more effectively access chromosomal DNA to trigger DNA cleavage and cell death. Such chemosensitization mechanism is consistent with that of certain histone deacetylase inhibitors, which can increase the lysine acetylation on histones H3 and H4 and sensitize drug-resistant cancer cells to chemotherapeutic agents [43, 44].
It is noteworthy that our findings were inconsistent with the notions that (1) autophagy induction is coupled to reduced H4K16ac [45, 46] and (2) CDDP treatment of ovarian cancer cells induces autophagy that contributes to chemoresistance [47]. Furthermore, GO/CDDP induced autophagy, but also diverted the autophagic flux and promoted the nuclear import of LC3 and CDDP that triggered atypical necrosis (instead of typical apoptosis). These discrepancies suggest that GO/CDDP treatment provoked autophagic events that are non-canonical to conventional autophagy [18], and killed Skov-3 cells in a non-conventional mechanism. In favor of our findings, it was also recently found that GO chemosensitizes cancer cells to doxorubicin and CDDP, but by inducing perturbations to the plasma membrane and cytoskeletal meshwork [48]. The crosstalk and correlation between the GO/CDDP- elicited non-canonical autophagy and plasma membrane damage await further investigation.
Conclusions
In summary, here we unraveled that GO/CDDP enhanced the nuclear trafficking of CDDP and elevated cell death via necrosis in not only colon cancer cells, but also in cells derived from various cancers such as ovarian (Skov-3 cells), cervical (HeLa) and prostate (Tramp-C1). We further deciphered the molecular mechanisms accounting for the enhanced chemosensitization effects. In particular, we unveiled a new role and correlation between autophagy, histone acetylation and epigenetic state of chromosome, which contribute to the improved killing of cancer cells. These findings shed light on how GO/CDDP chemosensitizes cancer cells and implicate the potential applications of GO/CDDP to treat multiple cancers, and may pave a new avenue to use nanomaterials as a new class of chemosensitizer.
Abbreviations
Cisplatin (CDDP: cisplatin; GO: graphene oxide; TLR: toll-like receptor; H4K16Ac: acetylation of histone H4 on lysine 16.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors acknowledge the support from the National Tsing Hua University (Toward World-Class University Project 106N526CE1, 105N526CE1, 104N2050E1) and Ministry of Science and Technology (MOST 103-2221-E-007-093-MY3).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323-35
2. Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256-69
3. Morelli E, Ginefra P, Mastrodonato V, Beznoussenko GV, Rusten TE, Bilder D. et al. Multiple functions of the SNARE protein Snap29 in autophagy, endocytic, and exocytic trafficking during epithelial formation in Drosophila. Autophagy. 2014;10:2251-68
4. Zhang L, Lu Z, Zhao Q, Huang J, Shen H, Zhang Z. Enhanced chemotherapy efficacy by sequential delivery of siRNA and anticancer drugs using PEI-grafted graphene oxide. Small. 2011;7:460-4
5. Zhang L, Xia J, Zhao Q, Liu L, Zhang Z. Functional graphene oxide as a nanocarrier for controlled loading and targeted delivery of mixed anticancer drugs. Small. 2010;6:537-44
6. Goenka S, Sant V, Sant S. Graphene-based nanomaterials for drug delivery and tissue engineering. J Control Release. 2014;173:75-88
7. Hu S-H, Chen Y-W, Hung W-T, Chen IW, Chen S-Y. Quantum-dot-tagged reduced graphene oxide nanocomposites for bright fluorescence bioimaging and photothermal therapy monitored in situ. Adv Mater. 2012;24:1748-54
8. Wang Y, Li Z, Hu D, Lin CT, Li J, Lin Y. Aptamer/graphene oxide nanocomplex for in situ molecular probing in living cells. J Am Chem Soc. 2010;132:9274-6
9. Chen G-Y, Pang DWP, Hwang SM, Tuan HY, Hu Y-C. A graphene-based platform for induced pluripotent stem cells culture and differentiation. Biomaterials. 2012;33:418-27
10. Yang K, Wan J, Zhang S, Tian B, Zhang Y, Liu Z. The influence of surface chemistry and size of nanoscale graphene oxide on photothermal therapy of cancer using ultra-low laser power. Biomaterials. 2012;33:2206-14
11. Tao Y, Ju E, Ren J, Qu X. Immunostimulatory oligonucleotides-loaded cationic graphene oxide with photothermally enhanced immunogenicity for photothermal/immune cancer therapy. Biomaterials. 2014;35:9963-71
12. Chen G-Y, Chen C-L, Tuan H-Y, Yuan P-X, Li K-C, Yang H-J. et al. Graphene oxide triggers toll-like receptors/autophagy responses in vitro and inhibits tumor growth in vivo. Adv Health Mater. 2014;3:1486-95
13. Chen G-Y, Yang H-J, Lu C-H, Chao Y-C, Hwang S-M, Chen C-L. et al. Simultaneous induction of autophagy and toll-like receptor signaling pathways by graphene oxide. Biomaterials. 2012;33:6559-69
14. Qu G, Liu S, Zhang S, Wang L, Wang X, Sun B. et al. Graphene oxide induces toll-like receptor 4 (TLR4)-dependent necrosis in macrophages. ACS Nano. 2013;7:5732-45
15. Zhou H, Zhao K, Li W, Yang N, Liu Y, Chen C. et al. The interactions between pristine graphene and macrophages and the production of cytokines/chemokines via TLR- and NF-κB-related signaling pathways. Biomaterials. 2012;33:6933-42
16. Chen G-Y, Meng C-L, Lin K-C, Tuan H-Y, Yang H-J, Chen C-L. et al. Graphene oxide as a chemosensitizer: diverted autophagic flux, enhanced nuclear import, elevated necrosis and improved antitumor effects. Biomaterials. 2015;40:12-22
17. Levy JMM, Thorburn A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol Therapeut. 2011;131:130-41
18. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1-222
19. Badding MA, Lapek JD, Friedman AE, Dean DA. Proteomic and functional analyses of protein-DNA complexes during gene transfer. Mol Ther. 2013;21:775-85
20. Ishiguro K, Ando T, Maeda O, Watanabe O, Goto H. Cutting edge: Tubulin alpha functions as an adaptor in NFAT-importin beta interaction. J Immunol. 2011;186:2710-3
21. Fullgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol. 2014;15:65-74
22. Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012;64:706-21
23. Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Et Biophy Acta-Mol Cell Res. 2013;1833:3448-59
24. Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin-induced cell death always produced by apoptosis? Mol Pharmacol. 2001;59:657-63
25. Cepeda V, Fuertes MA, Castilla J, Alonso C, Quevedo C, Perez JM. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med Chem. 2007;7:3-18
26. Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A. et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257
27. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P. et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573-7
28. Huang R, Xu Y, Wan W, Shou X, Qian J, You Z. et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57:456-66
29. Drake KR, Kang M, Kenworthy AK. Nucleocytoplasmic distribution and dynamics of the autophagosome marker EGFP-LC3. Plos One. 2010;5:e9806
30. Chook YM, Suel KE. Nuclear import by karyopherin-beta s: Recognition and inhibition. BBA-Mol Cell Res. 2011;1813:1593-606
31. Mahipal A, Malafa M. Importins and exportins as therapeutic targets in cancer. Pharmacol Ther. 2016;164:135-43
32. Kraft LJ, Manral P, Dowler J, Kenworthy AK. Nuclear LC3 associates with alowly diffusing complexes that survey the nucleolus. Traffic. 2016;17:369-99
33. Kraft LJ, Nguyen TA, Vogel SS, Kenworthy AK. Size, stoichiometry, and organization of soluble LC3-associated complexes. Autophagy. 2014;10:861-77
34. Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19:2092-100
35. Furuta S, Miura K, Copeland T, Shang WH, Oshima A, Kamata T. Light Chain 3 associates with a Sos1 guanine nucleotide exchange factor: its significance in the Sos1-mediated Rac1 signaling leading to membrane ruffling. Oncogene. 2002;21:7060-6
36. Nowak J, Archange C, Tardivel-Lacombe J, Pontarotti P, Pebusque MJ, Vaccaro MI. et al. The TP53INP2 protein is required for autophagy in mammalian cells. Mol Biol Cell. 2009;20:870-81
37. Martinez-Lopez N, Athonvarangkul D, Mishall P, Sahu S, Singh R. Autophagy proteins regulate ERK phosphorylation. Nat Commun. 2013;4:2799
38. Dou Z, Xu C, Donahue G, Shimi T, Pan J-A, Zhu J. et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527:105-9
39. Keck KM, Pemberton LF. Histone chaperones link histone nuclear import and chromatin assembly. Biochim Biophys Acta. 2012;1819:277-89
40. Baek SH, Il Kim K. Epigenetic control of autophagy: Nuclear events gain more attention. Mol Cell. 2017;65:781-5
41. Wilting RH, Dannenberg JH. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist Updat. 2012;15:21-38
42. Meisenberg C, Ashour ME, El-Shafie L, Liao CY, Hodgson A, Pilborough A. et al. Epigenetic changes in histone acetylation underpin resistance to the topoisomerase I inhibitor irinotecan. Nucleic Acids Res. 2017;45:1159-76
43. Hajji N, Wallenborg K, Vlachos P, Fullgrabe J, Hermanson O, Joseph B. Opposing effects of hMOF and SIRT1 on H4K16 acetylation and the sensitivity to the topoisomerase II inhibitor etoposide. Oncogene. 2010;29:2192-204
44. Mottamal M, Zheng SL, Huang TL, Wang GD. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898-941
45. Fullgrabe J, Heldring N, Hermanson O, Joseph B. Cracking the survival code: autophagy-related histone modifications. Autophagy. 2014;10:556-61
46. Fullgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q. et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. 2013;500:468-71
47. Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem. 2014;289:17163-73
48. Zhu J, Xu M, Gao M, Zhang Z, Xu Y, Xia T. et al. Graphene oxide induced perturbation to plasma membrane and cytoskeletal meshwork sensitize cancer cells to chemotherapeutic agents. ACS Nano. 2017;11:2637-51
Author contact
![]() Corresponding author: Phone: (886)3-571-8245; FAX: (886)3-571-5408; Email: yuchennthu.edu.tw
Corresponding author: Phone: (886)3-571-8245; FAX: (886)3-571-5408; Email: yuchennthu.edu.tw