Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(9):2300-2312. doi:10.7150/thno.21670 This issue Cite
Research Paper
Companion Diagnostic 64Cu-Liposome Positron Emission Tomography Enables Characterization of Drug Delivery to Tumors and Predicts Response to Cancer Nanomedicines
Helen Lee1*, Daniel Gaddy1*, Manuela Ventura2, Nicholas Bernards2, Raquel de Souza2*, Dmitri Kirpotin1*, Thomas Wickham1*, Jonathan Fitzgerald1*, Jinzi Zheng2,3 ![]() , Bart S. Hendriks1*
, Bart S. Hendriks1*
1. Merrimack Pharmaceuticals, Inc.
2. TECHNA Institute for the Advancement of Technology for Health, University Health Network, Toronto, Ontario, Canada.
3. Institute of Biomaterials and Biomedical Engineering, University of Toronto, Ontario, Canada.
*Affiliation at the time of study.
Received 2017-6-28; Accepted 2018-2-25; Published 2018-3-21
Abstract

Deposition of liposomal drugs into solid tumors is a potentially rate-limiting step for drug delivery and has substantial variability that may influence probability of response. Tumor deposition is a shared mechanism for liposomal therapeutics such that a single companion diagnostic agent may have utility in predicting response to multiple nanomedicines.
Methods: We describe the development, characterization and preclinical proof-of-concept of the positron emission tomography (PET) agent, MM-DX-929, a drug-free untargeted 100 nm PEGylated liposome stably entrapping a chelated complex of 4-DEAP-ATSC and 64Cu (copper-64). MM-DX-929 is designed to mimic the biodistribution of similarly sized therapeutic agents and enable quantification of deposition in solid tumors.
Results: MM-DX-929 demonstrated sufficient in vitro and in vivo stability with PET images accurately reflecting the disposition of liposome nanoparticles over the time scale of imaging. MM-DX-929 is also representative of the tumor deposition and intratumoral distribution of three different liposomal drugs, including targeted liposomes and those with different degrees of PEGylation. Furthermore, stratification using a single pre-treatment MM-DX-929 PET assessment of tumor deposition demonstrated that tumors with high MM-DX-929 deposition predicted significantly greater anti-tumor activity after multi-cycle treatments with different liposomal drugs. In contrast, MM-DX-929 tumor deposition was not prognostic in untreated tumor-bearing xenografts, nor predictive in animals treated with small molecule chemotherapeutics.
Conclusions: These data illustrate the potential of MM-DX-929 PET as a companion diagnostic strategy to prospectively select patients likely to respond to liposomal drugs or nanomedicines of similar molecular size.
Keywords: 64Cu-liposome PET, nanomedicine, tumor deposition
Introduction
Liposomes are a specific class of phospholipid nanoparticles used to encapsulate and deliver small molecule therapies. These drug delivery systems have been particularly useful in oncology, and provide a means to improve the toxicity profiles and therapeutic windows for small molecule chemotherapies by enabling long-circulating pharmacokinetics, tunable sustained release, as well as improved drug deposition and exposure in solid tumors [1]. Several liposomal therapeutics have been approved for cancer treatment, including Doxil®/Caelyx®, Myocet®, DaunoXome®, Marqibo®, ONIVYDE®, and many others are in clinical development.
The propensity for liposomes to deposit in solid tumors is largely governed by their size (typically 50-120 nm in diameter), systemic exposure (pharmacokinetics), which may be dictated by their physico-chemical properties, and the “leakiness” or permeability of the tumor vasculature. Known as the enhanced permeability and retention (EPR) effect, this phenomenon predicts that the large size of liposomes prevents extravasation from normal vasculature, resulting in longer systemic circulation and eventual deposition and retention in areas of functionally porous vasculature, such as areas of inflammation and leaky vasculature in some solid tumors [1-5].
It has been documented that liposomal tumor deposition is highly variable and a rate-limiting step for effective drug delivery and anti-tumor activity [6]. Vascular permeability varies across patients, across distinct tumors within an individual patient, and possibly by anatomical location and tumor of origin as well [7-9]. Using 111In-DTPA-labeled PEGylated liposomes, Harrington et al. demonstrated that liposome deposition ranged from undetectable to 53% injected dose/kg in patients with different tumor types [7]. Similarly, quantification of drug delivery in patient biopsies 72 h after administration of liposomal irinotecan demonstrated 38-fold variation in irinotecan delivery [9]. Moreover, the identification of liposomal tumor deposition as a rate-limiting step for drug delivery to tumor cells supports the theory that deposition variability may directly contribute to differential responses to therapeutics [6]. Additionally, preclinical studies have indicated a correlation between variable liposome deposition in rat xenograft models and tumor response to single-dose PLD treatment [10]. Perez-Medina et al. reported that mouse tumors with > 25 mg/kg liposomal doxorubicin deposition had better growth inhibition, as revealed by a co-injected 89Zr-nanoreporter as surrogate measurement of nanoparticle deposition in tumors [11]. These results suggest that identification of patients exhibiting increased tumor deposition may have potential to improve the overall response rate and clinical outcome of liposomal therapeutics.
We have previously described the development of a gradient-loadable chelator, 4-DEAP-ATSC, as a means to efficiently and stably incorporate copper-64 (64Cu, t1/2 = 12.7 h) into liposomal formulations, and its utility in labeling and tracking the biodistribution and deposition of drug-loaded liposomes in animal models, as well as human subjects [8,12,13]. In a Phase 1 study, we reported that tumor deposition of 64Cu-labeled HER2-targeted liposomal doxorubicin (64Cu-MM-302) varied 35-fold in metastatic HER2-positive breast cancer patients, which is likely driven by the variability of the EPR effect. Most importantly, an exploratory retrospective analysis suggested that high 64Cu-MM-302 tumor deposition, quantified using positron emission tomography (PET), was associated with more favorable treatment outcomes. The ability to radiolabel drug-loaded liposomes with a PET isotope such as 64Cu provides a valuable translational tool for obtaining quantitative biodistribution and deposition data for therapeutic agents. However, a drug-free, non-therapeutic companion diagnostic imaging agent would have the additional advantage of sparing patients from harmful side effects of chemotherapy-containing liposomes, as well as potentially identifying patients most likely to benefit from any liposomal therapeutic.
Here, we describe the development of a drug-free 64Cu-loaded liposomal PET agent, MM-DX-929. MM-DX-929 is a 64Cu-encapsulating, untargeted, PEGylated liposome that is designed to mimic the biodistribution and tumor deposition of similarly sized therapeutic agents, suitable for implementation as a companion diagnostic to prospectively select patients for liposomal therapeutics. Specifically, we aim to demonstrate that MM-DX-929 is an effective imaging surrogate for measurement of tumor deposition and intratumoral distribution of liposomal drugs in mouse xenograft models of human cancer. Furthermore, we show that quantification of MM-DX-929 tumor deposition by PET can successfully identify responders to three liposomal therapeutics in different xenograft models.
Materials and Methods
Liposome Preparation
PEGylated liposomal doxorubicin (PLD, Doxil®) (Janssen Products; Titusville, NJ) was obtained from a pharmacy. MM-302 was prepared as previously described [14]. MM-302 is composed of HSPC:cholesterol:PEG-DSPE (3:2:0.3 mol ratio) (hydrogenated soybean phosphatidylcholine:cholesterol:PEG-1,2-distearoyl-sn-glycero-3-phosphoethanolamine) with ammonium sulfate as the gradient for loading doxorubicin. The anti-HER2 F5-PEG-DSPE conjugates were subsequently inserted into the pre-formed doxorubicin-loaded liposome. Liposomal irinotecan (irinotecan liposome injection, ONIVYDE®, MM-398), prepared as previously described [15,16], is composed of DSPC:cholesterol:PEG-DSPE (3:2:0.015) with triethylammonium sucrose octasulfate (TEA-SOS) as the trapping agent for loading irinotecan. Fluorescently-labeled liposomes were prepared by incorporating lipophilic carbocyanine DiIC18(5)-DS (Life Technologies, D12730) during the liposome preparation procedure as previously described [14].
MM-DX-929 liposomes composed of DSPC:cholesterol:PEG-DSPE (3:2:0.3 mol ratio) were prepared using methods similar to that reported for liposomal irinotecan [15,16]. Briefly, lipid components dissolved in ethanol mixed with 0.43N TEA-SOS formed multilamellar vesicles, which were extruded to form unilamellar liposomes. Extraliposomal TEA-SOS was subsequently exchanged with HEPES-buffered saline (HBS; 10mM HEPES, 150 mM NaCl, pH 7.2) by diafiltration. The resulting liposomes have particle size of ~100 nm determined by dynamic light scattering. 3H-MM-DX-929 liposomes were prepared by incorporating [cholesteryl-1,2-3H(N)]-cholesteryl hexadexyl ether (3H-CDHE) into the lipid-ethanol mixture described above prior to extrusion.
Chelation of 64Cu to 4-DEAP-ATSC and Loading into MM-DX-929 Liposomes
64CuCl2 (Washington University; St. Louis, MO USA) was chelated to a loading agent (Diacetyl 4,4′-bis(3-(N,N-diethylamino)propyl)thiosemicarbazone, or 4-DEAP-ATSC) at 0.35 mCi/nmol ratio using a method as previously described [12]. 64Cu-chelator was added to MM-DX-929 liposomes (< 0.2% chelator:lipid mol ratio), heated to 65ºC for 10 min and then cooled to room temperature in an ice water bath. The efficiency of loading was measured by size exclusion chromatography (SEC) as previously described [12]. The 64Cu-loaded liposomes are referred to as MM-DX-929 hereafter.
In vitro and In vivo Stability of MM-DX-929
In vitro stability of MM-DX-929 was evaluated by incubation in human plasma at 37ºC, followed by SEC to separate liposomal 64Cu, free 64Cu, and 64Cu:4-DEAP-ATSC complex. The SEC procedure is described in detail elsewhere [12] and in Supplementary Fig. S1. In vivo stability of MM-DX-929 was evaluated by comparing the pharmacokinetics of 64Cu and 3H components in immunocompetent CD-1 mice (20 µmol lipid/kg). Mice were injected with 3H-MM-DX-929 intravenously; blood samples were collected via saphenous vein at 5 min, 1 h, 4 h, 24 h, and 48 h post-injection (h.p.i.). 64Cu and 3H in plasma were quantified by scintillation counting.
Animal Studies
All animal work carried out at University Health Network was approved by the institution's Animal Care Committee, and adhered to the Animal Welfare Act (AWA) and Animal Welfare Regulations (AWR). Animal studies performed at Merrimack Pharmaceuticals are also in compliance with guidelines established by the Institutional Animal Care and Use Committee.
PET/CT Image Acquisition and Analysis
Female nu/nu or NOD/SCID mice were inoculated with BT474-M3 n = 12 (orthotopic mammary fat pad; 10×106 cells), SUM190 n = 12 (orthotopic mammary fat pad; 10×106 cells), MDA-MB-231 n = 10 (orthotopic mammary fat pad; 4×106 cells), H520 n = 4 (subcutaneous; 5×106 cells), or HT-29 n = 9 (subcutaneous; 5×106 cells) cancer cells suspended in cell culture media (supplemented with 10% FBS, 1% penicillin-streptomycin). Once tumors were established (average tumor volume ~250 mm3), mice were injected with MM-DX-929 intravenously via the lateral tail vein (10-13 MBq/mouse, 20 µmol lipid/kg). For image acquisition, mice were anesthetized using 2% inhaled isoflurane. PET images were acquired on a Focus 120 or 220 microPET (Siemens; Malvern, PA). Images were acquired over a 45 min period. At the center field of view, the acquisition resolution was 1.4 mm. Data was reconstructed using a maximum a posteriori (MAP) algorithm with voxel size of 0.146 × 0.146 × 0.796 mm3, or 2D ordered subset expectation maximization (OSEM2D) algorithm with voxel size of 0.866 × 0.866 × 0.796 mm3 (HT-29 data only). Anatomical CT scans were obtained on a Locus Ultra microCT preclinical scanner (GE Healthcare; Pittsburgh, PA) operated at 80 kVp and 50 mA, or NanoSPECT/CT (Bioscan; Washington, DC).
PET/CT images were registered using a semi-automated rigid registration algorithm on Inveon Research Workplace (Siemens; Malvern, PA), or VivoQuant (inviCRO; Boston, MA) software. Regions of interest (ROIs) were drawn manually on PET/CT slices in each tissue of interest. A linear extrapolation algorithm was applied to connect the ROIs to generate tissue volumes for quantification.
Tracer kinetic modeling
Tracer kinetic modeling for describing liposome transport into and out of tumors was performed using a model identical in structure to that previously used [8] with parameters changed to reflect mouse physiology. Briefly, the pharmacokinetics of MM-DX-929 was represented with a single blood compartment, and clearance characterized by a first-order elimination rate constant, kel. The tumor was described in a semi-physiological manner with a vascular portion, and tissue portion consisting of cellular and interstitial space. The fractional volume of the tumor occupied by vasculature was described by vascular volume fraction (VVF). Blood flow rate into and out of the tumor (Q) was assumed constant at 0.212 L/kg/min [17]. Washout of particles from the tumor, either back into the blood or via lymphatic drainage, are lumped into a single process for simplicity. Deposition and washout of MM-DX-929 into and out of the tumor tissue space was assumed to follow first-order kinetics and were characterized by rate constants k1 and k-1, respectively. The following parameters were estimated directly from the kinetic data: kel, VVF, k1, k-1 using median values extracted from ROI from the PET images and associated tumor volume measurements. Additional information on the model parameters are provided in Supplementary Table S1. The model was implemented in MATLAB R2016b (The Mathworks, Natick, MA).
Tumor Deposition and Intratumoral Distribution of MM-DX-929 and Liposomal Drugs
BT474-M3 (n = 12) or HT-29 (n = 10) tumor-bearing xenografts were co-injected with MM-DX-929 and one of the liposomal drugs (DiI5-PLD at 3 mg/kg, DiI5-MM-302 at 3 mg/kg, liposomal irinotecan at 10 mg/kg) intravenously. At 24 h.p.i., mice were perfused with 10 mL of phosphate-buffered saline to remove residual agents in circulation, tumors were subsequently excised for gamma-scintillation counting, autoradiography, immunofluorescence (IF) imaging, or quantification of drug deposition using high performance liquid chromatography (HPLC). For autoradiography and IF, tumors were embedded in O.C.T. Tissue-Tek® (Sakura Finetek Europe; The Netherlands), and cut into 20 µm sections. Autoradiographic images were acquired using the Cyclone Plus Storage Phosphor System (Perkin Elmer Inc., MA); consecutive sections were mounted on glass slide for DiI5 detection using the Aperio Scanscope FL. Doxorubicin and irinotecan deposition in tumors were measured using previously reported HPLC methods [14,16].
Stratification with MM-DX-929 PET
At approximately 250 mm3 average tumor volume, BT474-M3 or HT-29 tumor-bearing xenografts were injected intravenously with MM-DX-929. PET/CT images of MM-DX-929 were acquired at 20-24 h.p.i. for quantification of tumor deposition. Immediately after PET/CT imaging, mice began treatment with the indicated therapeutics (MM-302, PLD, liposomal irinotecan, doxorubicin, or untreated) for up to 6 weekly cycles (q1w). Tumor volumes were measured twice per week using a digital caliper up to 50 days after beginning treatment. Animals were assigned to “high” and “low” deposition groups based on MM-DX-929 signal in tumors measured using PET, using median uptake of MM-DX-929 as the threshold for stratification.
Statistical Analysis
Statistical and pharmacokinetic analyses were performed using MATLAB or GraphPad Prism version 7.0 (GraphPad Software, San Diego, CA), respectively. Only nonparametric statistical tests were utilized, including Mann-Whitney, Kruskal-Wallis, or Spearman Correlation, wherever applicable. Data are shown as mean ± SEM unless otherwise indicated.
Results
Characterization of MM-DX-929 as a Liposomal Imaging Agent
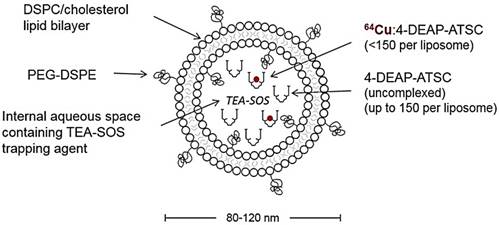
We have previously reported characterization of the 4-DEAP-ATSC chelator and its chelation chemistry [12]. In a recent Phase 1 clinical study, the 64Cu:4-DEAP-ATSC compound loaded into MM-302 administered to patients was well-tolerated and resulted in good quality PET images [8]. In the current study, the efficiency of 64Cu loading into MM-DX-929 liposomes was tested using various molar ratios of 4-DEAP-ATSC to phospholipid. Loading efficiency remained greater than 97% as the concentration of chelator increased relative to phospholipid concentration (up to 8000 chelator/liposome), illustrating a robust procedure capable of loading high levels of 64Cu into liposomes. The final 64Cu-loading protocol utilizes a molar excess of 4-DEAP-ATSC to 64Cu and liposome (~150 chelator/liposome) to ensure a high radiolabeling efficiency. High loading efficiency coupled with the observations that free 64Cu and 64Cu:4-DEAP-ATSC are rapidly cleared in vivo [12] obviate the need for any purification steps prior to use. A schematic representation of MM-DX-929 is shown in Fig. 1.
Schematic of MM-DX-929 - a liposomal PET imaging companion diagnostic agent. MM-DX-929 is a highly PEGylated (10 mol%) untargeted liposome, stably encapsulating 64Cu:4-DEAP-ATSC complex and excess uncomplexed 4-DEAP-ATSC chelators. PEG-DSPE = polyethylene glycol-1,2-distearoyl-sn-glycerol-3-phosphoethanolamine; TEA-SOS = triethylammonium sucrose octasulfate; DSPC = 1,2-distearoyl-sn-glycero-3-phosphocholine.
64Cu PET reporter is Stably Encapsulated in MM-DX-929 under Physiological Conditions
The stability of the MM-DX-929 was evaluated in vitro by incubation of MM-DX-929 in human plasma at 37°C for up to 48 h. Size exclusion chromatography (SEC) was performed to separate liposomal 64Cu from released/non-liposomal-64Cu. As shown in Fig. 2A and B, greater than 98% of 64Cu remained in the liposomal fraction at 0 h and after 48 h of incubation in human plasma at 37°C. This demonstrates that MM-DX-929 stably retains the 64Cu label over the useful imaging lifetime of 64Cu (radioactive t1/2 of 64Cu = 12.7 h) under physiological conditions.
In vivo and in vitro stability of 64Cu encapsulation in MM-DX-929. (A) Sepharose CL4B columns were characterized using fluorescently-labeled liposome or human plasma to determine the fractions at which liposome-bound or plasma-protein-bound 64Cu:4-DEAP-ATSC elute, respectively. Free 64Cu is retained within the column with < 3% of recovery from elution at 100 mL of cumulative volume. Following incubation with human plasma at 37ºC for 0 h and 48 h, aliquots of MM-DX-929/plasma mixture were loaded onto the columns to separate the liposomal 64Cu from non-liposomal 64Cu. (B) The percentage of 64Cu retained within the liposome was determined from (A) by dividing the 64Cu signal recovered from MM-DX-929 in the liposome fraction by the total radioactivity recovered. (C) Mice were injected with a single dose of MM-DX-929 or 3H-MM-DX-929. At designated time points up to 48 h.p.i., a blood sample was collected via saphenous vein puncture. 64Cu and 3H radioactivity in the plasma fraction was quantified using scintillation-counting. Data is decay-corrected. 3H serves as a stable tracer for the liposomal component of MM-DX-929 and was incorporated into the formulation as 3H-CDHE. (D) Ratio of 64Cu-to-3H in plasma derived from (C) was plotted as a function of time post-injection.
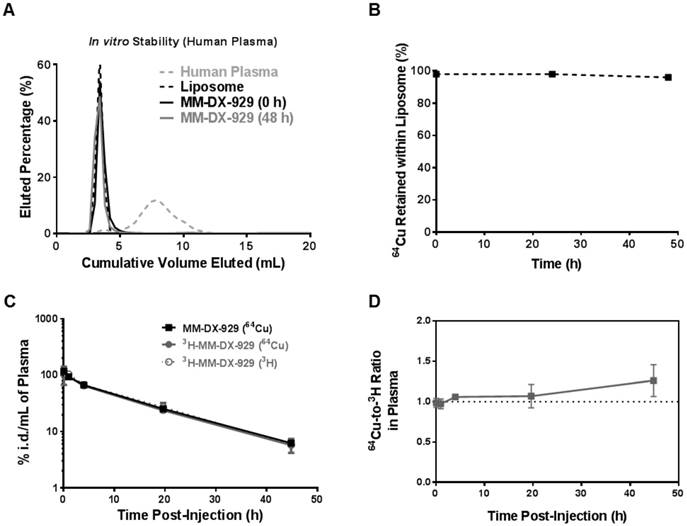
In order to confirm if the 64Cu remains associated with the liposome following intravenous injection, 3H-MM-DX-929 (i.e. dual-labelled with 3H and 64Cu) was prepared as a secondary in vivo tracer. The blood clearance of 3H (liposome shell) and 64Cu (PET reporter) in immunocompetent mice were comparable as shown in Fig. 2C and Table 1. There was no statistically significant difference in blood clearance rate detected among all groups (64Cu activity from MM-DX-929 group, 64Cu and 3H activity from 3H-MM-DX-929 group). These results indicate that incorporation of 3H-CDHE tracer into the liposomes did not affect the MM-DX-929 pharmacokinetics. Moreover, no significant difference was observed between pharmacokinetics measured by 64Cu and 3H activity in mice injected with 3H-MM-DX-929, with 64Cu and 3H remaining in an approximately 1:1 ratio for 48 h in vivo (Fig. 2D). This in vivo stability was further confirmed by examining mouse plasma collected from mice injected with MM-DX-929, where 64Cu detected in circulation at 24 h.p.i. remained primarily associated with the liposomes as determined by SEC (Supplementary Fig. S1A). The stability of the MM-DX-929 liposome is directly associated with PEGylation content, as reducing the PEG content from 10 mol% to 0.5 mol% resulted in the presence of non-liposomal, protein-bound 64Cu:4-DEAP-ATSC complex (Supplementary Fig. S1B).
Plasma pharmacokinetic parameters of MM-DX-929 (detected by 64Cu) and 3H-MM-DX-929 (detected by 64Cu or 3H) calculated using non-compartmental analysis.
Compound | Pharmacokinetics Parameters | |||
|---|---|---|---|---|
| t1/2 (h) | AUC(%i.d.h/mL) | CL (mL/h) | Vss (mL) | |
| MM-DX-929 (64Cu) | 12 ± 0.7 | 1500 ± 66 | 0.069 ± 0.003 | 1.2 ± 0.1 |
| 3H-MM-DX-929 (64Cu) | 11.6 ± 0.4 | 1400 ± 100 | 0.071 ± 0.006 | 1.1 ± 0.1 |
| 3H-MM-DX-929 (3H) | 11 ± 0.9 | 1300 ± 160 | 0.078 ± 0.010 | 1.2 ± 0.1 |
Tumor Deposition Imaging with MM-DX-929 PET
PET/CT imaging was performed in a panel of five mouse xenograft models injected intravenously with MM-DX-929 (Fig. 3), including three orthotopic mammary fat pad models of breast cancer (HER2+ve BT474-M3, inflammatory HER2+ve SUM190, and triple negative MDA-MB-231) and two subcutaneous models (non-small cell lung cancer H520, and colorectal cancer HT29). Fig. 3A illustrates an example of MM-DX-929 distribution kinetics in mice bearing bilateral HT-29 subcutaneous tumors. At 1 h.p.i., MM-DX-929 remains primarily in circulation. At 8-20 h.p.i., MM-DX-929 accumulated mainly in the liver and spleen, with significant signal detected in the tumors. The right panel in Fig. 3A shows the time-course quantification of MM-DX-929 in blood and tumors obtained from the PET/CT images. Over time MM-DX-929 was cleared from the blood while the amount of MM-DX-929 in tumors increased. This is indicative of liposome extravasation out of the vasculature and into the tumor interstitium via the EPR effect [1,2,4,5,18,19].
PET/CT imaging and quantification of MM-DX-929 deposition in xenograft models. (A) Representative maximum intensity projection of NOD-SCID mice bearing bilateral subcutaneous HT-29 tumors at 1, 8, and 20 h after MM-DX-929 injection via lateral tail vein. Quantification of MM-DX-929 tumor and blood signal as a function of time obtained from the images are provided on the right panel. (B) Quantification of MM-DX-929 tumor deposition at approximately 24 h.p.i. in multiple tumor xenograft models. Representative axial PET/CT images of BT474-M3 (orthotopic mammary fat pad), SUM190 (orthotopic mammary fat pad), MDA-MB-231 (orthotopic mammary fat pad), and H520 (subcutaneous) tumors are shown on the right panel. Images and data are decay-corrected.
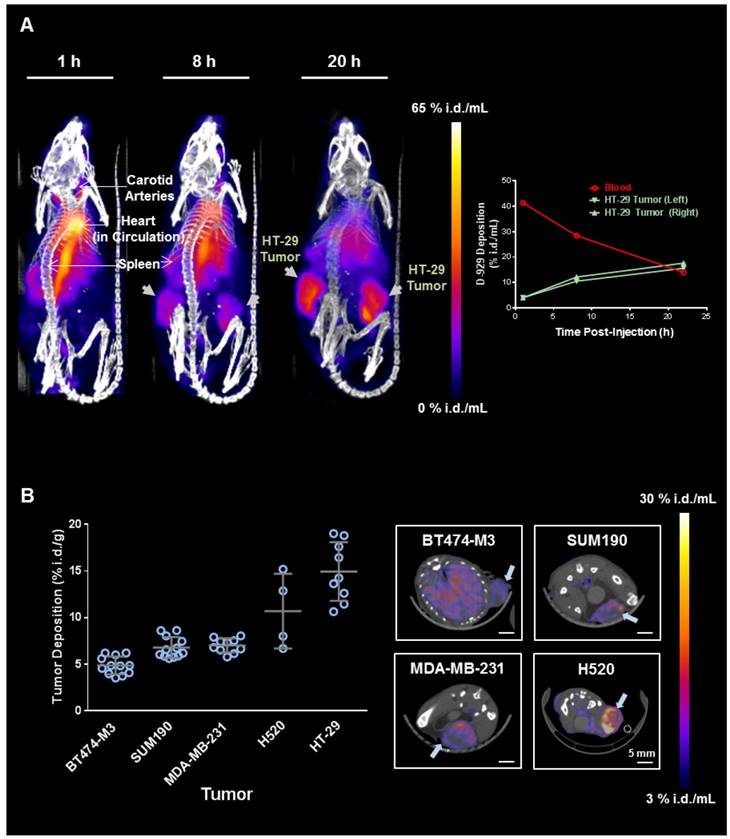
Tumor deposition of MM-DX-929 at 20-24 h.p.i. obtained from PET/CT images in the five xenograft models is illustrated in Fig. 3B. MM-DX-929 tumor deposition was found to be variable within each model and across models, ranging from 3.5 %i.d./g (percentage of injected dose per gram of tumor) to 19.0% i.d./g. Specifically, MM-DX-929 deposition in BT474-M3 and HT-29 tumors range from 3.5-6.2 %i.d./g and 10.7-19.0 %i.d./g, respectively. In a separate study where the drug deposition of MM-302 was assessed by HPLC measurement of encapsulated doxorubicin, drug payload detected in BT474-M3 and HT-29 tumors were found to range from 3.6-6.2 %i.d./g and 7.1-15.4 %i.d./g, respectively (data not shown). This demonstrates the feasibility of employying MM-DX-929 PET to quantitatively predict tumor deposition of liposomal drugs, and to effectively differentiate tumors with distinct permeability to liposomes.
Tracer Kinetic Modeling of MM-DX-929 Deposition
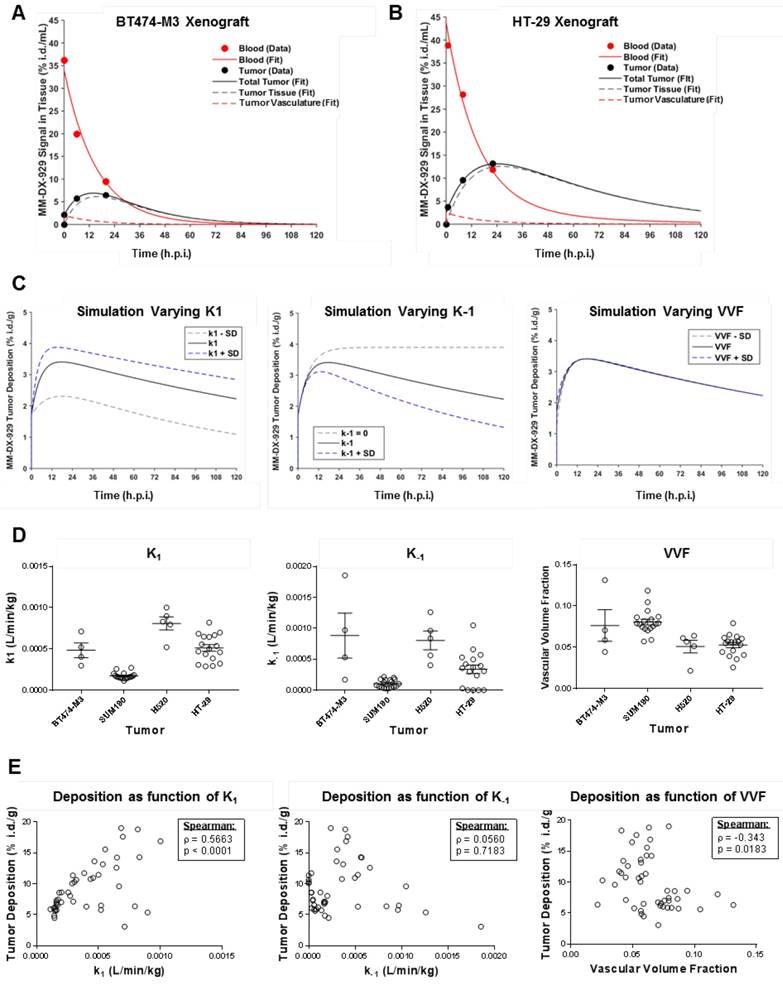
Previously, we reported a kinetic model for depicting the deposition (k1) and washout (k-1) kinetics of liposome deposition in a tumor [8]. Examples of blood and tumor deposition kinetics of MM-DX-929 in low (BT474-M3) and high (HT-29) deposition xenograft models, respectively, are shown in Fig. 4A and B. Kinetic modeling indicated that at 24 h.p.i., the MM-DX-929 PET signal detected in the tumors are primarily in tumor tissue, with minimal contribution from the tumor vascular signal. Simulation suggests that k1 and k-1 are more likely to affect liposome deposition or exposure in tumors compared to vascular volume fraction (VVF) (Fig. 4C). The ranking of kinetic parameters for each xenograft model (Fig. 4D: k1, k-1, VVF) did not seem to predict peak tumor accumulation of MM-DX-929 (Fig. 3B). However, a positive correlation (ρ = 0.57, p < 0.001) was observed between k1 and tumor deposition of individual tumors at 24 h.p.i., as shown in Fig. 4E, while no significant correlation was detected between k-1 or VVF and tumor deposition.
Modeling of MM-DX-929 transport kinetics and correlation with tumor deposition. (A) and (B) are representative examples of kinetics of MM-DX-929 in blood (solid red line) and tumor (solid black line) in BT474-M3 and HT-29 xenografts, respectively based on tracer kinetic modeling. Longitudinal experimental data for MM-DX-929 in the blood and tumor based on PET images are shown as red and black points, respectively. Simulated kinetics of contributions of tumor vasculature (dashed red line) and tumor interstitium (dashed black line) are also shown. (C) Simulated effect of k1, k-1, and VVF on MM-DX-929 tumor deposition kinetics (mean value (solid black line) ± one standard deviation (dashed lines)). (D) Transport kinetics parameters of MM-DX-929 (k1, k-1, VVF) for individual tumors obtained from PET/CT images of xenograft models injected with MM-DX-929. (E) Correlation of MM-DX-929 tumor deposition at approximately 24 h.p.i. with k1, k-1, and VVF. Data for tracer kinetic modeling are decay-corrected.
MM-DX-929 is Predictive of Deposition and Intratumoral Distribution of Liposomal Drugs
We hypothesize that liposome deposition via the EPR effect is a crucial rate-liming step for effective drug delivery to tumors (Fig. 5A). The EPR effect is a shared step in drug delivery, both for untargeted liposome as well as immunoliposomes targeting tumor cell antigens. For instance, as depicted in Fig. 5A, HER2-targeted liposomal doxorubicin (MM-302) accumulate in tumors in a similar manner as untargeted PLD. Following the deposition step, the HER2-targeting moiety directs MM-302 to be internalized by the HER2-overexpressing tumor cells for efficient intracellular delivery of the drug payload, while untargeted PLD are likely to be phagocytosed by macrophages [20]. In order to demonstrate that MM-DX-929 can be used as a universal imaging-based companion diagnostic tool for measuring tumor deposition of liposomal drugs, we sought to determine whether intratumoral distribution and deposition of MM-DX-929 is proportional to that of liposomal drugs of different properties.
Correlation of MM-DX-929 tumor deposition and intratumoral distribution with liposomal drugs. (A) Schematic depicting the mechanism in which liposomal drugs are delivered from circulation into the cellular target. Liposome deposition and transport into tumors from circulation via the EPR effect is a shared step for liposomal drugs with different physico-chemical properties. The intratumoral fate of the liposome and drug payload is determined after the deposition step depending on the design of the liposomal drug system. Mice bearing (B) HT-29 subcutaneous tumors and (C) BT474-M3 orthotopic mammary fat pad tumors were co-injected with MM-DX-929 and liposomal irinotecan (10 mg/kg) or MM-302 (3 mg/kg), respectively. At 24 h.p.i., mice were perfused with PBS and tumors were excised for quantification of 64Cu and drug content. Mice bearing BT474-M3 tumors were co-injected with MM-DX-929 and (D) DiI5-PLD (3 mg/kg) or (E) DiI5-MM-302 (3 mg/kg), respectively. Autoradiography was performed to obtain intratumoral distribution of MM-DX-929 at 24 h.p.i.; consecutive sections were used to obtain intratumoral distribution of DiI5-PLD or DiI5-MM-302 using Aperio ScanScope.
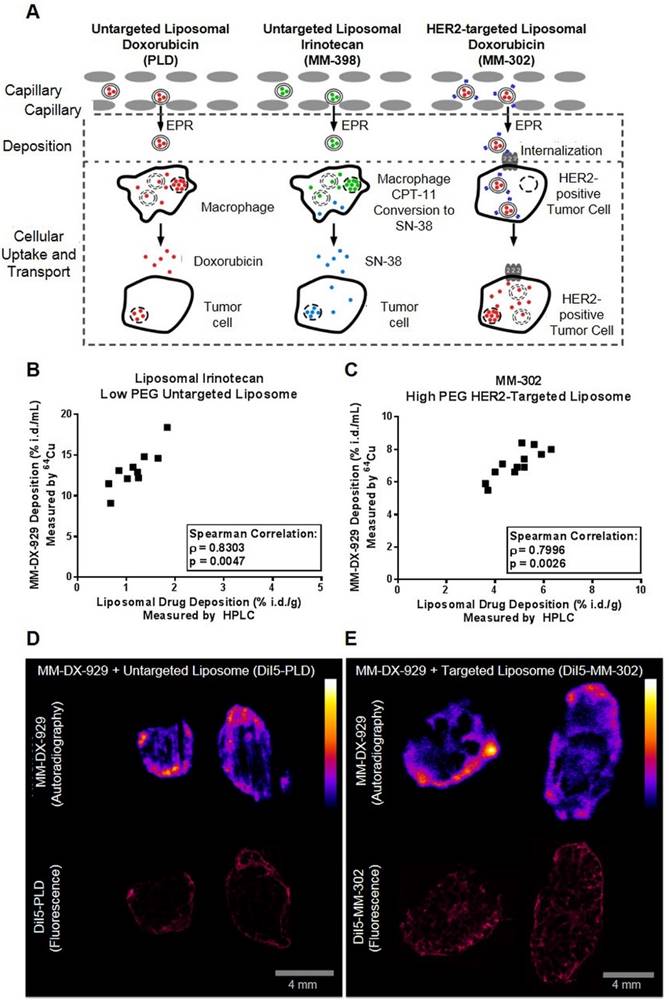
Using scintillation counting (64Cu for MM-DX-929) and HPLC quantification (drug uptake), tumor deposition of MM-DX-929 (10% PEG untargeted liposome) correlated with liposomal irinotecan (0.5% PEG untargeted liposome; Fig. 5B, Spearman Correlation ρ = 0.83, p = 0.047) and MM-302 (10% PEG HER2-targeted liposome; Fig. 5C, Spearman Correlation ρ = 0.80, p = 0.0026). Furthermore, the intratumoral distribution of MM-DX-929 qualitatively resembles that of PLD (Fig. 5D) and MM-302 (Fig. 5E), as determined by autoradiography (64Cu) and fluorescence microscopy (DiI5-labeled liposomes). These results demonstrate that MM-DX-929 accumulates in tumors through a similar mechanism as multiple other liposomal drugs as suggested in Fig. 5A.
MM-DX-929 Predicts Treatment Response of Liposomal Drugs but not Small Molecule Chemotherapeutics
We hypothesize that increased deposition of therapeutic liposomes in tumors will lead to improved treatment response, attributable to increased delivery of the liposomal drug contents to their molecular target within tumor cells. As a proof-of-concept, we administered MM-DX-929 intravenously in tumor-bearing mice as an imaging companion diagnostic agent, and acquired PET/CT images at approximately 24 h.p.i. to non-invasively quantify liposome deposition in tumors. Animals were then subjected to treatment of different liposomal therapeutics: MM-302 (3 mg/kg; Fig. 6A), PLD (3 mg/kg; Fig. 6B), or liposomal irinotecan (10 mg/kg; Fig. 6C). The median of tumor deposition within each group was selected as a stratification threshold. In three separate studies, mice with high tumor deposition (≥ median) of the MM-DX-929 imaging diagnostic had greater anti-tumor activity following multi-cycle liposomal therapeutic treatment, compared to those with low MM-DX-929 tumor deposition group (< median).
Retrospective stratification of treatment response using median of MM-DX-929 tumor deposition as a threshold. Mice bearing BT474-M3 or HT-29 tumors were injected with MM-DX-929 via lateral tail vein. At 24 h.p.i., PET/CT images were acquired for quantification of MM-DX-929 tumor deposition. Mice were then treated with (A) MM-302 (3 mg/kg, q1w, n = 17 tumors), (B) PLD (3 mg/kg, q1w, n = 20 tumors), (C) liposomal irinotecan (10 mg/kg, q1w, n = 17 tumors), (D) doxorubicin (6 mg/kg, q1w, n = 22 tumors), or (E) untreated (n = 21 tumors), for 3-6 cycles (as indicated by dotted vertical lines). Tumor volume change of low (< median, orange dotted line) and high (≥ median, black solid line) MM-DX-929 deposition groups were plotted as a function of days post-treatment. (F) Comparison of tumor volume change at study endpoint between low and high deposition groups.
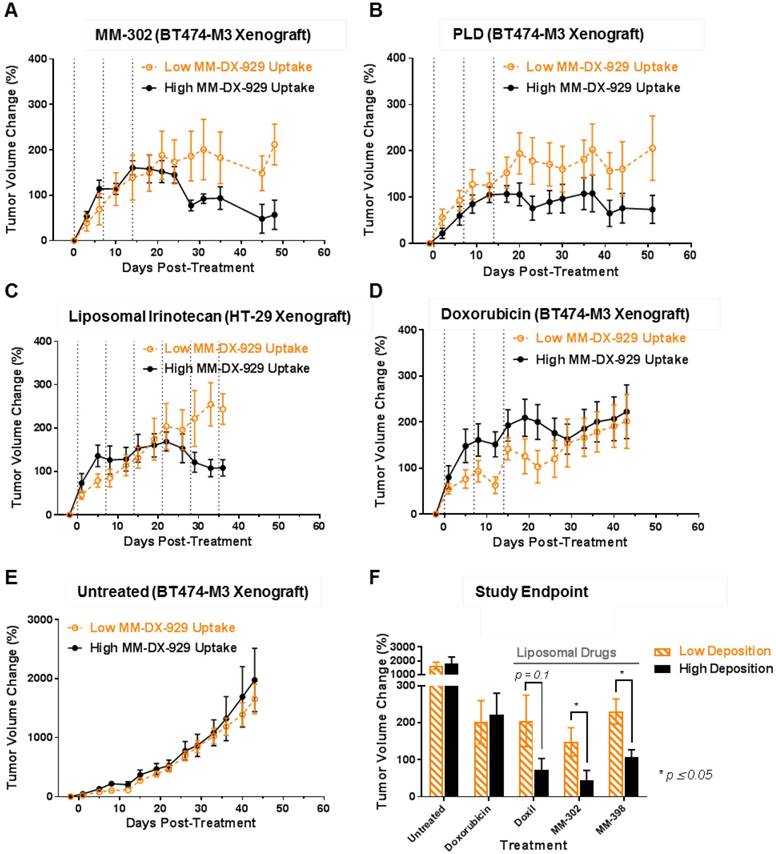
In contrast, MM-DX-929 tumor deposition did not differentiate treatment response of small molecule chemotherapeutics such as doxorubicin (6 mg/kg; Fig. 6D). Similarly, quantification of small molecule contrast enhancement (gadoteridol) in tumors using conventional dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) did not correlate with MM-DX-929 tumor deposition (Supplementary Fig. S2C & D). Furthermore, MM-DX-929 tumor deposition was not prognostic in tumor-bearing mice that did not receive any therapeutic intervention (Fig. 6E). Fig. 6F summarizes the change in tumor volume in high vs. low MM-DX-929 deposition groups at study endpoint; improvement in treatment response was only observed in the high MM-DX-929 deposition group when animals were treated with liposomal therapeutics (p ≤ 0.1).
Discussion
The rationale underlying the development of MM-DX-929 is based on the following key concepts: (1) deposition of liposomal drugs into solid tumors is a potentially rate-limiting step for drug delivery, (2) deposition of liposomal drugs into human tumors has substantial variability that may influence probability of response to therapy, and (3) tumor deposition is a shared mechanism for different liposomal drugs. As such, a single companion diagnostic agent may have utility in predicting response to multiple liposomal therapeutics, and possibly other nanomedicines of similar molecular size as well.
Tumor deposition is a complex product of multiple factors including circulating lifetime, degree of vascular density, vascular surface area, permeability, and interstitial fluid pressure. For this reason, functional measurements provide the simplest and most direct means to assess drug delivery. Kinetic modeling can yield additional insights into the contribution of some of these processes. Once long-circulating characteristics are achieved, liposome deposition or exposure in tumors was primarily dictated by the wash-in (k1) and washout (k-1) kinetics, which reflect the tumor permeability to nanoparticles. VVF determined by PET (analogous to microvessel density obtained from biopsies) was not predictive of liposome deposition as not all blood vessels are permeable for liposome extravasation (Fig. 4). The transport of molecules and macromolecules into tumors is highly dependent on molecular size [21,22]. As such, there is no gold standard for measuring the EPR effect of liposomes, and MM-DX-929 provides a quantitative measurement of this process. Conventional DCE-MRI is a powerful technique for measuring contrast dynamics, primarily for small molecule contrast agents. Multiple studies have demonstrated a lack of correlation between the kinetics of standard low molecular weight contrast agents (< 1 kDa, < 2 nm) and macromolecular albumin-conjugated agents (~90 kDa, ~6 nm) [23,24], suggesting that these existing contrast agents may not accurately reflect the kinetics of liposome transport (~100 nm) (Supplementary Fig. S2). Additionally, the k1 and k-1 kinetics of nanoparticles, such as liposomes, are significantly slower, requiring image acquisition on the timescale of days (Fig. 4A-C), rather than minutes as is done with DCE-MRI. Similarly, the current study demonstrated that liposomal PET agent MM-DX-929 deposition does not predict treatment response to small molecule doxorubicin (Fig. 6D). These evidence suggest that an imaging companion diagnostic agent of comparable size to the therapeutic agent of interest is required for accurately predicting tumor deposition and permeability. Our group and others have previously reported the use of ferumoxytol (750 kDa superparamagnetic iron oxide nanoparticles, 10-70 nm) and MRI as a companion diagnostic strategy for nanomedicine [9,25]. While this approach demonstrated correlation between ferumoxytol-MRI signal and nanomedicine treatment response, the clinical utility of MRI as a patient stratification tool may be limited due to its semi-quantitative nature and challenges in image acquisition for certain tissues such as lung and bone lesions. For these reasons, we believe that longitudinal imaging of a labeled liposomal agent such as MM-DX-929 and quantitative PET provide the best estimation of liposomal drug delivery in tumors.
MM-DX-929 was developed with the vision of a universal companion diagnostic agent for multiple liposomal therapeutics. In this study, we have demonstrated that MM-DX-929 can predict tumor deposition and resemble intratumoral spatial distribution of liposomal drugs with different physico-chemical properties (Fig. 5). For instance, it has been shown that varying levels of surface PEGylation affects liposome pharmacokinetic profiles, which is a key factor affecting liposome tumor deposition. In general, liposomes decorated with low amounts of PEG, such as liposomal irinotecan, are cleared faster in vivo compared to high-PEG liposomes [1]. The intratumoral distribution and tumor deposition of MM-DX-929, a highly PEGylated (10 mol%) untargeted liposome, was well correlated with PLD (10 mol% PEGylated untargeted liposomal doxorubicin), liposomal irinotecan (0.5 mol% PEGylation untargeted liposomal irinotecan), and MM-302 (PEGylated HER2-targeted liposomal doxorubicin) (Fig. 5B & C). These data support the use of MM-DX-929 as a universal imaging surrogate for measurement of liposomal drug deposition in tumors. Consequently, retrospective stratification by median MM-DX-929 tumor deposition measured by PET also results in improvement in treatment outcomes of all three liposomal drugs (Fig. 6A-C, F). It should be noted that MM-DX-929 PET was shown to have no prognostic value in untreated animals (Fig. 6D), nor predictive value for small molecule chemotherapeutics (Fig. 6E), suggesting that the differentiation of treatment outcome as a result of MM-DX-929 PET stratification is attributed to the variability in liposome deposition in tumors. All of the above support our hypothesis in which effective drug delivery and local drug concentration are major rate-limiting factors for response to liposomal drugs.
PET was selected as the optimal modality for our liposomal imaging agent due to its high sensitivity and quantitative nature, while 64Cu was identified as the PET reporter of choice because its radioactive half-life (t1/2 = 12.7 h) matches the transport kinetics of liposomes within their useful imaging window [26]. Another commercially available PET isotope, 89Zr (t1/2 = 78.4 h), would result in a much higher radiation absorbed dose and potentially undesirable radiation dosimetry as a liposome PET tracer [13]. Our Phase 1 clinical study demonstrated that 64Cu-MM-302 tumor signal peaks around 24-48 h.p.i. in patients, and determined that 24 h.p.i. is an optimal imaging time point for assessing liposome deposition in human tumors, without compromising image quality or overextending patients' time spent in the scanner [8], providing additional support for the clinical utility of a PET-based liposomal imaging agent.
Others have reported similar strategies in utilizing nanoimaging agents to predict single-dose short-term treatment efficacy of individual nanomedicines [10,11,25]. It is possible that therapeutic intervention may alter the tumor microenvironment and potentially result in different liposome deposition in tumors at subsequent treatment cycles [27]. In this work, a pre-treatment MM-DX-929 imaging session at 24 h.p.i. was found to be effective at predicting the multi-cycle treatment response to liposomal therapeutics in multiple preclinical xenograft models (Fig. 6). To the best of our knowledge, MM-DX-929 PET is the first nanoimaging agent that has been demonstrated to provide a longitudinal treatment outcome prediction after a single pre-treatment imaging session, and is translatable in the clinic as a companion diagnostic for liposomal therapeutics.
For clinical implementation, individual clinical trial and retrospective analysis may be required to determine an appropriate deposition threshold to identify responders for each liposomal therapeutic in different indications. While tumor deposition may be a strong indicator for treatment response to liposomal therapeutics, other factors such as chemosensitivity can potentially affect therapeutic outcome. A multi-factorial analysis combining tumor deposition with other biomarkers may provide further insights on the contribution of each factor, and develop a more accurate predictive algorithm in identifying patients who would truly benefit from nanotherapeutics.
In summary, we have described the development and characterization of MM-DX-929, and demonstrated preclinical proof-of-concept that a liposomal PET imaging companion diagnostic agent can predict the activity of liposomal therapies, providing support for clinical investigation.
Abbreviations
PET: Positron emission tomography; CT: computed tomography; PEG: polyethylene glycol; 4-DEAP-ATSC: Diacetyl 4,4′-bis(3-(N,N-di ethylamino)propyl)thiosemicarbazone; EPR: enhanced permeability effect; DTPA: diethylenetriaminepentacetate; PLD: PEGylated liposomal doxorubicin; HER2: Human epidermal growth factor receptor 2-positive; HSPC: Hydrogenated soybean phosphatidylcholine; TEA-SOS: triethylammonium sucrose octasulfate; SEC: size exclusion chromatography; VVF: vascular volume fraction; HPLC: high performance liquid chromatography; DCE-MRI: dynamic contrast enhanced magnetic resonance imaging.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Tom Voller from Washington University for the supply of 64Cu for the study. We would also like to thank inviCRO, LLC, Michael Dunne, Tianyu Tang for their technical support, as well as acknowledge Ulrik Nielsen, Daryl Drummond, Richard Huang, and David Jaffray for their input throughout the development of MM-DX-929.
Financial Support
Merrimack employees receive salaries and stock options from Merrimack Pharmaceuticals. This study was solely funded by Merrimack Pharmaceuticals. Post-doc salaries were in-part funded through the MITACS Accelerate and Elevate Postdoctoral Fellowship Programs, with financial contribution from Merrimack Pharmaceuticals and the Canadian Federal and the Ontario Provincial governments.
Authors affiliated with Merrimack Pharmaceuticals as indicated were employees of Merrimack Pharmaceuticals at the time of study. Jinzi Zheng is the Lead Investigator of an industry-sponsored research agreement funded by Merrimack Pharmaceuticals.
Competing Interests
Merrimack employees receive salaries and stock options from Merrimack Pharmaceuticals. This study was solely funded by Merrimack Pharmaceuticals. Post-doc salaries were in-part funded through the MITACS Accelerate and Elevate Postdoctoral Fellowship Programs, with financial contribution from Merrimack Pharmaceuticals and the Canadian Federal and the Ontario Provincial governments. Authors affiliated with Merrimack Pharmaceuticals as indicated were employees of Merrimack Pharmaceuticals at the time of study. Jinzi Zheng is the Lead Investigator of an industry-sponsored research agreement funded by Merrimack Pharmaceuticals.
References
1. Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51(4):691-743
2. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12):6387-92
3. Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73(8):2412-7
4. Greish K. Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. J Drug Target. 2007;15(7-8):457-64
5. Nehoff H, Parayath NN, Domanovitch L, Taurin S, Greish K. Nanomedicine for drug targeting: strategies beyond the enhanced permeability and retention effect. Int J Nanomedicine. 2014;9:2539-55
6. Hendriks BS, Reynolds JG, Klinz SG, Geretti E, Lee H, Leonard SC, Gaddy DF, Espelin CW, Nielsen UB, Wickham TJ. Multiscale kinetic modeling of liposomal Doxorubicin delivery quantifies the role of tumor and drug-specific parameters in local delivery to tumors. CPT pharmacometrics Syst Pharmacol. 2012;1(11):e15
7. Harrington KJ, Mohammadtaghi S, Uster PS, Glass D, Peters AM, Vile RG, Stewart JS. Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes. Clin cancer Res. 2001;7(2):243-54
8. Lee H, Shields AF, Siegel BA, Miller KD, Krop I, Ma CX, LoRusso PM, Munster PN, Campbell K, Gaddy DF, Leonard SC, Geretti E, Blocker SJ, Kirpotin DB, Moyo V, Wickham TJ, Hendriks BS. 64Cu-MM-302 Positron Emission Tomography Quantifies Variability of Enhanced Permeability and Retention of Nanoparticles in Relation to Treatment Response in Patients with Metastatic Breast Cancer. Clin Cancer Res. 2017;23(15):4190-4202
9. Ramanathan RK, Korn RL, Raghunand N, Sachdev JC, Newbold RG, Jameson G, Fetterly GJ, Prey J, Klinz SG, Kim J, Cain J, Hendriks BS, Drummond DC, Bayever E, Fitzgerald JB. Correlation Between Ferumoxytol Uptake in Tumor Lesions by MRI and Response to Nanoliposomal Irinotecan in Patients With Advanced Solid Tumors: A Pilot Study. Clin Cancer Res. 2017;23(14):3638-3648
10. Karathanasis E, Suryanarayanan S, Balusu SR, McNeeley K, Sechopoulos I, Karellas A, Annapragada AV, Bellamkonda RV. Imaging nanoprobe for prediction of outcome of nanoparticle chemotherapy by using mammography. Radiology. 2009;250(2):398-406
11. Pérez-Medina C, Abdel-Atti D, Tang J, Zhao Y, Fayad ZA, Lewis JS, Mulder WJ, Reiner T. Nanoreporter PET predicts the efficacy of anti-cancer nanotherapy. Nat Commun. 2016;7:11838
12. Lee H, Zheng J, Gaddy D, Orcutt KD, Leonard S, Geretti E, Hesterman J, Harwell C, Hoppin J, Jaffray DA, Wickham T, Hendriks BS, Kirpotin D. A gradient-loadable (64)Cu-chelator for quantifying tumor deposition kinetics of nanoliposomal therapeutics by positron emission tomography. Nanomedicine. 2015;11(1):155-165
13. Gaddy DF, Lee H, Zheng J, Jaffray DA, Wickham TJ, Hendriks BS. Whole-body organ-level and kidney micro-dosimetric evaluations of (64)Cu-loaded HER2/ErbB2-targeted liposomal doxorubicin ((64)Cu-MM-302) in rodents and primates. EJNMMI Res. 2015;5:24
14. Reynolds JG, Geretti E, Hendriks BS, Lee H, Leonard SC, Klinz SG, Noble CO, Lucker PB, Zandstra PW, Drummond DC, Olivier KI Jr, Nielsen UB, Niyikiza C, Agresta SV, Wickham TJ. HER2-targeted liposomal doxorubicin displays enhanced anti-tumorigenic effects without associated cardiotoxicity. Toxicol Appl Pharmacol. 2012;262(1):1-10
15. Drummond DC, Noble CO, Guo Z, Hong K, Park JW, Kirpotin DB. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006;66(6):3271-7
16. Kalra AV, Kim J, Klinz SG, Paz N, Cain J, Drummond DC, Nielsen UB, Fitzgerald JB. Preclinical activity of nanoliposomal irinotecan is governed by tumor deposition and intratumor prodrug conversion. Cancer Res. 2014;74(23):7003-13
17. Baxter LT, Zhu H, Mackensen DG, Jain RK. Physiologically based pharmacokinetic model for specific and nonspecific monoclonal antibodies and fragments in normal tissues and human tumor xenografts in nude mice. Cancer Res. 1994;54(6):1517-28
18. Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42(5):419-36
19. Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev. 2015;91:3-6
20. Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66(13):6732-40
21. Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153(3):198-205
22. Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55(17):3752-6
23. Daldrup H, Shames DM, Wendland M, Okuhata Y, Link TM, Rosenau W, Lu Y, Brasch RC. Correlation of dynamic contrast-enhanced MR imaging with histologic tumor grade: comparison of macromolecular and small-molecular contrast media. AJR Am J Roentgenol. 1998;171(4):941-9
24. Su MY, Wang Z, Carpenter PM, Lao X, Mühler A, Nalcioglu O. Characterization of N-ethyl-N-nitrosourea-induced malignant and benign breast tumors in rats by using three MR contrast agents. J Magn Reson Imaging. 1999;9(2):177-86
25. Miller MA, Gadde S, Pfirschke C, Engblom C, Sprachman MM, Kohler RH, Yang KS, Laughney AM, Wojtkiewicz G, Kamaly N, Bhonagiri S, Pittet MJ, Farokhzad OC, Weissleder R. Predicting therapeutic nanomedicine efficacy using a companion magnetic resonance imaging nanoparticle. Sci Transl Med. 2015;7(314):1-13
26. Hansen AE, Petersen AL, Henriksen JR, Boerresen B, Rasmussen P, Elema DR, af Rosenschold PM, Kristensen AT, Kjær A, Andresen TL. Positron Emission Tomography Based Elucidation of the Enhanced Permeability and Retention Effect in Dogs with Cancer Using Copper-64 Liposomes. ACS Nano. 2015;9(7):6985-95
27. Geretti E, Leonard SC, Dumont N, Lee H, Zheng J, De Souza R, Gaddy DF, Espelin CW, Jaffray DA, Moyo V, Nielsen UB, Wickham TJ, Hendriks BS. Cyclophosphamide-Mediated Tumor Priming for Enhanced Delivery and Antitumor Activity of HER2-Targeted Liposomal Doxorubicin (MM-302). Mol Cancer Ther. 2015;14(9):2060-71
Author contact
![]() Corresponding author: Jinzi Zheng, University Health Network, 101 College Street, 7th Floor, Toronto, Ontario, Canada M5G 1L7. (P): 416-581-7790; (F): 416-506-1828; (E): jinzi.zhenguhn.on.ca
Corresponding author: Jinzi Zheng, University Health Network, 101 College Street, 7th Floor, Toronto, Ontario, Canada M5G 1L7. (P): 416-581-7790; (F): 416-506-1828; (E): jinzi.zhenguhn.on.ca