Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(7):2031-2043. doi:10.7150/thno.24385 This issue Cite
Research Paper
α-Viniferin Improves Facial Hyperpigmentation via Accelerating Feedback Termination of cAMP/PKA-Signaled Phosphorylation Circuit in Facultative Melanogenesis
Cheong-Yong Yun1, Seon Mi Ko1, Yong Pyo Choi1, Beom Joon Kim2, Jungno Lee3, Jae Mun Kim3, Ju Yeon Kim4, Jin Yong Song1, Song-Hee Kim1, Bang Yeon Hwang1, Jin Tae Hong1, Sang-Bae Han1, Youngsoo Kim1 ![]()
1. College of Pharmacy, Chungbuk National University, Cheongju 28160, Korea.
2. Department of Dermatology, Chung-Ang University Hospital, Seoul 06973, Korea.
3. Bio-convergence R&D center, Coseedbiopharm Corporation, Cheongju 28161, Korea.
4. R&D Division, Celltrion Inc., Incheon 22014, Korea
Received 2017-12-16; Accepted 2018-1-24; Published 2018-2-16
Abstract

Rationale: cAMP up-regulates microphthalmia-associated transcription factor subtype M (MITF-M) and tyrosinase (Tyro) in the generation of heavily pigmented melanosomes. Here, we communicate a therapeutic mechanism of hyperpigmented disorder by α-viniferin, an active constituent of Caragana sinica.
Methods: We used cAMP-elevated melanocyte cultures or facial hyperpigmented patches for pigmentation assays, and applied immunoprecipitation, immunobloting, RT-PCR or reporter gene for elucidation of the antimelanogenic mechanism.
Results: C. sinica or α-viniferin inhibited melanin production in α-melanocyte-stimulating hormone (α-MSH)-, histamine- or cell-permeable cAMP-activated melanocyte cultures. Moreover, topical application with C. sinica containing α-viniferin, a standard in quality control, decreased melanin index on facial melasma and freckles in patients. As a molecular basis, α-viniferin accelerated protein kinase A (PKA) inactivation via the reassociation between catalytic and regulatory subunits in cAMP-elevated melanocytes, a feedback loop in the melanogenic process. α-Viniferin resultantly inhibited cAMP/PKA-signaled phosphorylation of cAMP-responsive element-binding protein (CREB) coupled with dephosphorylation of cAMP-regulated transcriptional co-activator 1 (CRTC1), thus down-regulating expression of MITF-M or Tyro gene with decreased melanin pigmentation.
Conclusion: This study assigned PKA inactivation, a feedback termination in cAMP-induced facultative melanogenesis, as a putative target of α-viniferin in the treatment of melanocyte-specific hyperpigmented disorder. Finally, C. sinica containing α-viniferin was approved as an antimelanogenic agent with topical application in skin hyperpigmentation.
Keywords: α-viniferin, Caragana sinica, PKA inactivation, feedback loop, melanogenesis
Introduction
Skin pigmentation represents a complex process of melanogenesis that synthesizes melanin biopolymers, black-brown eumelanin and yellow-red pheomelanin, in the melanosomes of epidermal melanocytes and transfers pigmented melanosomes to keratinocytes in the overlaying epidermis [1, 2]. Tyrosinase (Tyro), with dual activities of tyrosine hydroxylase and dopa oxidase, catalyzes a rate-limiting step in the biosynthetic pathways of eumelanin and pheomelanin [3, 4]. Tyro-related protein 1 (TYRP1) and dopachrome tautomerase (DCT) are required in the production of eumelanin but not pheomelanin [3, 4].
Skin disorders such as melasma and freckles are hyperpigmented with excess melanin biopolymers. cAMP plays as a key second messenger in the generation of heavily pigmented melanosomes [5]. α-Melanocyte-stimulating hormone (α-MSH) or histamine stimulate melanin production in epidermal melanocytes via G protein-coupled receptors (GPCRs)-mediated activation of adenylate cyclase [6]. cAMP binds to the regulatory (R)-subunit within inactive PKA holoenzyme, concomitantly unleashing the catalytic (C)-subunit for activation [7, 8]. On the other hand, phosphodiesterases (PDEs) hydrolyze cAMP in free form or bound to the R-subunit of PKA, and the C-subunit reassociates with the cAMP-free R-subunit in the signal termination [8, 9, 10]. The C-subunit of PKA phosphorylates cAMP-responsive element (CRE)-binding protein (CREB) at Ser-133 or salt-inducible kinase 2 (SIK2) at Ser-587 [11, 12]. Phospho (p)-SIKs are less active than dephospho-SIKs in the kinase activity on cAMP-regulated transcriptional co-activator 1 (CRTC1) at Ser-151 and -298 or CREB-binding protein (CBP) at Ser-89 [11, 13]. On the other hand, protein phosphatase 1 (PP1), PP2A and PP2B remove the phosphate groups on p-CREB, p-SIKs or p-CRTC1 [12, 14]. Cytoplasmic CRTC1 that is not labeled with phosphate tags translocates into the nucleus to reinforce CREB occupancy over CRE motif in the promoter regions of target genes, but p-CRTC1 is sequestered in the cytoplasm by protein 14-3-3 [12, 15]. The nuclear complex of p-CREB, with its co-activators CBP/p300 and dephospho-CRTC1, recruits the transcription machinery containing RNA polymerase II onto the core promoter of microphthalmia-associated transcription factor subtype M (MITF-M) gene, a master transcription factor in the control of melanogenesis [11, 16, 17]. In turn, MITF-M upregulates expression of Tyro, TYRP1 or DCT gene in melanocytes [4, 18].
α-Viniferin is an oligostilbene constituent of Caragana sinica, a plant of the Leguminosae family, whose roots are used as an alternative medicine in the treatment of inflammatory disorders. An extract of C. sinica inhibits dopa auto-oxidation, and α-viniferin interrupts lipid peroxidation via scavenging free radicals and suppresses expression of inflammatory genes in macrophages [19, 20, 21]. Numerous antioxidants or anti-inflammatory agents ameliorate melasma, a hyperpigmented disorder in the skin [22, 23]. In the current study, we investigated the antimelanogenic potential of C. sinica or its active constituent α-viniferin in cAMP-elevated melanocyte cultures or facial hyperpigmented patients, and then elucidated a molecular basis of its action.
Results
C. sinica or α-viniferin inhibited melanin production in cAMP-elevated melanocytes
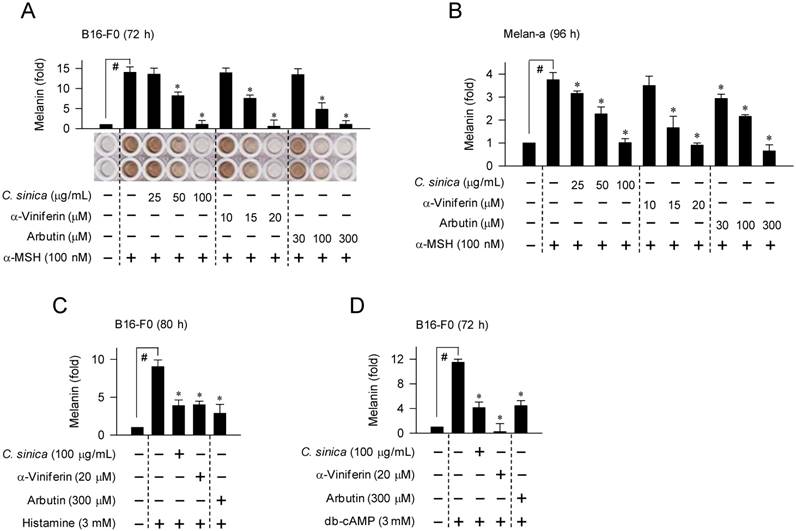
We first determined cAMP-induced melanin pigmentation in B16-F0 melanoma cell line and Melan-a melanocyte cell line that were employed as model melanocytes. Upon exposure to α-MSH alone, B16-F0 or Melan-a cells markedly increased melanin production over the unstimulated (basal) levels (Figure 1A-B). Treatment with C. sinica or α-viniferin dose-dependently inhibited α-MSH-induced melanin production, as did arbutin, in which α-viniferin was more effective (> 5-fold) than arbutin in the comparison with IC50 values (Figure 1A-B). C. sinica or α-viniferin also inhibited histamine- or N6,2'-O-dibutyryl (db)-cAMP-induced melanin production in B16-F0 cells (Figure 1C-D). Arbutin is a skin whitener targeting the substrate-binding site in the inhibition of Tyro activity, and db-cAMP is a cell-permeable cAMP agonist [24]. However, C. sinica or α-viniferin at the concentrations with antimelanogenic activity did not disturb the viability or proliferation of melanocytes (Figure S1A-B), excluding nonspecific cytotoxicity. The results suggested that C. sinica or α-viniferin could inhibit facultative melanogenesis in cAMP-elevated melanocytes.
Effect of C. sinica or α-viniferin on melanin production in melanocyte cultures. B16-F0 cells were stimulated with α-MSH (A), histamine (C) or db-cAMP (D) for 72-80 h in the presence of C. sinica or α-viniferin. (B) Melan-a cells were stimulated with α-MSH for 96 h in the presence of C. sinica or α-viniferin. Melanin pigments were quantified by measuring absorbance values at 405 nm, and are represented as a relative fold. Data are mean ± SEM. #p < 0.05 vs. medium alone-added group. *p < 0.05 vs. α-MSH, histamine or db-cAMP alone-stimulated group.
C. sinica containing α-viniferin as active constituent improved facial hyperpigmentation in patients
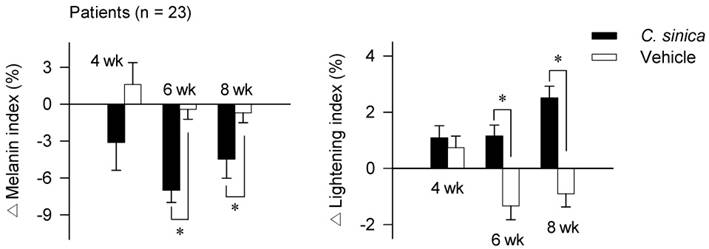
To understand whether the in vitro outcomes of C. sinica or α-viniferin could be translated into the hyperpigmented disorders in patients, a clinical investigation was designed as a randomized, double-blind, vehicle-controlled, split-face trial. Twenty-three patients, who had melasma and freckles on both sides of the face, were topically applied with C. sinica cream on one half of the face and vehicle cream on the other half in a twice-daily regimen for 8 consecutive weeks (wk). All of the patients completed the clinical investigation without any claims such as skin itching or other adverse discomforts. The C. sinica-treated group exhibited a significantly decreased melanin index on the face with hyperpigmented patches, as well as increased lightening index at 6-8 wk over the vehicle group (Figure 2; Figure S2; Table S1A-B). Formulations of the vehicle or C. sinica cream are described in Table S2, and contents of α-viniferin within C. sinica extract in Figure S3. On the basis of clinical satisfaction, C. sinica containing α-viniferin, a standard in quality control, was approved as a new antimelanogenic agent by the Korean Ministry of Food and Drug Safety.
Effect of C. sinica cream on facial hyperpigmentation in patients. The clinical investigation was designed as a randomized, double-blind, vehicle-controlled, split-face trial. Patients (n = 23), who had melasma and freckles on both sides of the face, were topically applied with C. sinica cream on one half of the face and vehicle cream on the other half in a twice-daily regimen for 8 consecutive wk. Melanin index or lightening index on hyperpigmented patches in the face were measured using a mexameter or chromameter. Primary outcomes are represented as percentage (%) change (Δ) of melanin index or lightening index from the baseline (0 time) to 4 wk, 6 wk or 8 wk. Data are mean ± SEM. *p < 0.05 vs. vehicle cream alone-applied group. The difference was determined using the Student's t-test.
C. sinica or α-viniferin suppressed Tyro induction in α-MSH-activated melanocytes
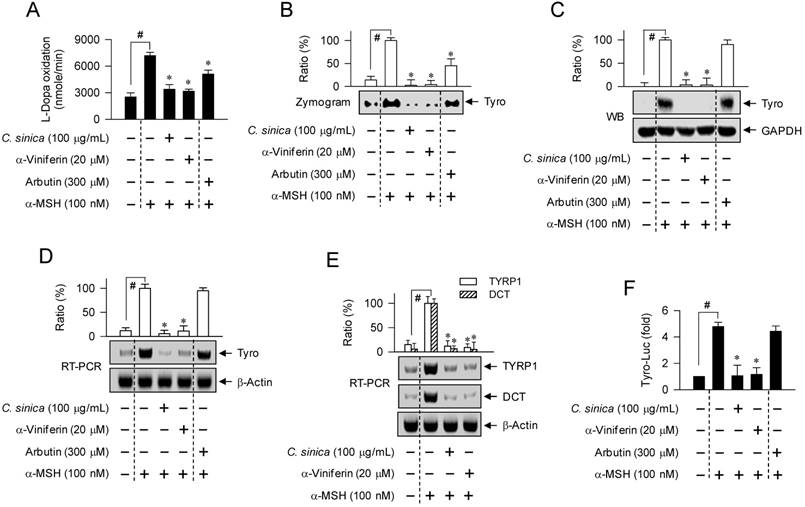
To determine an antimelanogenic mechanism, we first examined whether C. sinica or α-viniferin could affect cellular levels and/or catalytic activity of Tyro, a key enzyme in melanin biosynthesis. Treatment with C. sinica or α-viniferin decreased L-dopa oxidation activity in α-MSH-activated B16-F0 cells, as did arbutin (Figure 3A-B). C. sinica or α-viniferin also normalized α-MSH-induced protein and mRNA levels of Tyro to the basal states, in which arbutin was not effective (Figure 3C-D). Moreover, C. sinica or α-viniferin attenuated α-MSH-induced mRNA levels of TYRP1 or DCT gene (Figure 3E). Transcriptional regulation of Tyro gene by C. sinica or α-viniferin was further demonstrated by promoter activity-dependent reporter assay. B16-F0 cells were transfected with Tyro-Luc, a construct encoding the promoter (-2236/+59) of Tyro gene fused to firefly luciferase gene as a reporter. Upon exposure to α-MSH alone, B16-F0 cells harboring Tyro-Luc construct increased luciferase expression over the basal levels (Figure 3F). Treatment with C. sinica or α-viniferin inhibited α-MSH-induced expression of luciferase gene, reporting promoter activity of Tyro gene (Figure 3F). Tyro protein was then treated with C. sinica or α-viniferin in cell-free conditions and the velocity of L-dopa oxidation was determined. C. sinica or α-viniferin did not directly affect the catalytic activity of cell-free Tyro, whereas arbutin inhibited its activity (Figure S4). The results suggested that C. sinica or α-viniferin could suppress the expression of Tyro gene in α-MSH-activated melanocytes, but arbutin did not at all. Therefore, C. sinica or α-viniferin might have a distinct mechanism from arbutin.
Effect of C. sinica or α-viniferin on expression of Tyro gene. B16-F0 cells were pretreated with C. sinica or α-viniferin for 2 h and stimulated with α-MSH for 48 h (A-C) or 20 h (D, E) in the presence of C. sinica or α-viniferin. Cell lysates were prepared with phosphate buffer, and cell extracts with RIPA buffer. (A) Cell lysates were reacted with 1 mM L-dopa, and the velocity of increasing absorbance values at 475 nm was immediately measured. Tyro activity is represented as the initial velocity of L-dopa oxidation (nmol/min). (B) Cell lysates were resolved on non-denaturing acrylamide gels (without 2-mercaptoethanol) by electrophoresis, and subjected to zymography with soaking of the gels in 1 mM L-dopa. (C) Cell extracts were resolved on SDS-acrylamide gels by electrophoresis, and subjected to Western blot (WB) analysis with anti-Tyro or anti-GAPDH antibody. (D, E) Total RNAs were subjected to RT-PCR analysis of Tyro, TYRP1 or DCT with β-actin as an internal control, and resolved on agarose gels by electrophoresis. (F) B16-F0 cells were transfected with Tyro (-2236/+59)-Luc reporter construct in combination with Renilla control vector. The transfected cells were pretreated with C. sinica or α-viniferin for 2 h and stimulated with α-MSH for 18 h in the presence of C. sinica or α-viniferin. Firefly luciferase activity, a reporter of the promoter activity of Tyro gene, is represented as a relative fold after normalizing to Renilla activity, a reference of transfection efficiency. Data are mean ± SEM. #p < 0.05 vs. medium alone-added group. *p < 0.05 vs. α-MSH alone-stimulated group.
C. sinica or α-viniferin down-regulated cAMP-induced expression of MITF-M gene at the transcription level
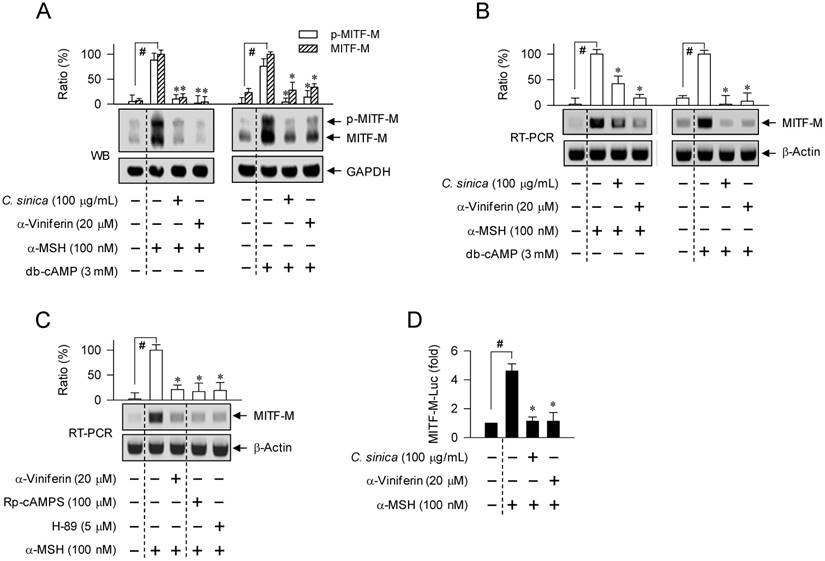
Melanocyte-specific MITF-M is also inducible in response to cAMP, and regulates expression of Tyro, TYRP1 or DCT gene [4, 17, 18]. Upon exposure to α-MSH or db-cAMP alone, B16-F0 cells markedly increased protein levels of MITF-M and p-MITF-M (Figure 4A). Treatment with C. sinica or α-viniferin attenuated not only α-MSH-induced protein levels of MITF-M and p-MITF-M but also db-cAMP-dependent induction of the proteins (Figure 4A). Accordingly, C. sinica or α-viniferin suppressed mRNA levels of MITF-M gene, as did the positive control agents such as Rp-adenosine 3',5'-cyclic phosphorothioate (Rp-cAMPS) and H-89 (Figure 4B-C). Rp-cAMPS, a cell-permeable cAMP antagonist, interrupts dissociation of the C-subunit from PKA holoenzyme, and H-89 inhibits the kinase activity of the C-subunit of PKA by targeting the ATP-binding site but not dissociation [25, 26]. Moreover, C. sinica or α-viniferin inhibited α-MSH-induced promoter activity of MITF-M gene, as determined using B16-F0 cells transfected with MITF-M-Luc, a construct encoding the promoter (-2200/+95) of MITF-M gene fused to firefly luciferase gene as a reporter (Figure 4D). The results suggested that C. sinica or α-viniferin could down-regulate cAMP-induced expression of MITF-M gene at the transcriptional level.
Effect of C. sinica or α-viniferin on expression of MITF-M gene. B16-F0 cells were pretreated with C. sinica or α-viniferin for 2 h and stimulated with α-MSH (A-C) or db-cAMP (A, B) for 4 h (A) or 2 h (B, C) in the presence of C. sinica or α-viniferin. (A) Cell extracts were resolved on SDS-acrylamide gels by electrophoresis, and subjected to Western blot (WB) analysis with anti-MITF-M or anti-GAPDH antibody. (B, C) Total RNAs were subjected to RT-PCR analysis of MITF-M with β-actin as an internal control, and resolved on agarose gels by electrophoresis. (D) B16-F0 cells were transfected with MITF-M (-2200/+95)-Luc reporter construct in combination with Renilla control vector. The transfected cells were pretreated with C. sinica or α-viniferin for 2 h and stimulated with α-MSH for 18 h in the presence of C. sinica or α-viniferin. Firefly luciferase activity, a reporter of the promoter activity of MITF-M gene, is represented as a relative fold after normalizing to Renilla activity. Data are mean ± SEM. #p < 0.05 vs. medium alone-added group. *p < 0.05 vs. α-MSH or db-cAMP alone-stimulated group.
α-Viniferin inhibited α-MSH- or db-cAMP-induced CREB phosphorylation and CRTC1 dephosphorylation
CREB-responsive CRE motifs appear to be essential for maximal transcription of MITF-M gene in cAMP-elevated melanocytes, even though the promoter region of MITF-M gene contains several cis-acting elements [17, 27]. Phosphorylation of CREB at Ser-133 is important in the recruitment of CBP/p300, acetylating nucleosomal histones for disassembly [11, 16, 28]. Dephospho-CRTC1, but not p-CRTC1, translocates into the nucleus and reinforces CREB occupancy over CRE motifs for expression of MITF-M gene in the melanogenic process [12, 17, 29].
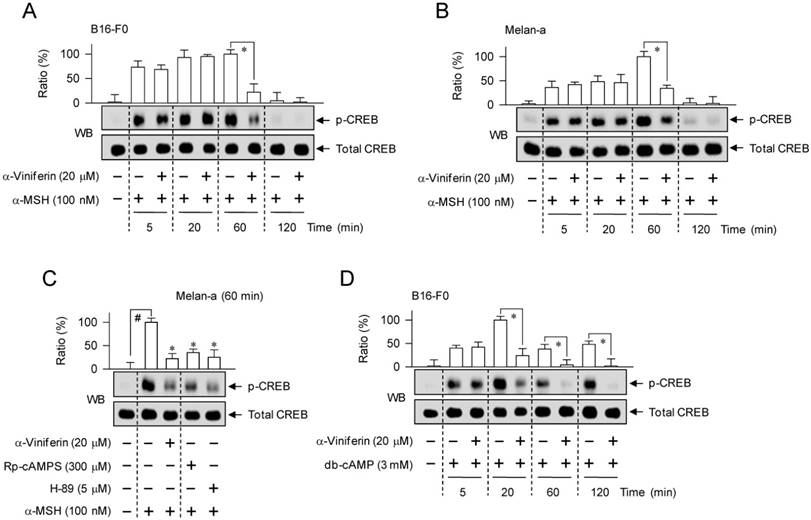
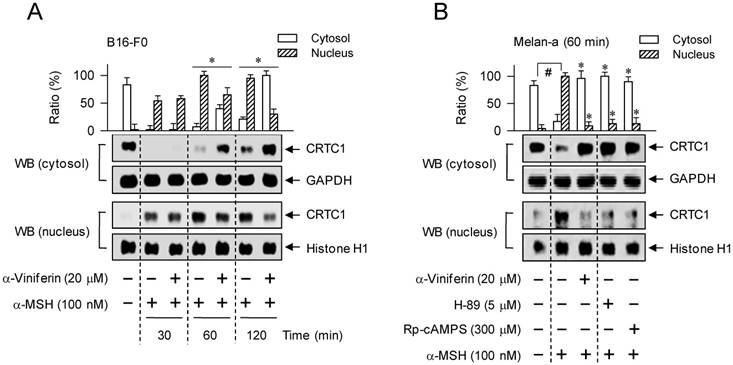
To understand whether the mechanism of α-viniferin (down-regulating transcription of MITF-M gene) could be correlated to CREB phosphorylation or CRTC1 dephosphorylation in the upstream pathway, melanocytes were pretreated with α-viniferin for 2 h and stimulated with α-MSH or db-cAMP for the indicated time points in the presence of α-viniferin. B16-F0 or Melan-a cells increased the phosphorylation of CREB at 5 min to 1 h after exposure to α-MSH, and at 5 min to 2 h after exposure to db-cAMP (Figure 5). Treatment with α-viniferin attenuated α-MSH-induced p-CREB levels at the time points of 1 h but not 5-20 min, as did Rp-cAMPS and H-89 at 1 h (Figure 5A-C). Treatment with α-viniferin also decreased db-cAMP-induced p-CREB levels at 20 min to 2 h but not 5 min, as did Rp-cAMPS and H-89 at 20 min (Figure 5D-E). On the other hand, B16-F0 or Melan-a cells markedly decreased p-CRTC1 levels upon exposure to α-MSH or db-cAMP (Figure 6). Treatment with α-viniferin restored p-CRTC1 levels at 1-2 h but not 5-20 min after stimulation with α-MSH (Figure 6A-B), as well as at 20 min to 2 h but not 5 min after stimulation with db-cAMP, as did Rp-cAMPS and H-89 at 20 min (Figure 6C-D). These results suggested that α-viniferin could inhibit not only CREB phosphorylation but also CRTC1 dephosphorylation at the same time points. Furthermore, α-viniferin inhibited nuclear translocation of CRTC1 in B16-F0 cells at the time points of 1-2 h but not 30 min after stimulation with α-MSH (Figure 7A), and also at 1 h in α-MSH-activated Melan-a cells, as did Rp-cAMPS and H-89 (Figure 7B).
Effect of α-viniferin on CREB phosphorylation. B16-F0 cells (A, D) or Melan-a cells (B, C, E) were pretreated with α-viniferin for 2 h and stimulated with α-MSH (A-C) or db-cAMP (D, E) for indicated time points in the presence of α-viniferin. Cell extracts were resolved on SDS-acrylamide gels by electrophoresis, and subjected to Western blot (WB) analysis with anti-p-CREB or anti-CREB antibody. Data are mean ± SEM. *p < 0.05 vs. α-MSH or db-cAMP alone-stimulated group. #p < 0.05 vs. medium alone-added group.
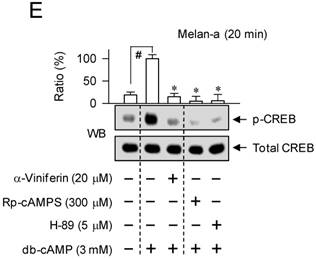
Effect of α-viniferin on CRTC1 dephosphorylation. B16-F0 cells (A, C) or Melan-a cells (B, D) were pretreated with α-viniferin for 2 h and stimulated with α-MSH (A, B) or db-cAMP (C, D) for indicated time points in the presence of α-viniferin. Cell extracts were resolved on SDS-acrylamide gels by electrophoresis, and subjected to Western blot (WB) analysis with anti-p-CRTC1 or anti-CRTC1 antibody. Data are mean ± SEM. *p < 0.05 vs. α-MSH or db-cAMP alone-stimulated group. #p < 0.05 vs. medium alone-added group.
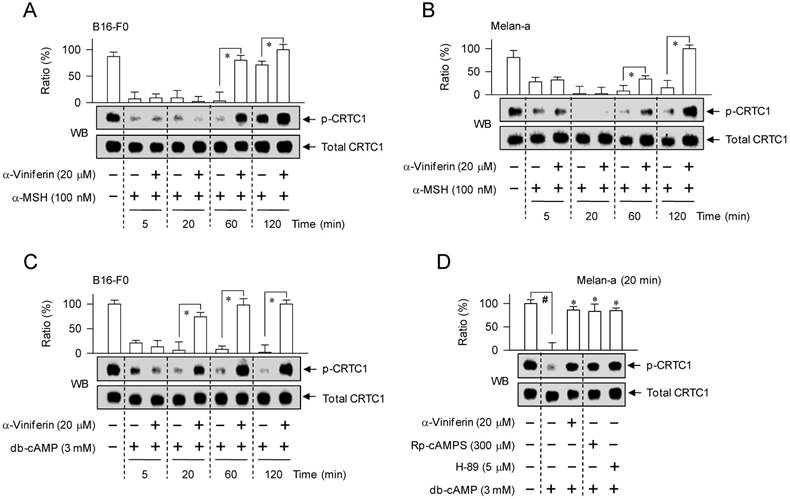
Effect of α-viniferin on nuclear translocation of CRTC1. B16-F0 cells (A) or Melan-a cells (B) were pretreated with α-viniferin for 2 h and stimulated with α-MSH for indicated time points in the presence of α-viniferin. Cell extracts were separated into fractions of cytosol or nucleus. Proteins in each compartment were resolved on SDS-acrylamide gels by electrophoresis, and subjected to Western blot (WB) analysis with anti-CRTC1, anti-GAPDH or anti-histone H1 antibody. Data are mean ± SEM. *p < 0.05 vs. α-MSH alone-stimulated group. #p < 0.05 vs. medium alone-added group.
To delineate which kinase or phosphatase could switch the phosphorylation circuit acting on the phosphorylation of CREB coupled with the dephosphorylation of CRTC1, B16-F0 cells were pretreated with specific inhibitors for 2 h and stimulated with α-MSH for 1 h or db-cAMP for 20 min in the presence of each inhibitor. PKA inhibitors (Rp-cAMPS, H-89) or p38 MAPK inhibitors (SB202190, SB203580) attenuated α-MSH- or db-cAMP-induced p-CREB levels, as did α-viniferin (Figure S5). However, an EPAC inhibitor (ESI-09), MEK1/2 inhibitors (U0126, PD98059), GSK-3α/β inhibitors (6BIO, SB216763), dual inhibitors of PP1 and PP2A (okadaic acid, calyculin A) or PP2B inhibitors (cyclosporin A, deltamethrin) did not affect the cAMP-induced phosphorylation of CREB (Figure S5). On the other hand, the PKA inhibitors (Rp-cAMPS, H-89) or the PP2B inhibitors (cyclosporin A, deltamethrin) restored p-CRTC1 levels in α-MSH- or db-cAMP-activated B16-F0 cells, but the dual inhibitors of PP1 and PP2A or the p38 MAPK inhibitors did not (Figure S6). In addition, all of the pharmacological agents except the MEK1/2 inhibitors attenuated melanin pigmentation in α-MSH- or db-cAMP-activated B16-F0 cells (Figure S7). The results suggested that PKA inhibitors (Rp-cAMPS, H-89) could regulate not only CREB phosphorylation but also CRTC1 dephosphorylation in cAMP-induced melanogenic process, as did α-viniferin.
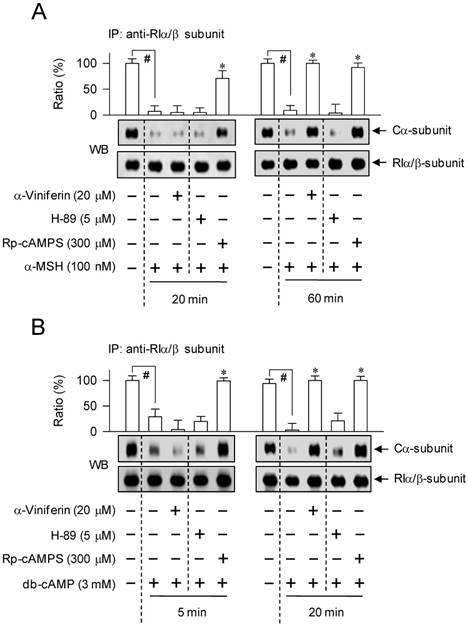
α-Viniferin accelerated the reassociation between C-subunit and R-subunit of PKA
We further asked whether α-viniferin could inhibit the dissociation of C-subunit from PKA holoenzyme or accelerate the reassociation between C-subunit and R-subunit to generate inactive holoenzyme, since it mimicked Rp-cAMPS in the modulation of CREB phosphorylation coupled with CRTC1 dephosphorylation. B16-F0 cells were pretreated with α-viniferin for 2 h and stimulated with α-MSH for 20 min or 1 h, and with db-cAMP for 5 min or 20 min in the presence of α-viniferin. The time points were selected according to the previous results that α-viniferin inhibited the cAMP/PKA-signaled phosphorylation circuit acting on CREB and CRTC1 at 1 h but not 20 min after exposure to α-MSH, and at 20 min but not 5 min after exposure to db-cAMP. Cell extracts were subjected to an immunoprecipitation (IP) with antibody against the RIα/β-subunit of PKA, and the IP complex was then probed with another antibody against the Cα-subunit of PKA to detect the co-precipitates. Upon exposure to α-MSH or db-cAMP alone, B16-F0 cells drastically dissociated the Cα-subunit of PKA from RIα/β-subunit (Figure 8). Treatment with α-viniferin restored the holoenzyme complex between Cα-subunit and RIα/β-subunit at the time points of 1 h but not 20 min after stimulation with α-MSH (Figure 8A), as well as at 20 min but not 5 min in db-cAMP-activated B16-F0 cells (Figure 8B). However, Rp-cAMPS inhibited the dissociation of Cα-subunit from RIα/β-subunit of PKA at all time points, but H-89 did not at all, as expected (Figure 8A-B). The results suggested that α-viniferin could accelerate the reassociation between C-subunit and R-subunit of PKA in melanocytes, a feedback loop in the cAMP-induced facultative melanogenesis.
Effect of α-viniferin on dissociation or reassociation of PKA holoenzyme. B16-F0 cells were pretreated with α-viniferin for 2 h and stimulated with α-MSH for 20 min or 1 h (A) or db-cAMP for 5 min or 20 min (B) in the presence of α-viniferin. Cell extracts were subjected to an immunoprecipitation (IP) with antibody against the RIα/β-subunit of PKA. IP complexes were then subjected to Western blot (WB) analysis with another antibody against the Cα-subunit of PKA to detect the co-precipitates. Data are mean ± SEM. #p < 0.05 vs. medium alone-added group. *p < 0.05 vs. α-MSH or db-cAMP alone-stimulated group.
Discussion
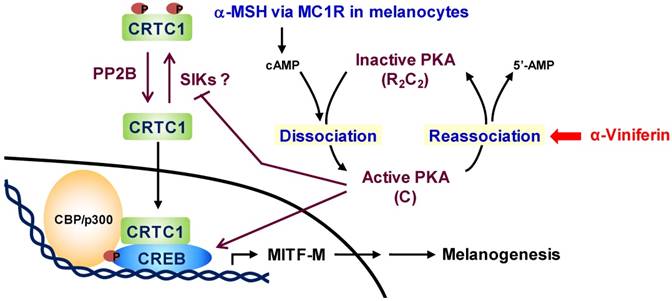
Chemical-based regulation of hyperpigmented disorders in the skin has been a long-standing goal for cosmetic and pharmaceutical applications. Numerous strategies have been attempted to discover chemicals that inhibit the catalytic activity of Tyro, the synthesis of melanin biopolymer or the transfer of pigmented melanosome in cell-based assays [30, 31]. However, a large number of the antimelanogenic candidates have met with limited or no success in the translation from the in vitro outcomes to patients [30, 32]. In the current study, we communicate a therapeutic pipeline of facial hyperpigmented disorders. C. sinica or its active constituent α-viniferin not only inhibited melanin pigmentation in α-MSH-, histamine- or db-cAMP-activated melanocytes but also decreased melanin index on facial melasma and freckles in patients. As a mechanism, α-viniferin accelerated the reassociation between C-subunit and R-subunit of PKA, becoming an inactive holoenzyme, in cAMP-elevated melanocytes. α-Viniferin resultantly decreased p-CREB levels, increased p-CRTC1 levels, and inhibited nuclear translocation of CRTC1, thus down-regulating transcription of MITF-M gene in the cAMP-induced facultative melanogenesis, as proposed in Figure 9.
A proposed antimelanogenic mechanism of α-viniferin. α-MSH increases cAMP levels via melanocortin 1 receptor (MC1R)-mediated activation of adenylate cyclase in melanocytes. cAMP binds to the regulatory (R)-subunit within PKA holoenzyme (R2C2), concomitantly unleashing the catalytic (C)-subunit for activation. On the other hand, the C-subunit of PKA reassociates with cAMP-free R-subunit, becoming an inactive complex, in the signal termination. α-Viniferin, an active constituent of C. sinica, primarily accelerates PKA inactivation via reassociation between C-subunit and R-subunit. This mechanism resultantly inhibits not only CREB phosphorylation, CRTC1 dephosphorylation but also nuclear translocation of CRTC1, thus down-regulating expression of MITF-M gene, a master transcription factor in cAMP-induced facultative melanogenesis.
A two-hit model (phosphorylation of CREB coupled with dephosphorylation of CRTCs) has been proposed to explain why some signals that promote CREB phosphorylation alone are not sufficient for the expression of target genes, since p-CREB in concert with dephospho-CTRCs are required in the efficient recruitment of CBP/p300, acetylating nucleosomal histones for disassembly, and then the transcription machinery containing RNA polymerase II [11, 16, 29]. B16-F0 cells express three isoforms of CRTCs, in which CRTC1 is predominant, and decreases ultraviolet (UV)-induced transcription of MITF-M gene in the melanogenic process upon overexpression of dominant-negative CRTC1 [13]. On the other hand, CRTC2 and CRTC3 are assigned as antimelanogenic targets of adiponectin, a stimulator of AMP-activated protein kinase (AMPK), in forskolin (an activator of adenylate cyclase) plus 3-isobutyl-1-methylxanthine (an inhibitor of PDE)-activated NHM or Mel-ab melanocytes [33].
In the current study, we examined CREB phosphorylation coupled with CRTC1 dephosphorylation, two critical events in the transcriptional control of MITF-M gene, in α-MSH- or db-cAMP-activated B16-F0 and Melan-a cells. In the basal states, dephospho-CREB was predominantly localized in the nucleus but p-CRTC1 in the cytoplasm. Upon exposure to cAMP elevators, melanocytes markedly phosphorylated CREB at Ser-133, removed the phosphate group of p-CRTC1 at Ser-151, and increased nuclear translocation of CRTC1. Treatment with α-viniferin or PKA inhibitors (Rp-cAMPS, H-89) decreased melanin pigmentation in cAMP-elevated melanocytes via inhibiting not only CREB phosphorylation but also CTRC1 dephosphorylation, suggesting a notable implication of cAMP/PKA-signaled phosphorylation circuit acting on CREB and CRTC1 in the antimelanogenic action. However, p38 MAPK inhibitors (SB202190, SB203580) affected CREB phosphorylation only, PP2B inhibitors (cyclosporin A, deltamethrin) on CRTC1 dephosphorylation only, and the inhibitors of EPAC, GSK-3α/β, PP1 or PP2A bypassed both CREB phosphorylation and CRTC1 dephosphorylation in the inhibition of cAMP-induced facultative melanogenesis.
As a primary target, α-viniferin accelerated the reassociation between Cα-subunit and RIα/β-subunit of PKA in cAMP-elevated melanocytes, a feedback loop in the cAMP/PKA-signaled melanogenic process. However, Rp-cAMPS, a control agent used in this study, interrupted the dissociation of Cα-subunit from RIα/β-subunit of PKA. Small molecules such as bosabolangelone, diphenylmethylene hydrazinecarbothioamide and 4-hydroxy-3-methoxycinnamaldehyde also inhibit melanin production in cAMP-elevated melanocytes via mimicking Rp-cAMPS, a PKA holoenzyme-directed cAMP antagonist [34, 35, 36]. Therefore, the antimelanogenic mechanism of α-viniferin is distinct from Rp-cAMPS or other cAMP antagonists, and has not been assigned to any cutaneous agents in the treatment of hyperpigmented disorders.
Little is known about the molecular basis in the feedback loop of PKA inactivation via the reassociation between C-subunit and R-subunit. The C-subunit released from PKA holoenzyme via cAMP migrates into the nucleus in a passive process, but the remaining R-subunit is sequestered in the cytoplasm by tethering with A-kinase anchoring proteins [8, 37]. PKA inhibitor peptide (PKI), a heat-stable protein expressed in many tissues, exports the C-subunit of PKA out of the nucleus via unmasking nuclear export signal on the C-subunit [38]. Cytoplasmic C-subunit of PKA is then reassociated with the cAMP-free R-subunit, becoming inactive holoenzyme [8]. PDEs hydrolyze cAMP in free form or bound to the R-subunit of PKA [9, 10]. In particular, PDE4D3 is upregulated in forskolin-activated melanocytes, which plays an important role in the feedback control of MITF-M-dependent melanocyte differentiation [39]. Moreover, PDE4 inhibitors (rolipram, Ro 20-1724) synergize with forskolin in the melanin pigmentation of dorsal skins in mice [39].
In conclusion, this study suggested that C. sinica containing α-viniferin, a standard in quality control, improved facial hyperpigmentation in patients via a unique mechanism, accelerating feedback termination of cAMP/PKA-signaled expression of MITF-M gene in melanocytes.
Materials and Methods
Drugs and chemicals
Pharmacological agents were α-viniferin (Sigma-Aldrich, SMB00074) as a test sample; arbutin (Sigma-Aldrich, A4256) as a skin lightening agent; Rp-cAMPS (Sigma-Aldrich, A165) or H-89 (Sigma-Aldrich, B1427) as PKA inhibitors; ESI-09 (Sigma-Aldrich, SML0814) as an EPAC inhibitor; U0126 (Sigma-Aldrich, U120) or PD98059 (Sigma-Aldrich, P215) as MEK1/2 inhibitors; 6BIO (Sigma-Aldrich, B1686) or SB216763 (Sigma-Aldrich, S3442) as GSK-3α/β inhibitors; SB202190 (Sigma-Aldrich, S7067) or SB203580 (Sigma-Aldrich, S8307) as p38 MAPK inhibitors; okadaic acid (Sigma-Aldrich, O9381) or calyculin A (Sigma-Aldrich, C5552) as dual inhibitors of PP1 and PP2A; and cyclosporine A (Sigma-Aldrich, C1822) or deltamethrin (Sigma-Aldrich, D9315) as PP2B inhibitors. All of the drugs and chemicals were > 99.5% in purity. The roots of C. sinica were extracted three times with 95% ethyl alcohol at room temperature and evaporated under reduced pressure to become a powder. Contents of α-viniferin within C. sinica extract were determined by HPLC analysis.
Cell culture
B16-F0 (ATCC, CRL-6322), a murine melanoma cell line, was seeded in DMEM (Sigma-Aldrich, D2902) supplemented with 10% FBS (Corning, 35-015-CV) and antibiotic-antimycotic cocktail (GIBCO, 15240062), and incubated under an atmosphere of 5% CO2 at 37 °C. Melan-a (a gift from D. C. Bennet at St George's University of London), a murine melanocyte cell line, was cultured in RPMI 1640 (GIBCO, 31800089) supplemented with 10% FBS, antibiotic-antimycotic cocktail and 200 nM 12-O-tetradecanoylphorbol 13-acetate (Sigma-Aldrich, P8139) under an atmosphere of 5% CO2 at 37 °C.
Melanin quantification
B16-F0 cells were stimulated with 100 nM α-MSH (Sigma-Aldrich, M4125), 3 mM histamine (Sigma-Aldrich, H7125) or 3 mM db-cAMP (Sigma-Aldrich, D0627) for 72-80 h, and Melan-a cells with 100 nM α-MSH for 96 h. Extracellular melanin in culture supernatants and intracellular melanin in cell pellets were determined by measuring absorbance values at 405 nm.
Clinical trial
Twenty-three patients participated in the IRB-approved protocol (P1602-59) of Chung-Ang University Hospital in order to determine whether C. sinica extract containing α-viniferin, a standard in quality control, could improve facial hyperpigmentation. The protocol was designed as a randomized, double-blind, vehicle-controlled, split-face trial. The patients were Korean women aged 24-55 years who had melasma and freckles on both sides of the face but were not undergoing pregnancy, lactation or receiving medications at least 1-3 months before enrollment. The patients were topically applied with C. sinica cream on one half of the face and vehicle cream on the other half in a twice-daily regimen (morning, night) for 8 consecutive wk. Primary outcomes from the baseline (0 time) to 4 wk, 6 wk or 8 wk were measured as the change (Δ) in melanin index or lightening index on hyperpigmented patches in the face using a mexameter or chromameter. All clinical investigations were conducted according to the Declaration of Helsinki principles. A written informed consent was received from participants prior to the clinical investigation.
L-Dopa oxidation assay
B16-F0 cells were stimulated with α-MSH for 48 h. Cell lysates were prepared with phosphate buffer (pH 6.5), and the initial velocity was measured by increasing absorbance values at 475 nm upon addition of 1 mM L-dopa (Sigma-Aldrich, D9628), a substrate of Tyro. Catalytic activity was represented as the velocity of L-dopa oxidation (nmol/min). Cell lysates were also resolved on 10% acrylamide gels (without 2-mercaptoethanol and SDS) by electrophoresis, and subjected to zymography with soaking of the gels in 1 mM L-dopa.
Western blot analysis
Cell extracts were prepared with RIPA buffer (iNtRON, IBS-BR002), resolved on SDS-acrylamide gels by electrophoresis, and transferred to polyvinylidene difluoride membranes (Roche 03010040001). The blots were incubated with either 5% non-fat milk (Becton-Dickinson, 232100) or 5% bovine serum albumin (BSA, Affymetrix, 10857) in Tris-buffered saline containing 0.05% Tween 20 (TBST) at room temperature for 1-3 h. After washing three times with TBST, the blots were reacted with primary antibody at 4 °C overnight followed by secondary antibody at room temperature for 2-4 h, in which primary antibody was diluted in 3-5% BSA and secondary antibody in 5% non-fat milk. Immune complex on the blots was visualized after reacting with an enhanced chemiluminescence reagent (GE Healthcare, RPN2232). We used primary antibody against Tyro (Santa Cruz Biotechnology, sc-7833; 1:1000), MITF-M (Abcam, ab12039; 1:2000), CREB (Cell Signaling Technology, 9197; 1:1000), p-CREB (Cell Signaling Technology, 9198; 1:1000), CRTC1 (Cell Signaling Technology, 2587; 1:1000), p-CRTC1 (Cell Signaling Technology, 3359; 1:1000), Cα-subunit of PKA (Cell Signaling Technology, 4782; 1:1000), RIα/β-subunit of PKA (Santa Cruz Biotechnology, sc-28893; 1:1000), GAPDH (Santa Cruz Biotechnology, sc-25778; 1:1000) or histone H1 (Santa Cruz Biotechnology, sc-8030; 1:1000). Secondary antibodies were rabbit anti-goat IgG labeled with horseradish peroxidase (HRP) (Thermo Fisher Scientific, A27011; 1:5000), goat anti-rabbit IgG labeled with HRP (Thermo Fisher Scientific, 31460; 1:5000), and goat anti-mouse IgG labeled with HRP (Thermo Fisher Scientific, 31430; 1:5000).
RT-PCR analysis
Total RNAs were prepared with NucleoZOL reagent (Macherey-Nagel, 1609), and subjected to RT-PCR analysis of Tyro, TYRP1, DCT or MITF-M with β-actin as an internal control in order to determine mRNA levels. Nucleotide sequences of PCR primers are described in Table S3. Briefly, total RNAs were reversely transcribed at 42 °C for 1 h using a premix reagent with oligo-dT as a primer (iNtRON, 25087). Single-stranded cDNAs were subjected to 27-32 cycles of PCR with a master premix reagent (Bioneer, K2018), in which one cycle consisted of denaturation at 94 °C for 30 s, annealing at 54-62 °C for 30 s, and DNA extension at 72 °C for 1 min. RT-PCR products were resolved on agarose gels by electrophoresis and stained with EcoDye (Biofact, ES301-1000).
Luciferase reporter assay
B16-F0 cells were transiently transfected with each reporter construct, Tyro (-2236/+59)-Luc or MITF-M (-2200/+95)-Luc, in combination with Renilla control vector using a lipofectamine reagent (Invitrogen, 11668). The transfected cells were stimulated with 100 nM α-MSH for 18 h. Cell extracts were subjected to dual luciferase assays (Promega, E1910). Firefly luciferase activity, a reporter of the promoter activity of Tyro or MITF-M gene, was normalized to Renilla luciferase activity for transfection efficiency.
IP assay
Cell extracts were incubated with antibody against the RIα/β-subunit of PKA (Santa Cruz Biotechnology, sc-28893) at 4 °C overnight, and precipitated with protein G-sepharose beads (GE Healthcare, 17-0618-01) at 4 °C for another 4-5 h.
Statistical analysis
Results are expressed as the mean ± SEM. Data were analyzed with ANOVA followed by the Student's t-test. p values < 0.05 were considered statistically significant.
Abbreviations
CRE: cAMP-responsive element; CREB: CRE-binding protein; CBP: CREB-binding protein; CRTC: cAMP-regulated transcriptional co-activator; EPAC: exchange factor directly activated by cAMP; GSK: glycogen synthase kinase; IP: immunoprecipitation; MAPK: mitogen-activated protein kinase; MEK: MAPK kinase; MITF-M: microphthalmia-associated transcription factor subtype M; PDE: phosphodiesterase; PKA: protein kinase A; PP: protein phosphatase; RT-PCR: reverse transcription-polymerase chain reaction; Tyro: tyrosinase; WB: Western blot analysis.
Supplementary Material
Supplementary figures: effect of C. sinica or α-viniferin on cell viability (S1); facial digital photographs of individual patients (S2); HPLC-DAD chromatogram of C. sinica extract (S3); effect of C. sinica or α-viniferin on catalytic activity of cell-free Tyro (S4); effects of α-viniferin, EPAC inhibitor, protein kinase inhibitors or PP inhibitors on CREB phosphorylation (S5); effects of α-viniferin, PP inhibitors or protein kinase inhibitors on CRTC1 dephosphorylation (S6); and effects of α-viniferin, EPAC inhibitor, protein kinase inhibitors or PP inhibitors on melanin production (S7). Supplementary tables: melanin index or lightening index of individual patients (S1); composition of vehicle or C. sinica cream (S2); and nucleotide sequences of RT-PCR primers (S3).
Supplementary figures and tables.
Acknowledgements
This work was financially supported by grants (2015R1D1A1A01057043, 2016R1A6A3A11933508) or a MRC program (2017R1A5A2015541) from the National Research Foundation of Korea, by a grant (WISET 2017-519) from the Korean Ministry of Science and ICT, and by the foresting project of Osong academy-industry convergence from the Korean Ministry of Trade, Industry & Energy. We thank the clinical research team (Chung-Ang University Hospital, Korea) including J. H. Shin, J. M. Seok, A. R. Kim, H. N. Lee, S. E. Lee, T. J. Lee, I. A. Kim, Y. H. Kim, C. R. Seo, M. H. Park, E. K. Lee, and J. H. Kang for care of patients and collection of raw data; S. Shibahara (Tohoku University School of Medicine, Japan) for supply of MITF-M-Luc construct; R. Ballotti (INSERM, France) for supply of Tyro-Luc construct; and H.-S. Ryu (Coseedbiopharm Corporation, Korea) for assistance in manuscript preparation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bastonini E, Kovacs D, Picardo M. Skin pigmentation and pigmentary disorders: focus on epidermal/dermal cross-talk. Ann Dermatol. 2016;28:279-89
2. Tadokoro R, Takahashi Y. Intercellular transfer of organelles during body pigmentation. Curr Opin Genet Dev. 2017;45:132-8
3. Hearing VJ. Determination of melanin synthetic pathways. J Invest Dermatol. 2011;131:E8-E11
4. Kondo T, Hearing VJ. Update on the regulation of mammalian melanocyte function and skin pigmentation. Expert Rev Dermatol. 2011;6:97-108
5. D'Orazio J, Fisher DE. Central role for cAMP signaling in pigmentation and UV resistance. Cell Cycle. 2011;10:8-9
6. Yuan XH, Jin ZH. Paracrine regulation of melanogenesis. Br J Dermatol. 2017
7. Kim C, Cheng CY, Saldanha SA. et al. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell. 2007;130:1032-43
8. Torres-Quesada O, Mayrhofer JE, Stefan E. The many faces of compartmentalized PKA signalosomes. Cell Signal. 2017;37:1-11
9. Krishnamurthy S, Moorthy BS, Xin Xiang L. et al. Active site coupling in PDE:PKA complexes promotes resetting of mammalian cAMP signaling. Biophys J. 2014;107:1426-40
10. Moorthy BS, Gao Y, Anand GS. Phosphodiesterases catalyze hydrolysis of cAMP-bound to regulatory subunit of protein kinase A and mediate signal termination. Mol Cell Proteomics. 2011;10:M110 002295
11. Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141-51
12. Uebi T, Tamura M, Horike N. et al. Phosphorylation of the CREB-specific coactivator TORC2 at Ser(307) regulates its intracellular localization in COS-7 cells and in the mouse liver. Am J Physiol Endocrinol Metab. 2010;299:E413-25
13. Horike N, Kumagai A, Shimono Y. et al. Downregulation of SIK2 expression promotes the melanogenic program in mice. Pigment Cell Melanoma Res. 2010;23:809-19
14. Wadzinski BE, Wheat WH, Jaspers S. et al. Nuclear protein phosphatase 2A dephosphorylates protein kinase A-phosphorylated CREB and regulates CREB transcriptional stimulation. Mol Cell Biol. 1993;13:2822-34
15. Katoh Y, Takemori H, Min L. et al. Salt-inducible kinase-1 represses cAMP response element-binding protein activity both in the nucleus and in the cytoplasm. Eur J Biochem. 2004;271:4307-19
16. Clark MD, Kumar GS, Marcum R. et al. Molecular basis for the mechanism of constitutive CBP/p300 coactivator recruitment by CRTC1-MAML2 and its implications in cAMP signaling. Biochemistry. 2015;54:5439-46
17. Hartman ML, Czyz M. MITF in melanoma: mechanisms behind its expression and activity. Cell Mol Life Sci. 2015;72:1249-60
18. Vachtenheim J, Borovansky J. "Transcription physiology" of pigment formation in melanocytes: central role of MITF. Exp Dermatol. 2010;19:617-27
19. Lee KT, Kim BJ, Kim JH. et al. Biological screening of 100 plant extracts for cosmetic use (I): inhibitory activities of tyrosinase and DOPA auto-oxidation. Int J Cosmet Sci. 1997;19:291-8
20. Jin Q, Han XH, Hong SS. et al. Antioxidative oligostilbenes from Caragana sinica. Bioorg Med Chem Lett. 2012;22:973-6
21. Chung EY, Roh E, Kwak JA. et al. α-Viniferin suppresses the signal transducer and activation of transcription-1 (STAT-1)-inducible inflammatory genes in interferon-γ-stimulated macrophages. J Pharmacol Sci. 2010;112:405-14
22. Ogbechie-Godec OA, Elbuluk N. Melasma: an up-to-date comprehensive review. Dermatol Ther (Heidelb). 2017;7:305-18
23. Shaikh ZI, Mashood AA. Treatment of refractory melasma with combination of topical 5% magnesium ascorbyl phosphate and fluorescent pulsed light in Asian patients. Int J Dermatol. 2014;53:93-9
24. Maeda K, Fukuda M. Arbutin: mechanism of its depigmenting action in human melanocyte culture. J Pharmacol Exp Ther. 1996;276:765-9
25. Dostmann WR, Taylor SS, Genieser HG. et al. Probing the cyclic nucleotide binding sites of cAMP-dependent protein kinases I and II with analogs of adenosine 3',5'-cyclic phosphorothioates. J Biol Chem. 1990;265:10484-91
26. Engh RA, Girod A, Kinzel V. et al. Crystal structures of catalytic subunit of cAMP-dependent protein kinase in complex with isoquinolinesulfonyl protein kinase inhibitors H7, H8, and H89: structural implications for selectivity. J Biol Chem. 1996;271:26157-64
27. Shibahara S, Takeda K, Yasumoto K. et al. Microphthalmia-associated transcription factor (MITF): multiplicity in structure, function, and regulation. J Investig Dermatol Symp Proc. 2001;6:99-104
28. Naqvi S, Martin KJ, Arthur JS. CREB phosphorylation at Ser133 regulates transcription via distinct mechanisms downstream of cAMP and MAPK signalling. Biochem J. 2014;458:469-79
29. Takemori H, Kajimura J, Okamoto M. TORC-SIK cascade regulates CREB activity through the basic leucine zipper domain. FEBS J. 2007;274:3202-9
30. Solano F, Briganti S, Picardo M. et al. Hypopigmenting agents: an updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006;19:550-71
31. Gillbro JM, Olsson MJ. The melanogenesis and mechanisms of skin-lightening agents: existing and new approaches. Int J Cosmet Sci. 2011;33:210-21
32. Hong SD, Yoon DY, Lee S. et al. Antimelanogenic chemicals with in vivo efficacy against skin pigmentation in guinea pigs. Arch Pharm Res. 2014;37:1241-51
33. Bang S, Won KH, Moon HR. et al. Novel regulation of melanogenesis by adiponectin via the AMPK/CRTC pathway. Pigment Cell Melanoma Res. 2017;30:553-7
34. Roh E, Yun CY, Young Yun J. et al. cAMP-binding site of PKA as a molecular target of bisabolangelone against melanocyte-specific hyperpigmented disorder. J Invest Dermatol. 2013;133:1072-9
35. Shin H, Hong SD, Roh E. et al. cAMP-dependent activation of protein kinase A as a therapeutic target of skin hyperpigmentation by diphenylmethylene hydrazinecarbothioamide. Br J Pharmacol. 2015;172:3434-45
36. Roh E, Jeong IY, Shin H. et al. Downregulation of melanocyte-specific facultative melanogenesis by 4-hydroxy-3-methoxycinnamaldehyde acting as a cAMP antagonist. J Invest Dermatol. 2014;134:551-3
37. Hagiwara M, Brindle P, Harootunian A. et al. Coupling of hormonal stimulation and transcription via the cyclic AMP-responsive factor CREB is rate limited by nuclear entry of protein kinase A. Mol Cell Biol. 1993;13:4852-9
38. Wen W, Meinkoth JL, Tsien RY. et al. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463-73
39. Khaled M, Levy C, Fisher DE. Control of melanocyte differentiation by a MITF-PDE4D3 homeostatic circuit. Genes Dev. 2010;24:2276-81
Author contact
![]() Corresponding author: Youngsoo Kim, Ph.D. & Professor. College of Pharmacy, Chungbuk National University, Cheongju 28160, Republic of Korea, E-mail: youngsooac.kr, Tel: +82-43-261-2823, Fax: +82-43-268-2732
Corresponding author: Youngsoo Kim, Ph.D. & Professor. College of Pharmacy, Chungbuk National University, Cheongju 28160, Republic of Korea, E-mail: youngsooac.kr, Tel: +82-43-261-2823, Fax: +82-43-268-2732