Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(9):2348-2360. doi:10.7150/thno.22901 This issue Cite
Research Paper
A novel USP9X substrate TTK contributes to tumorigenesis in non-small-cell lung cancer
Xiangling Chen1,4*, Chengli Yu1,4*, Jing Gao1*, Hongwen Zhu1, Binghai Cui2,4, Tao Zhang3,4, Yanting Zhou1, Qian Liu1,4, Han He1, Ruoxuan Xiao1,4, Ruimin Huang3,4, Hua Xie3,4, Daming Gao2,4, Hu Zhou1,4 ![]()
1. Department of Analytical Chemistry and CAS Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
2. CAS Key Laboratory of Systems Biology, Innovation Center for Cell Signaling Network, CAS Center for Excellence in Molecular Cell Science, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200031, China
3. State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China
4. University of Chinese Academy of Sciences, Number 19A Yuquan Road, Beijing 100049, China
* Xiangling Chen, Chengli Yu and Jing Gao contributed equally to this work.
Received 2017-7-20; Accepted 2018-2-26; Published 2018-3-22
Abstract

The X-linked deubiquitinase, USP9X, is implicated in multiple cancers by targeting various substrates. Increased expression of USP9X is observed in non-small-cell lung cancer (NSCLC) and is correlated with poor prognosis. However, the molecular mechanism for USP9X regulation of tumor cell survival and tumorigenesis in NSCLC is less defined.
Methods: In this study, chemical labeling, quantitative proteomic screening was applied to analyze A549 cells with or without USP9X RNA interference. Functional in vitro and in vivo experiments were performed to confirm the oncogenic effects of USP9X in NSCLC and to investigate the underlying mechanisms.
Results: The resulting data suggested that dual specificity protein kinase TTK is a potential substrate of USP9X. Further experimental evidences confirmed that USP9X stabilized TTK via direct interaction and efficient deubiquitination of TTK on K48 ubiquitin chain. Moreover, knockdown of USP9X or TTK inhibited cell proliferation, migration and tumorigenesis, and the immunohistochemical analysis of clinical NSCLC samples showed that the protein expression levels of USP9X and TTK were significantly elevated and positively correlated in tumor tissues.
Conclusions: In summary, our data demonstrated that the USP9X-TTK axis may play a critical role in NSCLC, and could be considered as a potential therapeutic target.
Keywords: non-small cell lung cancer (NSCLC), quantitative proteomics, deubiquitinase, USP9X, TTK
Introduction
Lung cancer is one of the leading causes of cancer death worldwide [1]. Incidence and mortality attributed to lung cancer have risen steadily since the 1930s [2]. If diagnosed early, survival of patients suffering from lung cancer could be improved primarily due to advances in treatments. NSCLC accounts for ~85% of all types of lung cancers, but is often diagnosed at the late or advanced stage and has poor prognosis [3]. Therefore, further study is necessary to discover biomarkers and therapeutic targets for NSCLC prognosis and therapy.
Ubiquitination, as a major post-translational modification involved in protein activation, localization and degradation, participates in various biological processes [4]. Protein ubiquitination is a reversible process catalyzed by ubiquitin ligases and deubiquitinases (DUBs). DUBs catalyze the release of ubiquitin from substrate proteins and oppose the action of ubiquitin ligases. In humans, DUBs are subdivided into five families: USPs, OTUs, UCHs, Josephins and JAMMs [5]. DUBs have been extensively studied due to their important roles in diverse biological processes including DNA repair [6], chromatin remodeling [7], cell-cycle control [8], embryonic stem cells stabilization [9] and particular functions in tumorigenesis [10-14]. DUBs regulating processes associated with cell proliferation and apoptosis are considered candidate targets for cancer therapeutics [15].
Among ~90 DUBs, the X-linked deubiquitinase, USP9X, regulates the components of multiple signaling pathways and can act as an oncogene or a tumor suppressor in different cancers. For example, USP9X is considered a tumor suppressor in pancreatic ductal adenocarcinoma [16, 17]. In contrast, in melanoma [18], esophageal squamous cell carcinoma [16, 19], and lung cancer [20, 21], overexpression of USP9X correlates with poor prognosis, which indicates its oncogenic role. Therefore, in-depth study of USP9X is important to demonstrate the mechanism of USP9X involvement in tumorigenesis and further cancer therapy.
Dual specificity protein kinase TTK, also known as Mps1, plays crucial roles in spindle pole assembly, spindle checkpoint, genotoxic stress response, cytokinesis and meiosis [22]. The expression level of TTK is tightly regulated during cell-cycle progression [22]. TTK mRNA levels are elevated in breast cancer [23], lung cancer [24] and thyroid carcinoma [25]. TTK is degraded by the ubiquitin-proteasome pathway [26]. The ubiquitin ligases anaphase-promoting complex/ cyclosome (APC-c), including APC-cCdc20 and APC-cCdh1, have been indicated to target TTK in a cell cycle-dependent manner [26]. And, another previous study also found that ubiquitin ligase Ufd2 is required for efficient degradation of TTK [27]. However, little is known about the deubiquitinases of TTK.
The protein interaction partners of DUBs and their substrates can be identified by mass spectrometry-based methods. The DUBs binding ubiquitinated substrates complexes can be enriched and identified using tagging affinity-based approaches followed by tandem mass spectrometry analysis. For example, this approach was successfully applied to identify CEP131 [28] as a substrate of USP9X. Another useful strategy for discovering the substrates of DUBs is to investigate the changed proteins in the cells with knockdown/overexpression of the DUBs by global proteomics. Poulsen et al. applied the SILAC-based quantitative proteomic approach to compare wild-type Saccharomyces cerevisiae to 20 DUBs knock-out strains, and demonstrated that Sec28p is a novel substrate of Ubp3p [29]. In this study, we utilized a chemical labelling, quantitative proteomic approach to identify the potential substrates of the deubiquitinase USP9X. In particular, TTK was identified as a potential substrate of USP9X. We demonstrated that USP9X directly interacted with and stabilized TTK. Knockdown of USP9X increased TTK ubiquitination, decreased TTK protein level, and suppressed cell proliferation, migration, invasion and tumor growth, demonstrating a phenotype similar to TTK depletion. Consistently, significant correlation between USP9X and TTK expression levels was observed in human NSCLC tissues, where they are over-expressed compared with matched adjacent normal tissues. Taken together, these results suggested that the USP9X-TTK axis could be considered as a potential therapeutic target for NSCLC.
Results
Identification of TTK as a candidate substrate for USP9X
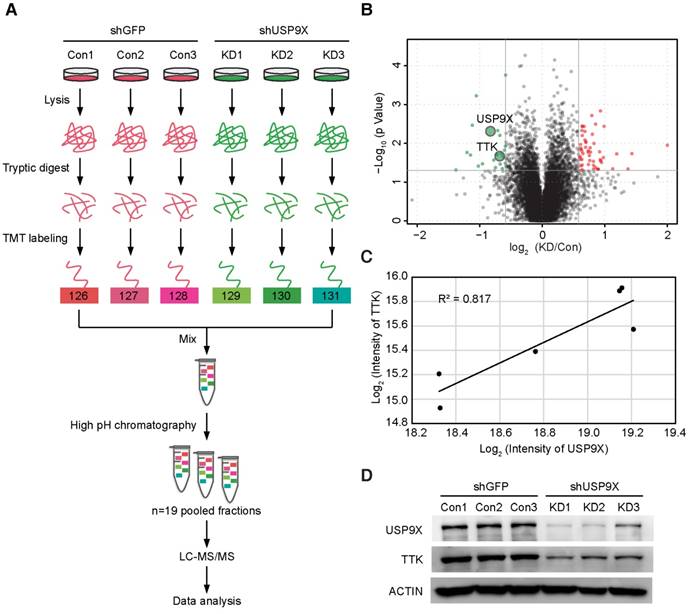
To identify the candidate substrates of USP9X, we performed a proteome-wide screening experiment. Briefly, USP9X was knocked down in A549 cells with 3 shRNAs targeting different regions of the USP9X transcript. Expressions of endogenous proteins treated with shUSP9X were compared to the control shRNA-treated cells using a tandem mass tags (TMT)-based quantitative proteomic approach (Figure 1A). As a result, a total of 7471 proteins were identified in the proteomic experiment, and 22 proteins were remarkably down regulated in USP9X knockdown cells (fold change > 1.5, students' t test P value < 0.05; Figure 1B and Table S1), which could be the candidate substrates of deubiquitinase USP9X. Effective knockdown of USP9X was verified by Western blotting (Figure 1D). Moreover, the down-regulation of USP9X in the shUSP9X-treated group was further confirmed by the TMT-based proteomic quantification results. Among these significantly down-regulated proteins, TTK was selected as a potential substrate of USP9X (the ratio between the protein intensities of the shUSP9X and control shRNA samples was 0.62, P value = 0.02, Table S1). In addition, the expression of TTK showed a significant correlation with that of USP9X, indicating that down-regulation of TTK was strongly correlated with decreased USP9X expression (Figure 1C). Several cell cycle-associated proteins, including CLASPIN, XIAP, SURVIVIN, and centrosome protein CEP131, have recently been reported as the substrates of USP9X [28, 30-32]. These results indicated that TTK, as one of the spindle checkpoint proteins, can also be considered as a potential substrate of USP9X.
TMT-based quantitative proteomics identifies TTK as a candidate substrate of USP9X. (A) Flow diagram of the TMT-based quantitative proteomics platform applied to identify the substrates of USP9X. A549 cells were stably transfected with 3 different shRNAs targeting USP9X (KD1, KD2, KD3) or control shRNA (Con1, Con2, Con3) and the whole cellular proteins were extracted and quantified. Following trypsin digestion of equal amount of proteins, the resolved peptides were labeled with 6-plex TMT reagents, fractionated by HPLC and analyzed by mass spectrometry. (B) Summary of the TMT labeling assay results. 7471 proteins identified by TMT assay are plotted in the volcano plot, in which the logarithmic ratio of protein intensities in the shUSP9X/control shRNA samples are plotted against negative logarithmic P values of the t-test performed from three replicates. 22 proteins were significantly down-regulated (green), 53 proteins were up-regulated (red) (fold change > 1.5, students' t test P value < 0.05). (C) Decreased USP9X expression correlates with decreased TTK protein level. (D)Validation of protein expression of USP9X and TTK in stably expressing A549 cells by immunoblotting.
USP9X stabilizes the TTK protein by deubiquitination
To test the hypothesis that TTK is a direct substrate for USP9X, we first examined the interaction between USP9X and TTK. By constructing a flag tagged-TTK construct, a transfection/co-immunoprecipitation experiment was performed in 293T cells, which revealed that TTK interacts with USP9X (Figure 2A). We also detected the interaction of endogenous USP9X and endogenous TTK by performing immunoprecipitation experiments using USP9X antibody or IgG control from 293T cell protein extracts (Figure 2B). Furthermore, in order to validate the correlation between USP9X and TTK in NSCLC cells, we exploited siRNA-mediated depletion of USP9X in A549 cells using two different siRNAs targeting USP9X mRNAs. Both USP9X siRNAs markedly reduced USP9X protein levels after 48 h transfection, and also resulted in decreased TTK levels (Figure 2C). Moreover, the effect of USP9X depletion on TTK levels was not only found in A549 cells but also observed in other NSCLC cell lines, including NCI-H1975, NCI-H125 and NCI-H3122 (Figure S2A-C). And, examination of the protein expression levels of USP9X and TTK in multiple lung cancer cell lines showed a positive correlation (Figure S2D). The effect of USP9X reduction on the level of TTK was observed in HeLa cells as well (Figure 2D).
USP9X is functionally linked to the stability of TTK. (A) GFP, empty vector control (EV), and flag-TTK were transfected into 293T cells. The cells were extracted for immunoprecipitation with the anti-flag agarose and the proteins were analyzed by immunoblotting. (B) Immunoprecipitated proteins with anti-USP9X antibodies or control IgG from 293T extracts were analyzed by western blotting. Endogenous TTK interacting with USP9X was detected using an anti-TTK antibody. (C) A549 cells were transfected with either two different siRNAs targeting USP9X or a control siRNA. After 72 h, cells were harvested and proteins were analyzed by immunoblotting. (D) The effect of USP9X depletion on TTK in HeLa cells as in (C). (E) Flag-TTK, HA-Ub, siRNA or siUSP9X were co-transfected into 293T cells. The cells were treated with or without MG132 as indicated. Then, cells were extracted for immunoprecipitation with anti-flag agarose and analyzed by western blotting. (F) Flag-TTK, ubiquitin K48-only plasmids, siRNA or siUSP9X were co-transfected into 293T cells. The indicated cells were treated with MG132 overnight. Then, cells were extracted for immunoprecipitation with anti-flag agarose and analyzed by western blotting. (G) A549 cells were transfected with either control or USP9X siRNAs. After 48 h, 200 μg/mL CHX was added and cells were harvested at the indicated times. Protein samples were analyzed by immunoblotting. Quantification of TTK levels relative to β-actin are presented. (H) Half-life analysis of TTK in constructed stable A549 cells. (I-J) HeLa (I) or 293FT (J) cells were transfected with plasmids expressing functional V5-USP9X (WT), catalytically dead C1566S (CS) USP9X or empty vector (EV). After 36 h, 200 μg/mL CHX was added and cells were harvested for immunoblotting.
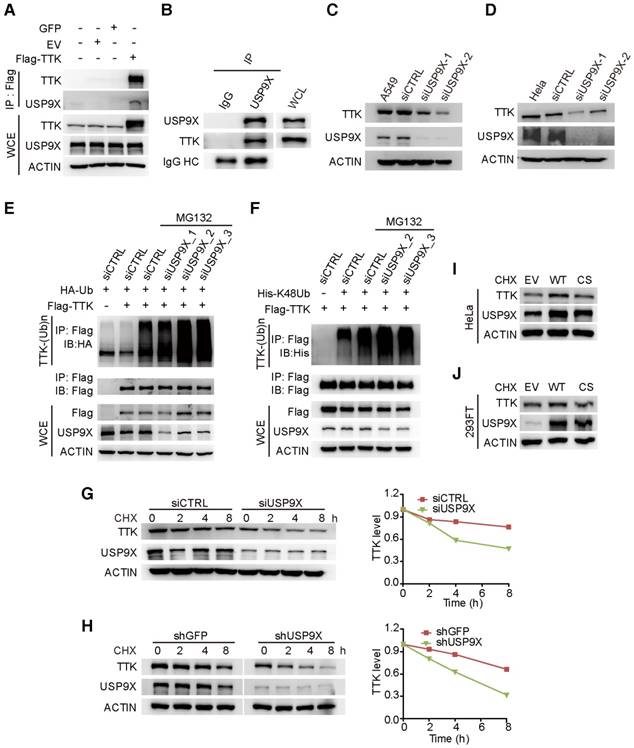
To test whether USP9X regulates TTK through deubiquitination, haemegglutinin (HA)-Ub, Flag-TTK, and siRNAs targeting USP9X or control siRNAs (siCTRL) were co-transfected into 293T cells followed by treatment with the proteasome inhibitor MG132. The results suggested that knockdown of USP9X increased the ubiquitination of TTK protein (Figure 2E). Furthermore, to test whether USP9X regulates TTK by deubiquitinating ubiquitin chains through K48 or K63 linkages, ubiquitin mutant vectors K48 and K63 (containing arginine substitutions of all of its lysine residues except those at positions 48 and 63, respectively), Flag-TTK, and siRNAs targeting USP9X or control siRNAs (siCTRL) were co-transfected into 293T cells followed by treatment with the proteasome inhibitor MG132. USP9X-mediated polyubiquitination of TTK was significantly increased in the presence of ubiquitin K48 plasmids, which indicated USP9X could efficiently remove the Lys 48 rather than Lys 63 type of the ubiquitin chain on TTK (Figure 2F and Figure S2E). To consolidate the finding that USP9X protects TTK from ubiquitination-dependent proteasomal degradation, we directly used the proteasome inhibitor Bortezomib to treat cells transfected with either control siRNA or USP9X-targeting siRNA. Although endogenous USP9X was effectively decreased after siRNA transfection as indicated, TTK protein level was still increased in both of the transfected cells in a manner very similar to p27, which is a known substrate of the proteasomal degradation pathway (Figure S2F) [33].
To further explore whether USP9X enhances TTK stability, we transfected either control siRNA or USP9X-targeting siRNA into A549 cells to determine the half-lives of TTK. After 48 h transfection, the protein synthesis inhibitor cycloheximide (CHX) was added and cells were harvested at the indicated time points for immunoblotting. As expected, following the depletion of USP9X, TTK protein levels rapidly decreased (Figure 2G). Similar results were obtained in A549 cells stably expressing shGFP and shUSP9X (Figure 2H). We then went on to test whether stabilization of TTK by USP9X is achieved through the deubiquitination enzymatic activity of USP9X [30]. Briefly, wild type USP9X or catalytically inactive C1566S mutant were transfected in HeLa cells and cycloheximide was added 36 h post-transfection. Overexpression of wild-type USP9X rather than the C1566S USP9X inactive mutant led to an increase in TTK protein level (Figure 2I). Similar results were obtained in 293FT cells (Figure 2J). The results outlined above suggested that USP9X interacts with and deubiquitinates TTK for its stabilization.
USP9X and TTK regulate cell migration, invasion, and colony formation
Overexpression of USP9X has been reported in NSCLC, resulting in tumorigenesis and poor prognosis [20, 34]. To gain insight into the functional connection between USP9X and TTK on lung cancer migration and invasion, A549 cells were transfected with either control siRNA or two different siRNAs targeting USP9X, and subjected to wound-healing assays 48 h post-transfetion. Knockdown of USP9X/ TTK significantly delayed the cells from recovering the wounded area (Figure 3A-B). Western blotting experiments confirmed that TTK was significantly knocked down by the specific siRNAs (Figure S3A). We further performed invasion assays with A549 cells stably expressing shUSP9X and shTTK, respectively. Similarly, decreased expression of either USP9X or TTK remarkably reduced the number of invaded cells (Figure 3C). Therefore, the data above supported a functional consistency of USP9X and TTK in cell migration and invasion. We next performed colony formation assays to determine the impact of USP9X and TTK reduction. As shown in Figure 3D and Figure S3B, D, knockdown of USP9X or TTK dramatically decreased the colony numbers in A549 cells or Hela cells treated with shUSP9X or shTTK.
Knock down of USP9X or TTK inhibits cell migration and invasion. (A) A549 cells were transfected with either two different siRNAs targeting USP9X or a control siRNA. After 48 h, cell migration was measured by a wound-healing assay. The wound edges are indicated by black lines. Representative images are shown. The quantitative results are shown on the right. The y-axis represents the percentage of wound closure. Data are represented as mean ± s.d. (n = 3, *** P < 0.001, t-test). Scale bars, 300 µM. (B) The migration ability of A549 cells with depletion of TTK examined via the wound-healing assay as in (A). Data are represented as mean ± s.d. (n = 3, *** P < 0.001, t-test). Scale bars, 300 µM. (C) Stable knockdown of either USP9X or TTK decreased A549 cells' invasion ability, which was measured by a transwell assay. The blue dye indicates the transwell cells. The quantitative results by counting cell numbers are shown on the right. Data are represented as mean ± s.d. (n = 3, *** P < 0.001, t-test). Scale bars, 100 µM. (D) Stable knockdown of either USP9X or TTK decreased A549 cells' colony forming ability, which was measured by a colony formation assay. The quantitative results by counting cell numbers are shown on the right. Data are represented as mean ± s.d. (n = 3, *** P < 0.001, t-test). (E) WP1130, an inhibitor of DUBs, could decrease A549 cells' colony forming ability. A549 cells were treated with the indicated drug concentration for the colony formation assay. After two weeks, cells were subjected to crystal violet staining. Data are represented as mean ± s.d. (n = 3, ** P < 0.01, t-test). (F) HeLa cells were induced into mitosis by treating with nocdazole, then the cells were treated with or without WP1130 as the indicated times and the cells were harvested for immunoblotting. Quantification of TTK levels relative to β-actin are shown. (G) Depletion of USP9X or TTK increased A549 cells' sensitivity to WP1130. A549 cells stably expressing shGFP, shUSP9X or shTTK were seeded into 96-well plates and then treated with different concentrations of WP1130 as indicated. The optical density (OD) at 490 nm for each well was detected. Data are represented as mean ± s.d. (n = 5, *** P < 0.001, t-test).
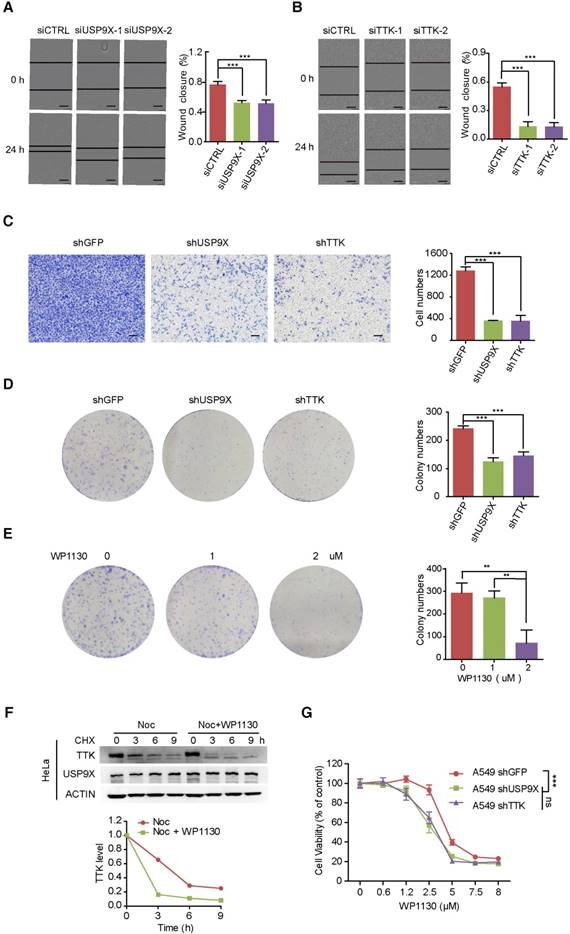
We next investigated the effect of WP1130, a partially selective inhibitor of deubiquitinating enzymes, directly inhibiting DUB activity of USP9X, USP5, USP14, and UCH37 [35], in regulating TTK expression and cell proliferation. As expected, WP1130 decreased the colony forming ability of A549 cells in a dose-dependent manner (Figure 3E). Furthermore, we treated A549 cells with WP1130 and found that TTK expression levels were affected in a dose- and time-dependent manner (Figure S3C-D). To obtain better insights into the role of USP9X in TTK stability, HeLa cells were arrested in mitosis with nocodazole. Cycloheximide was then added to prevent further protein synthesis in the presence or absence of the USP9X inhibitor WP1130 and cells were harvested at the indicated time points. The results illuminated that WP1130 treatment destabilized the TTK proteins in mitosis cells (Figure 3F). Consistently, A549 cells became more sensitive to WP1130 with USP9X or TTK knockdown (Figure 3G).
USP9X and TTK modulate A549 cell proliferation and tumorigenesis
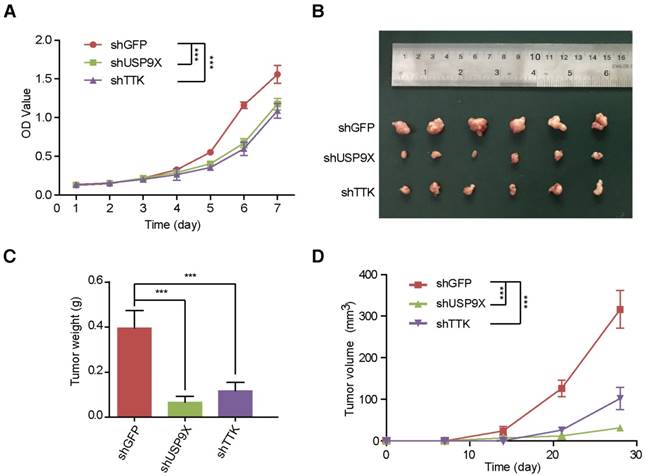
In order to uncover the function of USP9X/TTK in NSCLC carcinogenesis, we developed A549 cells stably expressing shUSP9X or shTTK (Figure S4A-B). Firstly, we examined whether USP9X or TTK could influence A549 cell proliferation. We seeded A549 cells with stable knockdown of USP9X or TTK, and measured the cell proliferation daily. Both USP9X-deficient and TTK-deficient A549 cells exhibited a much slower growth rate compared with shGFP A549 cells (Figure 4A). Then, to further figure out how USP9X/TTK functions in NSCLC tumorigenesis, A549 cells with stable knockdown of USP9X or TTK were injected into the right flank of nude mice, and the size of the resulted tumors was measured weekly. The measurements of the xenograft tumor size (Figure 4B) and weight (Figure 4C) demonstrated that the knockdown of USP9X or TTK suppressed tumor growth. The tumor proliferation curve showed that tumors originating from shUSP9X or shTTK-stable cells grew significantly slower than shGFP cells (Figure 4D). Finally, to test whether USP9X promoted A549 cell proliferation through TTK, we transiently overexpressed TTK in the shUSP9X cells. Our results demonstrated that transient overexpression of TTK could rescue cell proliferation suppression induced by USP9X knockdown (Figure S4C-D). Taken together, these results suggested that USP9X-mediated TTK stabilization plays an important role in tumorigenesis.
USP9X and TTK knockdown partially suppres cell proliferation and NSCLC. (A) A549 cells stably expressing shGFP, shUSP9X or shTTK were subjected to a cell proliferation assay. Each point represents the mean ± s.d. for biological quintuplicate experiments (n = 5, *** P < 0.001, two-way ANOVA). (B) Tumor images of nude mice (BALB/c; Slac Laboratory Animal, Shanghai, China) with subcutaneous injection of 5×106 A549 cells stably transfected with shGFP, shUSP9X or shTTK at day 28 after implantation. (C) Weights of tumors in (B) were measured. Each bar represents the mean ± s.d. for different mice groups (n = 6, *** P < 0.001, one-way ANOVA). (D) Tumor volumes were measured weekly. Each bar represents the mean ± s.d. for different mice groups (n = 6, *** P < 0.001, two-way ANOVA).
The expressions of USP9X and TTK are elevated and correlated in human NSCLC
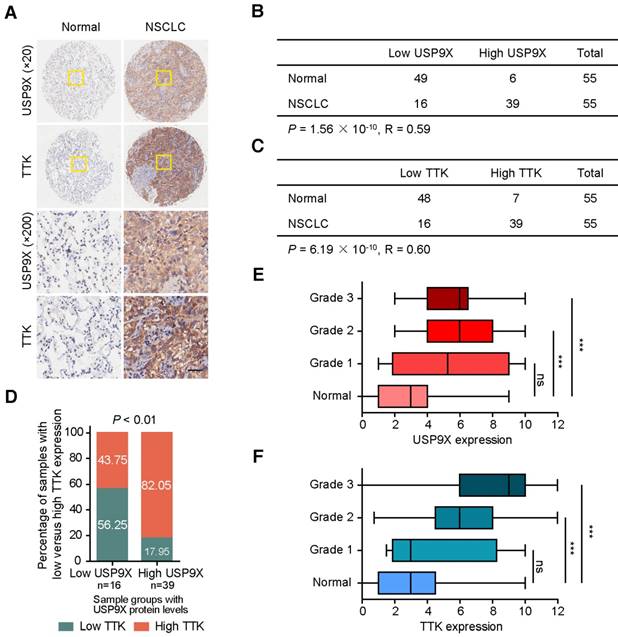
Owing to its tumor promotion nature, we postulated that USP9X and TTK were both abnormally expressed in human cancers. To test this hypothesis, we examined the protein levels of USP9X and TTK in a tissue microarray by immunohistochemical staining (Figure 5A). High staining scores of USP9X and TTK were obtained in 39 out of the 55 (71%) NSCLC tumor samples. While only 11% (6 out of 55) for USP9X and 13% (7 out of 55) for TTK in the matched normal tissues showed high expression, respectively (Figure 5B-C). Statistical analysis revealed that there is a significantly positive correlation between USP9X/TTK expression and NSCLC. Meanwhile, about 56% of tumor samples with low USP9X (n =16) showed low TTK expression, while 82% of the high USP9X samples (n =39) exhibited high TTK expression (Figure 5D). Moreover, we found a trend between USP9X/TTK expression and pathologic differentiation. Although USP9X and TTK were not elevated in stage I tumors compared to normal tissues, they were elevated in tumors ranging from stage II to III (Figure 5E-F). These results suggested that the progressive NSCLC were highly associated with USP9X and TTK expressions.
The expression levels of USP9X and TTK are increased and correlated in NSCLC. (A) Representative immunohistochemical staining for USP9X in NSCLC tissues (n = 55) and matched adjacent normal tissues (n = 55). Scale bar, 50 μm. (B) USP9X expression levels in tumors and normal tissues. (C) TTK expression levels in tumors and normal tissues. Statistical significance in (B-C) was calculated by the chi-square test. R is the Pearson correlation coefficient. (D) Positive correlation between USP9X and TTK expression levels in NSCLC (** P < 0.01, chi-square test). (E) Correlation between USP9X protein expression level and the clinical stages of NSCLC. (F) Correlation between TTK protein expression level and the clinical stages of NSCLC (*** P< 0.001, one-way ANOVA).
Discussion
USP9X has been reported to be an oncogene or a tumor suppressor in different cancers primarily based on the context-specific function of its substrates [36]. To shed light on the functional role of USP9X in lung cancer, we carried out TMT-based quantitative proteomics to identify substrate candidates of USP9X. Compared with a TMT quantitative siUSP9X proteome dataset in MCF-7 cells presented by Wang et al. [37], we identified 94% of the total proteins in their reported dataset, and there were 3265 more proteins identified in our study (Figure S1A-B). Consistent with the MCF-7 proteome, we identified the known USP9X substrate protein CEP55. Moreover, 96 additional down-regulated proteins (using criterion adapted from Wang et al.) were uniquely represented in our dataset. Therefore, the quantitative proteomic method in this study was highly sensitive and reproducible, thus allowing for identification of the low abundance substrate proteins of USP9X, such as kinases and transcriptional factors.
Among these down-regulated proteins, TTK was verified to have the same downregulation trend as USP9X (Figure 1B-D). Depletion of USP9X by siRNA led to reduced protein level and stability of TTK (Figure 2C-D, G-H). Immunoprecipitation assays showed that there was a direct interaction between USP9X and TTK (Figure 2A-B). Our experiments also demonstrated that a reduction in USP9X also led to a reduction of TTK stability in a proteasome-dependent manner (Figure 2E). Furthermore, we evidenced that depletion of USP9X only increased TTK K48 linkage poly-ubiquitination and had minimal effect on TTK K63 linkage poly-ubiquitination, further supporting that USP9X protects TTK from ubiquitination-mediated proteasome degradation (Figure 2F and Figure S2E).
We noted that previous studies reported overexpression of USP9X in human NSCLC [20, 21]. TTK mRNA level was also reported to be elevated [24], and TTK was considered as a tumor marker and therapeutic target for lung cancer (Y. N, Y. D, S. N, patent EP2102360A2). Therefore, we believed that the link between USP9X and TTK was one of multiple pathways that play important roles in cancer cells. We hypothesized that overexpression of USP9X elevates TTK levels, which in turn promotes progression of NSCLC. Although USP9X has been reported to have multiple substrates and participate in many cellular processes, our experimental results reveal that TTK is another substrate of USP9X and contributes to the function of USP9X. Suppression of USP9X or TTK expression caused significant proliferation, migration and growth inhibition of A549 cells (Figure 3A-E and Figure 4A). These results suggested that USP9X may regulate cell proliferation, migration and growth through its control of TTK degradation. Transiently overexpressed TTK on shUSP9X cells could rescue the USP9X knockdown-induced cell proliferation suppression (Figure S4C-D), demonstrating that TTK could contribute to the effect of USP9X depletion.
The partially selective DUB inhibitor WP1130 triggers aggresome formation and directly inhibits DUB activity of USP9X, USP5, USP14 and UCH37 to increase tumor cell apoptosis [35]. WP1130 treatment suppressed USP9X expression, resulting in transcription factor E-twenty-sis related gene (ERG) degradation and growth inhibition of ERG-positive prostate tumors [38]. Previous studies stated that WP1130 increases cisplatin sensitivity in estrogen receptor-negative breast cancer cells through inhibition of USP9X [39]. WP1130 becomes an effective tool in investigation of DUBs function in cancer. In our study, we confirmed that WP1130 reduced the effect of USP9X to stabilize TTK by cycloheximide-chase assays (Figure 3F). We additionally found that USP9X inhibitor WP1130 decreased cell proliferation of A549 cells (Figure 3E). Meanwhile, we observed that A549 cells depleted of either USP9X or TTK showed increased sensitivity towards WP1130 treatments (Figure 3G), which indicates the impact of TTK on the function of USP9X. To further investigate the role of USP9X/TTK in tumorigenesis of NSCLC, we transplanted USP9X- or TTK-depleted A549 cells into the right flank region of nude mice, and observed greatly suppressed tumor growth in nude mice with A549 cells expressing lower levels of USP9X or TTK (Figure 4B-D).
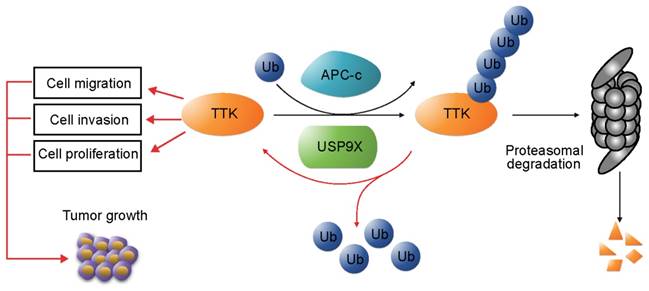
In addition, we examined the protein levels of USP9X and TTK in clinical samples of NSCLC. Our results revealed that both USP9X and TTK expressions were elevated in NSCLC tissues and positively correlated to NSCLC progression. In conclusion, this study identified USP9X as a deubiquitinase of TTK, and USP9X promotes tumorigenesis of NSCLC through stabilizing TTK (Figure 6).
USP9X-mediated stabilization of TTK promotes tumorigenesis in NSCLC. TTK is targeted for proteasome-dependent degradation by E3 ubiquitin ligase APC-c. USP9X decreases ubiquitination of TTK, and thereby enhances its half-life. Overexpression of USP9X would increase TTK levels and subsequently promote cell proliferation, migration and invasion. Immunohistochemistry experiments on clinical samples of NSCLC showed that elevated USP9X in NSCLC patients dramatically increases TTK levels and thus promotes tumor growth.
Future studies are warranted to investigate the physiological functions of USP9X and its synergy with other DUBs to control TTK in vivo. Sufficient elucidation of TTK function in cell proliferation and tumor growth is also valuable for understanding the post-translational regulatory spectrum in cancer cells.
Materials and methods
Plasmids, siRNAs and antibodies
Plasmids expressing the full length USP9X (pEF-DEST51 V5-tagged mouse USP9X) or the catalytically inactive mutant (C1566S) were kindly provided by professor Stephen Wood, Eskitis Institute for Cell and Molecular Therapies, Griffith University, Brisbane, Queensland, Australia. Human TTK was cloned into pCMV-Flag. All siRNA sequences were synthesized by GenePharma (Shanghai, China). Two out of three siRNAs of USP9X were previously described [30]: 5'- AGAAAUCGCUGGUAUAAAUUU -3' (siUSP9X_1), 5'- GCAGUGAGUGGCUGGAAGUTT -3' (siUSP9X_2). The other one is 5'- GGACUUCUUUGAAAGUAAUTT -3' (siUSP9X_3). The siRNA target sequences of TTK are: 5'- GGAUUUAAGUGGCAGAGAATT -3' (siTTK_1), 5'- GGUCGUUACAGUCAAGCAATT - 3' (siTTK_2) and 5'- CCAGAGGACAGACUACUAATT -3' (siTTK_3). Plasmids and siRNAs were transfected with Lipofectamine 3000 (Thermo, L3000015). All siRNAs were used at 60 nM. The following antibodies were used: β-ACTIN (Cell Signaling, 3700), USP9X (Cell Signaling, 14898), TTK (Millipore, 05-682), p27 (Cell Signaling, 3686), HA (Sigma-Aldrich, H6908), Flag (Sigma-Aldrich, SAB4301135), His (Sigma-Aldrich, H1029), IgG (Cell Signaling, 2729).
Cell culture and drug treatments
A549, 293T, HeLa, NCI-H3122, NCI-H125, NCI-H1975 and 293FT cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China), and A549, 293T, HeLa and 293FT were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gibco, 26400044) and 1% penicillin/streptomycin. NCI-H3122, NCI-H125 and NCI-H1975 were maintained in Roswell Park Memorial Institute 1640 (RPMI 1640). MG132 (Sigma-Aldrich, C2211) was used at 10 μM, cycloheximide (CHX; Targetmol, T1225) was used at 100 μg/mL unless otherwise specified, nocodazole (Noc; Sigma-Aldrich, M1404) at 100 ng/mL, WP1130 (SelleckChem, S2243) at 5 μM unless otherwise specified, and Bortezomib (SelleckChem, S1013) at 10 μM. For synchronization in mitosis, A549 and HeLa cells were treated with nocodazole for 16 h.
shRNA constructs and Lentiviral production
The target sequences of USP9X shRNAs: 5'- CGACCCTAAACGTAGACATTA - 3' (shUSP9X_1), 5'-CGATTCTTCAAAGCTGTGAAT - 3' (shUSP9X_2) and 5' -CCACCTCAAACCAAGGATCAA -3' (shUSP9X_3). The target sequences of TTK shRNAs: 5' -GCACAATTTGAACTGTCACAA -3' (shTTK_1) and 5' -GCCAACTTGTTGGTCTGAATT - 3' (shTTK_2). The shRNAs of USP9X and TTK were constructed in pLKO.1 vector. Plasmid shRNAs in the PLKO.1 vector were co-transfected into 293T cells along with two assistant vectors containing the vsvg and Δ8.9 genes. Lentivirus was harvested at 24 h and 48 h after transfection and subsequently clarified by a 0.45 μm filter. 10 μg/mL polybrene was added to the viruses before infecting cells, and stable cell lines were selected with treatment of 2 μg/mL puromycin for one week.
Cell viability and proliferation analysis
Cell viability was analyzed using Cell Counting Kit-8 (CCK-8; Beyotime, C0038). Cells were seeded into 96-well plates (7000 cells/well). After treatment with different concentrations of WP1130, the optical density (OD) at 490 nm for each well was obtained using a microplate reader (BIO-RAD Model 3550). Cell proliferation was also analyzed with CCK-8 assay. Cells were seeded into 96-well plates (500 cells/well), then incubated at 37 °C for the next 6 days. From day 1 to day 7, CCK-8 assay was performed conforming to the manufacturer's instructions. The OD value at 450 nm for each well was measured.
Immunoprecipitation and immunoblotting
Immunoprecipitation and immunoblotting were performed as described previously [40].
Cohort and immunohistochemistry
The immunohistochemistry was done by Shanghai Outdo Biotech Company, Shanghai, China. 55 pairs of NSCLC tissues with matched adjacent normal tissues were analyzed. Incubation with antibodies against USP9X (1:100 dilution, Cell Signaling, #14898) or TTK (1:1000 dilution, Merck Millipore, MABE571) was carried out at 4 ºC overnight. Photomicrographs were taken with the ScanScope XT (Leica, CA, USA). Slides were scored by a certified pathologist blinded to the clinicopathological data of the samples. Each sample was assigned a score according to the intensity of the staining (no staining = 0, weak staining = 0-1, moderate staining = 1-2, strong staining = 2-3) and the percentage of positive cells (0% = 0, 1-24% = 1, 25-49% = 2, 50-74% = 3, 75-100% = 4) as previously reported [41]. The final score was obtained by multiplying the intensity score with the percentage of positive cells. Samples exhibiting scores < 6 were called USP9X/TTK low, and samples showing scores 6-12 were summarized as USP9X/TTK high. The correlation between TTK and USP9X, and the correlation of TTK or USP9X with tissue type were verified by the chi-square test.
Colony formation assay
A549 cells stably expressing shRNAs targeting USP9X or TTK were plated onto 6-well plates (500 cells/well). After the cells had incubated for 14 days and their colonies were visible to the naked eye, each well was washed three times with PBS, fixed with methanol for 10 min, and then stained with 1% crystal violet solution for 15 min. The numbers of colonies per well were manually counted. The experiments were independently triplicated. For the WP1130 treatment assay, the indicated concentration of WP1130 was added to the cells after plating (500 cells/well). After 14 days, the cells were washed with PBS, fixed with methanol, stained with 1% crystal violet and counted.
Cell migration and invasion assay
A549 cells treated with different siRNAs targeting USP9X or TTK were plated in 96-well plates for wound-healing assays. The plates were photographed by IncuCyte ZooM (Essen BioScience, Michigan, USA). For the transwell assays (Costar, #3422), A549 cells stably expressing shRNAs were placed on the upper layer of a cell-permeable membrane and media containing 10% FBS was placed in the lower chamber. After incubation for 20 h, the cells that had migrated through the membrane were stained and counted.
Tumor xenografts
The animal experiment was carried out by the animal experimental center in Shanghai University of Traditional Chinese Medicine. A549 cells stably expressing shRNAs (5×106) were suspended in 100 μL PBS and subcutaneously injected into the right flank region of nude mice (4-6 weeks old). Tumor sizes were measured every 7 days with a caliper and tumor volumes were determined with the formula: L × W2 × 0.5, where L is the longest diameter and W is the shortest diameter. 4 weeks later, all mice were sacrificed and the tumors were harvested, photographed and weighed. All treatments were administered according to the guidelines of the Institution Animal Care and Use Committee and all protocols were approved by the Ethics Committee of Shanghai University of Traditional Chinese Medicine.
Protein extraction and filter-aided sample preparation
The A549 cells were washed three times with cold PBS and the cells were lysed in SDT lysis buffer (0.2% SDS (m/v), 100 mM DTT, 100 mM Tris, pH=7.6). The lysates were homogenized with sonication and centrifuged at 15,000 ×g for 30 min. The supernatants were collected, and protein concentration was determined by tryptophan fluorescence emission assay as described previously [42]. FASP was performed as described previously with slight modifications [43], using 50 mM triethylammonium bicarbonate (TEAB) instead of ABC buffer for compatibility with TMT labeling.
TMT labeling
Peptides were reconstituted in 100 mM TEAB and their concentration determined by the BCA assay. 3 shGFPs control samples and 3 USP9X shRNAs knockdown samples (50 μg, each) were labelled by TMT 6-plex reagents (0.8 mg, each) (Thermo Fisher Scientific) with reporters at m/z = 126, 127, 128, 129, 130, 131, respectively. Each sample containing 50 μg of peptide in 50 μL volume of TEAB buffer was combined with 41 μL of its respective 6-plex TMT reagent and incubated for 1 h at room temperature. Then, 8 μL of 5% hydroxylamine was added to the sample and incubated for 15 min to quench the reaction. Equal amounts of each TMT-labelled sample were combined in new microcentrifuge tubes and dried down using a SpeedVac.
High pH fractionation
Half of the TMT-labeled peptides mixture was fractionated using a Waters XBridge BEH130 C18 3.5 μm 2.1 × 150 mm column on an Agilent 1290 HPLC operating at 0.2 mL/min. Buffer A consisted of 10 mM ammonium formate and buffer B consisted of 10 mM ammonium formate with 90% acetonitrile; both buffers were adjusted to pH 10 with ammonium hydroxide as described previously [44]. A CBS-B programed multifunction automatic fraction-collecting instrument (Huxi instrument, Shanghai, China) was coupled to the HPLC and used to collect eluted peptides. The total number of fractions collected was 36 and concatenated to 19 (pooling equal interval RPLC fractions). Ammonium hydroxide and/or ammonium formate were evaporated in a SpeedVac.
Nanoflow liquid chromatography tandem mass spectrometry
All experiments were performed on an Orbitrap Fusion Lumos mass spectrometer with an ancillary nanoLC (EASY-nLC II; Thermo Fisher Scientific). Peptides were loaded on a self-packed column (75 μm × 150 mm, 3 μm ReproSil-Pur C18 beads, 120 Å, Dr. Maisch GmbH, Ammerbuch, Germany) and separated with a 120 min gradient for fractionation of samples and 180 min gradient for total samples at a flow rate of 300 nL/min. Solvent A was 100% H2O, and 0.1% formic acid; solvent B was 100% acetonitrile, and 0.1% formic acid. The Orbitrap Fusion Lumos was programmed in the data-dependent acquisition mode. An MS1 survey scan of 350-1500 m/z in the Orbitrap at a resolution of 60,000 was collected with an AGC target of 400,000 and maximum injection time of 20 ms. Precursor ions were filtered according to monoisotopic precursor selection, charge state (+ 2 to + 7), and dynamic exclusion (40 s with a ±10 ppm window). Then, the most intense precursors were subjected to HCD fragmentation with a duty cycle of 3 s. The instrument parameters were set as follows: 32% normalized collision energy, 30,000 resolution, 50,000 AGC target, 50 ms maximum injection time, 100 Da first mass, inject ions for all available parallelizable time enabled, 1.6 m/z isolation width.
MS raw data
The mass spectrometry proteomics data have been deposited to the ProteomeXchange [45] Consortium via the PRIDE [46] partner repository with the dataset identifier PXD007726.
Abbreviations
APC-c: anaphase-promoting complex/ cyclosome; B: Bortezomib; CHX: protein synthesis inhibitor cycloheximide; D: DMSO; DMEM: Dulbecco's modified Eagle's medium; DUB: deubiquitinase; JAMMs: JAB1/MPN/Mov34 metalloenzymes; Noc: Nocodazole; NSCLC: non-small cell lung cancer; OTUs: Ovarian tumour proteases; RPMI 1640: Roswell Park Memorial Institute 1640; SILAC: stable isotope labeling with amino acids in cell culture; TTK: Dual specificity protein kinase TTK; UCHs: ubiquitin C-terminal hydrolases; USPs: ubiquitin-specific proteases; WT: wild type.
Acknowledgements
We thank Professor Stephen Wood for kindly providing the plasmids containing the full length USP9X or the catalytically inactive mutant (C1566S). This research was supported by Grant No. 2017YFC1700200 of the National Key Research and Development Program from the Ministry of Science and Technology of China, by Grant No. 21375138 and 31570830 from National Natural Science Foundation of China, by “one hundred talent program” of Chinese Academy of Sciences, by the Strategic Priority Research Program of the Chinese Academy of Sciences, “Personalized Medicines-Molecular Signature-based Drug Discovery and Development” (Grant No. XDA12030203), and by the Innovation Project of Instrument and Equipment Function Development (Grant No. YZ201542 etc.) from the Bureau of Goods, Chinese Academy of Sciences. We thank the Institutional Technology Service Center of Shanghai Institute of Materia Medica, Chinese Academy of Sciences for technical support.
Author contributions
XLC, CLY and JG performed the experiments. CLC, CLY and HZ wrote the paper. HWZ and YTZ participated in mass spectrometry data analysis. BHC, QL and RXX contributed to plasmid construction. HH prepared the fractionation-MS samples. TZ and HX contributed to the wound-healing assays and invasion assays. RMH contributed to the co-IP and TTK rescue experiments. HZ and DMG conceived the project and supervised the experiments.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Global Burden of Disease Cancer C, Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M. et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015;1:505-27
2. Ridge CA, McErlean AM, Ginsberg MS. Epidemiology of lung cancer. Semin Intervent Radiol. 2013;30:93-8
3. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367-80
4. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203-29
5. Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550-63
6. Li Y, Luo K, Yin Y, Wu C, Deng M, Li L. et al. USP13 regulates the RAP80-BRCA1 complex dependent DNA damage response. Nat Commun. 2017;8:15752
7. Martin Y, Cabrera E, Amoedo H, Hernandez-Perez S, Dominguez-Kelly R, Freire R. USP29 controls the stability of checkpoint adaptor Claspin by deubiquitination. Oncogene. 2015;34:1058-63
8. Song L, Rape M. Reverse the curse-the role of deubiquitination in cell cycle control. Curr Opin Cell Biol. 2008;20:156-63
9. Jin J, Liu J, Chen C, Liu Z, Jiang C, Chu H. et al. The deubiquitinase USP21 maintains the stemness of mouse embryonic stem cells via stabilization of Nanog. Nat Commun. 2016;7:13594
10. Luise C, Capra M, Donzelli M, Mazzarol G, Jodice MG, Nuciforo P. et al. An atlas of altered expression of deubiquitinating enzymes in human cancer. PloS One. 2011;6:e15891
11. Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene. 2012;31:2373-88
12. Qin J, Zhou Z, Chen W, Wang C, Zhang H, Ge G. et al. BAP1 promotes breast cancer cell proliferation and metastasis by deubiquitinating KLF5. Nat Commun. 2015;6:8471
13. Xiao H, Tian Y, Yang Y, Hu F, Xie X, Mei J. et al. USP22 acts as an oncogene by regulating the stability of cyclooxygenase-2 in non-small cell lung cancer. Biochem Biophys Res Commun. 2015;460:703-8
14. Li X, Stevens PD, Yang H, Gulhati P, Wang W, Evers BM. et al. The deubiquitination enzyme USP46 functions as a tumor suppressor by controlling PHLPP-dependent attenuation of Akt signaling in colon cancer. Oncogene. 2013;32:471-8
15. D'Arcy P, Wang X, Linder S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol Ther. 2015;147:32-54
16. Perez-Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL. et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature. 2012;486:266-70
17. Toloczko A, Guo F, Yuen HF, Wen Q, Wood SA, Ong YS. et al. Deubiquitinating Enzyme USP9X Suppresses Tumor Growth via LATS kinase and Core Components of the Hippo pathway. Cancer Res. 2017;77:4921-33
18. Potu H, Peterson LF, Kandarpa M, Pal A, Sun H, Durham A. et al. Usp9x regulates Ets-1 ubiquitination and stability to control NRAS expression and tumorigenicity in melanoma. Nat Commun. 2017;8:14449
19. Peng J, Hu Q, Liu W, He X, Cui L, Chen X. et al. USP9X expression correlates with tumor progression and poor prognosis in esophageal squamous cell carcinoma. Diagn Pathol. 2013;8:177
20. Wang Y, Liu Y, Yang B, Cao H, Yang CX, Ouyang W. et al. Elevated expression of USP9X correlates with poor prognosis in human non-small cell lung cancer. J Thorac Dis. 2015;7:672-9
21. Peddaboina C, Jupiter D, Fletcher S, Yap JL, Rai A, Tobin RP. et al. The downregulation of Mcl-1 via USP9X inhibition sensitizes solid tumors to Bcl-xl inhibition. BMC cancer. 2012;12:541
22. Liu X, Winey M. The MPS1 family of protein kinases. Annu Rev Biochem. 2012;81:561-85
23. Yuan B, Xu Y, Woo JH, Wang Y, Bae YK, Yoon DS. et al. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin Cancer Res. 2006;12:405-10
24. Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A. et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PloS One. 2008;3:e1651
25. Salvatore G, Nappi TC, Salerno P, Jiang Y, Garbi C, Ugolini C. et al. A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma. Cancer Res. 2007;67:10148-58
26. Cui Y, Cheng X, Zhang C, Zhang Y, Li S, Wang C. et al. Degradation of the human mitotic checkpoint kinase Mps1 is cell cycle-regulated by APC-cCdc20 and APC-cCdh1 ubiquitin ligases. J Biol Chem. 2010;285:32988-98
27. Liu C, van Dyk D, Choe V, Yan J, Majumder S, Costanzo M. et al. Ubiquitin ligase Ufd2 is required for efficient degradation of Mps1 kinase. J Biol Chem. 2011;286:43660-7
28. Li X, Song N, Liu L, Liu X, Ding X, Song X. et al. USP9X regulates centrosome duplication and promotes breast carcinogenesis. Nat Commun. 2017;8:14866
29. Poulsen JW, Madsen CT, Young C, Kelstrup CD, Grell HC, Henriksen P. et al. Comprehensive profiling of proteome changes upon sequential deletion of deubiquitylating enzymes. J Proteomics. 2012;75:3886-97
30. McGarry E, Gaboriau D, Rainey MD, Restuccia U, Bachi A, Santocanale C. The Deubiquitinase USP9X Maintains DNA Replication Fork Stability and DNA Damage Checkpoint Responses by Regulating CLASPIN during S-Phase. Cancer Res. 2016;76:2384-93
31. Engel K, Rudelius M, Slawska J, Jacobs L, Ahangarian Abhari B, Altmann B. et al. USP9X stabilizes XIAP to regulate mitotic cell death and chemoresistance in aggressive B-cell lymphoma. EMBO Mol Med. 2016;8:851-62
32. Vong QP, Cao K, Li HY, Iglesias PA, Zheng Y. Chromosome alignment and segregation regulated by ubiquitination of survivin. Science. 2005;310:1499-504
33. Dobashi Y, Tsubochi H, Minegishi K, Kitagawa M, Otani S, Ooi A. Regulation of p27 by ubiquitin ligases and its pathological significance in human lung carcinomas. Hum Pathol. 2017;66:67-78
34. Kushwaha D, O'Leary C, Cron KR, Deraska P, Zhu K, D'Andrea AD. et al. USP9X inhibition promotes radiation-induced apoptosis in non-small cell lung cancer cells expressing mid-to-high MCL1. Cancer Biol Ther. 2015;16:392-401
35. Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010;70:9265-76
36. Murtaza M, Jolly LA, Gecz J, Wood SA. La FAM fatale: USP9X in development and disease. Cell Mol Life Sci. 2015;72:2075-89
37. Wang Q, Tang Y, Xu Y, Xu S, Jiang Y, Dong Q. et al. The X-linked Deubiquitinase USP9X Is an Integral Component of Centrosome. J Biol Chem. 2017;292:12874-84
38. Wang S, Kollipara RK, Srivastava N, Li R, Ravindranathan P, Hernandez E. et al. Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc Natl Acad Sci U S A. 2014;111:4251-6
39. Fu P, Du F, Liu Y, Yao M, Zhang S, Zheng X. et al. WP1130 increases cisplatin sensitivity through inhibition of usp9x in estrogen receptor-negative breast cancer cells. Am J Transl Res. 2017;9:1783-91
40. Inuzuka H, Gao D, Finley LW, Yang W, Wan L, Fukushima H. et al. Acetylation-dependent regulation of Skp2 function. Cell. 2012;150:179-93
41. Yuan L, Lv Y, Li H, Gao H, Song S, Zhang Y. et al. Deubiquitylase OTUD3 regulates PTEN stability and suppresses tumorigenesis. Nat Cell Biol. 2015;17:1169-81
42. Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11:319-24
43. Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359-62
44. Wang Y, Yang F, Gritsenko MA, Wang Y, Clauss T, Liu T. et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics. 2011;11:2019-26
45. Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T. et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017;45:D1100-D6
46. Vizcaino JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I. et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:D447-56
Author contact
![]() Corresponding author: Hu Zhou, Email: zhouhuac.cn, Tel: +86-21-50806706
Corresponding author: Hu Zhou, Email: zhouhuac.cn, Tel: +86-21-50806706