Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2018; 8(7):2061-2063. doi:10.7150/thno.24454 This issue Cite
Editorial
Resistant to Targeted Therapy - Aim for Metabolic Liabilities
André Lima Queiroz, Helin Vakifahmetoglu-Norberg, Erik Norberg ![]()
Department of Physiology and Pharmacology, Karolinska Institutet, Nanna Svartz väg 2, SE-171 77, Stockholm, Sweden
Received 2017-12-19; Accepted 2017-12-20; Published 2018-2-20
Commentary-article in Theranostics, Volume 8, 1808
Abstract
The advent of targeted therapies generated much optimism when discovered. Targeted therapies, are however associated with rapid acquisition of resistance. In a recent study by Dong et al. (Theranostics 2018; 8(7):1808-1823. doi:10.7150/thno.23177) it was shown that lung tumors resistant to the EGFR-inhibitor (Erlotinib), reprogram their metabolism and acquire a pro-survival dependency on Phosphoglycerate Dehydrogenase (PHGDH) that can be targeted to eliminate resistant tumors.
Keywords: EGFR, Lung Cancer, Non-small Cell Lung Carcinomas, Lung Adenocarcinomas, PHGDH, Tumor Metabolism, Serine
Lung cancer remains the leading cause of cancer mortality worldwide. Non-small cell lung carcinomas (NSCLC) accounts for approximately 80% of all lung cancers, the most common histological form of NSCLC is lung adenocarcinomas. These tumors display large heterogeneity and are often too advanced when diagnosed. Hence, first-line platinum-based chemotherapies display modest effect on improving survival of these patients [2, 3].
The Epidermal Growth Factor Receptor (EGFR) family of tyrosine kinases belongs to the HER family receptors, which also comprises HER2-4. Upon ligand binding, these receptors undergo a series of conformational changes and triggers phosphorylation events leading to the activation downstream pathways that regulate cellular processes including, proliferation, survival, apoptosis and metabolism. Activating mutations and over-expression of these receptor family members has been shown to contribute to tumorigenesis. Accordingly, EGFR is overexpressed in approximately 50% of NSCLC patients and its expression correlates with poor prognosis. Constitutive activation of EGFR is caused by gene mutations, mainly in the adenosine triphosphate (ATP)-binding pocket of the tyrosine kinase domain of EGFR, or by gene amplification. This provided a rationale to develop targeted therapies against EGFR. In particular, Gefinitib (Iressa®) or Erlotinib (Tarceva®), were designed to be competitive inhibitors of the ATP-binding at the active site of the EGFR tyrosine kinase. Although, these drugs were approved in 2003 and 2004, respectively, for the treatment of NSCLCs, they showed only a partial response in approximately 10% of the NSCLC patients, which rapidly developed resistance to these inhibitors. The acquired drug resistance developed within 6-12 months of the initial therapy, which heavily limits the ability of these drugs to prolong the survival of patients [2, 3]. Established phenotypes of EGFR inhibitor resistance includes that the inhibitors lose their capacity to reduce glucose uptake and the activation of the AKT/mTOR pathway, which leads to enhanced tumor cell survival (Figure 1). Accordingly, activation of mTORC1 signaling was identified as a mechanism of resistance to EGFR inhibition in vivo. Hence, Rapamycin, an allosteric inhibitor of mTORC1 could reverse the resistance [4-7]. This emphasizes that a deeper understanding of the acquired dependencies and alternative therapeutic strategies may have a tremendous impact to eliminate resistant tumors.
Tyrosine Kinase-Inhibitor (TKI) -sensitive and -resistant Tumors and the Cellular Consequences. Both TKI-inhibitor sensitive (left panel) and resistant (right panel) tumors display an activating mutation in the EGFR. TKI-inhibitors reduce glucose uptake and suppresses pro-survival signals from PI3K-AKT-mTOR or Ras-Raf-MEK payhways, leading to tumor elimination in TKI-inhibitor sensitive tumors (left panel). On the contrary, tumor can become resistant to EGFR-inhibitors due to a mutation in the ATP-binding site of the EGFR kinase (the target of the inhibitors). The resistant tumors display enhanced glucose uptake and glycolysis. Dong et al now show that PHGDH is upregulated on the gene expression and protein level, which produces serine and replenish antioxidants, nucleotides and DNA damage repair proteins. These events combined promote tumor growth in the EGFR-resistant tumors.
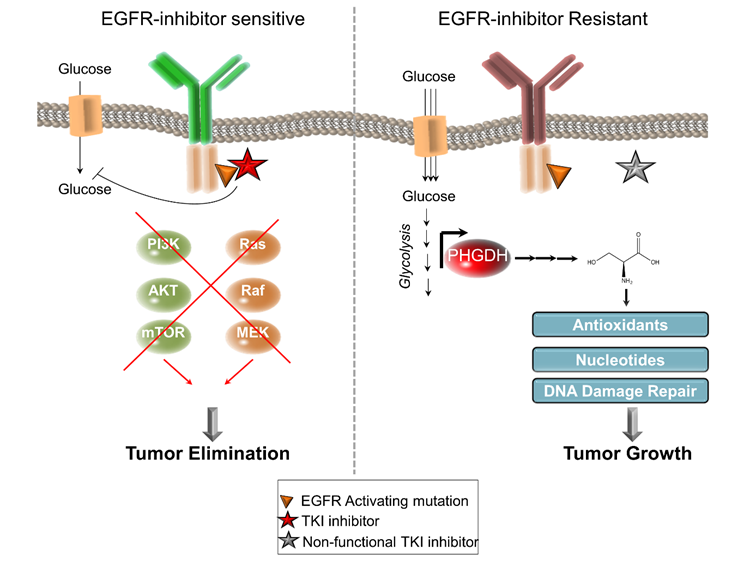
A recent study by Dong et al. [1] aims to enhance the efficacy and to reverse the resistance to the EGFR inhibitor Erlotinib in lung adenocarcinomas displaying EGFR-activating mutations. Interestingly, resistant cells displayed an elevation of PHGDH, the rate-limiting enzyme in the de novo biosynthesis of serine on both the mRNA and the protein level and also greater incorporation of glucose-derived carbons into serine (Figure 1). Although, PHGDH has been associated to human cancers for more than 30 years [8], more recently, the Vander Heiden lab [9] and the Sabatini lab [10] identified PHGDH as often amplified or overexpressed in breast and melanoma tumors. Consequently, the cancer cells expressing high levels of PHGDH display a more rapid proliferation, larger incorporation of glucose-derived carbons into serine and is associated with a poor prognosis in melanoma, breast cancer [9, 10] and lung cancer [11, 12].
Dong et al. now demonstrates that genetic depletion of PHGDH causes a suppression of clonogenic survival and leads to cell death selectively in the resistant cells, while Erlotinib-sensitive and the normal non-transformed HBE cells were not affected by PHGDH depletion in terms of clonogenic growth, mitochondrial metabolism or glycolysis. The study further reveals that chemical inhibition of PHGDH restored sensitivity to Erlotinib in cell lines and in vivo. Conversely, ectopic expression of PHGDH in sensitive cell lines confers resistance to Erlotinib, establishing high levels of PHGDH as a prerequisite for Erlotinib-resistance in lung adenocarcinomas.
In the search for a mechanistic basis for their observation, the authors performed RNA sequencing analysis to identify transcripts that were differentially expressed upon PHGDH inhibition. Among the more than 1000 genes that were identified to be differentially expressed, many downstream pathways of serine metabolism were included such as DNA replication, cell cycle, mismatch repair and purine/pyrimidine metabolism and the p53-signaling pathway. Serine is important for several biosynthetic pathways including synthesis of glycine, cysteine and methionine, phospholipids, glutathione, one-carbon metabolism and purine/pyrimidine biosynthesis [13]. Consequently, genetic depletion of PHGDH has been shown to cause significant DNA damage due to a suppression of the purine/pyrimidine biosynthesis [12]. Additionally, serine can contribute to replenishing the intracellular levels of glutathione [11, 12]. Therefore, genetic knockdown or inhibition of PHGDH may cause an elevation of the intracellular reactive oxygen species (ROS), as a consequence of reducing the antioxidant glutathione. Accordingly, Dong et al. found that the PHGDH-inhibitor NCT503 [14] causes significant DNA damage selectively in the Erlotinib-resistant cells, antioxidants could blunt these effects (Figure 1).
This study illustrates several important aspects of resistance to targeted therapies. First, it demonstrates quantitative metabolic differences between tumors being either sensitive or resistant to EGFR inhibitor. Further, it emphasizes that upon acquiring resistance to targeted therapies, the metabolic profile is re-wired and consequently new metabolic dependencies are engaged. In line with the fact that resistant cells can maintain a high glucose uptake, it can divert carbons into serine biosynthetic pathway driven by PHGDH. Second, it highlights that the re-wired metabolism in resistant cells is targetable to reduce tumor growth. Third, it demonstrates that overexpression of a metabolic enzyme (i.e. PHGDH) can transform EGFR-inhibitor-sensitive tumor to be more resistant. It also highlights the importance of metabolic profiling during the disease development as well as in the acquisition of resistance, to allow an understanding of the preferred metabolism under different scenarios. Combined, these findings suggest that the acquired metabolic liabilities could potentially allow stratification of additional NSCLC subpopulations that may benefit from therapy targeting their specific metabolism.
It remains to be seen if acquired resistant metabolic phenotypes may be relevant across different cancer types treated with the same therapy, or whether the cell of origin of tumors may be relevant in this context. It would also be interesting to see if the resistance to one drug can lead to the development of distinct metabolic liabilities even within the same tumor type. This may be particularly relevant to NSCLCs that displays huge molecular heterogeneity on multiple levels including metabolism. Human NSCLC tumors have recently been shown to display a heterogeneous glucose metabolism between human tumors that also has the capacity to oxidize several types of nutrients in vivo [15]. Regardless of the underlying mechanism, it is encouraging that molecular profiling can lead to the discovery of acquired metabolic vulnerabilities. Therefore, further analyses on the effect of acquired resistance to targeted therapies are needed, which may uncover additional metabolic liabilities.
Acknowledgements
The authors are supported by grants from the Swedish Research Council (VR), The Swedish Association for Medical Research (SSMF), The Malin and Lennart Philipson Foundation, The Ragnar Söderberg Foundation, The Jeanssons Foundation.
References
1. Dong JK, Lei HM, Liang Q, Tang YB, Zhou Y, Wang Y, Zhang S, Li WB, Tong Y, Zhuang G, Zhang L, Chen HZ, Zhu L, Shen Y. Overcoming erlotinib resistance in EGFR mutation-positive lung adenocarcinomas through repression of phosphoglycerate dehydrogenase. Theranostics. 2018;8(7):1808-1823 doi:10.7150/thno.23177
2. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S. et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med. 2017;376:2109-21
3. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169-81
4. Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Magyar C, Braas D. et al. Targeted Inhibition of EGFR and Glutaminase Induces Metabolic Crisis in EGFR Mutant Lung Cancer. Cell Rep. 2017;18:601-10
5. Su H, Bodenstein C, Dumont RA, Seimbille Y, Dubinett S, Phelps ME. et al. Monitoring tumor glucose utilization by positron emission tomography for the prediction of treatment response to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res. 2006;12:5659-67
6. Forloni M, Gupta R, Nagarajan A, Sun LS, Dong Y, Pirazzoli V. et al. Oncogenic EGFR Represses the TET1 DNA Demethylase to Induce Silencing of Tumor Suppressors in Cancer Cells. Cell Rep. 2016;16:457-71
7. Pirazzoli V, Nebhan C, Song X, Wurtz A, Walther Z, Cai G. et al. Acquired resistance of EGFR-mutant lung adenocarcinomas to afatinib plus cetuximab is associated with activation of mTORC1. Cell Rep. 2014;7:999-1008
8. Snell K. Enzymes of serine metabolism in normal, developing and neoplastic rat tissues. Adv Enzyme Regul. 1984;22:325-400
9. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ. et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869-74
10. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K. et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346-50
11. DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D. et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47:1475-81
12. Zhang B, Zheng A, Hydbring P, Ambroise G, Ouchida AT, Goiny M. et al. PHGDH Defines a Metabolic Subtype in Lung Adenocarcinomas with Poor Prognosis. Cell Rep. 2017;19:2289-303
13. Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer. 2016;16:650-62
14. Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, Swier LJ. et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol. 2016;12:452-8
15. Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J. et al. Metabolic heterogeneity in human lung tumors. Cell. 2016;164:681-94
Author contact
![]() Corresponding author: Erik Norberg, Department of Physiology and Pharmacology, Karolinska Institutet, Nanna Svartz väg 2, SE-171 77, Stockholm, Sweden. E-mail: Erik.Norbergse
Corresponding author: Erik Norberg, Department of Physiology and Pharmacology, Karolinska Institutet, Nanna Svartz väg 2, SE-171 77, Stockholm, Sweden. E-mail: Erik.Norbergse