Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(9):2447-2458. doi:10.7150/thno.23002 This issue Cite
Research Paper
Soluble CD146 is a predictive marker of pejorative evolution and of sunitinib efficacy in clear cell renal cell carcinoma
Maeva Dufies1, Marie Nollet2, Damien Ambrosetti3, Wael Traboulsi2, Julien Viotti4, Delphine Borchiellini5, Renaud Grépin1, Julien Parola6, Sandy Giuliano1, Dominique Helley-Russick7, Karim Bensalah8, Alain Ravaud9, Jean-Christophe Bernhard10, Renaud Schiappa4, Nathalie Bardin2, Françoise Dignat-George2, Nathalie Rioux-Leclercq11, Stephane Oudard7, Sylvie Négrier12, Jean-Marc Ferrero5, Emmanuel Chamorey4, Marcel Blot-Chabaud2*, Gilles Pagès6 ![]() *
*
1. Centre Scientifique de Monaco, Biomedical Department, 8 Quai Antoine Ier, MC-98000 Monaco, Principality of Monaco.
2. Aix Marseille Univ, INSERM 1263, INRA 1260, C2VN, Marseille, France.
3. University Cote d'Azur, Nice University Hospital, Department of Pathology,
4. Centre Antoine Lacassagne, Statistic department, Nice, France
5. Centre Antoine Lacassagne, Clinical research department Nice, France
6. University Cote d'Azur, Institute for research on cancer and aging of Nice, CNRS UMR 7284; INSERM U1081, Centre Antoine Lacassagne, France.
7. European Hospital Georges Pompidou, Paris, France
8. Centre Hospitalier Universitaire (CHU) de Pontchaillou Rennes, service d'urologie
9. Bordeaux University, Bordeaux University Hospital (CHU), Medical Oncology department, Bordeaux, France.
10. Bordeaux University, Bordeaux University Hospital (CHU), Urology department, Bordeaux, France.
11. Rennes University, Rennes University Hospital, Department of Pathology, Rennes, France
12. Centre Léon Bérard, Lyon, France
*MBC and GP co-directed the work
Received 2017-9-25; Accepted 2017-12-14; Published 2018-3-28
Abstract

The objective of the study was to use CD146 mRNA to predict the evolution of patients with non-metastatic clear cell renal cell carcinoma (M0 ccRCC) towards metastatic disease, and to use soluble CD146 (sCD146) to anticipate relapses on reference treatments by sunitinib or bevacizumab in patients with metastatic ccRCC (M1).
Methods: A retrospective cohort of M0 patients was used to determine the prognostic role of intra-tumor CD146 mRNA. Prospective multi-center trials were used to define plasmatic sCD146 as a predictive marker of sunitinib or bevacizumab efficacy for M1 patients.
Results: High tumor levels of CD146 mRNA were linked to shorter disease-free survival (DFS) and overall survival (OS). ccRCC patients from prospective cohorts with plasmatic sCD146 variation <120% following the first cycle of sunitinib treatment had a longer progression-free survival (PFS) and OS. The plasmatic sCD146 variation did not correlate with PFS or OS for the bevacizumab-based treatment. In vitro, resistant cells to sunitinib expressed high levels of CD146 mRNA and protein in comparison to sensitive cells. Moreover, recombinant CD146 protected cells from the sunitinib-dependent decrease of cell viability.
Conclusion: CD146/sCD146 produced by tumor cells is a relevant biological marker of ccRCC aggressiveness and relapse on sunitinib treatment.
Keywords: sCD146, clear cell renal cell carcinoma, sunitinib, predictive marker, plasma
Introduction
Metastatic clear cell renal cell carcinomas (ccRCC) are highly angiogenic tumors bearing a mutation, deletion or methylation in the VHL gene. Inactivation of VHL leads to over-expression of VEGF. Therefore, anti-angiogenic therapies targeting the VEGF/VEGFR pathway represent a paradigm for the treatment of ccRCC. Bevacizumab in combination with interferon alpha (IFN) was the first anti-angiogenic to be used [1] but sunitinib, a multi kinase inhibitor (TKI) targeting the VEGF, PDGF, CSF1 receptors, c-KIT, FLT3 and RET is currently the treatment of reference in the first-line [2]. However, patients ineluctably relapse. At progression on sunitinib, patients receive other TKI such as axitinib [3], pazopanib [4, 5], cabozantinib [6], inhibitors of immune checkpoints such as nivolumab [7] or mTOR inhibitors [7]. The efficacy of sunitinib is very heterogeneous. Some patients are refractory and die rapidly, most of them show a transient response and a minority of patients are responders for a very long period of time [1, 2]. These results are probably linked to the huge heterogeneity of ccRCC [8]. The necessity of an early predictive marker of sunitinib efficacy represents a therapeutic challenge to rapidly adapt treatment and propose alternative treatments among those available.
CD146 (or MUC-18, MCAM) was recently described as a new factor involved in tumor angiogenesis [9]. It is a membrane glycoprotein present on endothelial cells but is also neo-expressed in several tumors including lung, melanoma, pancreas, prostate, breast, stomach and renal tumors [10-12]. In prostate cancer, neo-expression results from hypermethylation of the CD146 promoter [13]. CD146 was described as a co-receptor for VEGFR2 in tumor angiogenesis [14, 15], suggesting a synergistic role of both molecules in the development of tumor vascularization. In addition to the membrane- anchored form of CD146, we identified a soluble form (sCD146), which is generated by the shedding of the membrane-associated form [16, 17]. This soluble form is secreted by tumors expressing CD146 and displays both autocrine effects on proliferation and survival of cancer cells, and paracrine effects on tumor angiogenesis [18]. These effects are mediated through binding of sCD146 to the p80 isoform of angiomotin [18]. This sCD146 receptor is expressed on endothelial and tumor cells [19, 20] and inhibits the YAP oncoprotein [21]. Plasmatic sCD146 concentrations are increased in several cancers [18, 22], indicating its major role in the development of the pathology.
We hypothesized that relapses on sunitinib occurring in ccRCC may involve an increase in membrane CD146 and consequently an increase in sCD146 production. Therefore, the detection of increased levels of plasmatic sCD146 could represent an early predictive marker of sunitinib failure, allowing a rapid switch to a second-line treatment before visualization of relapse by imaging.
Materials and Methods
Description of the patients
The studies were approved by the ethics committee at each participating Center and were in agreement with the International Conference on Harmonization of Good Clinical Practice Guideline. The management of non-metastatic (M0) and metastatic (M1) patients is summarized in Figure S1.
M0 patients for qPCR analysis
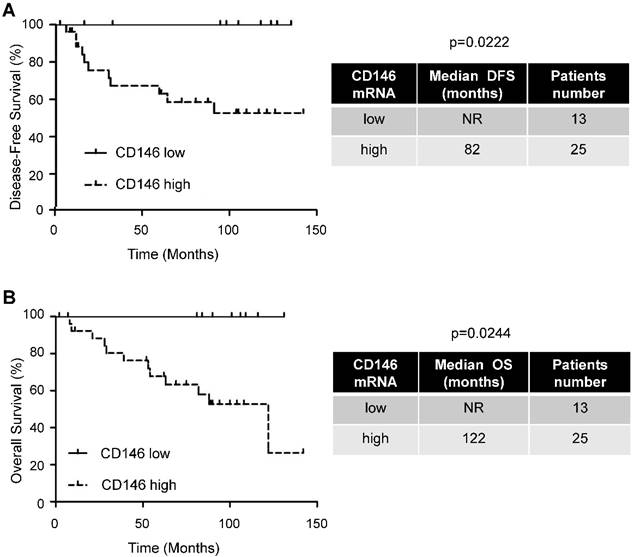
Primary tumor samples (tumor section) of M0 ccRCC patients were obtained from the Rennes University hospital [23]. The disease-free survival (DFS) and overall survival (OS) were calculated from patient subgroups with CD146 mRNA levels that were less or greater than the first quartile value (Figure 1 and Table S1).
The amount of intra-tumor CD146 mRNA correlated with pejorative evolution of M0 patients. Kaplan-Meier analysis of DFS (A) or OS (B) of M0 patients. DFS and OS were calculated from patient subgroups with CD146 mRNA levels that were less or greater than the first quartile. Statistical significance (p value) is indicated (see Table S1).
Neo-adjuvant patients for qPCR analysis
Samples (tumor section) were obtained from Nice, Bordeaux and Monaco Hospitals. The patients' characteristics have already been described [24]. Patients were treated for at least two months before surgery (Figure 4 and Table S3).
Patients included in clinical trials
Eligible patients for SUVEGIL, TORAVA and PREINSUT trials were at least 18 years of age and had metastatic ccRCC confirmed by histology, with the presence of measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Patients had not received previous systemic therapy for ccRCC and were eligible for sunitinib or bevacizumab combined with IFN treatment in the first-line setting. Patients were ineligible if they had symptomatic or uncontrolled brain metastases, an estimated lifetime less than three months, uncontrolled hypertension or clinically significant cardiovascular events (heart failure, prolongation of the QT interval), or a history of another primary cancer. All patients gave written informed consent (Table 1, Figures 2 and 3).
Characteristics of the patients included in the clinical trials. (See Figures 2 and 3).
| COHORT Treatment | Prospective cohort Sunitinib | Prospective cohort Bevacizumab | Prospective cohort of validation Sunitinib neoadjuvant |
|---|---|---|---|
| Trials (number of patients) | SUVEGIL (24) TORAVA (12) | TORAVA (34) | PREINSUT (13) |
| Number of patients | 36 | 34 | 13 |
| Gender | |||
| Female | 6 (16.7%) | 9 (26.5%) | 3 (23.1%) |
| Male | 30 (83.3%) | 25 (73.5%) | 10 (76.9%) |
| PRIMARY TUMOR | |||
| Fuhrman grade | |||
| 1 | 1 (2.8%) | 2 (5.9%) | 0 (0%) |
| 2 | 9 (25%) | 8 (23.5%) | 0 (0%) |
| 3 | 17 (47.2%) | 14 (41.2%) | 6 (46.1%) |
| 4 | 9 (25%) | 10 (29.4%) | 4 (30.8%) |
| X | 0 (0%) | 0 (0%) | 3 (23.1%) |
| pT | |||
| 1 | 8 (22.2%) | 5 (14.7%) | 1 (7.7%) |
| 2 | 10 (27.8%) | 5 (14.7%) | 1 (7.7%) |
| 3 | 16 (44.4%) | 18 (52.9%) | 8 (61.5%) |
| X | 2 (5.6%) | 6 (17.6%) | 3 (23.1%) |
| pN | |||
| 0 | 15 (41.7%) | 0 (0%) | 4 (30.8%) |
| 1 | 5 (13.9%) | 0 (0%) | 2 (15.4%) |
| 2 | 1 (2.7%) | 0 (0%) | 1 (7.7%) |
| X | 15 (41.7%) | 34 (100%) | 6 (46.1%) |
| BEGINNING OF THE TREATMENT | |||
| Age at therapy initiation (yr) | 60.4 (9.7) | 58.7 (11.3) | 63.5 (9.8) |
| Metastatic from the diagnosis | 18 (50%) | 14 (41.2%) | 8 (61.5%) |
| Time from diagnosis to treatment | |||
| < 1 yr | 23 (63.8%) | 20 (58.8%) | 9 (69.2%) |
| ≥ 1 yr | 13 (36.1%) | 14 (41.2%) | 4 (30.8%) |
| Number of metastatic sites | |||
| 1 | 17 (47.2%) | 17 (50%) | 6 (46.1%) |
| 2 | 13 (36.1%) | 10 (29.4%) | 6 (46.1%) |
| ≥ 3 | 6 (16.7%) | 7 (20.6%) | 1 (7.7%) |
| Risk factor (MSKCC) | |||
| Good | 10 (27.8%) | 14 (41.2%) | 0 (0%) |
| Intermediate | 10 (27.8%) | 18 (52.9%) | 0 (0%) |
| Bad | 8 (22.2%) | 0 (0%) | 0 (0%) |
| X | 8 (22.2%) | 2 (5.9%) | 13 (100%) |
| Median followed (month) | 34.5 (18.2-NR) | 24 (23-NR) | NA |
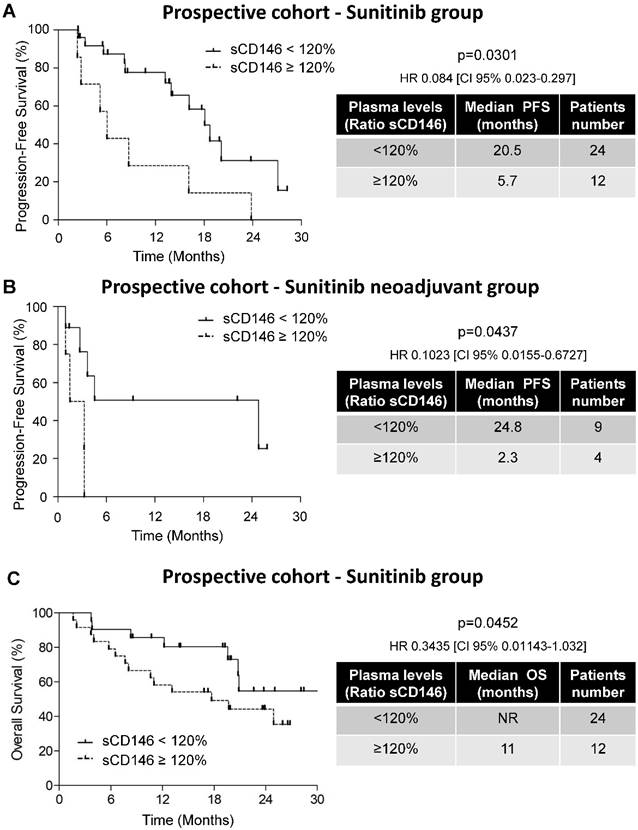
Variations in the sCD146 plasmatic levels were indicative of PFS and OS for patients treated with sunitinib. (A and B) Kaplan-Meier analysis of PFS: the PFS was calculated from patient subgroups with a ratio of plasmatic levels for sCD146 obtained between diagnosis and after the first cycle, which was less or greater than a cut-off ratio of 120%, for SUVEGIL and TORAVA trial - sunitinib group (A) or for an independent cohort of patients treated in a neoadjuvant setting (B). (C) Kaplan-Meier analysis of OS: OS was calculated from patient subgroups with a ratio of plasmatic levels for sCD146 obtained between diagnosis and after the first cycle, which was less or greater than a cut-off ratio of 120%, for SUVEGIL and TORAVA trial - sunitinib group. Statistical significance (p value) and the time of PFS and OS are indicated (see Figure S4 and Table S2).
Variation in the sCD146 plasmatic levels did not correlate with the PFS and OS of patients treated with bevacizumab. Kaplan-Meier analysis of PFS (A) or OS (B) of patients with ccRCC. The PFS and OS were calculated from patient subgroups with a ratio of plasmatic levels for sCD146 obtained between the diagnosis and after the first cycle, which was less or greater than a cut-off ratio of 120%, for TORAVA trial - bevacizumab group. Statistical significance (p value) and the time of PFS and OS are indicated (see Figure S4).
Clinical trial design (SUVEGIL and TORAVA trial)
The prospective cohort includes patients from the SUVEGIL and TORAVA trials.
The SUVEGIL trial (clinicaltrial.gov, NCT00943839) was a multi-center prospective single-arm study. The goal of the trial was to determine whether a link exists between the effectiveness of therapy with sunitinib malate and development of blood biomarkers in patients with ccRCC. 24 patients received oral sunitinib (50 mg/day) once daily for four weeks (on days 1 to 28), followed by two weeks without treatment. Courses were repeated every 6 weeks in the absence of disease progression or unacceptable toxicity.
The TORAVA trial (clinicaltrial.gov, NCT00619268) was a randomized prospective study. Patient characteristics and results have been previously described [25]. Briefly, patients aged 18 years or older with untreated metastatic ccRCC were randomly assigned (2:1:1). 34 patients received the combination of bevacizumab (10 mg/kg iv every 2 weeks) and temsirolimus (25 mg iv weekly), or the combination of bevacizumab (10 mg/kg iv every 2 weeks) and IFN (9 mIU iv trice per week), and 12 patients receive one of the standard treatments: sunitinib (50 mg/day orally for 4 weeks followed by 2 weeks off) [25]. The patients' characteristics and pathological parameters are summarized in Table 1.
Blood samples were collected during the inclusion visit (baseline) and at the end of the four weeks of sunitinib administration or two weeks of bevacizumab-based therapies.
Prospective cohort of validation (PREINSUT trial)
The PREINSUT trial (clinicaltrial.gov, NCT00930345) was a multi-center prospective single-arm study part of the PREDICT program (FP7 with France and UK). Sunitinib was given as first-line therapy (2 cycles of 50 mg 4 weeks ON/2 weeks OFF) before cyto-reductive nephrectomy. The Primary Renal Tumor (PRT) response prior to surgery (decrease > 10%), the progression-free survival (PFS) and OS were assessed. Plasmatic VEGF-A, SDF-1 and VEGFR-1 and -2 were prospectively studied by ELISA at the beginning and the end of each 4 week sunitinib treatment. Correlation between sunitinib pharmacokinetics at the end of the first cycle (C1D28) and biomarker change were assessed. Thirteen patients from this trial were assessed for sCD146 plasmatic levels (Table 1).
Biochemical analysis
Blood samples were centrifuged (10000 ×g for ten minutes) and the plasma collected and conserved at -80°C. Plasmatic levels of sCD146 were determined by ELISA using a CD146 ELISA Kit (Biocytex, Marseille, France).
Cell culture
786-0 (786) and HUVEC were purchased from the American Tissue Culture Collection. RCC10 cells were a kind gift of Dr WH Kaelin (Dana Farber Cancer Institute Boston, MA). 786-O cells resistant to sunitinib (786R) were generated in the laboratory as previously described [26]. Primary cells resistant to sunitinib were already described [27].
Tumor fragments were treated with collagenase overnight at 37°C and/or mechanically disaggregated with scalpels. Tumor cells were suspended in cell culture medium specific for renal cells (PromoCell, Heidelberg Germany).
Gene expression microarray analysis
Normalized RNA sequencing (RNA-Seq) data produced by The Cancer Genome Atlas (TCGA) were downloaded from cbioportal (www.cbioportal.org, TCGA Provisional; RNA-Seq V2). Different parameters were available for 503 ccRCC tumor samples. The results published here are in whole or in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/ [28, 29].
Quantitative real-rime PCR (qPCR) experiments
For tumor sample, total RNA was extracted with RNeasy FFPE Kit (QIAGEN, Hilden, Germany). For cells, total RNA was extracted with the RNeasy Mini Kit (QIAGEN, Hilden, Germany). The amount of RNA was evaluated with NanoDrop™ spectrophotometers (ThermoFisher Scientific, Waltham, MA USA). One microgram of total RNA was used for reverse transcription, using the QuantiTect Reverse Transcription kit (QIAGEN, Hilden, Germany), with a blend of oligo (dT) and random primers to prime first-strand synthesis. SYBR master mix plus (Eurogentec, Liege, Belgium) was used for qPCR and specific oligonucleotides (Sigma Aldrich), to assess mRNA expression for CD146 total (forward: GGCTAATGCCTCAGATCGATG; reverse: AATATGGTGTTGAATCTGTCTTG), CD146 short form (forward: CCACTGGCCTCAGCACTTCC; reverse: CTACTCACCTTTCTGGACAG) and CD146 long form (forward: TGGTTTGTACACCTTGCAGAGTATTC; reverse: TGGGCAGCCGGTAGTTG). mRNA levels were normalized to a housekeeping mRNA coding for ribosomal protein large P0 (RPLP0; forward: CAGATTGGCTACCCAACTGTT; reverse: GGCCAGGACTCGTTTGTACC).
Cell viability (XTT)
5×103 cells were incubated in a 96-well plate with different effectors for the times indicated in the figure legends. Fifty microliters of sodium 3′-[1-phenylaminocarbonyl)-3,4-tetrazolium]-bis(4-methoxy-6-nitro) benzenesulfonicacid hydrate (XTT) reagent was added per well. The assay was based on the cleavage of the yellow tetrazolium salt XTT to form an orange formazan dye by metabolically active cells. The absorbance of the formazan product, reflecting cell viability, was measured at 490 nm. Each assay was performed in quadruplicate.
Anti-soluble CD146 antibody
The M2J-1 mAb (AC CD146) was used to inhibit the effects of soluble CD146. This antibody was produced in our laboratory (18) and is specific for the soluble form of the molecule since it does not recognize the membrane forms.
Cell proliferation
Human Umbilical Vein Endothelial Cells (HUVEC) or 786-O/786-R cells were seeded on 96-well plates (5.103 cells/well) and cultured in EGM-2 medium or renal cells culture medium, respectively. Cell proliferation was assessed using the BrdU Labeling and Detection Kit III (Roche Corporation) as indicated by the manufacturer. In experiments performed in the presence of the anti-soluble CD146 antibody, it was added at a final concentration of 1µg/mL. Experiments were performed at least in triplicate.
Immunoblotting
Cells were lyzed in buffer containing 3% SDS, 10% glycerol and 0.825 mM Na2HPO4. 30-50 μg of proteins was separated on 10% SDS-PAGE, transferred onto a PVDF membrane (Immobilon, Millipore) and then exposed to the appropriate antibodies. Proteins were visualized with the ECL system using horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies.
Flow cytometry
After stimulation, cells were washed with PBS and stained with CD146-PE antibody (Biocytex, Marseille, France) according to the manufacturer's procedure. Fluorescence was measured using the FL2 of a fluorescence-activated cell sorter apparatus (FACS-Calibur cytometer).
Statistical analysis
For in vitro analysis
Results are expressed as the mean ± the standard error (SEM). Statistical significance and p values were determined by the two-tailed Student's t-test. One way ANOVA was used for statistical comparisons. Data were analyzed with Prism 5.0b (GraphPad Software) by one-way ANOVA with Bonferroni post hoc.
For patient analysis
DFS was defined as the time from surgery to the appearance of metastasis. PFS was defined as the time between surgery and subsequent blood sampling and progression, or death from any cause, censoring live patients and progression free at last follow-up. OS was defined as the time from blood sample collection to the date of death from any cause, censoring those alive at last follow-up. The Kaplan Meier method was used to produce survival curves and analyses of censored data were performed using Cox models.
Smoothing splines curves for hazard ratio were used to determine cut-offs for censored data for sunitinib or bevacizumab treatments. All analyses were performed using R software, version 3.2.2 (Vienna, Austria, https://www.r-project.org/).
Results
The level of CD146 mRNA was predictive of DFS and OS in non-metastatic ccRCC patients
Despite the major role of CD146 in the aggressiveness of several tumors, our goal was to correlate CD146 to the disease progression of M0 patients. These patients have variable outcomes and relevant biological markers may improve their surveillance. We showed by IHC that CD146 is expressed by ccRCC cells (Figure S2). Analysis of online available data showed that tumors expressing the highest levels of CD146 mRNA are ccRCC (Figure S3). Moreover, high levels of CD146 mRNA in the tumors of M0 patients (first quartile cut-off, n=25/38) correlated with a shorter DFS (82 months versus not reached, p = 0.0222) and OS (122 months versus not reached, p = 0.0244; Figure 1 and Table S1).
An increase in the plasmatic level of sCD146 was predictive of PFS and OS in metastatic ccRCC patients treated with sunitinib
The purpose of our study was to correlate sCD146 with survival in a prospective cohort of patients treated with sunitinib (SUVEGIL + TORAVA trials). Their median PFS was 15.4 months and their median OS was 24.6 months (Figures S4A and B). sCD146 levels were highly variable and did not correlate with outcome. Hence, we analyzed sCD146 in patients that had an objective response (category 1), a stable (category 2) or a progressive disease (category 3) according to RECIST 1.1 after the first cycle of treatment with sunitinib. Patients of categories 1 and 2 had decreased or stable levels of plasmatic sCD146, whereas patients of category 3 presented with an increase in sCD146 levels (median value of up-regulation ≥ 120%; see Materials and Methods for the definition of this threshold). According to the definition of this threshold, the modulation of sCD146 levels between the diagnosis and the first cycle of sunitinib correlated with PFS. Patients with an increase in the sCD146 plasmatic level ≥ 120%, had a shorter PFS (5.7 months, n=12/36) compared to patients with a threshold increase < 120% (some of these patients had a decreased or stable plasmatic sCD146 level, 20.5 months, n=24/36, p = 0.0301, HR 0.084 (CI 95% 0.023-0.297), Figure 2A and Figure S5).
To confirm these results, we analyzed sCD146 in the plasma of ccRCC patients treated with sunitinib in a neo-adjuvant setting (PREINSUT clinical trial). In this independent cohort, patients with an increase in sCD146 plasmatic levels > 120%, had a shorter PFS (2.3 months, n=4/13) compared to patients with a threshold increase < 120% (24.8 months, n=9/13, p = 0.0437, HR 0.1023 (CI 95% 0.0155-0.6727), Figure 2B).
The variations in the sCD146 levels (inferior or superior to 120%) and clinical parameters of patients (Fuhrman grade, pT, pM or MSKCC score, the standard score used for patient evaluation in clinical practices) did not correlate (Table S2A). The biological and clinical parameters were then analyzed in a multivariate Cox regression model for PFS. Variations in the sCD146 levels were identified as an independent prognostic parameter for PFS (p = 0.001, HR 8.298 (CI 95% 2.221 - 31); Table S2B). Therefore, the difference in sCD146 levels between the diagnosis and the first cycle of sunitinib can be considered as an independent marker for PFS.
By using the same threshold, we then analyzed the correlation between variations in sCD146 levels at diagnosis and after the first cycle of sunitinib to OS. The median OS of patients with sCD146 < 120% included in the prospective cohort was not reached (NR, n=24/36) whereas patients with sCD146 ≥ 120% had a median OS of 11 months (n=12/36, p = 0.0452, Figure 2C).
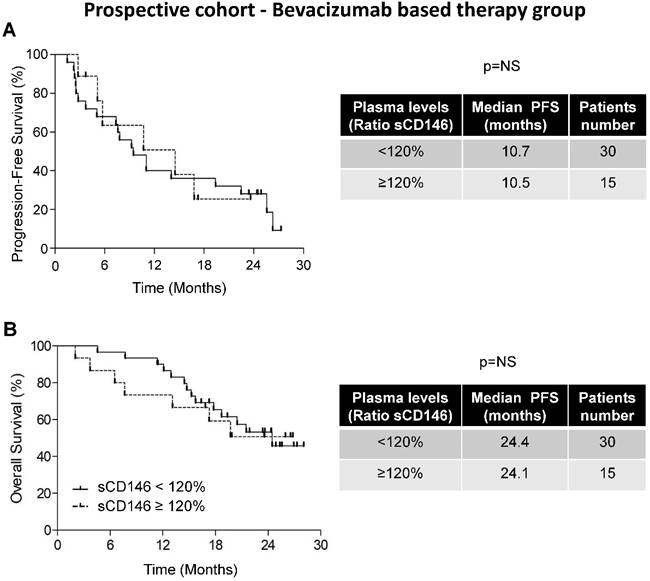
Correlation between sCD146 plasmatic levels and PFS and OS in the bevacizumab-based therapy group
To determine the predictive role of sCD146 for sunitinib efficacy, we tested the modulation of sCD146 levels between the diagnosis and the first cycle of treatment in a subset of patients of the TORAVA clinical trial that were treated with bevacizumab plus IFN or temsirolimus. Their median PFS was 10.6 months and their median OS was 24.1 months (Figures S4C and D). By using spline curves as mentioned in the Materials and Methods section, no specific cut-off defined populations that relapsed or did not relapse on bevacizumab. Therefore, the cut-off used for sunitinib was chosen for homogeneity of the results.
The difference in median PFS of patients with sCD146 < 120% (10.7 months, n=25/34) and of patients with sCD146 ≥ 120% (10.5 months, n=9/34) was not significant (p = 0.6467, Figure 3A). An equivalent trend was obtained for OS (24.4 months for patients with sCD146 < 120% and 24.1 months for patients with sCD146 ≥ 120%, p = 0.3525, Figure 3B). These results strongly suggest that the 120% increase in the sCD146 level is predictive of sunitinib- but not of bevacizumab-based therapy efficacy.
Sunitinib stimulated CD146 mRNA expression in ccRCC patients
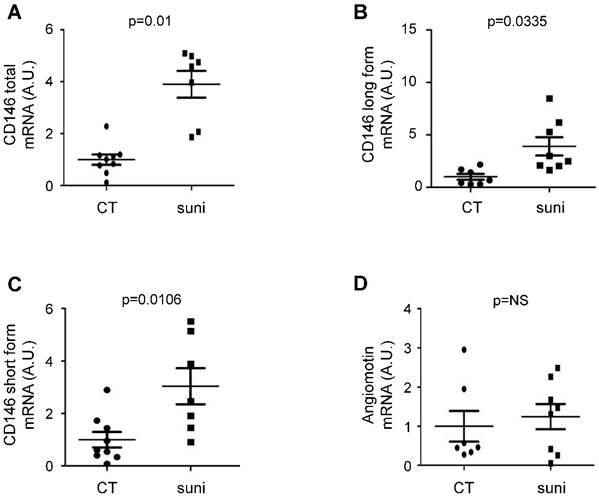
To establish a correlation between sunitinib treatment and CD146, we compared CD146 and angiomotin mRNA levels in tumors from untreated patients and in tumors from patients that received sunitinib in a neoadjuvant setting. Although tumor samples were not paired before and after sunitinib treatment, this method represents a first approach to understand the relationship between sCD146 and survival (Figure 4). These samples were independent of those of the PREINSUT trial for which only blood samples were available. These new tumor samples were already analyzed for another parameter [24]. In these tumor samples, mRNA of total CD146 (Figure 4A) and the long (Figure 4B) and short (Figure 4C) CD146 isoforms were significantly increased in sunitinib-treated patients. No modification in angiomotin mRNA expression was observed between the two groups (Figure 4D).
Sunitinib stimulated CD146 expression in patients treated with sunitinib in a neoadjuvant setting. Tumors from untreated ccRCC patients and tumors from patients treated with sunitinib in a neoadjuvant setting were compared (see Table S3). The levels of CD146 total (short + long form, A), CD146 long (B), CD146 short (C) and angiomotin (D) mRNA were determined by qPCR.
Sunitinib stimulated CD146/sCD146 expression in sunitinib-resistant ccRCC cells
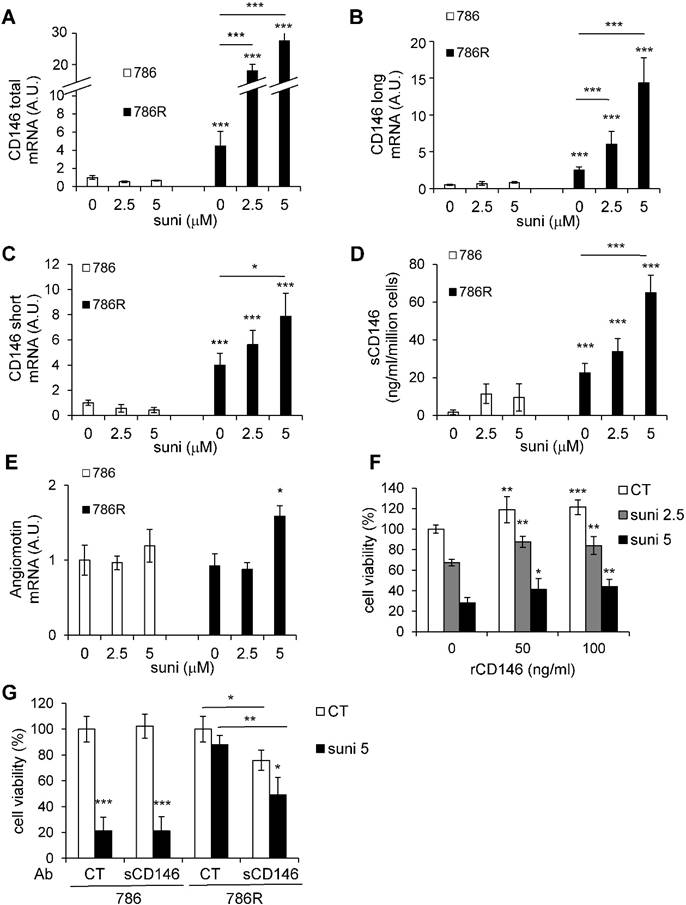
We hypothesized that the increase in CD146 levels, responsible for the resistance to sunitinib, resulted from a direct effect on tumor cells. Therefore, we used ccRCC cells that are respectively sensitive (786) and resistant (786R) to sunitinib. According to previous results, sunitinib resistance is dependent on lysosomal trapping [26]. Therefore, the resistance may be transient in part. Hence, 786R cells were left sunitinib-free for three days then re-challenged with different concentrations of the drug. Whereas sunitinib had no effect on the expression of total CD146, long and short forms in 786 cells, it further stimulated CD146 expression at the mRNA (Figures 5A to C) and protein levels (Figure S6A-C) in 786R cells. In addition, it also stimulated soluble CD146 secretion, as observed by ELISA (Figure 5D) or western-blot (Figure S6D) in cell culture supernatants. Moreover, the increase of CD146 level in 786R in basal conditions relies on activation of the c-Jun N-terminal Kinase (JNK) pathway (Figure S7). Indeed, inhibition of JNK by SP600125 (Figure S7A) decreased CD146 expression at the mRNA (Figure S7B-D) and protein levels (membrane CD146 analyzed by FACS (Figure S7E), sCD146 analyzed by ELISA (Figure S7F)). These results strongly suggest that the CD146 produced by tumor cells is directly involved in resistance to sunitinib. They also mimic the increment of sCD146 that we observed in relapsed patients. The basal levels of angiomotin mRNA were not different in 786 and 786R cells but were increased in 786R cells stimulated with a high sunitinib concentration, suggesting that an autocrine amplification loop on tumor cells may occur in resistant patients (Figure 5E). An increase in soluble CD146 did not occur when 786R cells were treated with bevacizumab (Figure S8A) and cell viability was not modified (Figure S8B). In addition, when HUVEC were treated with sunitinib, at two concentrations that reduced by 30% cell proliferation, or by bevacizumab, CD146, sCD146 and angiomotin were not modified (Figures S9A-D).
Basal CD146 expression is higher in 786R cells and is further stimulated by sunitinib. (A to E) 786 and 786R cells were treated with 2.5 or 5 µM sunitinib for 48 h. The mRNA levels of CD146 total (A), long (B) and short forms (C) and angiomotin (E) were evaluated by qPCR. Results are represented as the mean of three independent experiments ± SEM. (D) The sCD146 protein in cell supernatants was evaluated by ELISA. (F) 786 cells were treated with sunitinib, in the presence of recombinant sCD146 for 48 h. Cell viability was measured with a XTT assay. Results are represented as the mean of three independent experiments ± SEM. (G) 786 and 786R cells were treated with sunitinib (5 µM), in the presence of 1 µg/mL of irrelevant (CT Ab) or anti-sCD146 (sCD146 Ab) antibodies for 48 h. Cell viability was measured by XTT assays. Results are represented as the mean of three independent experiments ± SEM. * p<0.05, ** p<0.01, *** p<0.001.
The link between expression of CD146 mRNA and production of sCD146 following sunitinib was confirmed on primary cells resistant to sunitinib. Their IC50 for the drug is 10 µM (equivalent to the IC50 of 786R cells; Figure S10A). Their mRNA (total, long, short forms, Figure S10B-D) and protein (membrane CD146 analyzed by FACS (Figure S10E), sCD146 analyzed by ELISA (Figure S10F)) levels are expressed to a higher extent as compared to control 786 cells and are further stimulated by sunitinib.
To analyze the effect of an autocrine loop involving CD146 and angiomotin on sunitinib resistance, we then analyzed the effect of sCD146 (50 and 100 ng/mL) on the viability of 786 cells treated with sunitinib (2.5 or 5 µM). sCD146 increased cell viability in a dose-dependent manner in 786 cells. Moreover, it also reduced the sunitinib-dependent decreased cell viability (Figure 5F). sCD146 also prevented the sunitinib-dependent decreased cell viability in another ccRCC cell line (RCC10, Figure S8C). sCD146 stimulated the proliferation of HUVEC but did not protect HUVEC from sunitinib (Figure S9E).
In view of the large increase in sCD146 secretion in 786-R cells, we tested the effect of the anti-sCD146 antibody, M2J-1 mAb, on cell viability. Anti-sCD146 antibody decreased 786R but not 786 viability (Figure 5G). This result was confirmed in proliferation experiments. 786-R cell proliferation was significantly decreased by M2J-1 mAb whereas no effect of the control IgG was observed (Figure S11). Altogether, these results strongly suggest the involvement of CD146 in ccRCC cell resistance to sunitinib.
Discussion
No curative treatment exists for metastatic ccRCC. Therefore, despite new therapeutic options, it remains a disease with poor prognosis. Two types of patients are currently diagnosed: i) M0 patients that have good prognosis although some patients progress toward a M1 phase without any robust method to predict such a pejorative evolution, and ii) M1 patients that are treated with sunitinib in the first-line. The efficacy in tumor control varies from a few days to several years [30]. Unfortunately, relapse is synonymous with ineluctable death. However, ccRCC is probably the only disease that has led to the development of more than ten treatments in the last years. Therefore, the identification of predictive marker(s) of M0/M1 evolution and of sunitinib efficacy may allow a more accurate survey of at risk patients and a rapid switch to a second-line treatment before the detection of relapse by imaging using, for example, CT scans. Such markers must be detectable: i) with material sampled from patients by a non-invasive technique (urine, blood), or ii) by a simple and sensitive method easily transposable to clinical practices. The presence of high amounts of mRNA in tumors of patients that relapsed toward a M1 phenotype must be confirmed in plasma of M0 patients. An increase in plasmatic sCD146 ≥ 120% between the diagnosis of a metastatic disease and the first cycle of sunitinib may represent such a relevant marker of relapse. This simple detection predicts a pejorative evolution that may occur after several cycles of treatment. This detection may highlight a relapse earlier than those detected by CT scans that are generally performed after the second cycle of sunitinib.
sCD146 is generated by the shedding of the membrane form of CD146 through an unidentified process that might involve matrix metalloproteases [31]. CD146 is expressed on endothelial and several cancer cells including ccRCC cells. We observed a large increase in the long and short forms of the CD146 mRNA in tumors, which may be related to up-regulation on tumor and/or tumor-associated endothelial cells. The role of both isoforms in cancer cells is unknown. However, the long CD146 isoform is essentially involved in structural functions at the cell-cell junction, whereas the short CD146 isoform is involved in angiogenic functions in endothelial cells. Further studies will be necessary to delineate the roles of these forms in ccRCC cells. To discriminate between the effects on cancer and endothelial cells, we performed in vitro experiments with HUVEC and 786 and 786R cells. Whereas no effect of sunitinib was observed on CD146 mRNA expression in HUVEC and a trend of decreased expression in 786 cells, a huge increase of total, long and short forms was observed in 786R cells. This effect on mRNA was associated with an increase in sCD146 production and in cell viability of 786R cells. These results were also consistent with increased plasmatic levels above the threshold of 120% observed in patients that relapsed on sunitinib. Thus, these experiments strongly suggest that one of the mechanisms of resistance to sunitinib may depend on a huge increase in CD146/sCD146 expression. sCD146 stimulates VEGF production leading to enhanced proliferation and survival of cancer cells expressing CD146 [18]. Indeed, sCD146 regulates numerous proteins involved in cell survival, cell cycle, cellular stress and nuclear proteins acting as transcription factors such as BCL-XL and the oncoprotein c-MYC, which is phosphorylated in response to sCD146 [18]. Finally, CD146 is involved in epithelial-mesenchymal transition (EMT) [32, 33]. Since this phenomenon plays a major role in metastatic dissemination, we hypothesize that increased CD146 expression induced by sunitinib stimulates EMT and consequently accelerates metastatic development at relapse. A recapitulative schema is shown is Figure S12. In addition to its role as a predictive marker of relapse on sunitinib, sCD146 may also serve as a therapeutic target. For this purpose, we recently developed a neutralizing antibody that specifically binds and inhibits the effects of sCD146 [18]. This antibody displayed inhibitory effects on two models of xenografts in nude mice. Indeed, the antibody reduced tumor growth and angiogenesis. Further experiments are ongoing to test the beneficial role of the sCD146 antibody at progression on sunitinib in experimental models of ccRCC.
Conclusion
Our study defines sCD146 as a relevant predictive marker of pejorative evolution of M0 patients and of response to sunitinib in metastatic ccRCC. We established that an increase ≥ 120% of sCD146 in patients after sunitinib is indicative of a high risk of relapse. These results must be confirmed with a larger number of patients. The measurement of sCD146 could then be introduced into clinical practices. It would then be used to monitor the M0 patients and to propose alternative treatments to sunitinib in patients with a high risk of relapse according to CD146 level. In addition, our study suggests combining sunitinib with a CD146/sCD146-directed antibody or using it upon relapse on sunitinib is a relevant therapeutic approach. This strategy deserves testing in phase I/II clinical trials.
Abbreviations
ccRCC: clear cell renal cell carcinoma; DFS: disease-free survival; IFN: interferon alpha; IHC: immunohistochemistry; M0: non-metastatic; M1: metastatic; OS: overall survival; PFS: progression-free survival; sCD146: soluble CD146; TKI: tyrosine kinase inhibitor.
Acknowledgements
This work was supported by the French association for cancer research (ARC, GP financial supports), the Fondation de France (GP financial supports), the French National Institute for Cancer Research (INCA, GP financial supports), Inserm (MBC financial supports), Aix-Marseille University (MBC financial supports), MSD Avenir grant (MBC financial supports) and FX Mora Foundation. This study was conducted as part of the Centre Scientifique de Monaco Research Program, funded by the Government of the Principality of Monaco. We thank the Department of Pathology, especially Arnaud Borderie and Sandrine Destree, for technical help.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Escudier B, Bellmunt J, Negrier S, Bajetta E, Melichar B, Bracarda S. et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol. 2010;28:2144-50
2. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S. et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584-90
3. Motzer RJ, Escudier B, Tomczak P, Hutson TE, Michaelson MD, Negrier S. et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013;14:552-62
4. Escudier B, Porta C, Bono P, Powles T, Eisen T, Sternberg CN. et al. Randomized, controlled, double-blind, cross-over trial assessing treatment preference for pazopanib versus sunitinib in patients with metastatic renal cell carcinoma: PISCES Study. J Clin Oncol. 2014;32:1412-8
5. Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J. et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med. 2013;369:722-31
6. Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F. et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1814-23
7. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S. et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma: final results and analysis of prognostic factors. Cancer. 2010;116:4256-65
8. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E. et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883-92
9. Yan X, Lin Y, Yang D, Shen Y, Yuan M, Zhang Z. et al. A novel anti-CD146 monoclonal antibody, AA98, inhibits angiogenesis and tumor growth. Blood. 2003;102:184-91
10. Zeng P, Li H, Lu PH, Zhou LN, Tang M, Liu CY. et al. Prognostic value of CD146 in solid tumor: A Systematic Review and Meta-analysis. Sci Rep. 2017;7:4223
11. Wang Z, Yan X. CD146, a multi-functional molecule beyond adhesion. Cancer Lett. 2013;330:150-62
12. Ouhtit A, Gaur RL, Abd Elmageed ZY, Fernando A, Thouta R, Trappey AK. et al. Towards understanding the mode of action of the multifaceted cell adhesion receptor CD146. Biochim Biophys Acta. 2009;1795:130-6
13. Liu JW, Nagpal JK, Jeronimo C, Lee JE, Henrique R, Kim MS. et al. Hypermethylation of MCAM gene is associated with advanced tumor stage in prostate cancer. Prostate. 2008;68:418-26
14. Jiang T, Zhuang J, Duan H, Luo Y, Zeng Q, Fan K. et al. CD146 is a coreceptor for VEGFR-2 in tumor angiogenesis. Blood. 2012;120:2330-9
15. Wellbrock J, Fiedler W. CD146: a new partner for VEGFR2. Blood. 2012;120:2164-5
16. Stalin J, Harhouri K, Hubert L, Garrigue P, Nollet M, Essaadi A. et al. Soluble CD146 boosts therapeutic effect of endothelial progenitors through proteolytic processing of short CD146 isoform. Cardiovasc Res. 2016;111:240-51
17. Boneberg EM, Illges H, Legler DF, Furstenberger G. Soluble CD146 is generated by ectodomain shedding of membrane CD146 in a calcium-induced, matrix metalloprotease-dependent process. Microvasc Res. 2009;78:325-31
18. Stalin J, Nollet M, Garigue P, Fernandez S, Vivancos L, Essaadi A. et al. Targeting soluble CD146 with a neutralizing antibody inhibits vascularization, growth and survival of CD146-positive tumors. Oncogene. 2016;35:5489-500
19. Lv M, Shen Y, Yang J, Li S, Wang B, Chen Z. et al. Angiomotin Family Members: Oncogenes or Tumor Suppressors? Int J Biol Sci. 2017;13:772-81
20. Troyanovsky B, Levchenko T, Mansson G, Matvijenko O, Holmgren L. Angiomotin: an angiostatin binding protein that regulates endothelial cell migration and tube formation. J Cell Biol. 2001;152:1247-54
21. Adler JJ, Johnson DE, Heller BL, Bringman LR, Ranahan WP, Conwell MD. et al. Serum deprivation inhibits the transcriptional co-activator YAP and cell growth via phosphorylation of the 130-kDa isoform of Angiomotin by the LATS1/2 protein kinases. Proc Natl Acad Sci U S A. 2013;110:17368-73
22. Ilie M, Long E, Hofman V, Selva E, Bonnetaud C, Boyer J. et al. Clinical value of circulating endothelial cells and of soluble CD146 levels in patients undergoing surgery for non-small cell lung cancer. Br J Cancer. 2014;110:1236-43
23. Kammerer-Jacquet SF, Brunot A, Pladys A, Bouzille G, Dagher J, Medane S. et al. Synchronous Metastatic Clear-Cell Renal Cell Carcinoma: A Distinct Morphologic, Immunohistochemical, and Molecular Phenotype. Clin Genitourin Cancer. 2017;15:e1-e7
24. Dufies M, Giuliano S, Ambrosetti D, Claren A, Ndiaye PD, Mastri M. et al. Sunitinib Stimulates Expression of VEGFC by Tumor Cells and Promotes Lymphangiogenesis in Clear Cell Renal Cell Carcinomas. Cancer Res. 2017;77:1212-26
25. Negrier S, Gravis G, Perol D, Chevreau C, Delva R, Bay JO. et al. Temsirolimus and bevacizumab, or sunitinib, or interferon alfa and bevacizumab for patients with advanced renal cell carcinoma (TORAVA): a randomised phase 2 trial. Lancet Oncol. 2011;12:673-80
26. Giuliano S, Cormerais Y, Dufies M, Grepin R, Colosetti P, Belaid A. et al. Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy. 2015;11:1891-904
27. Grepin R, Ambrosetti D, Marsaud A, Gastaud L, Amiel J, Pedeutour F. et al. The relevance of testing the efficacy of anti-angiogenesis treatments on cells derived from primary tumors: a new method for the personalized treatment of renal cell carcinoma. PLoS ONE. 2014;9:e89449
28. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
29. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
30. Gore ME, Szczylik C, Porta C, Bracarda S, Bjarnason GA, Oudard S. et al. Final results from the large sunitinib global expanded-access trial in metastatic renal cell carcinoma. Br J Cancer. 2015;113:12-9
31. Bardin N, Blot-Chabaud M, Despoix N, Kebir A, Harhouri K, Arsanto JP. et al. CD146 and its soluble form regulate monocyte transendothelial migration. Arterioscler Thromb Vasc Biol. 2009;29:746-53
32. Liu WF, Ji SR, Sun JJ, Zhang Y, Liu ZY, Liang AB. et al. CD146 expression correlates with epithelial-mesenchymal transition markers and a poor prognosis in gastric cancer. Int J Mol Sci. 2012;13:6399-406
33. Imbert AM, Garulli C, Choquet E, Koubi M, Aurrand-Lions M, Chabannon C. CD146 expression in human breast cancer cell lines induces phenotypic and functional changes observed in Epithelial to Mesenchymal Transition. PLoS One. 2012;7:e43752
Author contact
![]() Corresponding author: gpagesfr
Corresponding author: gpagesfr