Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusions
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(9):2459-2476. doi:10.7150/thno.23880 This issue Cite
Research Paper
Endogenous IgG-based affinity-controlled release of TRAIL exerts superior antitumor effects
Hao Yang1*, Yanru Feng 1*, Huawei Cai2, Dianlong Jia1,4, Heng Li1, Ze Tao1, Yi Zhong3, Zhao Li1, Qiuxiao Shi1, Lin Wan1, Lin Li2, Xiaofeng Lu1 ![]()
1. Key Lab of Transplant Engineering and Immunology, MOH;
2. Department of Nuclear Medicine;
3. Proteomics and Metabolomics Laboratory, West China-Washington Mitochondria and Metabolism Research Center; West China Hospital, Sichuan University, Chengdu, 610041, China
4. Present address: College of Pharmacy, Liaocheng University, Liaocheng, Shandong, 252000, China
*These authors contribute equally to this work.
Received 2017-11-16; Accepted 2018-2-8; Published 2018-3-28
Abstract

The inefficiency of recombinant tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-based clinical regimens has been dominantly attributed to the short half-life of TRAIL. Affinity-controlled release using endogenous long-acting proteins, such as IgG and albumin, as carriers is extremely attractive for improving the pharmacokinetics of TRAIL. Up to now, it is unclear whether IgG-binding is efficient for affinity-controlled release of TRAIL.
Methods: An IgG-binding affibody, IgBD, was genetically fused to the N-terminus of TRAIL to produce IgBD-TRAIL.The IgG-binding ability, cytotoxicity, serum half-life, and in vivo antitumor effect of IgBD-TRAIL were compared with that of TRAIL. In addition, an albumin-binding affibody, ABD, was fused to TRAIL to produce ABD-TRAIL. The cytototoxicity, serum half-life, and antitumor effect of IgBD-TRAIL and ABD-TRAIL were compared.
Results: IgBD fusion endowed TRAIL with high affinity (nM) for IgG without interference with its cytotoxicity. The serum half-life of IgBD-TRAIL is 50-60 times longer than that of TRAIL and the tumor uptake of IgBD-TRAIL at 8-24 h post-injection was 4-7-fold that of TRAIL. In vivo antitumor effect of IgBD-TRAIL was at least 10 times greater than that of TRAIL. Owing to the high affinity (nM) for albumin, the serum half-life of ABD-TRAIL was 80-90 times greater than that of TRAIL. However, after binding to albumin, the cytotoxicity of ABD-TRAIL was reduced more than 10 times. In contrast, binding to IgG had little impact on the cytotoxicity of IgBD-TRAIL. Consequently, intravenously injected IgBD-TRAIL showed antitumor effects superior to those of ABD-TRAIL.
Conclusions: Endogenous long-acting proteins, particularly IgG-based affinity-controlled release, prolonged the serum half-life as well as significantly enhanced the antitumor effect of TRAIL. IgBD-mediated endogenous IgG binding might be a novel approach for the affinity-controlled release of other protein drugs.
Keywords: drug delivery, affinity-controlled release, biopharmaceuticals, immunoglobulin, albumin
Introduction
Elucidation of the molecular pathways of pathogenesis allows us to accurately develop biotherapeutics using a disease-associated innate process. Although small molecule drugs remain valuable, biopharmaceuticals have dominated the worldwide revenue generated by the pharmaceutical market in recent years. The market value of biopharmaceuticals is expected to increase from $151.9 billion in 2013 to $222.7 billion by 2019. Except for the 246 biopharmaceuticals approved in the United States and European Union, over 900 biopharmaceutical candidates are currently in clinical trials [1-3]. Compared to small molecule drugs, biopharmaceuticals with high molecular mass show advantages of higher specificity and potency. However, most non-antibody biopharmaceuticals, i.e., therapeutic proteins and peptides, exhibit short half-lives. To maintain the therapeutic level, frequent injections are typically required for protein and peptide drugs, which might encounter problems of patient compliance and tolerance [4]. Consequently, improving pharmacokinetics is key for the development of biopharmaceuticals [5]. In particular, different proteins might have different physicochemical and biological properties; therefore, protein-specific strategies might be required for pharmacokinetics improvement [6].
Controlled release is a popular strategy for pharmacokinetics improvement. Conventional release technologies, such as drug entrapment within degradation-controlled polymeric matrices (bulk or surface erosion) [7], swelling-controlled hydrogels [8], and macro/nanoparticles [9,10], have been demonstrated to be efficient for improving the pharmacokinetics of small molecule drugs. However, the tertiary structures required by specificity and potency typically make proteins more fragile than small molecule drugs. Exposure to harsh processing conditions, such as organic solvent, shear force, sonication, lyophilization, and high temperature, might be detrimental to protein structure and function. Consequently, due to the involvement of harsh conditions in the preparation of the delivery system, the controlled release strategies designed for small molecule drugs do not consistently work well for protein drugs. In addition, burst release is a common problem for these delivery systems, as the release of drug from the carrier is governed by the diffusion rate of the entrapped drug and enzymatic degradation of the carrier [2,3,11,12]. To overcome the challenges in the controlled release of protein drugs, increasing attention has been paid to affinity-controlled release in recent years.
Here, affinity refers to the preferred noncovalent interaction between two binding partners, such as protein-protein, protein-peptide, protein-aptamer, or protein-polymer interactions [3]. Strategies for the affinity-controlled release of drugs were initially inspired by the extracellular matrix (ECM)-controlled release of cytokines under physiological conditions. Previous studies have shown that cytokines containning heparin-binding domains could accumulate in the heparin-rich ECM, followed by slow diffusional release, which was predominantly attributed to the transient interaction between the heparin-binding domain of cytokines and heparin in the ECM. These results suggest that affinity-controlled release of heparin-binding proteins might be achieved by using heparinized delivery systems. In fact, natural heparin-rich ECM, heparinized polymers and micro/nanoparticles have been successfully used as affinity carriers for the delivery of heparin-binding cytokines [11]. Since the interaction between binding partners occurs under physiological conditions, the protein drug for affinity-controlled release could be efficiently loaded onto the carrier by simply mixing these components together, which has few risks for inducing protein denaturation and loss of biological activity [3]. Compared to the delivery under harsh conditions, affinity-controlled release is particularly attractive for protein drug delivery.
However, according to the conventional concept, constructing an affinity-controlled release system requires challenging studies including identification of proper binding partners to achieve the required release profile and preparation of affinity carriers with a high loading rate. Previous studies have focused on natural binding partners, such as heparin/heparin-binding domain [11], collagen/ collagen-binding domain [13], and SH3 domain/SH3-binding domain [14], etc. These natural binding partners are limited by their low affinities (μM). Recently, several classes of artificial high-affinity (nM) molecules, such as affibodies [15], anticalins [16], adnectins [17], and aptamers [18], have emerged as novel binding partners. To prepare affinity carriers, binding molecules have typically been immobilized onto insoluble carriers, such as ECM, polymer, or micro/nanoparticles in previous studies [2,11,12]. The clinical translation of these exogenous insoluble carriers might be limited by their biocompatibility and immunogenicity. Considering the risk of these exogenous insoluble carriers, it is better to use endogenous soluble long-acting proteins as carriers for affinity-controlled release [19].
Owing to neonatal Fc receptor (FcRn)-mediated recycling, plasma proteins, such as albumin and IgG, exhibit an extraordinarily long (2-4 weeks) serum half-life [20]. In addition, albumin and IgG are the most abundant plasma proteins, making these molecules attractive as endogenous drug carriers for affinity-controlled release. Frankly, two marketed diabetes drugs have been produced by incorporating albumin-binding fatty acids to insulin (Levemir®) and glucagon-like peptide-1 (GLP-1) agonist (Victoza ®), respectively [21]. In addition to fatty acids, numerous albumin-binding domains (ABD) have been used as binding partners for the controlled release of peptides and proteins [22]. Similar to albumin, IgG is also abundant in plasma, accounting for approximately 10-20% of total plasma proteins. The serum half-life of IgG [~23 days] is longer than that of albumin (~19 days) in humans. Importantly, the efficiency of FcRn-mediated recycling for IgG is higher than that for albumin [23]. A limited number of previous studies demonstrated that binding to IgG elongated the circulation time of recombinant proteins [24,25]. As more IgG binding domains (IgBDs) are identified [26,27], the potential use of endogenous IgG as a carrier for the affinity-controlled release of protein drugs should be evaluated.
Recombinant tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces apoptosis in a variety of tumor cells at nanomolar concentrations but spares normal cells in vitro. The superior activity and specificity of TRAIL in preclinical experiments prompted its testing in clinical trials in cancer patients. However, the promising in vitro antitumor effect of TRAIL was not observed in Phase I/II clinical trials, which was predominantly attributed to the poor pharmacokinetics of TRAIL [28]. Albumin fusion [29], Fc-fusion [30], PEGylation [31], and entrapment by nanoparticles [32,33] have been used to extend the serum half-life of TRAIL. In our previous work, to achieve the affinity-controlled release of TRAIL using endogenous albumin as a carrier, an affibody ABD was fused to the N-terminus of TRAIL to produce the albumin binding ABD-TRAIL. Intravenously injected ABD-TRAIL showed a prolonged serum half-life and enhanced in vivo antitumor effects. Nevertheless, the steric hindrance between albumin and TRAIL drastically (4-6 times) reduced its cytotoxicity in tumor cells [34]. Since IgG is comparable to albumin in serum half-life, we attempt to evaluate the potential use of endogenous IgG as a carrier for the affinity-controlled release of TRAIL.
In this study, we first produced the fusion protein IgBD-TRAIL by genetically fusing a Fab-specific IgBD to the N-terminus of TRAIL. Subsequently, after measurement of the IgG binding ability of IgBD-TRAIL, we determined the contributeon of IgBD-mediated endogenous IgG binding to the serum half-life extension and in vivo antitumor effect enhancement of TRAIL. Finally, we compared the efficacy of IgBD-mediated endogenous IgG binding and ABD-mediated endogenous albumin binding in improving the pharmacokinetics and enhancing the in vivo antitumor effect of TRAIL.
Materials and methods
Protein expression and purification
To endow TRAIL with IgG-binding ability, an IgBD, SPGC3FabRR, was genetically fused to the N-terminus of TRAIL to produce the fusion protein IgBD-TRAIL. The gene encoding SPGC3FabRR was designed according to its amino acid sequence [35] and optimized for recombinant expression in E. coli. The TRAIL gene was cloned in our previous study [36]. A flexible linker (G4S)3 was inserted between IgBD and TRAIL. The gene encoding IgBD-TRAIL was synthesized and sub-cloned into pQE30 to produce the expression plasmid pQE30-IgBD-TRAIL by Convenience Biology Inc. (Changzhou, China). To reduce the hepatic accumulation of the fusion protein, the 6His-tag in the original pQE30 plasmid was replaced by an (HE)3-tag according to the description of Hofstrom et al. [37]. To prepare IgBD-TRAIL, the expression plasmid pQE30-IgBD-TRAIL was transformed into the competent cells of E. coli M15, followed by induction at 25 °C overnight with isopropyl β-D-thiogalactoside (IPTG, 0.1 mM). The cells collected from a 1 L culture were resuspended in 50 mL of lysis buffer (50 mM phosphate, pH 8.0, 300 mM NaCl, 10 mM β-mecaptoethanol, 1 mM Phenylmethylsulfonylfluoride, and 5 mM imidazole) and processed 4-5 times with a high pressure homogenizer (80-100 MPa). The recombinant proteins in the supernatant were recovered by affinity chromatography with Ni-NTA super flow (Qiagen, CA). The purity and molecular weight of IgBD-TRAIL were determined by 12.75% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The purified proteins were dialyzed against phosphate-buffered saline (PBS, 10 mM Na2HPO4, 137 mM NaCl, 2.68 mM KCl, and 2 mM KH2PO4, pH 7.4) overnight for further use. To produce ABD-TRAIL, the ABD domain was genetically fused at the N-terminus of TRAIL as previously described [34]. The preparation of TRAIL has been described in a previous study [36].
Enzyme-linked immunosorbent assays
Enzyme-linked immunosorbent assay (ELISA) was performed according to Strok et al. [38] with some modification. To determine the IgG-binding ability of IgBD-TRAIL, IgG (100 nM in PBS) derived from mouse (mIgG) or human (hIgG) was coated onto the wells of medium-binding ELISA plates (BIO BASIC, CA). After 4 washes with phosphate-buffered saline (PBST, 10 mM Na2HPO4, 500 mM NaCl, 2.68 mM KCl, 2 mM KH2PO4, and 0.075% Tween 20, pH 7.4), the wells were blocked with 1% bovine serum albumin (BSA) at 37 °C for 1 h. IgBD-TRAIL at different concentrations (0-2.5 nM) was added into the wells followed by incubation at 37 °C for 1 h. Biotin-labeled antibody against TRAIL was used to detect IgBD-TRAIL bound to the wells. TRAIL was used as a control. ELISA for albumin binding of ABD-TRAIL was performed as previously described [34].
To compare the FcRn binding of IgG in the presence or absence of IgBD-TRAIL, hIgG was labeled with biotin by incubating hIgG with biotin at a molar ratio of 1:10 at room temperature for 1 h. To remove the unconjugated biotin, the mixture was dialyzed against PBS (pH 6.0) overnight. The biotin-labeled hIgG was incubated with or without IgBD-TRAIL (calculated as trimer) in a molar ratio of 1:1 at room temperature for 0.5 h, followed by addition into wells of high-binding ELISA plates (BIO BASIC, CA) coated with FcRn protein (R&D, MN). After 3 washes with PBS (pH 6.0) supplemented with 0.075% Tween 20, the bound IgG was detected by using horseradish peroxidase (HRP)-labeled streptavidin.
Size exclusion chromatography
Size exclusion chromatography (SEC) was performed on AKTA Pure with a Superdex 200 10/30 column (GE Healthcare, PA) as previously described [34]. To determine the IgG binding, IgBD-TRAIL (calculated as trimmer) was incubated with IgG (molar ratio of 2:1, 1:1, or 1:2) at room temperature for 0.5 h followed by loading onto the column. Phosphate buffer (20 mM phosphate, pH 7.4, 500 mM NaCl) was used as the eluent at a flow rate of 0.5 mL/min. Protein peak was recorded by monitoring absorbance at 280 nm, and the components of each protein peak were identified by SDS-PAGE. Protein peaks containing both IgG and IgBD-TRAIL reflected the formation of the IgG-IgBD-TRAIL complex. Albumin binding of ABD-TRAIL was also determined in the same manner. Briefly, after the incubation of ABD-TRAIL (calculated as trimer) with albumin at a molar ratio of 1:1, the mixture was separated on a Superdex 75 10/30 column. A protein peak with a molecular size larger than that of albumin and ABD-TRAIL reflects the formation of the albumin-ABD-TRAIL complex.
Protein-protein interaction
Biolayer interferometry performed on Octet® systems (Pall ForteBio LLC, CA) was used to reveal protein-protein interactions. For IgG binding assay, IgG was immobilized on a protein A-coated sensor. Subsequently, the sensor was inserted into a solution containing different concentrations (0-2 μM) of IgBD-TRAIL for 300 s to enable association, followed by disassociation in PBS for 200 s. The association constant ka, disassociation constant kd, and affinity KD (KD=kd/ka) were calculated by using software for Octet® systems according to 1:1 binding model. For death receptor binding assay, DR4-Fc or DR5-Fc (R&D, MN) were captured by a protein A-coated sensor, followed by the insertion of the sensor into solutions containing different concentrations of TRAIL fusion proteins. For albumin binding assays, ABD-TRAIL was captured by using a His-tag antibody-coated sensor, followed by the insertion of the sensor into a series of albumin solutions.
Cytotoxicity and apoptosis
Cell lines, including COLO 205, LS174T, and HCT 116, were purchased from American Type Culture Collection (ATCC, VA) and cultured in 1640 or DMEM supplemented with 10% fetal bovine serum at 37 °C in a 5% CO2 humidified atmosphere. To measure the cytotoxicity of protein, 1-2×104 cells were inoculated in the wells of a 96-well plate, followed by the addition of different concentrations of protein. After treatment overnight, the viable cells were counted by using CCK-8. PBS was used as negative control with 100% viability. To estimate the influence of IgG and albumin binding on the cytotoxicity of TRAIL fusion proteins, IgG or albumin were mixed with IgBD-TRAIL or ABD-TRAIL at a molecular ratio of 1:1 and incubated at room temperature for 0.5 h, followed by addition into the cells. Apoptosis in cells was revealed by caspase activity detection and TdT-mediated dUTP nick-end labeling (TUNEL) as previously described [34].
Cellular uptake and recycling of protein
Human umbilical vein endothelial cells (HUVEC) were seeded (1×104 cells/well) onto a 20 mm glass-bottomed cell culture dish (Nest, Wuxi, China) and cultured in DMEM overnight. To monitor the cellular uptake, TRAIL proteins were labeled with Alex Flour-488 (green) and hIgG was labeled with Alex Flour-610 (red) according to Sockolosky et al. [25]. The hIgG was mixed with IgBD-TRAIL or TRAIL (200 nM) at a molar ratio of 1:1. After incubation at room temperature for 0.5 h, the mixture was added into the cells followed by incubation for 1 h at 37 °C. After two washes with PBS, the cells were observed under a multi-photon laser scanning confocal microscope (Nikon, Tokyo, Japan). Cellular localization of IgBD-TRAIL with transferrin was performed according to Gobel et al. [39].
To monitor the recycling, TRAIL proteins (50-200 nM, 200 μL) were mixed with hIgG at a molar ratio of 1:1 and incubated at room temperature for 0.5 h, followed by addition to HUVECs (5×10 5 cells). After incubation at 37 °C for 2 h, the cells were washed three times with PBS. Subsequently, 120 μL of fresh serum-free 1640 medium was added to the cells. After further incubation at 37 °C for 4 h, the medium was collected for cytotoxicity assay. Compared to the cytotoxicity of the medium from cells treated with a mixture of TRAIL and hIgG, an increase in the cytotoxicity of the medium from cells treated with IgBD-TRAIL preincubated with hIgG reflects the IgG-mediated recycling of IgBD-TRAIL.
In vivo pharmacokinetics and biodistribution
Blood clearance of TRAIL proteins was first measured by monitoring the time-dependent decrease in cytotoxicity of residual proteins in blood. Balb/c mice (n=3 for each time point) were intravenously injected with TRAIL proteins (10 mg/kg). The blood samples of mice were collected at different times (1 min to 96 h) post-injection. The plasma was diluted at different ratios, followed by cytotoxicity assays in COLO 205 cells. The speed of the cytotoxicity decrease reflects the protein clearance rate.
In addition, a traditional radioactive method was also used to monitor the blood clearance of TRAIL proteins and IgG. I131-labeling was performed according to Fan et al. [40]. To monitor the blood clearance, normal Balb/c mice (n=3 for each time point) were intravenously injected with I131-labeled TRAIL proteins or IgG preincubated with or without IgBD-TRAIL at a molar ratio of 1:1 (100 μL, 27.5 kBq/g body weight). The mice were sacrificed at different times (1 min to 96 h) post-injection and the radioactivity in the blood was measured using a FJ-2008PS Gamma counter. The serum half-lives of TRAIL proteins or IgG were calculated using DAS software version 2.11 according to Wang et al. [41].
The biodistribution of TRAIL proteins and IgG preincubated with or without IgBD-TRAIL were determined in normal mice or nude Balb/c mice (n=3 for each time point) bearing COLO 205 tumor grafts. After intravenous injection, the mice were sacrificed at different times post-injection, and the radioactivity in tumor grafts and other normal organs/tissues was measured.
Optical imaging
TRAIL proteins were labeled with the near-infrared fluorescence dye CF750, succinimidyl ester (CF750, Sigma, CA) according to Fan et al. [40]. Balb/c nude mice bearing COLO 205 tumor grafts (~100 mm3) were intravenously injected with CF750-labeled proteins, followed by dynamic scanning with SPECTRAL Lago and Lago X Imaging Systems (Spectral, AZ).
In vivo antitumor effect
Approximately 2×10 6 cells (COLO 205, HCT 116, or LS174T) were subcutaneously injected into the back between the head and neck of Balb/c nude mice. Once the tumor grafts reached the indicated size, the mice were randomly divided into several groups followed by the intravenous injection of TRAIL proteins (100 μL) at a dose of 5 mg/kg. The mice in the control group were injected with the same amount of PBS. Tumor growth was monitored by measuring the longitudinal (L) and transverse (W) diameters of the tumor grafts daily. The tumor volume (V) was calculated using the formula V=L×W2/2. The tumor grafts were removed and weighed at the end of the experiment.
Short term acute toxicity
Balb/c mice (n=10) were intravenously injected with TRAIL proteins at 10 mg/kg every other day for a total of 8 injections. The mice in the control group were injected with the same volume of PBS. The body weights of mice were recorded every day. At the end of this experiment, the mice were sacrificed. The blood samples were collected for white blood cells (WBCs) counting and biochemical indicator measuring. Biochemical indicators reflecting the function of liver (glutamic-oxaloacetic transaminase, AST; glutamic-pyruvic transaminase, ALT) and kidney (urea; creatinine; and blood urea nitrogen, BUN) were analyzed. The cellular structures in paraffin-sectioned tissues derived from liver, kidney, spleen, heart, lung, muscle, and brain were analyzed by H&E staining.
Statistical analysis
For multiple comparisons, one-way analysis of variance (ANOVA) test was performed using SPSS software version 17.0. The significance level was defined as P<0.05. The results are expressed as mean ± standard error of the mean (SEM).
Results
IgBD fusion endows TRAIL with IgG-binding ability
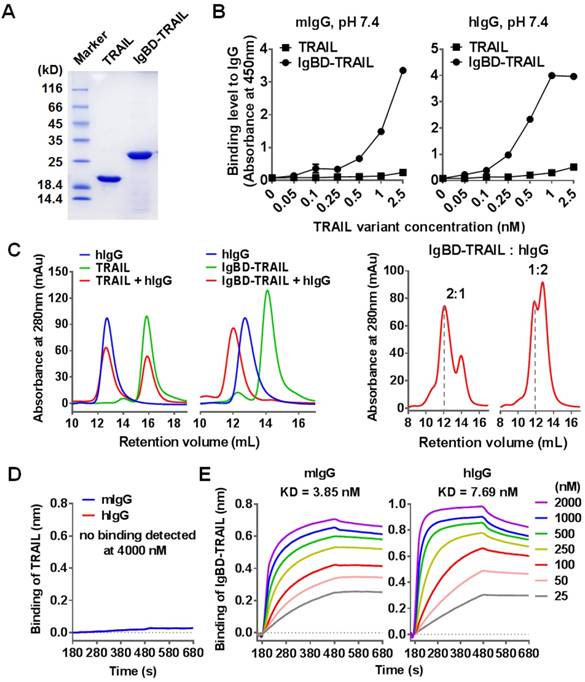
According to the molecular design, IgBD was introduced to the N-terminus of TRAIL to produce the fusion protein IgBD-TRAIL. SDS-PAGE revealed IgBD-TRAIL as a single protein band with an expected molecular weight of approximately 28 KD in the presence of β-mercaptoethanol (2-ME) (Figure 1A). On the SEC column, recovered IgBD-TRAIL was separated as a single protein peak with a molecular weight of approximately 80 KD in the absence of 2-ME (Figure S1A), indicating that IgBD-TRAIL existed as a homogenous trimer in PBS. SDS-PAGE and SEC demonstrated that TRAIL also existed as a homogenous trimer. ELISA performed at pH 7.4 demonstrated that IgBD-TRAIL, but not TRAIL, showed dose-dependent binding to wells precoated with mIgG or hIgG (Figure 1B). In addition, when TRAIL (calculated as trimer) was mixed with hIgG at a molar ratio of 1:1, two protein peaks representing TRAIL and hIgG were detected by SEC (Figure 1C). In contrast, only one protein peak larger than IgBD-TRAIL and hIgG was detected by SEC when IgBD-TRAIL and hIgG were mixed at the same molar ratio. Subsequent SDS-PAGE revealed hIgG and IgBD-TRAIL in this protein peak (Figure S1B), indicating the formation of hIgG-IgBD-TRAIL complex. Further protein-protein interaction analysis verified that TRAIL did not bind to mIgG and hIgG (Figure 1D). In contrast, IgBD-TRAIL showed high affinity for both mIgG (KD=3.85 nM) and hIgG (KD=7.69 nM) (Figure 1E). These results demonstrated that IgBD-TRAIL, but not TRAIL, could bind IgG. When the molar ratio of IgBD-TRAIL to hIgG varied from 1:1 to 2:1 or 1:2, excessive IgBD-TRAIL or hIgG was detected by SEC (Figure 1C and Figure S1C). In addition, no novel complex larger than IgG-IgBD-TRAIL was detected when the molar ratio of IgBD-TRAIL to hIgG was increased to 1:4 or 1:6 (Figure S1C). Dynamic light scattering analysis demonstrated that the average diameters of pure IgBD-TRAIL and hIgG were 5.7±2.3 nm and 18±5.9 nm, respectively. The average diameters of the particles in the mixture of IgBD-TRAIL and hIgG at molar ratios of 1:1 and 1:2 were 24.7±8.1 nm and 24.2±9.6 nm, respectively (Figure S1D). These results indicated that one IgBD-TRAIL trimer could only bind one hIgG, suggesting that IgBD-mediated binding of TRAIL to IgG would not induce IgG aggregation.
IgG binding of IgBD-TRAIL. (A) SDS-PAGE of purified proteins. (B) ELISA for IgG binding at pH 7.4. IgBD-TRAIL or TRAIL was added to wells precoated with mIgG or hIgG. The bound proteins were measured by using antibody against TRAIL. (C) SEC of TRAIL proteins preincubated with or without IgG at pH 7.4. IgBD-TRAIL (calculate as trimer) was preincubated with hIgG at a molar ratio of 1:1, 2:1 or 1:2. TRAIL was used as control. (D, E) Affinity of TRAIL (D) and IgBD-TRAIL (E) for IgG.
IgBD-mediated IgG binding shows little impact on the cytotoxicity of TRAIL
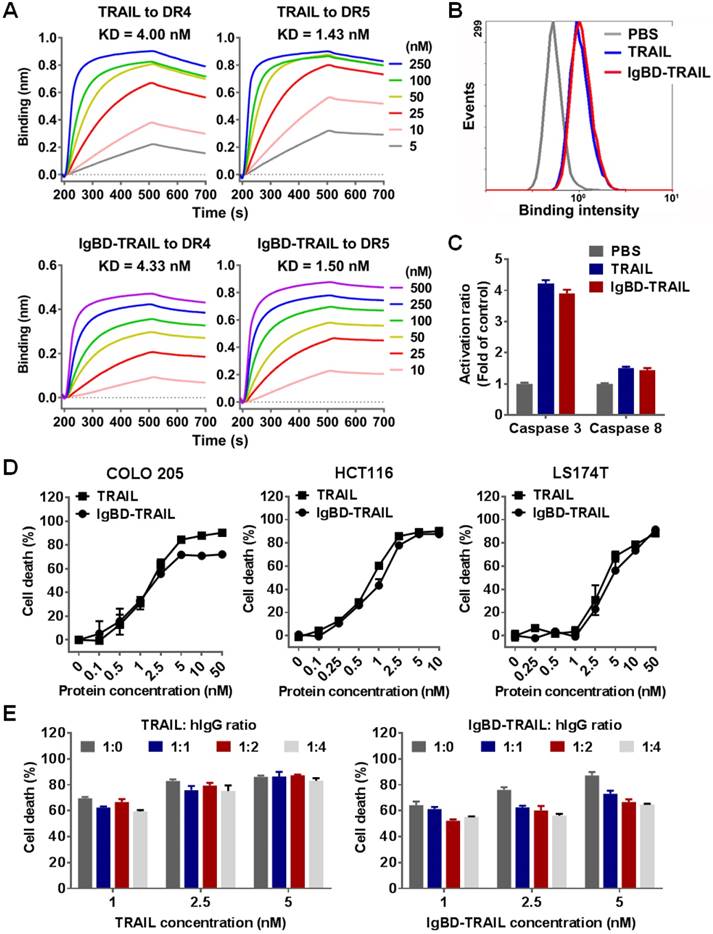
Death receptor binding is crucial for TRAIL to exert cytotoxicity in tumor cells. Since the fusion of TRAIL to IgBD might induce steric hindrance and thus reduce the death receptor binding of TRAIL, we first determined the binding of IgBD-TRAIL to DR4-Fc and DR5-Fc by biolayer interferometry. Figure 2A shows that both IgBD-TRAIL and TRAIL could bind to DR4-Fc and DR5-Fc. The affinities of IgBD-TRAIL for DR4-Fc (4.33 nM) and DR5-Fc (1.50 nM) were comparable to those of TRAIL for DR4-Fc (4.00 nM) and DR5-Fc (1.43 nM). Moreover, IgBD-TRAIL and TRAIL showed similar cell binding (Figure 2B), caspase activation (Figure 2C) and cytotoxicity (Figure 2D) in tested tumor cells, indicating that IgBD fusion did not reduce the death receptor binding and cytotoxicity of TRAIL. In addition, IgG binding might induce steric hindrance between IgG and IgBD-TRAIL, thus reducing the cytotoxicity of TRAIL. To evaluate the impact of this steric hindrance on TRAIL, the cytotoxicities of IgBD-TRAIL preincubated with or without IgG were compared. As shown in Figure 2E, IgBD-TRAIL preincubated with hIgG at a molar ratio of 1:1, 1:2, and 1:4 showed a similar, but limited (less than 20%) reduction in cytotoxicity, indicating that the IgG binding had little impact on the cytotoxicity of TRAIL.
Death receptor binding and cytotoxicity of IgBD-TRAIL. (A) Affinity of TRAIL and IgBD-TRAIL for death receptors. (B) Flow cytometry of COLO 205 tumor cells preincubated with FAM-labeled TRAIL proteins. (C) Activity of caspase in COLO 205 tumor cells treated with TRAIL proteins. PBS was used as a control. (D) Cytotoxicity of TRAIL proteins in tumor cells in the absence of IgG. (E) Cytotoxicity of TRAIL proteins in HCT116 tumor cells in the presence of IgG. TRAIL proteins (calculated as trimer) were preincubated with increasing concentration of hIgG prior to addition into the cells.
IgBD-mediated IgG binding contributes to recycling of TRAIL in endothelial cells
FcRn-mediated recycling contributes to the long serum half-life of IgG. After entering the cells, IgG accumulates in early endosomes and binds to FcRn at an acidic pH (<6.5), which protects IgG from lysosomal degradation. After exocytosis, IgG disassociates from FcRn at neutral pH (>7.2) and re-enters circulation [42,43]. To exploit the recycling pathway of IgG, IgBD-TRAIL must bind IgG at acidic pH without interfering with its FcRn binding. As shown in Figure 3A, a unique protein peak representing the hIgG-IgBD-TRAIL complex was revealed by SEC after preincubation of IgBD-TRAIL and hIgG at a molar ratio of 1:1 at pH 6.0. ELISA also showed dose-dependent IgG binding of IgBD-TRAIL at pH 6.0 (Figure S2A). These results suggested that IgBD-TRAIL could bind IgG in acidic endosomes. Moreover, hIgG preincubated with or without IgBD-TRAIL showed similar FcRn binding (Figure 3B and Figure S2B), indicating that IgG binding to IgBD-TRAIL preserved FcRn binding.
IgG binding-mediated intracellular transportation and recycling of IgBD-TRAIL in endothelial cells. (A) SEC of TRAIL proteins preincubated with or without hIgG at pH 6.0. (B) FcRn binding of hIgG preincubated with or without IgBD-TRAIL. FcRn was coated on the wells of ELISA plates, followed by addition of biotin-labeled hIgG preincubated with or without IgBD-TRAIL. After washing with PBS, the bound hIgG was measured by using HRP-streptavidin. (C) Uptake and localization of IgBD-TRAIL preincubated with hIgG in endothelial cells. Alex Flour-488 (green)-labeled IgBD-TRAIL was preincubated with Alex Flour-610 (red)-labeled hIgG followed by addition into HUVEC. TRAIL was used as a control. To co-localize IgBD-TRAIL with transferrin, Alex Flour-488 (green)-labeled IgBD-TRAIL preincubated with hIgG and Alex Flour-610 (red)-labeled transferrin was added to cells. (D) Recycling of TRAIL proteins preincubated with IgG. IgBD-TRAIL or TRAIL was preincubated with hIgG at a molar ratio of 1:1 prior to addition into HUVEC. After incubation for 2 h at 37 °C, the medium was replaced by fresh serum-free medium. Approximately 4 h later, the medium was collected for cytotoxicity assays in COLO 205 tumor cells.
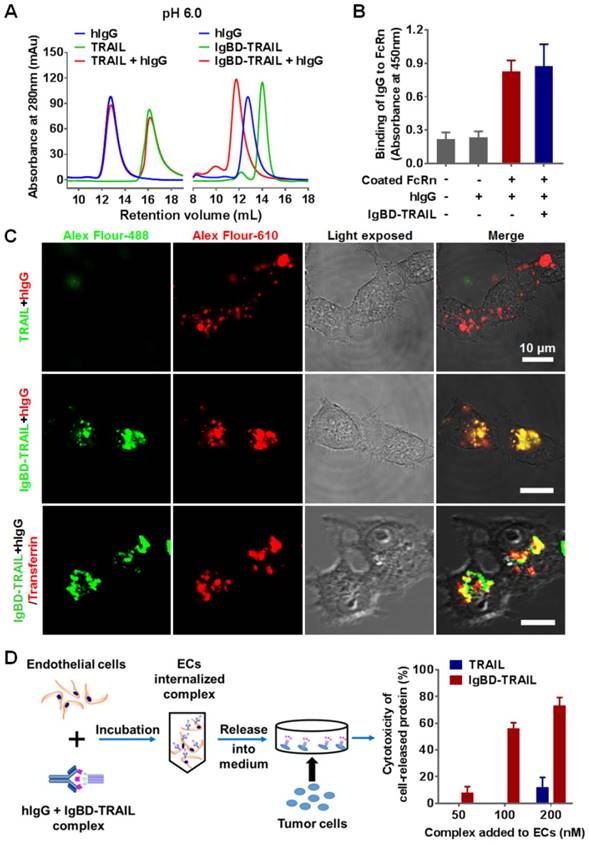
Intracellular transport of the IgG-IgBD-TRAIL complex was monitored by fluorescence microscopy. Alex Flour-488-labeled IgBD-TRAIL and Alex Flour-610-labeled hIgG were mixed at a molar ratio of 1:1, followed by addition to endothelial cells. The results showed that IgBD-TRAIL preincubated with hIgG accumulated more than TRAIL preincubated with IgG (Figure 3C and Figure S3A) and IgBD-TRAIL preincubated without IgG (Figure S3B), indicating that IgBD-mediated IgG binding facilitated the cell entry of TRAIL. Moreover, IgBD-TRAIL preincubated with hIgG colocalized with transferrin in the cell (Figure 3C), suggesting that IgBD-TRAIL accumulated within endosomes. To monitor the IgG binding-mediated recycling, different amounts of IgBD-TRAIL were preincubated with hIgG, followed by addition to the cells. After 2 h incubation, the culture medium was replaced by fresh medium, followed by further incubation for 4 h. The medium was collected for cytotoxicity assay. As shown in Figure 3D, the cytotoxicity of the medium collected from cells incubated with IgBD-TRAIL was obviously stronger than that of medium collected from cells incubated with TRAIL, indicating that IgBD-mediated IgG binding increased the cellular uptake and recycling of TRAIL.
IgBD-mediated IgG binding prolongs serum half-life, thus increasing tumor uptake of TRAIL
Given that IgBD-TRAIL bound to IgG in vitro with a high affinity, we determined the blood clearance of 131I-labeled IgBD-TRAIL and 131I-labeled IgG preincubated in a 1:1 molar ratio with IgBD-TRAIL by monitoring the time-dependent decrease of radioactivity in the blood of mice. As shown in Figure 4A, the radioactivity of 131I-labeled TRAIL was reduced by 3 logs within 48 h post-injection. However, the radioactivity of 131I-labeled IgBD-TRAIL was only reduced by 2 logs at 72 h post-injection, indicating that the blood clearance of IgBD-TRAIL was much slower than that of TRAIL. The calculated serum half-life of IgBD-TRAIL was15.7±0.39 h, which was 50-60 times longer than that of TRAIL (15.3±1.5 min). The prolonged circulation of IgBD-TRAIL was also verified by a time-dependent decrease in the cytotoxicity of residual protein in the blood. As shown in Figure 4B, after the blood samples collected at 0.5 h post-injection were diluted 25 times, no obvious cytotoxicity of residual TRAIL was detectable in COLO 205 tumor cells. However, IgBD-TRAIL retained in blood samples collected at 96 h post-injection induced at least 60% cell death, even after these samples were diluted 100 times. Since IgBD-TRAIL and TRAIL showed similar cytotoxicities in tumor cells (Figure 2D), a slower decrease in cytotoxicity confirmed that the circulation time of IgBD-TRAIL was longer than that of TRAIL. These results demonstrated that IgBD-mediated IgG binding significantly prolonged the serum half-life of TRAIL. Surprisingly, the blood clearance curves of 131I-labeled IgG preincubated with or without IgBD-TRAIL (Figure S4A-B) were similar. These results indicated that IgBD-mediated IgG binding extended the circulation time of IgBD-TRAIL, but did not accelerate the blood clearance of IgG.
Pharmacokinetics and biodistribution of IgBD-TRAIL. (A) Time-dependent clearance of TRAIL proteins. (B) Time-dependent decrease of cytotoxicity of TRAIL proteins. The blood samples were collected at different times (1 min to 96 h) post-injection. After 25-500 times dilution, blood samples were used for cytotoxicity assays in COLO 205 tumor cells. (C) Tumor uptake of TRAIL proteins visualized by optical imaging. CF750-labeled IgBD-TRAIL or TRAIL was intravenously injected into Balb/c nude mice bearing COLO 205 tumor grafts (arrow indicated) followed by kinetic scanning with an optical imaging system. (D) Biodistribution of TRAIL proteins. 131I-labeled IgBD-TRAIL or TRAIL was intravenously injected into Balb/c nude mice bearing COLO 205 tumor grafts. The mice were sacrificed at 8 h or 24 h for measuring the radioactivity in normal tissues and tumor tissues. Insertion indicates the tumor uptake of IgBD-TRAIL (fold of TRAIL).
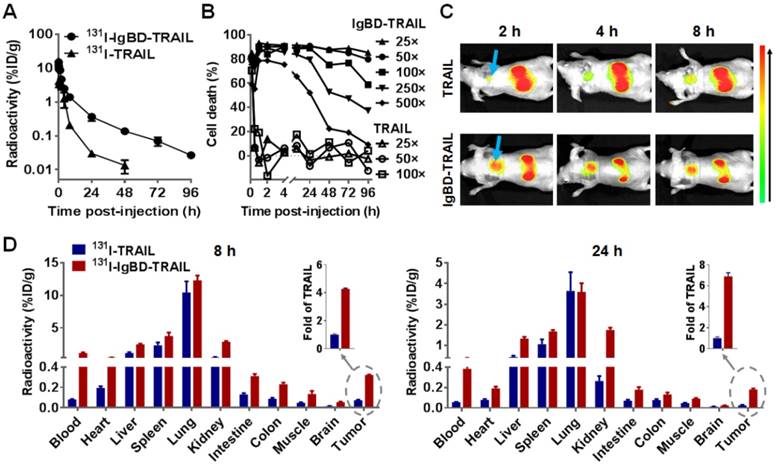
Since prolonged serum half-life might increase the tissue uptake of protein, we further determined the biodistribution of IgBD-TRAIL and IgG preincubated with or without IgBD-TRAIL. Tumor uptake of IgBD-TRAIL was first monitored by using optical imaging after intravenous injection of CF750-labeled protein into mice bearing COLO 205 tumor grafts. As shown in Figure 4C, high-contrast images of subcutaneous tumor grafts were obtained at 2-8 h after the injection of IgBD-TRAIL but not TRAIL. The tumor uptake of IgBD-TRAIL at 8 and 24 h post-injection was approximately 4-7 times higher than that of TRAIL. Accordingly, the blood concentration of IgBD-TRAIL at 8 and 24 h post-injection were approximately 7-14 times higher than that of TRAIL (Figure 4D), indicating that IgBD-mediated IgG binding increased tumor uptake of IgBD-TRAIL by prolonging circulation. However, 131I-labeled mIgG preincubated with or without IgBD-TRAIL exhibited similar tumor uptake at 8 h post-injection (Figure S4C), indicating that IgBD-TRAIL binding did not induce accumulation of IgG in tumor grafts. In addition, in Balb/c mice, 131I-labeled mIgG (Table S1) and hIgG (Table S2) preincubated with or without IgBD-TRAIL showed similar tissue uptake profiles, demonstrating that IgBD-TRAIL bound to IgG did not alter the biodistribution of IgG.
IgBD-mediated IgG binding enhances the in vivo antitumor effect of TRAIL
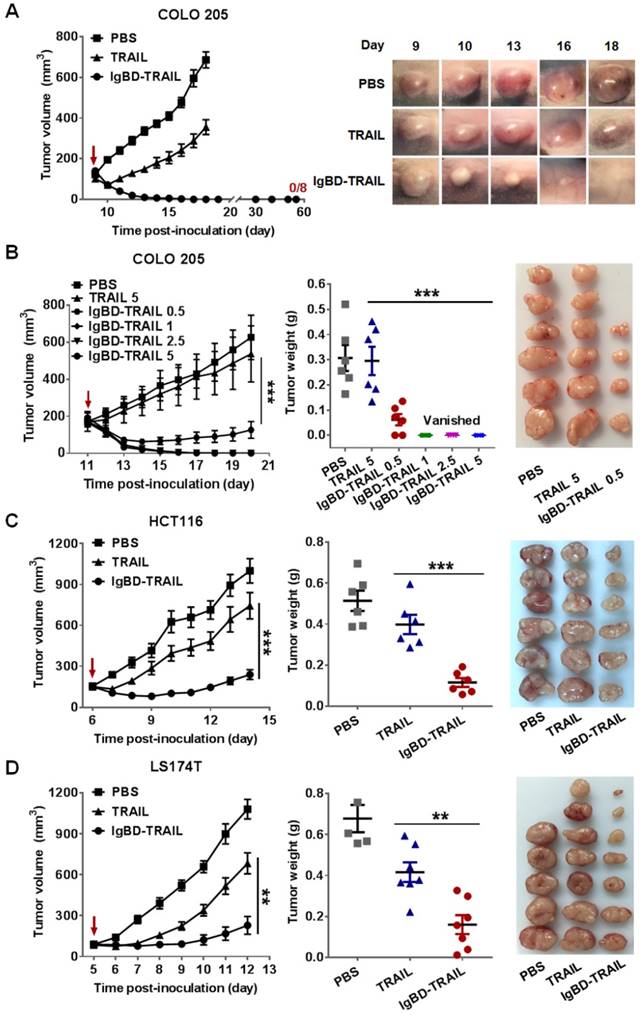
In vivo tumor growth suppression mediated by IgBD-TRAIL and TRAIL was first compared in mice bearing COLO 205 tumor grafts. Once the tumor volumes reached 100-150 mm3, the mice were intravenously injected with a single dose (5 mg/kg) of IgBD-TRAIL or TRAIL. As shown in Figure 5A, compared to PBS, injection of TRAIL retarded the tumor growth. However, injection of IgBD-TRAIL induced shrinking of tumor grafts from the second day post-injection. All tumor grafts in these mice (n=8) were eradicated within one week, and no tumor recurrence was observed in these mice within 50 days post-injection. In addition, all tumor grafts in mice injected with 2.5 and 1 mg/kg IgBD-TRAIL were also eradicated. The growth rate of tumor grafts in mice injected with 0.5 mg/kg IgBD-TRAIL was much slower than that of tumor grafts in mice treated with 5 mg/kg TRAIL (Figure 5B), indicating that the antitumor effect of IgBD-TRAIL was at least ten times greater than that of TRAIL. In mice bearing HCT 116 tumor grafts, tumor growth rate in IgBD-TRAIL-treated mice was significantly (p<0.001) slower than that of TRAIL-treated mice. At the end of this experiment, the average tumor volume and tumor mass of IgBD-TRAIL-treated mice were 238 ± 35 mm3 and 0.114 ± 0.022 g, compared to 743 ± 97 mm3 and 0.398 ± 0.047 g for TRAIL-treated mice (Figure 5C). Similarly, IgBD-TRAIL exerted greater tumor growth suppression than TRAIL in mice bearing LS174T tumor grafts (Figure 5D). These results demonstrated that IgBD-mediated IgG binding enhanced the antitumor effect of TRAIL.
In vivo antitumor effect of IgBD-TRAIL in Balb/c nude mice bearing COLO 205 (A, B), HCT116 (C), or LS174T (D) tumor grafts. Cells were subcutaneously implanted into Balb/c nude mice. Once the average tumor volume reached 100-150 mm3, mice were intravenously injected with IgBD-TRAIL or TRAIL (5 mg/kg) at the indicated times (A, C, D), or the mice were treated with 0.5, 1, 2.5 and 5 mg/kg IgBD-TRAIL, or 5 mg/kg TRAIL (B). PBS was used as control. The tumor volume was measured every day. At the end of the experiment, all tumor grafts were removed and weighed. Since COLO 205 tumor grafts of mice treated with IgBD-TRAIL were eradicated, representative photographs of the same tumor graft at different times are provided in (A). **p<0.01, *** p<0.001.
ABD-mediated albumin binding increases the in vivo antitumor effect of TRAIL
In addition to IgG, albumin is another long-acting protein in blood. To compare the efficacy of IgG binding and albumin binding in enhancing the antitumor effect of TRAIL, we also produced ABD-TRAIL by fusing albumin-binding affibody ABD at the N-terminus of TRAIL. As shown in Figure S5, ABD-TRAIL was expressed in E. coli and purified to homogeneity by affinity chromatography. ELISA demonstrated that ABD-TRAIL, but not TRAIL, showed dose-dependent binding to wells precoated with HSA or MSA (Figure 6A). SEC revealed a major protein peak that was larger than that observed with HSA and ABD-TRAIL in the mixture of ABD-TRAIL and HSA (Figure 6A). In contrast, two obvious protein peaks corresponding to HSA and TRAIL were illustrated in the mixture of TRAIL and HSA. These results indicated that ABD-TRAIL, but not TRAIL, bound HSA and formed the HSA-ABD-TRAIL complex. Biolayer interferometry revealed that the affinity of ABD-TRAIL for HSA was 1.68 nM (Figure 6B). These results demonstrated that ABD-TRAIL, but not TRAIL, could bind HSA with high affinity.
Albumin binding, pharmacokinetics, and antitumor effect of ABD-TRAIL. (A) Albumin binding of ABD-TRAIL analyzed by SEC and ELISA. (B) Affinity of ABD-TRAIL for HSA measured by biolayer interferometry. ABD-TRAIL was captured by anti-His tag antibody-coated biosensor prior to association in solution containing increasing concentration of HSA. (C) FcRn binding of HSA preincubated with ABD-TRAIL and time-dependent clearance of ABD-TRAIL. (D)Time-dependent decrease of cytotoxicity of ABD-TRAIL. (E) Affinity of ABD-TRAIL for DR4-Fc and DR5-Fc. (F) In vivo antitumor effect. ABD-TRAIL (5 mg/kg) was intravenously into mice bearing COLO 205 tumor grafts at the indicated times. The tumor volume was measured every day. At the end of the experiment, tumor grafts were removed and weighed.
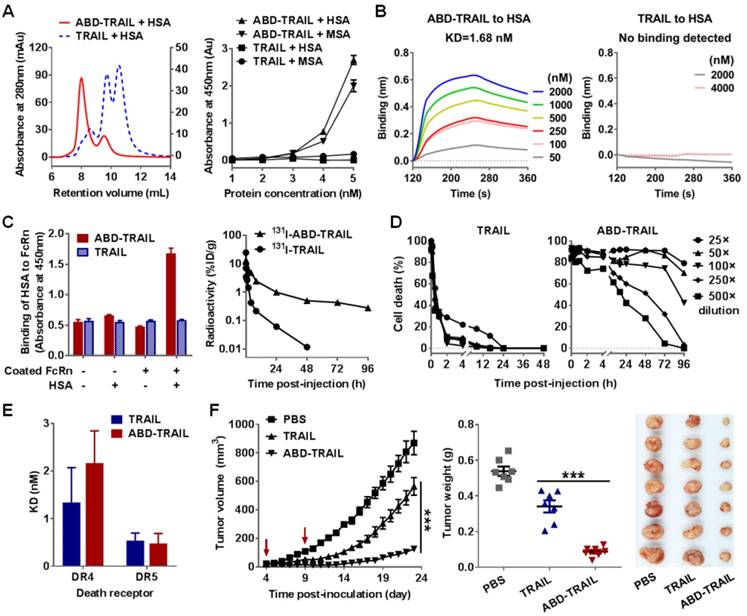
Since FcRn binding contributed to the long circulation of albumin, the FcRn binding of albumin bound to ABD-TRAIL was measured by ELISA. As shown in Figure 6C, in the presence of HSA, ABD-TRAIL, but not TRAIL, could bind FcRn, indicating that ABD-TRAIL bound to HSA did not interfere with the FcRn binding of HSA. Moreover, both radioactivity quantitation (Figure 6C) and cytotoxicity assays (Figure 6D) demonstrated that the blood clearance rate of ABD-TRAIL was definitely slower than that of TRAIL. The serum half-life of ABD-TRAIL was approximately 29.6±2.4 h, compared to 18.5±2.2 min for TRAIL. Accordingly, tumor grafts in mice administered ABD-TRAIL grew slower than those in mice treated with TRAIL. At the end of the experiment, the average tumor mass of ABD-TRAIL- and TRAIL-treated mice were 0.088±0.028 g and 0.34± 0.089 g, respectively (Figure 6F). These results demonstrated that ABD-mediated albumin binding extended the serum half-life and thus enhanced the in vivo antitumor effect of TRAIL. In fact, ABD-TRAIL and TRAIL showed similar receptor binding (Figure 6E and Figure S6A), cytotoxicity (Figure S6B) and caspase activation ability (Figure S6C), suggesting that the enhanced antitumor effect of ABD-TRAIL was predominantly attributed to albumin binding-mediated serum half-life prolongation.
IgBD-mediated IgG binding is superior to ABD-mediated albumin binding in enhancing the in vivo antitumor effect of TRAIL
The antitumor effects of IgBD-TRAIL and ABD-TRAIL administered at different times post- inoculation were compared in mice bearing COLO 205 tumor grafts. As shown in Figure 7A, when injected at an early time post-inoculation (tumor volume ~ 50 mm3), ABD-TRAIL showed greater tumor suppression than TRAIL. At the end of the experiment, the average tumor mass of ABD-TRAIL-treated mice was 0.039 ± 0.015 g, which was significantly (P<0.05) lower than that of TRAIL-treated mice (0.173 ± 0.047 g). Nevertheless, the injection of ABD-TRAIL only retarded the growth of tumor grafts in these mice. However, all tumor grafts in mice (n=6) were eradicated within one week after injection of the same amount of IgBD-TRAIL. Moreover, when injected at a later time (tumor volume ~150 mm3), ABD-TRAIL and TRAIL showed similar tumor growth suppression. At the end of this experiment, the average tumor masses of ABD-TRAIL- and TRAIL-treated mice were 0.272 ± 0.038 g and 0.267 ± 0.071 g, respectively (Figure 7B). However, all tumor grafts in mice (n=6) were eradicated after injection of the same amount of IgBD-TRAIL. These results demonstrated that the in vivo antitumor effect of intravenously injected IgBD-TRAIL is superior to that of ABD-TRAIL.
IgBD-TRAIL is superior to ABD-TRAIL for in vivo antitumor effect. (A, B) Tumor suppression mediated by TRAIL proteins (5 mg/kg) injected at an early time (A) or a late time (B). (C) Comparison of calculated serum half-lives. (D) Comparison of tumor uptake measured by optical imaging. (E) Comparison of apoptosis induction illustrated by TUNEL. (F) Cytotoxicity of IgBD-TRAIL and ABD-TRAIL preincubated with or without hIgG or HSA. TRAIL proteins were preincubated with or without hIgG or HSA at a molar ratio of 1:1 for 0.5 h at room temperature prior to addition into COLO 205 tumor cells. (G) Comparison of affinity for death receptor between IgG-IgBD-TRAIL complex and HSA-ABD-TRAIL complex.
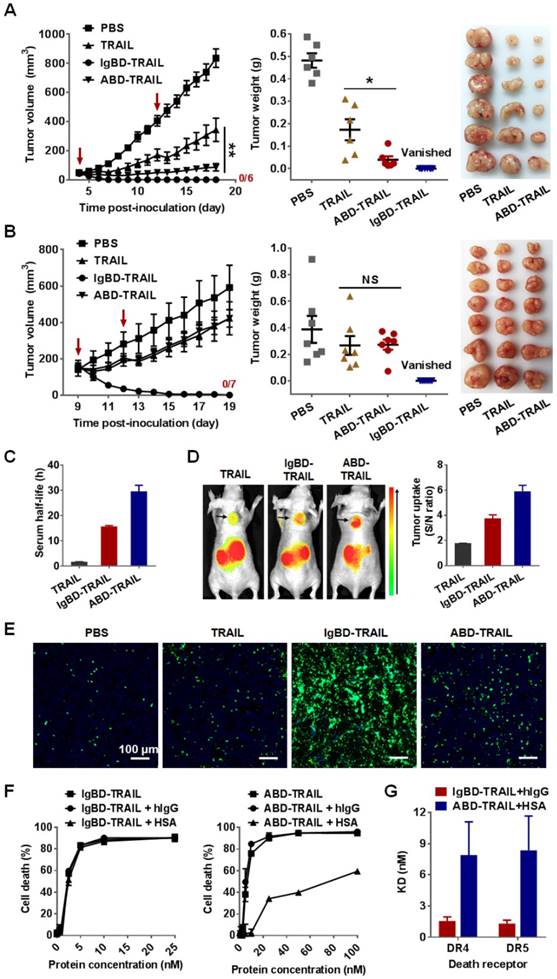
The difference between IgBD-TRAIL and ABD-TRAIL for in vivo antitumor effects might be predominantly attributed to their difference in serum half-life and cytotoxicity. As shown in Figure 7C, the calculated serum half-life of ABD-TRAIL (29.6±2.4 h) is longer than that of IgBD-TRAIL (15.7±0.39 h), indicating that ABD-mediated albumin binding is more efficient than IgBD-mediated IgG binding in extending circulation of TRAIL. Accordingly, optical imaging demonstrated that the tumor uptake of ABD-TRAIL was higher than that of IgBD-TRAIL (Figure 7D). Unexpectedly, intravenously injected ABD-TRAIL induced less apoptosis than did IgBD-TRAIL (Figure 7E and Figure S7), suggesting that the cytotoxicity of ABD-TRAIL might be weaker than that of IgBD-TRAIL. As shown in Figure 7F, in the absence of albumin and IgG, IgBD-TRAIL and ABD-TRAIL exerted similar cytotoxicity in COLO 205 tumor cells. IgBD-TRAIL preincubated with or without IgG exhibited similar cytotoxicity. However, the cytotoxicity of ABD-TRAIL preincubated with albumin was 10 times lower than that of ABD-TRAIL preincubated without albumin (Figure 7F), suggesting that these TRAIL proteins might exert cytotoxicity in complex form. In fact, the affinity of the HSA-ABD-TRAIL complex for death receptors and cell binding ability were lower than those of the IgG-IgBD-TRAIL complex (Figure 7G and Figure S8-9). These results demonstrated that steric hindrance between HSA and TRAIL exhibited a greater reduction in the death receptor binding and cytotoxicity of TRAIL than the hindrance between IgG and TRAIL.
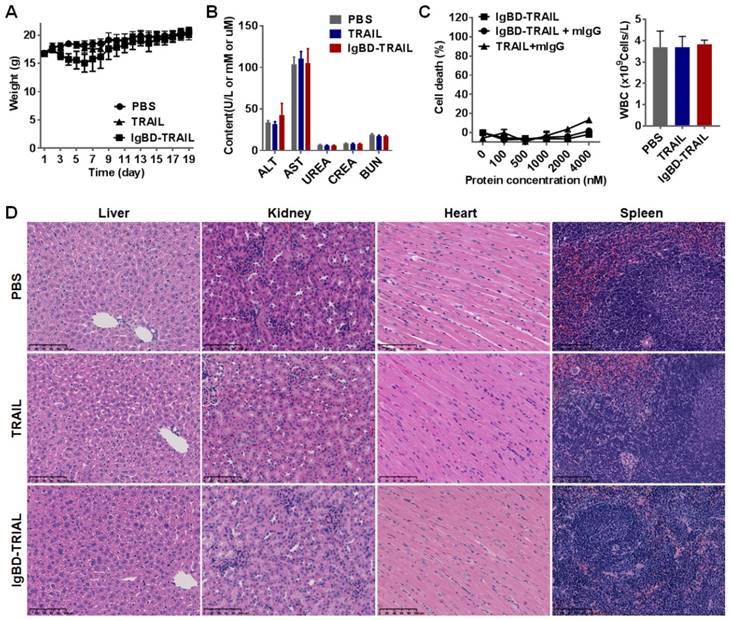
IgBD-TRAIL had no obvious acute toxicity
As shown in Figure 8A, intravenously injected IgBD-TRAIL only induced a slight and transient decrease in body weights. The biochemical indicators for function of liver (ALT, AST) and kidney (urea, creatinine, BUN) of IgBD-TRAIL-treated mice were similar to that of TRAIL- and PBS-treated mice (Figure 8B). In addition, no obvious abnormal structures were observed in the liver, kidney, heart, spleen, lung, muscle, and brain derived from IgBD-TRAIL-treated mice (Figure 8D and Figure S10). The number of WBCs of IgBD-TRAIL-treated mice was similar to that of TRAIL- and PBS-treated mice (Figure 8C, right). IgBD-TRAIL preincubated with mIgG showed no obvious cytotoxicity to mouse WBCs even at a high concentration of 4000 nM (Figure 8C, left). These results demonstrated that IgBD-TRAIL bound to IgG had no acute toxicity in normal WBCs and other normal organs.
Evaluation of the short term acute toxicity of IgBD-TRAIL in mice. Mice were intravenously injected with 10 mg/kg IgBD-TRAIL or TRAIL every other day for a total eight injections. PBS was used as control. (A) The body weight of mice. (B) Biochemical indicators for function of liver (AST, ALT), kidney (urea, creatinine, BUN). (C) In vitro (left) and in vivo (right) cytotoxicity of IgBD-TRAIL. To measure the in vitro cytotoxicity, IgBD-TRAIL or TRAIL preincubated with mIgG was added into mouse WBCs followed by measuring the surviving cells using CCK-8. To monitor the in vivo cytotoxicity, WBCs from mice injected with IgBD-TRAIL or TRAIL were counted at the end of this experiment. (D) Histochemistry of tissues derived from mice injected with IgBD-TRAIL or TRAIL.
Discussion
Owing to their high specificity and efficacy, many biopharmaceuticals have been developed in recent years. However, the clinical application of non-antibody proteins is typically limited by their short serum half-lives. Controlled release is an important strategy for improving the pharmacokinetics of drugs. Due to the involvement of harsh conditions that might be detrimental for the structure and function of proteins, conventional controlled release systems designed for small molecule drugs are typically improper for protein drug delivery. Affinity-controlled release is an attractive strategy, as proteins containing binding partners could easily be loaded onto their affinity carrier by mixing under physiological conditions [2,12]. The identification of proper binding partners and preparation of the affinity carrier are crucial for the construction of an affinity-controlled release system [3]. In previous studies, natural binding partners combined with their affinity carriers have been widely used for protein delivery [11]. However, these systems might be limited by the low affinity (μM) of binding partners. In addition, the biocompatibility of exogenous carriers should be considered. To construct a novel affinity-controlled release system, it might be better to use high-affinity (nM) binding partners and endogenous carriers.
Due to FcRn-mediated recycling, albumin and IgG exhibit extraordinarily long (~3 weeks) serum half-lives in humans [23]. Recently, numerous affibodies with high affinity (nM) for albumin and IgG have been identified [22,26]. Once incorporated into a protein, these affibodies might mediate binding of the protein to endogenous albumin or IgG followed by slow affinity-controlled release. In a previous study, we fused an albumin-specific affibody ABD to the N-terminus of TRAIL to produce the fusion protein ABD-TRAIL. It was found that intravenously injected ABD-TRAIL could bind endogenous albumin, thus exhibiting a prolonged serum half-life and an enhanced antitumor effect, indicating that endogenous albumin could be used as affinity carrier for affinity-controlled release of protein [34]. However, the amplitude of enhancement of the antitumor effect was limited by the steric hindrance between albumin and TRAIL, which reduced the cytotoxicity of TRAIL by 4-6 times. Surprisingly, in a preliminary study, we found that binding to IgG had little impact on the cytotoxicity of TRAIL, which prompted our interest in evaluating the potential use of endogenous IgG as an affinity carrier for the affinity-controlled release of TRAIL.
To construct the IgG-based affinity-controlled release system of TRAIL, an IgG-binding domain, IgBD, was genetically fused at the N-terminus of TRAIL. The resulting fusion protein IgBD-TRAIL exhibited high affinity for both mIgG and hIgG (Figure 1B, E). SEC (Figure 1C) and SDS-PAGE (Figure S1B) verified that IgBD-TRAIL formed a complex with IgG once mixed together at pH 7.4. Notably, the IgG-IgBD-TRAIL complex could be formed within 5 min after mixing at room temperature, suggesting that intravenously injected IgBD-TRAIL could rapidly bind endogenous IgG. In addition, IgBD-TRAIL could also form a complex with IgG at pH 6.0 (Figure 3A and Figure S2A) without interfering with the FcRn-binding ability of IgG (Figure 3B and Figure S2B). After incubation with endothelial cells, IgBD-TRAIL bound to IgG was co-localized with transferrin (Figure 3C), an indicator of endosomes [39], suggesting that IgBD-TRAIL could bind FcRn in endosomes by bound IgG, thus escaping lysosome degradation. Accordingly, recycling of IgBD-TRAIL was observed in an endothelial cell model (Figure 3D). These results demonstrated the involvement of FcRn-mediated recycling in the cellular transport of IgBD-TRAIL bound to IgG. Consequently, compared to TRAIL without IgG binding ability, intravenously injected IgBD-TRAIL exhibited a prolonged (50-60 times) serum half-life (Figure 4A-B), increased (4-7 times) tumor uptake (Figure 4C-D), and enhanced antitumor effect (Figure 5). These results demonstrated that the affinity-controlled release of TRAIL could be achieved by using endogenous IgG as an affinity carrier.
Furthermore, we compared the efficacy of IgG- and albumin-based affinity-controlled release in improving the pharmacokinetics and enhancing the in vivo antitumor effect of TRAIL. According to a previous study [34], an albumin binding affibody ABD was fused at the N-terminus of TRAIL to produce ABD-TRAIL (Figure S5). As expected, owing to its high affinity for albumin (Figure 6A-B), intravenously injected ABD-TRAIL showed a prolonged serum half-life (Figure 6C-D) and enhanced in vivo antitumor effect (Figure 6F). These results demonstrated that the affinity-controlled release of TRAIL could be achieved by using endogenous albumin as an affinity carrier. Unexpectedly, intravenously injected ABD-TRAIL only retarded the growth of COLO 205 tumor grafts, whereas all tumor grafts were eradicated by the same amount of IgBD-TRAIL (Figure 7A-B). In fact, the serum half-life of ABD-TRAIL was approximately 2 times longer than that of IgBD-TRAIL (Figure 7C). The tumor uptake of ABD-TRAIL was greater than that of IgBD-TRAIL (Figure 7D). Although the IC50 of ABD-TRAIL was comparable to that of IgBD-TRAIL (Figure 7F), the IC50 of HSA-ABD-TRAIL was more than 10 times lower than that of IgG-IgBD-TRAIL. The difference between ABD-TRAIL and IgBD-TRAIL suggested that these TRAIL proteins might exert cytotoxicity in complex form. Further pharmacokinetics assay demonstrated that 131I-labeled IgBD-TRAIL was cleared faster than 131I-labeled mIgG (Figure S11), suggesting that the IgBD-TRAIL could be released, but at a lower rate, from endogenous IgG. Definitely, in HSA-ABD-TRAIL complex, the steric hindrance between HSA and ABD-TRAIL was detrimental to the receptor and cell binding of TRAIL (Figure 7G and Figure S9). However, IgG binding had little impact on the biological activity of IgBD-TRAIL (Figure 7F-G and Figure S9), which might be determental to the superior antitumor effect of intravenously injected IgBD-TRAIL.
Since IgG plays an important role in immunoreactions, the impact of bound IgBD-TRAIL on IgG should be considered. Since IgBD-TRAIL exists as a trimer under physiological conditions, it is important to determine whether binding to IgBD-TRAIL would induce aggregation of IgG, which might accelerate the blood clearance of IgG. As shown in Figure 1C, when IgBD-TRAIL (calculated as trimer) was mixed with hIgG at a molar ratio of 1:1, a unique novel protein peak was observed on the column of SEC. Once the ratio was varied from 1:1 to 2:1 or 1:2, excessive IgBD-TRAIL or hIgG was detectable. These results demonstrated that only one hIgG molecule was bound to one trimer of IgBD-TRAIL, suggesting that intravenously injected IgBD-TRAIL would not induce aggregation of endogenous IgG. A pharmacokinetics assay demonstrated that the blood clearance curves of 131I-labeled IgG preincubated with or without IgBD-TRAIL were highly similar (Figure S4A-B), suggesting that intravenously injected IgBD-TRAIL would not accelerate the elimination of endogenous IgG. In addition, 131I-labeled IgG preincubated with or without IgBD-TRAIL exhibited a similar biodistribution profile (Table S1-2), suggesting that intravenously injected IgBD-TRAIL would not induce accumulation of IgG in normal organs and tissues. Moreover, the Fab domain of an antibody is responsible for antigen binding. Since IgBD is Fab-specific [35], we determined the impact of IgBD-TRAIL on antigen binding of antibodies. As shown in Figure S12, the positive rates of endothelial cells stained with antibody against CD31 preincubated with or without IgBD-TRAIL were nearly identical (99.8% vs. 99.5%), suggesting that intravenously injected IgBD-TRAIL had little impact on the antigen binding of endogenous antibody. These results demonstrated that using endogenous IgG as an affinity carrier prolonged the serum half-life of TRAIL without altering the function and metabolism of IgG.
In addition to albumin and IgG binding, fusion to albumin [29] or the Fc domain of IgG [30] has also been used to prolong the serum half-life of TRAIL. The serum half-life of IgBD-TRAIL (15.7±0.39 h) was comparable to that of HSA-TRAIL (~15 h) [29] in mice. As the affinity of the Fc domain for FcRn is much lower than that of intact antibody [44], the serum half-life of TRAIL-Fc (~1.5 h) [30] was approximately 10 times shorter than that of IgBD-TRAIL (15.7±0.39 h). Most importantly, TRAIL fused to albumin or Fc domain was typically limited by the low yield (<1 mg/L) of recombinant proteins required for expression in mammalian cells. However, TRAIL fused to the small affibodies has the advantage of high yield (20-30 mg/L) in E. coli. These results demonstrated that small IgBD-mediated IgG binding is a simple but efficient strategy for affinity-controlled release of TRAIL. The release profile of TRAIL might be easily tuned by using IgBDs with different affinity for IgG.
Conclusions
IgBD fusion endows TRAIL with high affinity for IgG. Once this fusion protein enters the vasculature, fused IgBD mediates binding of TRAIL to endogenous IgG followed by a slow release. Consequently, TRAIL fused to IgBD exhibited a prolonged serum half-life and thus exerted superior in vivo antitumor effects, demonstrating that using endogenous IgG as affinity carrier is a simple but efficient strategy for the controlled release of TRAIL. Since most non-antibody protein drugs are limited by their short half-lives, endogenous IgG-based affinity-controlled release might be a novel approach for improving their pharmacokinetics.
Supplementary Material
Supplementary figures and tables.
Abbreviations
TRAIL: tumor necrosis factor-related apoptosis-inducing ligand; IgG: immunoglobulin G; hIgG: human immunoglobulin G; mIgG: mouse immunoglobulin G; IgBD: IgG-binding affibody; ABD: albumin-binding affibody; ECM: extracellular matrix; IPTG: isopropyl β-D-thiogalactoside; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; ELISA: enzyme-linked immunosorbent assay; PBS: phosphate-buffered saline; FcRn: neonatal Fc receptor; HRP: horseradish peroxidase; GLP-1: glucagon-like peptide-1; ATCC: american type culture collection; SEC: size exclusion chromatography; HUVEC: human umbilical vein endothelial cell; WBC: white blood cell; AST: glutamic-oxaloacetic transaminase; ALT: glutamic-pyruvic transaminase; BUN: blood urea nitrogen; ANOVA: one-way analysis of variance; SEM: standard error of the mean. 2-ME: β-mercaptoethanol; HSA: human serum albumin; MSA: mouse serum albumin.
Acknowledgements
This study was funded by the National Natural Science Foundation of China (81273419 and 81573336) and the National Key Clinical Program.
Ethical approval
All applicable institutional guidelines for the care and use of animals were followed.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wash G. Biopharmaceutical benchmarks 2014. Nat Biotech. 2014;32:992-1000
2. Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov. 2014;13:655-72
3. Pakulska MM MS, Shoichet MS. Designer protein delivery: from natural to engineered affinity-controlled release systems. Science. 2016;351:aac4750
4. Vaishya R, Khurana V, Patel S, Mitra AK. Long-term delivery of protein therapeutics. Expert Opin Drug Deliv. 2015;12:415-40
5. Shah DK. Pharmacokinetic and pharmacodynamic considerations for the next generation protein therapeutics. J Pharmacokinet Pharmacodyn. 2015;42:553-71
6. Yun YH, Lee BK, Park K. Controlled drug delivery: historical perspective for the next generation. J Control Release. 2015;219:2-7
7. Doppalapudi S, Jain A, Domb AJ, Khan W. Biodegradable polymers for targeted delivery of anti-cancer drugs. Expert Opin Drug Deliv. 2016;13:891-909
8. Siepmann JS. et al. Swelling controlled drug delivery systems. In Fundamentals and Applications of Controlled Release Drug Delivery; Siepmann, J, Siegel, R A, Rathbone, M J, Eds; Springer: New York. 2012:153-70
9. Martinez Rivas CJ, Tarhini M, Badri W, Miladi K, Greige-Gerges H, Nazari QA. et al. Nanoprecipitation process: from encapsulation to drug delivery. Int J Pharm. 2017;532:66-81
10. Bahrami B, Hojjat-Farsangi M, Mohammadi H, Anvari E, Ghalamfarsa G, Yousefi M. et al. Nanoparticles and targeted drug delivery in cancer therapy. Immun Lett. 2017;190:64-83
11. Vulic K, Shoichet MS. Affinity-based drug delivery systems for tissue repair and regeneration. Biomacromolecules. 2014;15:3867-80
12. Delplace V, Obermeyer J, Shoichet MS. Local affinity release. ACS Nano. 2016;10:6433-6
13. An B, Lin YS, Brodsky B. Collagen interactions: drug design and delivery. Adv Drug Deliv Rev. 2016;97:69-84
14. Vulic K, Pakulska MM, Sonthalia R, Ramachandran A, Shoichet MS. Mathematical model accurately predicts protein release from an affinity-based delivery system. J Control Release. 2015;197:69-77
15. Stahl S, Graslund T, Eriksson Karlstrom A, Frejd FY, Nygren PA, Lofblom J. Affibody molecules in biotechnological and medical applications. Trends Biotechnol. 2017;35:691-712
16. Richter A, Eggenstein E, Skerra A. Anticalins: exploiting a non-Ig scaffold with hypervariable loops for the engineering of binding proteins. FEBS Lett. 2014;588:213-8
17. Lipovsek D. Adnectins: engineered target-binding protein therapeutics. Protein Eng Des Sel. 2011;24:3-9
18. Wu X, Chen J, Wu M, Zhao JX. Aptamers: active targeting ligands for cancer diagnosis and therapy. Theranostics. 2015;5:322-44
19. Park K. Enhanced antitumor effects of hTRAIL by binding to endogenous albumin. J Control Release. 2016;228:206
20. Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol. 2011;22:868-76
21. Larsen MT, Kuhlmann M, Hvam ML, Howard KA. Albumin-based drug delivery: harnessing nature to cure disease. Mol Cell Ther. 2016;4:3
22. Nilvebrant J, Hober S. The albumin-binding domain as a scaffold for protein engineering. Comput Struct Biotechnol J. 2013;6:e201303009
23. Kontermann RE. Strategy to extending half-life of recombinant antibody. BioDrugs. 2009;23:93-109
24. Hutt M, Farber-Schwarz A, Unverdorben F, Richter F, Kontermann RE. Plasma half-life extension of small recombinant antibodies by fusion to immunoglobulin-binding domains. J Biol Chem. 2012;287:4462-9
25. Sockolosky J, Kivimäe S, Szoka FC. Fusion of a short peptide that binds immunoglobulin G to a recombinant protein substantially increases its plasma half-life in mice. PloS One. 2014;9:e102566
26. Mouratou B, Behar G, Pecorari F. Artificial affinity proteins as ligands of immunoglobulins. Biomolecules. 2015;5:60-75
27. Kruljec N, Bratkovic T. Alternative affinity ligands for immunoglobulins. Bioconjug Chem. 2017;28:2009-30
28. Micheau O, Shirley S, Dufour F. Death receptors as targets in cancer. Br J Pharmacol. 2013;169:1723-4
29. Muller N, Schneider B, Pfizenmaier K, Wajant H. Superior serum half life of albumin tagged TNF ligands. Biochem Biophys Res Commun. 2010;396:793-9
30. Wang H, Davis JS, Wu X. Immunoglobulin Fc domain fusion to TRAIL significantly prolongs its plasma half-life and enhances its antitumor activity. Mol Cancer Ther. 2014;13:643-50
31. Chae SY, Kim TH, Park K, Jin CH, Son S, Lee S. et al. Improved antitumor activity and tumor targeting of NH(2)-terminal-specific PEGylated tumor necrosis factor-related apoptosis-inducing ligand. Mol Cancer Ther. 2010;9:1719-29
32. De Miguel D, Gallego-Lleyda A, Ayuso JM, Erviti-Ardanaz S, Pazo-Cid R, del Agua C. et al. TRAIL-coated lipid-nanoparticles overcome resistance to soluble recombinant TRAIL in non-small cell lung cancer cells. Nanotechnology. 2016;27:185101
33. Nair PM, Flores H, Gogineni A, Marsters S, Lawrence DA, Kelley RF. et al. Enhancing the antitumor efficacy of a cell-surface death ligand by covalent membrane display. Proc Natl Acad Sci U S A. 2015;112:5679-84
34. Li R, Yang H, Jia D, Nie Q, Cai H, Fan Q. et al. Fusion to an albumin-binding domain with a high affinity for albumin extends the circulatory half-life and enhances the in vivo antitumor effects of human TRAIL. J Control Release. 2016;228:96-106
35. Unverdorben F, Hutt M, Seifert O, Kontermann RE. A Fab-selective immunoglobulin-binding domain from streptococcal protein G with improved half-life extension properties. PloS One. 2015;10:e0139838
36. Jia D, Yang H, Tao Z, Wan L, Cheng J, Lu X. Preparation and characterization of a novel variant of human tumor necrosis factor-related apoptosis-inducing ligand from the rhesus monkey, Macaca mulatta. Appl Micro Biotech. 2016;100:3035-47
37. Hofstrom C, Orlova A, Altai M, Wangsell F, Graslund T, Tolmachev V. Use of a HEHEHE purification tag instead of a hexahistidine tag improves biodistribution of affibody molecules site-specifically labeled with (99m)Tc, (111)In, and (125)I. J Med Chem. 2011;54:3817-26
38. Stork R, Muller D, Kontermann RE. A novel tri-functional antibody fusion protein with improved pharmacokinetic properties generated by fusing a bispecific single-chain diabody with an albumin-binding domain from streptococcal protein G. Protein Eng Des Sel. 2007;20:569-76
39. Goebl NA, Babbey CM, Datta-Mannan A, Witcher DR, Wroblewski VJ, Dunn KW. Neonatal Fc receptor mediates internalization of Fc in transfected human endothelial cells. Mol Biol Cell. 2008;19:5490-505
40. Fan Q, Cai H, Yang H, Li L, Yuan C, Lu X. et al. Biological evaluation of 131I- and CF750-labeled Dmab(scFv)-Fc antibodies for xenograft imaging of CD25-positive tumors. BioMed Res Int. 2014;2014:459676
41. Wang F, Chen L, Zhang R, Chen Z, Zhu L. RGD peptide conjugated liposomal drug delivery system for enhance therapeutic efficacy in treating bone metastasis from prostate cancer. J Control Release. 2014;196:222-33
42. Rath T, Baker K, Dumont JA, Peters RT, Jiang H, Qiao SW. et al. Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Crit Rev Biotechnol. 2015;35:235-54
43. Pyzik M, Rath T, Lencer WI, Baker K, Blumberg RS. FcRn: The architect behind the immune and nonimmune functions of IgG and albumin. J Immunol. 2015;194:4595-603
44. Unverdorben F, Richter F, Hutt M, Seifert O, Malinge P, Fischer N. et al. Pharmacokinetic properties of IgG and various Fc fusion proteins in mice. MAbs. 2016;8:120-8
Author contact
![]() Corresponding author: Xiaofeng Lu (xiaofengluedu.cn)
Corresponding author: Xiaofeng Lu (xiaofengluedu.cn)