Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(7):1808-1823. doi:10.7150/thno.23177 This issue Cite
Research Paper
Overcoming erlotinib resistance in EGFR mutation-positive lung adenocarcinomas through repression of phosphoglycerate dehydrogenase
Jiang-Kai Dong1,2,#, Hui-Min Lei1,2,#, Qian Liang1,2,#, Ya-Bin Tang1,2, Ye Zhou1,2, Yang Wang1,2, Shengzhe Zhang3, Wen-Bin Li1, Yunguang Tong4,5, Guanglei Zhuang3, Liang Zhang1,2, Hong-Zhuan Chen1,2, ![]() , Liang Zhu1,2,
, Liang Zhu1,2, ![]() , Ying Shen1,2,
, Ying Shen1,2, ![]()
1. Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China
2. Shanghai Universities Collaborative Innovation Center for Translational Medicine, Shanghai 200025, China
3. State Key Laboratory of Oncogenes and Related Genes, Renji-Med X Clinical Stem Cell Research Center, Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200127, China
4. Basic Medical College, Xinxiang Medical University, Xinxiang 453003, Henan, China
5. Department of Medicine, Cedars-Sinai Medical Center, University of California Los Angeles School of Medicine, Los Angeles, CA 90048, United States
#These authors contributed equally to the manuscript
Received 2017-10-5; Accepted 2018-1-9; Published 2018-2-12
Abstract

How to improve the efficacy and reverse the resistance to epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs), such as erlotinib, remains a major challenge in the targeted therapy of lung adenocarcinoma with EGFR-activating mutation. Phosphoglycerate dehydrogenase (PHGDH) is the key enzyme of de novo serine biosynthesis over-expressed in various types of cancer including lung cancer. Elevated PHGDH expression is correlated with a worse overall survival in clinical lung adenocarcinoma patients. Here we investigated the role of PHGDH in lung adenocarcinoma with the acquisition of resistance to erlotinib.
Methods: The necessary genes required for the acquired erlotinib resistance in lung adenocarcinoma cells were screened out by RNA-Seq analysis. Then the protein and mRNA levels of PHGDH were confirmed by immunoblotting and qRT-PCR in the erlotinib resistant cells. The effects of PHGDH inhibition or overexpression on erlotinib resistance were examined using cell culture and tumor xenograft mouse models respectively. To explore mechanism, the ROS level and DNA damage marker, γH2AX, were tested by DCFH-DA staining and immunofluorescence after PHGDH inhibition.
Results: We found that PHGDH level was significantly increased in the lung adenocarcinoma PC9ER4 and HCC827ER9 cells that acquired resistance to erlotinib. Perturbation of PHGDH by siPHGDH transfection or NCT-503, a small molecular PHGDH inhibitor, synergistically augmented the tumoricidal effect and restored sensitivity to erlotinib in cell lines and xenografts. Over-expression of PHGDH caused xenografts resistant to erlotinib. Furthermore, multiple DNA damage repair pathways related genes were changed by PHGDH depletion specifically in erlotinib resistant cells. ROS stress and DNA damage marker γH2AX were enhanced by siPHGDH and NCT-503, which was reversed by NAC.
Conclusion: Our study indicated that PHGDH inhibition has potential therapeutic value in lung adenocarcinoma with the acquired resistance to EGFR-TKIs.
Keywords: phosphoglycerate dehydrogenase, erlotinib resistance, metabolic activity, epidermal growth factor receptor
Introduction
Lung cancer, classified into non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC) subtypes, is the first leading cause of mortality with the five-year survival rate as low as 8-15%. Lung adenocarcinoma is the most common form of NSCLC, which represents about 50% of lung cancer cases [1, 2]. Epidermal growth factor receptor (EGFR) is a predominant driver oncogene and therapeutic target mutated in higher frequencies at about 60% of lung adenocarcinoma in East Asian countries, than at 35% in Western Caucasian population.
The first generation of epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs), such as erlotinib, gefitinib and afatinib, have been shown remarkable clinical benefit and consequently been approved as first-line precision therapies in advanced-stage NSCLC, especially in lung adenocarcinoma patients harboring somatic activating EGFR mutations mainly including the deletion of exon 19 (Del19) and the L858R missense mutation of exon 21 [3]. However, even if it is effective in the early treatment, the develop of drug resistance is inevitable after 4 to 12 months therapy then leads to tumor recurrence, which is obstacle to the clinical application of EGFR-TKIs targeted therapy in EGFR mutation-positive lung adenocarcinoma.
Tremendous efforts have been made to discover multiple mechanisms of resistance to EGFR-targeted therapies, including but not limited to a secondary mutation in EGFR (encoding a p.T790M alteration), bypass signaling caused by c-MET/HGF or HER2 amplification, and histological transformation NSCLC to SCLC and epithelial-mesenchymal transition (EMT) [4, 5]. Unfortunately, the intrinsic mechanisms of EGFR-TKIs resistance remain unclear for up to 30% of lung adenocarcinoma patients.
Along this line, multiple inhibitors targeted the key effectors responsible for the acquired resistance to EGFR-TKIs have been proposed. The 3rd generation of EGFR-TKIs such as AZD9291, has been approved for the treatment of NSCLC patients harboring EGFR T790M [6]. Unfortunately, a new resistance mutation EGFR C797S was identified from about 40% of patients after AZD9291 treatment for various periods then caused tumor relapse [7]. A selective c-MET small molecular inhibitor, INC280, combined with gefitinib treatment, has shown well-tolerated and encouraging clinical activity in EGFR-TKIs resistant NSCLC patients with MET overexpression [8]. Zakowski et al. reported disease progression of patients with stage IV lung adenocarcinoma after the treatment of erlotinib, the patients were diagnosed as SCLC by lung biopsy. There was no diminution after standard SCLC chemotherapy regimen or combined with EGFR-TKIs treatment [9]. It is necessary to develop more effective strategies to overcome the EGFR-TKIs resistance and improve their clinical outcomes.
According to well-known “Warburg effect” characterized by increased glycolysis and lactate production regardless of oxygen availability in malignant tumor cells compared with adjacent normal cells, the tumor cells with acquired drug resistance also seem to involve metabolic adaptation under the drug stress. Targeting the peculiar metabolic pathways and key enzymes might be considered as potential therapeutic strategies in tumors resistant to EGFR-TKIs. Phosphoglycerate dehydrogenase (PHGDH) is a gatekeeper of serine biosynthesis pathway by diverting the glycolytic intermediate 3-phosphoglycerate to phosphohydroxypyruvate, then contributing to the cellular redox balance, energy demand and nuclear one-carbon biosynthesis. PHGDH is over-expressed in NSCLC, breast cancer and many other types of cancer. Zhu JH et al. reported that high expression of PHGDH predicts poor prognosis in NSCLC, especially in lung adenocarcinoma [10]. Norberg lab also found that PHGDH defines a metabolic subtype in lung adenocarcinoma with poor prognosis [11].
Multiple research groups have reported PHGDH inhibitors with good anti-tumor activity. CBR-5884 is the first reported PHGDH non-competitive inhibitor by disrupting the enzyme oligomerization state, and thus specifically inhibits the proliferation of melanoma and breast cancer cells with high levels of serine synthesis activity [12]. NCT-503, another PHGDH inhibitor, has been shown to reduce the production of glucose-derived serine and the synthesis of nucleotides through decreasing the one-carbon units from glucose-derived and exogenous serine [13]. Thereby NCT-503 suppresses the growth of PHGDH-dependent MDA-MB-468 cancer cells both in vitro and in mice bearing orthotopic xenograft tumors [13]. Wang et al. reported the first PHGDH allosteric regulatory inhibitor, PKUMDL-WQ-2201, inhibits the proliferation of breast cancer cells and the tumor growth [14].
Here, we aim to explore whether PHGDH could potentially facilitate lung adenocarcinoma cells resistant to erlotinib treatment, and whether erlotinib treatment could benefit from simultaneous suppression of PHGDH.
Results
PHGDH is up-regulated in the acquired erlotinib-resistant NSCLC cells. To identify the necessary genes required for the acquired erlotinib resistance in NSCLC cells, PC9 and HCC827 cells were chronically treated with increasing concentrations of erlotinib. Treatment of all cells in dose-response assays with erlotinib demonstrated that erlotinib had IC50 values of about 25 μM for the PC9ER1, PC9ER3, PC9ER4 and HCC827ER9 cells, 500-fold to 1000-fold higher than those for their parental cells respectively (Fig. 1A, B). The SNP sequencing results confirmed that EGFR T790M mutation in exon 20 was negative in all the above erlotinib resistant cells, which indicated that the acquired resistance to erlotinib is independent of the EGFR secondary mutation in these cells (Table S1).
Acquired resistance to erlotinib requires higher PHGDH level to de novo synthesize glucose-derived serine in NSCLC cells. (A and B) Inhibition rate of cell viability in the indicated cells treated with various concentrations of erlotinib for 72h detected by CCK8 assays. (C) List of the top 13 genes up-regulated in PC9ER4 cells compared to PC9 cells. PC9ER4-s is a stable clone passaged in 5 μM erlotinib containing medium continuously. The mRNA (D) and protein (E) levels of PHGDH were determined by qRT-PCR and immunoblotting respectively. (F) Profiling of 20 amino acids consumption in the medium obtained from 72h cultured cells by LC-MS/MS. Serine is the top one amino acid expended by the erlotinib resistant cells. (G) The histogram of the serine consumed described above. (H) Intracellular serine concentration was quantified by LC-MS/MS in the cell extracts after 72h culture. (I) Concentration of serine secreted to Kreb's buffer from cells at various time-point was also detected by LC-MS/MS. (J) Intracellular serine (M+3) concentration was quantified by LC-MS/MS in the cell extracts after 6h culture. Results were shown as mean ± SEM of triplicates. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
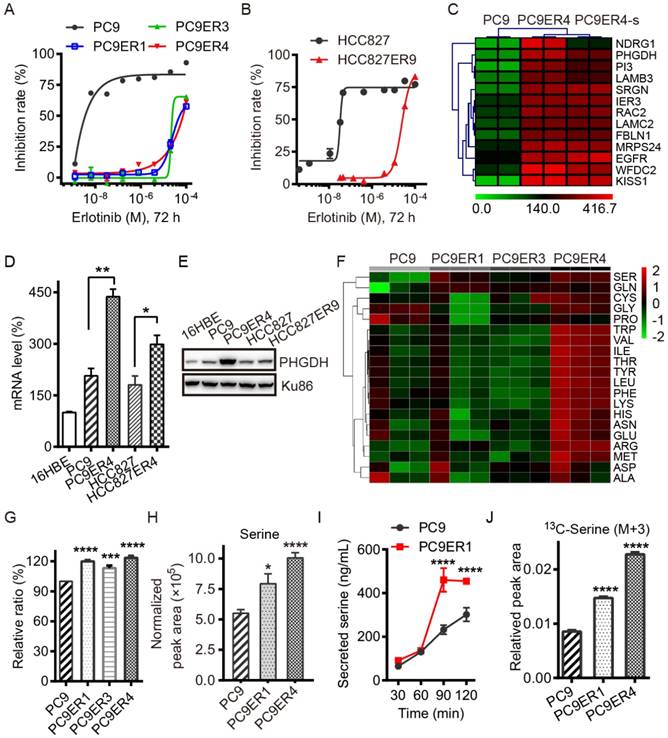
We screened out the top druggable candidates using RNA-Seq analysis, which showed that PHGDH was significantly high in the PC9ER4 cells compared with the parental PC9 cells (Fig. 1C). Thereafter, the increased PHGDH at the mRNA and protein levels was confirmed in the PC9ER1, PC9ER3, PC9ER4 and HCC827ER9 cells relative to their parental cells respectively, while weak signal was observed in normal human bronchial epithelial cells (16HBE) (Fig. 1D-E and Fig. S1).
As PHGDH is the key enzyme of de novo serine biosynthesis overexpressed in various types of cancer, we subsequently tested the level of serine in the erlotinib resistant NSCLC cells. The consumption of totally 20 amino acids was quantified in the three PC9ER cells and PC9 cells. The results of liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis showed that the serine was the top hit expended in the PC9ERs cells culture (Fig. 1F, G). The intracellular serine concentration extracted from cell lysates was determined by LC-MS/MS analysis. The result showed the significant increased serine level in PC9ER1 and PC9ER4 cells at (7.94±0.81)×105 and (10.07±0.42)×105 respectively compared to PC9 cells at (5.50±0.30)×105 normalized peak area (Fig. 1H). The concentration of serine secreted to Kreb's buffer was also higher in PC9ER1 cells with the time-course dependent manner (Fig. 1I). In order to see if the increased amounts of serine is from glucose-derived de novo serine synthesis pathway, we observed 13C incorporation from glucose into serine at 6 h (M+3, labeled on all three carbons). This analysis showed a higher flux in PC9ER1 and PC9ER4 cells compared to their parental PC9 cells (Fig. 1J). As significant up-regulation of PHGDH and serine level was typically observed in PC9ER4 cells as well as another two PC9ER cells, we observed the role of PHGDH in the PC9ER4 cells in vitro and in vivo in the following experiments.
Taken together, these results suggest that acquired resistance to erlotinib requires higher PHGDH level to de novo synthesize glucose-derived serine in NSCLC cells.
PHGDH is required for proliferation and metabolic adaptation of erlotinib resistant NSCLC cells. To minimize the possibility of off-target effects, three siPHGDH candidates, siPHGDH#2, siPHGDH#4, siPHGDH#5 were tested and knocked down the PHGDH at 20 nM for 72 h in 16HBE, PC9, PC9ER4, HCC827, HCC827ER9 cells (Fig. 2A). siPHGDH#2 with much less knockdown efficiency of PHGDH level still exerted more inhibition of clonogenic survival in PC9ER4 cells than in parental PC9 cells. When PHGDH was down-regulated to greater extent by siPHGDH#5, preferential inhibition on clonogenic survival in PC9ER4 cells (26% viable) relative to PC9 parental cells (46% viable) was observed (Fig. 2B, C). Similar inhibition on clonogenic survival preferentially in HCC827ER9 cells (23.2% and 3.67% viable) relative to HCC827 cells (57.4% and 60.3% viable) was observed by siPHGDH#4 and siPHGDH#5 transfection respectively (Fig. 2D, E). All three siPHGDHs had no significant role in 16HBE cells even at high concentration as 50 nM (Fig. S2). PHGDH depletion caused significantly more apoptosis in PC9ER4 cells (25.85% and 7.95%) than in PC9 cells (17.35% and 5.75%) by siPHGDH#4 and siPHGDH#5 transfection for 72 h respectively (Fig. 2F). Similarly, more apoptosis was induced in HCC827ER9 cells with siPHGDH#4 and siPHGDH#5 transfection (Fig. 2G).
PHGDH depletion exerts preferential toxicity in erlotinib resistant cells. (A) Knockdown efficiency of PHGDH by siPHGDH candidates was measured by immunoblotting. (B-E) Clonogenic assays were performed after cells transfected with 20 nM siPHGDHs as indicated. (F, G) Cells were transfected with 25 nM siPHGDH#4 for 72h, apoptotic cells were analyzed by annexinV-PI double staining assay. (H) Cell energy phenotype test. Knockdown of PHGDH restrained both ECAR and OCR in the PC9ER4 cells after 25 nM siPHGDHs transfection for 72 h. Error bars represent mean ± SEM of triplicates. *, p < 0.05; ****, p < 0.0001.
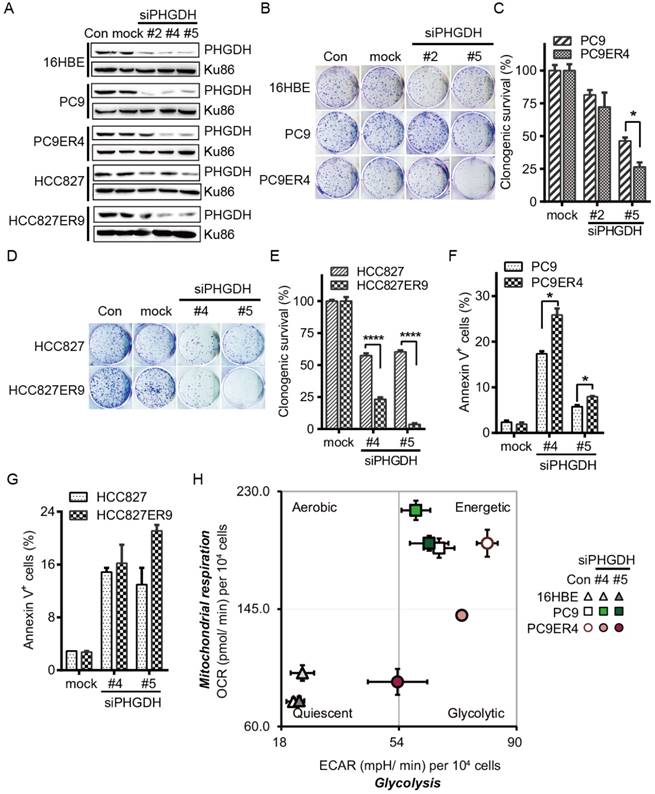
As PHGDH plays an essential role in metabolic adaptation via de novo serine biosynthesis pathway branching off the aerobic glycolytic pathway during tumorigenesis [15], we subsequently examined the effect of siPHGDHs on the overall metabolic profiles of 16HBE, PC9 and PC9ER4 cells by measuring their oxygen consumption rate (OCR), an indicator of mitochondrial respiration activity, and extracellular acidification rate (ECAR), an indicator of glycolytic activity [16]. As shown in Fig. 2H, PC9 and PC9ER4 cells stayed at bioenergetic status, while PC9ER4 cells showed higher ECAR and similar OCR level compared with PC9 cells. siPHGDH#4 and siPHGDH#5 significantly inhibited both ECAR and OCR levels in PC9ER4 cells. The OCR level was slightly increased in PC9 cells by siPHGDH treatment. Meanwhile, the normal 16HBE cells kept their quiescent status intact with siPHGDH transfection. ECAR levels were analyzed detailedly with sequential injections of glucose, oligomycin and 2-DG, which indicate the glycolytic activity including basal glycolysis, glycolytic capacity and glycolytic reserve [17]. As shown in Fig. S3A, the glycolytic activity was significantly enhanced in the indicated erlotinib resistant cells. PHGDH depletion decreased the glycolytic activity in the PC9ER4 and HCC827ER9 cells comparable to their parental cells (Fig. S3B, C). We also determined the ratio of ATP/ADP and NADH/NAD by LC-MS/MS after 72h transfection of 25 nM siPHGDH#4 and siPHGDH#5 respectively. Consistently, the ratio of ATP/ADP was significantly and preferentially decreased in PC9ER4 cells and HCC827ER9 cells (Fig. S4A). NADH/NAD ratio was significantly decreased in both PC9 and PC9ER4 cells, while it was increased in HCC827 cells and decreased in HCC827ER9 cells remarkably after siPHGDH#4 and siPHGDH#5 treatment (Fig. S4B).
All these results indicate that acquired erlotinib resistance harbors heightened requirement of PHGDH to regulate cell proliferation and metabolic adaptation in NSCLC cells.
PHGDH inhibition recovers the drug sensitivity of the erlotinib resistant cells. Since we proved that PHGDH benefits to growth of erlotinib resistant NSCLC cells, we were wondering whether PHGDH inhibition would re-sensitize the erlotinib resistant cells to erlotinib treatment. Consistently with the results of clonogenic assays described above, siPHGDH#4 inhibited the proliferation of PC9ER4 cells at around 35% inhibition of control after 72 h incubation (Fig. 3A). Combined with erlotinib treatment, the PC9ER4 cell growth was completely ablated (Fig. 3B). Similar synergistic inhibition was observed in HCC827ER9 cells (Fig. 3C).
PHGDH inhibition re-sensitizes the erlotinib resistant NSCLC cells to erlotinib treatment. (A) PC9ER4 cells was transfected with various concentrations of siPHGDH#4 for 72h. (B) PC9ER4 cells were treated with different doses of erlotinib and indicated siPHGDHs or negative siRNA (mock) at 5 nM for 72h. PC9 cells were exposed to erlotinib with transfection medium as control. (C) HCC827ER9 cells were co-treated with different doses of erlotinib and siPHGDH#4 (10 nM) or siPHGDH#5 (20 nM) for 72 h. HCC827 cells were exposed to erlotinib with transfection medium as control. (D) NCT-503, a PHGDH inhibitor, treated indicated cells for 72 h. (E and F) PC9ER4 and HCC827ER9 were co-treated with NCT-503 and erlotinib at various dosages for 72 h. The cell growth inhibition rate was analyzed by CCK8 assays after the treatment described above. Results were shown as mean ± SEM of triplicates. *, p < 0.05; **, p < 0.01.
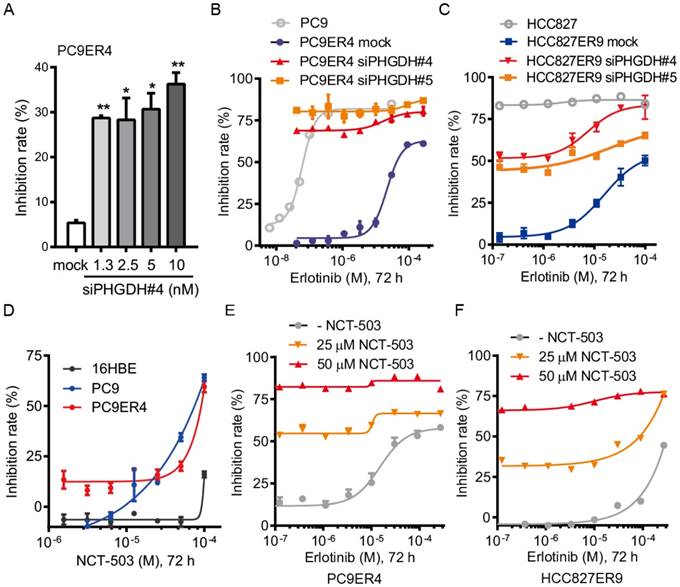
Besides the siPHGDHs knocking down PHGDH protein level, two small molecular inhibitors of PHGDH, NCT-503 and PKUMDL-WQ-2201 showed comparable effectiveness in suppressing the PC9ER4 cell growth (Fig. S5). NCT-503 inhibited the PC9 and PC9ER4 cells growth while minimally affecting the normal 16HBE cells (Fig. 3D). To test the synergistic effect of NCT-503 with erlotinib, we used NCT-503 at 25 μM and 50 μM, causing no more than 20% inhibition rate of PC9ER4 cell growth individually. As shown in Fig. 3E and 3F, NCT-503 significantly up-regulated the erlotinib sensitivity of PC9ER4 and HCC827ER9 cells. These results suggest that PHGDH inhibition re-sensitizes the resistant NSCLC cells to erlotinib treatment.
Durable depletion of PHGDH augments the tumoricidal effect of erlotinib in vivo. To confirm the validity of these results in vivo, we tested the combination therapy using PC9 and HCC827 cells conditionally expressing shPHGDH#4, PC9 pT-shPHGDH#4 and HCC827 pT-shPHGDH#4, in the murine subcutaneous models. The inducible depletion of PHGDH was observed by immunoblotting with 1 μg/mL doxycycline treatment for 72 h in the PC9 pT-shPHGDH#4 and HCC827 pT-shPHGDH#4 cells respectively (Fig. S6). To get the maximum tet-on effect in vivo, the mice, dedicated in doxycycline and combination treatment groups, were fed with doxycycline containing water 2 days before tumor cell implantation. Dosage escalation of erlotinib administration, from 5 to 50 mg/kg weight once per day (mpk opd) as indicated in Fig. 4A, was initiated once the tumor sizes reached 100 mm3 after implantation and the dosage was increased depending on the tumor progression, which caused xenografts adaptively resistant to erlotinib in vivo.
Conditional depletion of PHGDH suppresses the adaptive resistance to erlotinib treatment in vivo. (A) Tumor growth curve of subcutaneous HCC827 pT-shPHGDH#4 xenografts in nude mice. The xenografts harboring mice were gavaged with dosage escalation of erlotinib, from 5 to 50 mpk opd, or drinked doxycycline (2 mg/mL) containing water, or combination of erlotinib and doxycycline. (B) Weight of resected HCC827 pT-shPHGDH#4 tumors as indicated. (C) HE and IHC staining of PHGDH. (D) Tumor growth curve of subcutaneous PC9 pT-shPHGDH#4 xenografts in nude mice. Nude mice bearing PC9 pT-shPHGDH#4 tumors in the flank were treated as indicated. (E) The weight of PC9 pT-shPHGDH#4 tumor mass. Mean tumor volume ± SEM are shown in 5-6 mice per group. *, p < 0.05; **, p < 0.01; ***, p < 0.001, ****, p < 0.0001.
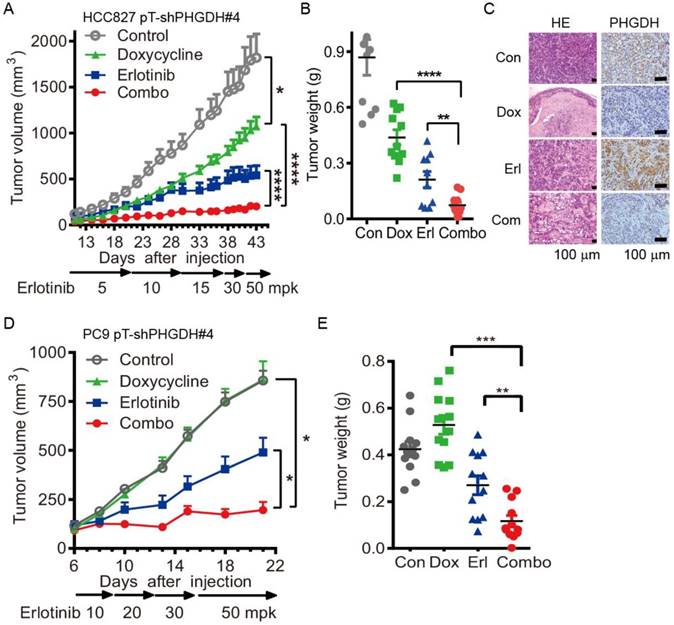
Under these conditions, the T/C ratio of doxycycline or erlotinib-treated HCC827 pT-shPHGDH#4 tumors were 51.5% and 26.6%, respectively. In comparison, tumors treated with both doxycycline and erlotinib showed a T/C ratio of 10.3% (Fig. 4A). The tumor weight was almost completely regressed after combined treatment (Fig. 4B). The hematoxylin and eosin (HE) staining results confirmed that doxycycline and erlotinib treatment induced much tumor necrosis in the xenografts (Fig. 4C). PHGDH staining by immunohistochemistry (IHC) showed that the up-regulation of PHGDH level by dose escalation of erlotinib treatment was depleted by doxycycline-induced shPHGDH#4 expression (Fig. 4C). These effects were near identical to those observed in the PC9 pT-shPHGDH#4 xenografts. The combination of doxycycline and erlotinib caused complete ablation, although doxycycline treatment had little potency in suppressing the growth of PC9 pT-shPHGDH#4 xenografts (Fig. 4D, E). Collectively, our results suggest that PHGDH inhibition augments the tumoricidal activity and eliminates adaptive resistance to erlotinib in vivo.
Over-expression of PHGDH causes NSCLC cells resistant to erlotinib in mouse xenografts. Since PHGDH inhibition restrains the adaptive resistance to erlotinib, we next tested the potency of PHGDH inhibition in restraining the acquired resistance using a mouse PC9ER4 pT-shPHGDH#4 subcutaneous model. PHGDH depletion was observed in doxycycline treated PC9ER4 pT-shPHGDH#4 cells (Fig. S6). The mice were fed with doxycycline containing water in doxycycline and doxycycline-erlotinib combined treatment group. Erlotinib treatment, from 30 mpk opd to high dosage of 75 mpk opd, was initiated with tumor sizes ≤100 mm3. The growth of PC9ER4 pT-shPHGDH#4 xenografts was modestly suppressed by high dose of erlotinib (T/C ratio of 61.75%) and doxycycline (T/C ratio of 84.66%) respectively. Combined treatment significantly dispelled the growth of PC9ER4 pT-shPHGDH#4 xenografts with T/C ratio of 23.39% (Fig. 5A). The results of tumor weight was consistent with the growth curve, which showed almost complete regression of PC9ER4 pT-shPHGDH#4 xenografts after combined treatment (Fig. 5B). These results suggest that PHGDH inhibition restrains the acquired resistance to erlotinib in vivo.
Over-expression of PHGDH causes NSCLC cells resistant to erlotinib in mouse xenografts. (A) Tumor growth curve of subcutaneous PC9ER4 pT-shPHGDH#4 xenografts in nude mice. The xenografts harboring mice were gavaged with erlotinib, from 30 to 75 mpk opd, or drinked doxycycline (2 mg/mL) containing water, or combination of erlotinib and doxycycline. (B) Weight of PC9ER4 pT-shPHGDH#4 tumors as indicated. (C) Tumor growth curve of subcutaneous HCC827 EV and HCC827 PHGDH xenografts in nude mice. The nude mice bearing tumors were dosed with erlotinib or vehicle as indicated. (D) Immunoblotting of PHGDH in the tumors, Ku86 as loading control. Mean tumor volume ± SEM are shown in 5-6 mice per group. (E) IHC staining of PHGDH and HE stain on the tissue microarray from 90 lung adenocarcinoma tissues. (F) Statistical analysis of PHGDH score according to the staining signal and intensity of PHGDH in the tumor and para-tumor sections in the (E). *, p < 0.05; **, p < 0.01; ****, p < 0.0001.
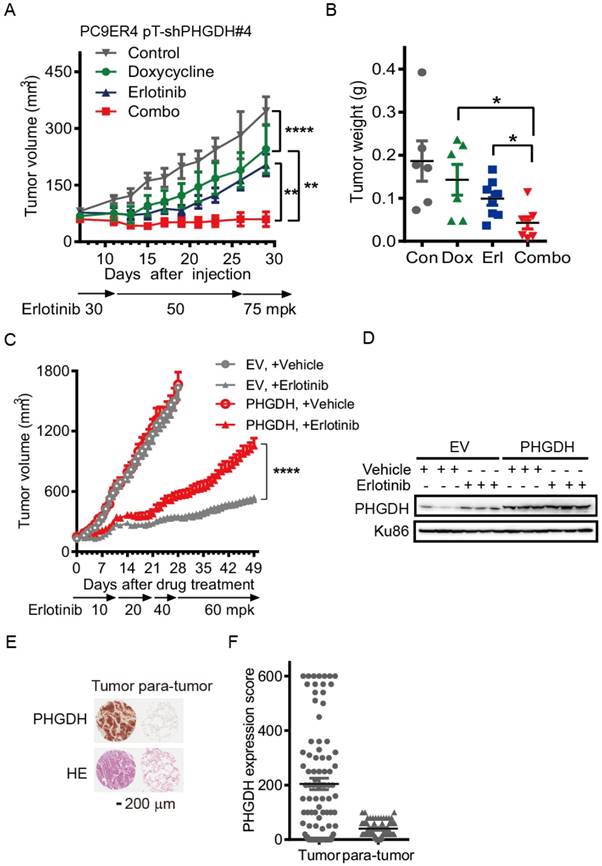
To further understand whether PHGDH accelerates erlotinib resistance during drug treatment, we over-expressed c-myc tagged PHGDH in HCC827 (HCC827-PHGDH) cells and performed the mouse subcutaneous model with dose escalation of erlotinib treatment. The expression of c-myc tagged PHGDH was tested by immunoblotting (Fig. S7). HCC827-PHGDH xenografts exhibited higher resistance to erlotinib treatment, although no significant difference of growth was observed compared to the empty vector (EV) control group under vehicle treatment (Fig. 5C). Immunoblotting analysis confirmed a significant elevation of PHGDH levels in erlotinib treated tumors compared to corresponding vehicle treated group, while over-expression of PHGDH was persistent in HCC827-PHGDH xenografts (Fig. 5D). Meanwhile, we performed patient derived tissue microarray analysis on 90 lung adenocarcinoma tissues and 90 matched adjacent normal tissues. IHC staining revealed that PHGDH expression is markedly elevated in tumor samples (Fig. 5E, F). In aggregate, high level of PHGDH accelerates erlotinib resistance in NSCLC cells.
PHGDH contributes to acquired resistance of erlotinib by regulating the transcripts associated with DNA damage repair and nucleotides metabolism in NSCLC cells. Since our results support that up-regulation of PHGDH is required to erlotinib resistance in NSCLC cells, we next performed RNA-Seq analysis to identify the transcripts that were differentially regulated in PC9ER4 cells and PC9 cells after PHGDH inhibition. Using the standard 2-fold change in expression as our threshold criterion, totally 1011 genes were specifically changed in PC9ER4 cells while 404 genes were commonly changed in PC9 and PC9ER4 cells (Fig. 6A). Bio-informative analysis data showed that the 1011 genes were closely associated with multiple DNA damage repair pathways including mismatch repair, nucleotide excision repair, homologous recombination and base excision repair, besides nucleotides metabolism including pyrimidine and purine (Fig. 6B). We then found that the level of γH2AX was selectively and extremely enhanced in PC9ER4 cells after siPHGDH#4 72h transfection, which indicates that PHGDH inhibition induces extensive DNA damage in erlotinib resistant cells (Fig. 6C). The accumulation of γH2AX foci was also significantly increased in PC9ER4 cells after siPHGDH#4 and siPHGDH#5 treatment (Fig. 6D).
PHGDH contributes to acquired resistance of erlotinib by regulating the transcripts associated with DNA damage repair and nucleotides metabolism in NSCLC cells. (A) RNA-Seq analysis. The transcripts were differentially regulated in PC9ER4 cells and PC9 cells after 20 nM siPHGDH#4 transfection for 72 h. (B) The KEGG pathway cluster analysis based on the 1011 genes significantly changed in PC9ER4 cells as described in (A). (C) Immunoblotting of γH2AX in the indicated cells after 20 nM siPHGDH#4 transfection for 72 h. M, negative siRNA as mock control; #4, siPHGDH#4. (D) Representative immunofluorescence staining. PC9ER4 cells and PC9 parental cells were stained for γH2AX after 72h transfection of 25 nM siPHGDH#4 and siPHGDH#5 or mock control. ×200 magnification. (E) PC9ER4 cells were transfected with 25 nM siPHGDH#4 and siPHGDH#5 for 36 h followed by 10 mM NAC for 6 h. The level of ROS was analyzed by DCFH-DA staining. (F) Immunoblotting of γH2AX and PHGDH after cells were treated by siPHGDH#4 and siPHGDH#5 for 72 h followed by NAC (10 mM) for 1 h. β-actin as loading control. (G) PC9ER4 cells were treated with NCT-503 (50 μM) for 1 h or pre-treated with NAC (10 mM) for 1 h, then the level of ROS was analyzed by DCFH-DA staining. (H) Immunoblotting of γH2AX after cells were treated as described in (G), Ku86 as loading control. (I) GSH/GSSG ratio of PC9 and PC9ER4 cells after treatment of NCT-503 (50 μM) for 1 h. Results were shown in mean ± SEM of triplicates. *, p < 0.05; **, p < 0.01; ****, p < 0.0001.
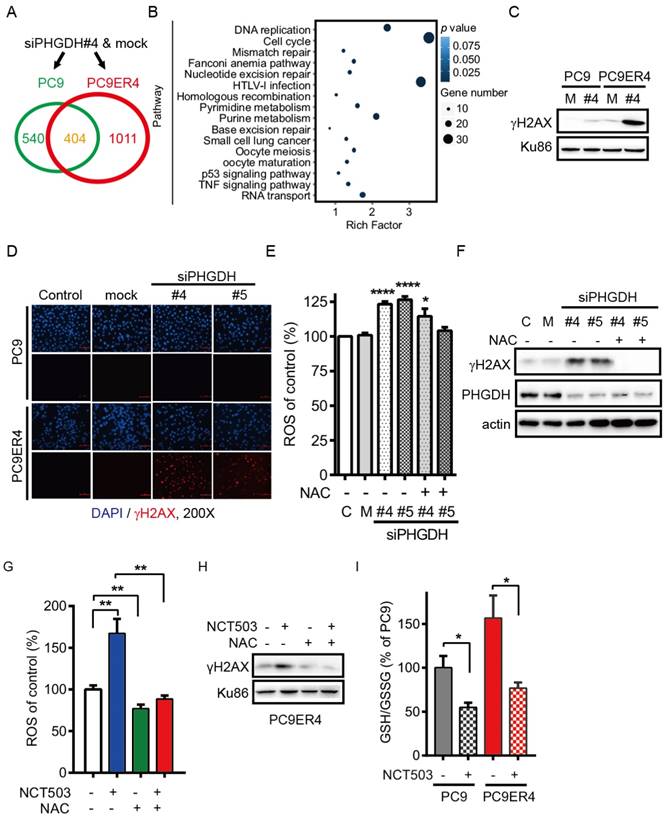
Previous reports showed that elevated intracellular levels of reactive oxygen species (ROS) causes damage to DNA, proteins and lipids [18]. GSH derived from PHGDH-mediated serine biosynthesis pathway counteracts chemotherapy-induced formation of ROS [19, 20]. Accordingly, we hypothesized that PHGDH is required for cellular redox balance and activation of adaptive response to continuous exposure to erlotinib. Consistent with our hypothesis, PHGDH depletion caused a high level of ROS by siPHGDH#4 and siPHGDH#5 treatment in PC9ER4 cells, which was reversed in the present of N-acetyl-l-cysteine (NAC), a ROS scavenger (Fig. 6E). Correspondingly, the up-regulation of γH2AX induced by both siPHGDHs was eliminated by NAC treatment (Fig. 6F). PHGDH inhibition by NCT503 also induced high level of ROS and γH2AX accumulation, which could be diminished by NAC treatment (Fig. 6G, H). The ratio of reduced glutathione/oxidized glutathione (GSH/GSSG) was higher in PC9ER4 cells compared with PC9 cells without drug treatment. NCT-503 decreased the ratio of GSH/GSSG promptly after 1h treatment in PC9 and PC9ER4 cells (Fig. 6I). These results suggest that PHGDH contributes to suppressing ROS and extensive DNA damage accumulation, resulting from serine derived GSH/GSSG status, during acquired resistance to erlotinib in NSCLC cells.
Discussion
Metabolism reprogramming has taken center stage as an emerging hallmark of cancer to sustain cell survival and rapid proliferation. Moreover, resistance to targeted therapies seems to involve dis-regulated metabolic pathways in tumor cells [21]. Here, we identified the PHGDH as a novel metabolic regulator of erlotinib resistance. Expression of PHGDH was up-regulated in the acquired erlotinib resistant cells. PHGDH inhibition by RNAi suppressed colony formation and induced cell apoptosis to a greater extent in erlotinib resistant cells than in their parental cells, which provided the evidence that acquired resistance to erlotinib retains oncogenic stress that requiring PHGDH compensation. Moreover, durable PHGDH depletion significantly augmented the tumoricidal effect of erlotinib and reversed the acquired resistance in vivo. Thus, co-targeting EGFR and PHGDH provides the opportunity for both prevention and treatment of TKIs resistance, which is a more effective combination strategy than EGFR-TKIs monotherapy.
Our previous study discovered that adaptive and acquired resistance to erlotinib treatment converged on the feedback reactivation of MAPK pathway, by MET receptor and CRAF or NRAS amplification respectively [22]. Metabolic and transcriptional profiling revealed downregulation of pyruvate dehydrogenase kinase 4 (PDK4) contributes to a glycolysis to oxidative phosphorylation (OXPHOS) shift, causing EMT and promoting acquired resistance to erlotinib in EGFR mutant lung cancer cells [23]. Thiagarajan et al. reported that cells undergoing early adaptive drug escape are in proliferative-metabolic quiescent, with enhanced EMT-ness and stem cell signaling, exhibiting global bioenergetics suppression including reverse Warburg effect [24].
Here, we found that the aerobic glycolysis was significantly elevated in the erlotinib resistant PC9ER1, PC9ER4 and HCC827ER9 cells compared with their parental PC9 and HCC827 cells respectively (Fig. S3A). PHGDH deletion by siPHGDHs led to significantly decreased metabolic activity in the erlotinib resistant cells. Conditional suppression of PHGDH expression by doxycycline treatment impaired tumor growth and combated erlotinib resistance, not only in the early adaptive resistant models established in PC9 pT-shPHGDH and HCC827 pT-shPHGDH xenografts exposed to increasing dose of erlotinib treatment, but also in the acquired resistant models in PC9ER4 pT-shPHGDH and HCC827ER9 pT-shPHGDH xenografts. Accordingly, PHGDH plays a common and central role like a performer for metabolic adaption in erlotinib resistance no matter different cells harboring multiple resistance mechanisms.
The mechanisms by which PHGDH regulates erlotinib resistance is not completely clear. PHGDH catalyzes the first, rate-limiting step in the serine biosynthetic pathway derived from glycolysis, which provides the necessary intermediates for the following processes like folate pool, amino acid and lipid biosynthesis. In addition, PHGDH amplification diverts fluxes from 3PG out of glycolysis confers several advantages for cell growth and the development of human cancer. These include limiting ATP production, oxidation of 3PG and generation of nicotinamide adenine dinucleotide phosphate (NADPH) and GSH to maintain appropriate cellular redox status, and the production of α-ketoglutarate (α-KG), a tricarboxylic acid (TCA) cycle intermediate, from glutamate, all of which are reported to benefit cell growth through multiple mechanisms [25].
Possemato and colleagues conducted an in vivo shRNAs library targeting 133 metabolic enzymes and metabolite transporters screen and identified PHGDH necessary for the growth of breast cancer xenografts with PHGDH amplification in mice, mainly dependent on the α-KG production for TCA cycle [26]. Zhang X and Bai W reported that the triple-negative breast cancer cells with PHGDH overexpression exposed to doxorubicin underwent metabolic remodeling, resulting in increased glucose flux for serine synthesis, then converted to GSH which countered doxorubicin induced formation of ROS. Repression of PHGDH caused high ROS level and increased doxorubicin sensitivity [19]. Repression of PHGDH by siPHGDHs also inhibited the generation of NADPH in one-carbon metabolism pathway for folate pool, then destroyed the cellular redox balance, induced cell apoptosis, and ultimately inhibited the enrichment breast cancer stem cell and lung metastasis [27].
Our findings revealed that decreasing PHGDH expression by siPHGDHs led to significantly increased γH2AX level, a marker of DNA damage, in erlotinib resistant cells. Consistently, NCT-503, a PHGDH inhibitor, treatment for 1 h immediately decreased GSH production, activated ROS accumulation that resulted in high level of γH2AX, which was reversed by the pretreatment of antioxidant NAC in PC9ER4 cells. Taken together, these findings point to a key role for the PHGDH in erlotinib resistance probably through enhancing cellular resistance to oxidative stress partially via increasing GSH production during cell metabolic adaption.
Additionally, the PHGDH loss by siPHGDHs impaired the proliferation of erlotinib resistant cells more effectively compared to the parental erlotinib sensitive cells cultured in the complete medium with adequately exogenous serine, whereas PHGDH inhibitors, like NCT-503 and PKUMDL-WQ-2201, didn't show such great selectivity. About 15% serine and glycine can be generated de novo from glycolysis through the oxidation of the metabolic intermediate 3-phosphoglycerate by PHGDH, while the 60-70% serine is transported into cells exogenously [28]. The observation raised the possibility that PHGDH may have serine synthesis-independent activity in addition to its role in glucose-derived serine metabolism. In support of this hypothesis, it was shown that PHGDH has promiscuous enzymatic activity and catalyzes the NADH-dependent reduction of α-KG to the oncometabolite D-2-hydroxyglutarate, which might explain its oncogenic function [29]. However, the detailed signaling pathway contributes to upregulating the level of PHGDH during erlotinib resistance still needs further investigation.
In conclusion, our study demonstrated that PHGDH inhibition has potential therapeutic value in EGFR mutation positive lung adenocarcinoma with the acquisition of resistance to EGFR-TKIs. Moreover, the inhibition reversed the sensitivity of erlotinib in the acquired resistant cells, which provided a promising clinical translation of the combination strategy to overcome the erlotinib resistance in lung adenocarcinoma.
Materials and Methods
Cell culture and reagents
Tumor cell lines were obtained from ATCC and were cultured in RPMI1640 (Invitrogen) with 10% FBS (Gemini). 16HBE and 293FT cells were cultured in DMEM (Invitrogen, CA) with 10% FBS. All cell lines were passaged at 37 °C with 5% CO2. Erlotinib (LC Laboratories, MA) was reconstituted in DMSO (Sigma-Aldrich) at a stock concentration of 10 mM. Doxycycline (Clontech) was dissolved at 1 mg/mL in PBS.
Erlotinib resistant PC9ER1, PC9ER3, PC9ER4, HCC827ER9 cells were established and maintained as described previously [22]. Briefly, the parental PC9 or HCC827 cells were grown in culture medium containing escalating dosages of erlotinib. After 5 months of passages, the remaining cells that could grow in 5 μM erlotinib were considered as resistant cells.
RNA-Seq analysis
Total RNA was extracted with RNeasy plus mini kit (Qiagen) according to the manufacturer's protocol from the indicated cell samples. RNA was subjected to RNA-Seq analysis on BGISEQ-500 system by Beijing Genomics Institute (BGI), China. In short, the RNA was sheared and reverse transcribed using random primers to obtain cDNA which was used for library construction. Sequencing was performed on prepared library [30]. All the generated raw sequencing reads were filtered to get clean reads stored as FASTQ format [31]. Bowtie2 and HISAT were used to map clean reads to reference gene and genome respectively [32, 33]. Gene expression level (FPKM) is quantified by RSEM [34]. NOISeq method was used to screen out differentially expressed genes between two groups with fold change ≥2 and diverge probability ≥0.8 [35]. Gene ontology (GO) and pathway annotation and enrichment analyses were based on the GO Database (http://www.geneontology.org/) and KEGG pathway database (http://www.genome.jp/kegg/), respectively.
LC-MS/MS analysis of amino acids
For each sample, 20 μL was added to 80 μL acetonitrile, follow by addition 300 μL cold acetonitrile (containing 0.1% formic acid). Samples were vortexed 30 s, centrifuged at 20000 × g for 10 min and the supernatant was transferred to glass HPLC vials for LC-MS/MS analysis. A 5 μL sample was loaded onto a BEH-HILIC (3 mm × 100 mm, 1.7 μm, Waters, MI) column. The mobile phase consisted of (A) 0.3% (v/v) formic acid-acetonitrile and (B) 0.3% (v/v) formic acid in water containing 5 mM ammonium formate. The acetonitrile composition was held for 2 min, ramped from 10% to 30% over 4 min, then to 50% over 3 min, held there for 2 min, subsequently returned to 10% acetonitrile in the mobile phase for column regeneration, and then held for 2 min. The total runtime for each sample was 13 min at a flow rate of 0.35 mL/min.
The study used a triple-quadrupole mass spectrometer (API 4000, AB Sciex, CA) equipped with a TurboIonSpray ionization source [an electrospray ionization interface (ESI)] operated in the positive ionization mode. The ESI ion source parameters were: temperature, 550°C; curtain gas, 35 psi; gas 1, 50 psi; gas 2, 50 psi; IonSpray voltage, 5500 V; and collision-activated dissociation (CAD) gas, 4 psi. Relative amounts of metabolites were calculated by the area under the curve using retention time, and normalized to the internal standard. Statistical difference in metabolite concentrations were examined by Pearson for repeated measurements (http://www.metaboanalyst.ca/).
LC-MS/MS analysis of intracellular serine
Sample preparation NSCLC cells were seeded in 6-well plates (1×106/well) in triplicates allowed to adhere overnight with normal media and glucose-free and media. Cells were cultured with 13C6-labelled glucose about 1 h and 6 h to detect intracellular levels of serine. All cell samples were extracted with 500 μL of MeOH : H2O mixture (8:2, v/v). Samples were centrifuged for 15 min at 20000 × g and 4 °C to settle any particulate matter. Supernatants were taken separately for LC-MS/MS analysis. For UPLC/MS samples, three biological replicates were analyzed.
LC-MS/MS analysis The analysis was performed on SCIEX ExionLC AC (UPLC System, consisting of vacuum degasser, auto-sampler with thermostat, and binary pump) coupled to SCIEX Triple Quad 6500. The separation of metabolites was carried out on Waters ACQUITY UPLC BEH Amide column (2.1 mm × 100 mm, 1.7 μm). The mobile phase consisted of a solvent A (0.1% formic acid and 10 mM ammonium formate in water) and a solvent B (0.1% formic acid and 10 mM ammonium formate in 95% acetonitrile and 5% water). A flow rate of 0.4 mL/min was used using a gradient elution of 5% A at 1.2 min , 5%-55% A between 1.2-4 min, maintained for 2 min at 55% A and back to 5% A for 6-6.3 min and a re-equilibration step to the initial solvent from 6.3 to 8 min. The metabolites were ionized using the electrospray ionization interface operating in negative ion mode. IonSpray voltage was set at -4500 V, curtain gas was kept at 35 psi, ion source temperature was 550 °C, nebulizing gas and drying gas were 55 psi. Multiple reaction monitoring (MRM) mode was used to collect mass spectral data of precursor and product ion transitions. Different fragmentor voltages were tried for each metabolite. The collision energies were also optimized with respect to individual analyte between 10 and 30 eV to maximize the analyte response. Data was acquired and processed using MultiQuant software version 3.0.1 (AB Sciex).
Determination of intracellular ATP, ADP, GSH, GSSG, NAD, NADH by LC-MS/MS
Sample preparation Cells were seeded in 6-well plates (2×105/well) in triplicates allowed to adhere overnight and cells were transfected with 25 nM siPHGDH#4 and siPHGDH#5 for 72 h to detect intracellular levels of ATP, ADP, GSH, GSSG, NAD, NADH. All cell samples were extracted with 1 mL of MeOH : H2O mixture (8:2, v/v). Samples were centrifuged for 15 minutes at 20000 × g and 4 °C to settle any particulate matter. Supernatants were taken separately for LC-MS/MS analysis. For UPLC/MS samples, three biological replicates were analyzed.
LC-MS/MS analysis The analysis was performed on Shimadzu Nexera UPLC System (consisting of vacuum degasser, auto-sampler with thermostat, and binary pump) coupled to TripleTOF™ 5600+ (AB Sciex). The separation of metabolites was carried out on Waters ACQUITY UPLC BEH Amide column (2.1 mm×100 mm, 1.7 μm). The mobile phase consisted of a solvent A (0.1% aqueous ammonia and 20 mM ammonium carbonate in water) and a solvent B (acetonitrile). A flow rate of 0.3 mL/min was used using a gradient elution of 5% A at 2 min, 5%-55% A between 2-9 min, maintained for 2 min at 55% A and the total run time was 11 min, including a 4 min equilibration time. The metabolites were ionized using the electrospray ionization interface operating in negative ion mode. IonSpray voltage was set at -4500 V, curtain gas was kept at 35 psi, ion source temperature was 500 °C, nebulizing gas and drying gas were 55 psi. The collision energies were optimized with respect to analyte between 15 and 45 eV to maximize the analyte response. Data was acquired and processed using MultiQuant software version 2.0 (AB Sciex).
Immunoblotting
Cells and tissue sections were lysed in the radio-immunoprecipitation assay (RIPA) buffer containing 1mM PMSF (Beyotime, China), then quantified by BCA Protein Assay Kit (Thermo). Equal amounts of proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electro-transferred to nitrocellulose membranes (Millipore). Membranes were blocked with TBS buffer containing 5% nonfat milk and 0.1% Tween 20 and incubated overnight at 4 °C with primary antibodies including PHGDH (Santa Cruz), γH2AX (Millipore), actin (Santa Cruz) and Ku86 (Santa Cruz) respectively. Ku86 or actin was used as an internal control for loading control. After incubated with corresponding secondary antibodies (Santa Cruz), the immunoblots were scanned with ECL (Thermo).
Reverse transcription and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cells with TRIzol reagent (Invitrogen) according to the manufacturer's instruction. Reverse transcription was carried out using a RevertAid First Strand cDNA Synthesis Kit (Invitrogen, CA). qRT-PCR was performed using LightCycler 480 II system (Roche, Switzerland). 20 μL reaction mixtures contained of SYBR Premix Ex Taq, 6 pmol of each primer, and cDNA. Primers for PHGDH were: 5′-TTC AGT CAC ATG CTG CTT CC-3′ (forward) and 5′-GGC TGC TGT CCT ACC AGA CT-3′ (reverse). GAPDH primers were used as internal controls: 5′-GTG AAG GTC GGA GTC AAC G-3′ (forward) and as 5′-TGA GGT CAA TGA AGG GGT C -3′ (reverse). The PCR program was as follows: denaturation at 96°C for 3 min 25 cycles of 96°C for 30 s, 58°C for 30 s, and 72 °C for 30 s followed by a 72 °C elongation step for 5 min. Relative quantification was performed by the ΔΔCT method.
Cell viability assays
Cells were seeded in a 96-well plate at a density of 5000 cells per well. 10-12 h later, siPHGDHs, different dosage of drugs or vehicle control were added to achieve indicated concentration and incubated for 72 h. Cell viability was measured by CCK8 assays (Dojindo, Japan) according to the instruction. The optical density (OD) was measured at 450 nm using a microplate reader, and the inhibition rate of cell growth was calculated as [1- (ODdrug treated-ODblank)/(ODvehicle control-ODblank)] × 100%.
RNA interference and clonogenic assay
PHGDH knockdown was performed by transfecting cells with independent siRNA candidates, siPHGDH#2, siPHGDH#4 and siPHGDH#5 (GenePharma, China) using Lipofectamine 3000 reagent (Invitrogen, CA) following the protocol, negative siRNA (GenePharma) as mock control. The sense sequences of siPHGDHs were as following: siPHGDH#2 (UGC CGG AAG AUC UUG CAA GTT), siPHGDH#4 (GGG AAC AGA GCU GAA UGG ATT), siPHGDH#5 (CUG ACC CUG UAG UAC AGC ATT). PHGDH expression were then detected by immunoblotting after 72 h transfection.
The transfected cells were replated on the 6-well plates at 500 cells/well, and cultured in complete medium for about 2 weeks. Cells were then stained with crystal violet and individual colonies (>50 cells) were counted.
Inducible TRIPZ lentiviral shPHGDH#4 transfection
pGIPZ shPHGDH#4, which the sense sequence was GGGAACAGAGCTGAATGGA, derived from CCSB-Broad Lentiviral Expression Library (GE, CA) was cloned into inducible TRIPZ lentiviral shRNA expression vector (Dharmacon), named pT-shPHGDH#4, according to the manufacture's instruction. Lentiviral particles were packaged with pMD2G and psPAX2 vectors (Addgene) in 293FT cells, then added to the indicated cells. After 48 hours the cells were incubated in complete medium containing puromycin (2 μg/mL) for 3 days in order to select transfacted cells. The knockdown efficiency of PHGDH was analyzed after 1 μM doxycycline treated cells for 72 h by immunoblotting.
PHGDH over-expression
Lentiviral c-myc tagged PHGDH expression clone (GeneCopoeia, MD) was packaged with pMD2G and psPAX2 vectors (Addgene) in 293FT cells, empty vector (EV) as negative transfection control. The lentiviral particles were transfected into HCC827cells and selected the positive clones by puromycin. The overexpression of PHGDH was analyzed by immunoblotting.
hematoxylin-eosin (HE) staining and immunohistochemistry (IHC)
The mice xenografts were formalin-fixed, paraffin embedded, then cut into 4 μm-thick sections for HE staining and IHC analysis. Briefly, deparaffinized slides were baked and passed through graded alcohols, then incubated for 5 min in Harry's hematoxylin before counterstained in 0.5% eosin for 1 min. For IHC, after antigen retrieval, the slides were incubated with PHGDH antibody (Santa Cruz) over 24 h at 4 °C followed by secondary antibody for30 min at 25 °C. Slides were then counterstained with hematoxylin, dehydrated, and cover slipped using mounting solution. Commercial tissue microarray (HLug-Ade180Sur-01, Outdo Biotech, China) including 90 cases of lung adenocarcinoma was used to analyze the PHGDH level in clinical samples by integrating intensity and distribution. Intensity fell into categories of 0, 1+, 2+, 3+, denoting no, weak, moderate and strong staining, respectively. Distribution of staining was referred as the percentage of positive tumor cells (0% to 100%). The final PHGDH expression score was obtained by multiplying two variables together.
Reactive oxygen species (ROS) generation assay
The ROS levels in the cells of the control and treatment groups were determined by the Reactive Oxygen Species Assay Kit (Beyotime, China) according to the manufacturer's instructions. PC9ER4 cells were plated in triplicates (1 × 105 cells/well) and treated with indicated drugs. Cells were stimulated with medium containing 10 μM DCFH-DA for 20 min at 37 °C and washed by serum-free medium. The fluorescence intensity was examined at an excitation wavelength of 498 nm and an emission wavelength of 522 nm byAccuri C6 flow cytometry (Becton Dickinson, NJ). The mean fluorescent intensity (MFI) of 20,000 cells was analyzed using triplicates for each experimental condition.
GSH/GSSG ratio
Cells were suspended at a density of 2.5 × 106 cells/mL in triplicates and dosed with indicated drugs. Cells were lysed in 10 mM HCl by two freeze/thaw cycles. Proteins were precipitated by addition of 1% 5-sulfosalicylic acid and pelleted by centrifugation at 8000 × g, 4 °C for 10 min. The ratio of reduced to oxidized glutathione (GSH/GSSG) in the supernatant was measured using the quantification kit for oxidized and reduced glutathione (Beyotime) according to the manufacturer's instructions.
Immunofluorescence staining
The cells at logarithmic growth phase were seeded onto the gelatin pre-coated coverslips in the 6-well plates. After 72 h transfection of siPHGDH#4 and siPHGDH#5, cells were then fixed with 4% para-formaldehyde for 20 min, blocked in 2% BSA/PBS for 30 min, and incubated in primary anti-γH2AX antibody (Millipore) overnight at 4 °C. Cells were washed three times in 2% BSA/ PBS, incubated in Alexa Fluor 594 secondary antibody (Invitrogen) for 1 h at room temperature. DAPI was added to stain nuclei. Cells were imaged on an upright fluorescence microscope (Nikon, Melville, NY).
Annexin V-PI double staining
The apoptosis of the cells was detected using FITC Annexin V apoptosis detection kit I (Dojindo) according to the instructions of the manufacturer.
Seahorse XF-96 metabolic flux analysis
The modalities of glucose metabolism in cells stimulated or not by siPHGDH for 72 h were explored using the Seahorse XF96 extracellular flux analyzer in combination with the XF glycolysis stress kit and cell energy phenotype test kit (Seahorse Biosciences, MA) according to the manufacturer's instruction.
In vivo subcutaneous xenografts
All animal studies were performed in accordance with the Animal Care and Use Rules at Shanghai Jiao Tong University Animal Care and Use Committee. Cells were injected subcutaneously into the nude mice (BALB/c nu/nu, female, 5 weeks old). Tumor volume was determined by vernier caliper and calculated based on the formula: (width)2 x length/2 every 2-3 days. When tumor size reached 100 mm3, mice were randomly allocated to four groups. For experiments involving doxycycline, mice were fed with 2 mg/mL doxycycline containing water 2 days before tumor implantation. Erlotinib was given by oral gavage once a day at indicated dosage. One group of mice was administrated with 0.5% methylcellulose and 0.2% Tween-80 as vehicle control. Relative tumor volume (RTV) was calculated by (RTV = TVt/TV0, where TV0 is the tumor volume measured when starting drug treatment). The anti-tumor effect of drug treatment was calculated by drug treated/control (T/C) ratio (T/C = RTVtreated/RTVcontrol × 100%). Mice were euthanized when tumor volume reached 2000 mm3 or tumors became ulcerated in accordance with our institutional guidelines for animal welfare and experimental conduct.
Statistical analysis
Student's t-test or one-way ANOVA with Bonferroni post-test was properly used to test statistical significance with GraphPad Prism 6.0 software (La Jolla, CA). A p-value less than 0.05 was considered statistically significant.
Abbreviations
EGFR-TKIs: epidermal growth factor receptor tyrosine kinase inhibitors; PHGDH: phosphoglycerate dehydrogenase; NSCLC: non-small cell lung cancer; SCLC: small cell lung cancer; EMT: epithelial-mesenchymal transition; 16HBE: normal human bronchial epithelial cells; LC-MS/MS: liquid chromatography tandem mass spectrometry; OCR: oxygen consumption rate; ECAR: extracellular acidification rate; mpk opd: mg/kg weight once per day; ROS: reactive oxygen species; NAC: N-acetyl-l-cysteine; GSH/GSSG: reduced glutathione/oxidized glutathione; PDK4: pyruvate dehydrogenase kinase 4; OXPHOS: oxidative phosphorylation; NADPH: nicotinamide adenine dinucleotide phosphate; α-KG: α-ketoglutarate; TCA: tricarboxylic acid; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; EV: empty vector; HE: hematoxylin and eosin; IHC: immunohistochemistry.
Acknowledgements
We are grateful to the grants from the National Natural Science Foundation of China (81773748, 81473232 and 81573018), Research Program of Shanghai Municipal Commission of Health and Family Planning (201440576), Science Foundation of Shanghai Jiao Tong University School of Medicine (14XJ10002).
Supplementary Material
Supplementary figures and tables.
Conflict of Interest Policy
The authors declare no conflict of interest.
References
1. Moro-Sibilot D, Audigier-Valette C, Merle P. et al. Non-small cell lung cancer recurrence following surgery and perioperative chemotherapy: Comparison of two chemotherapy regimens (IFCT-0702: A randomized phase 3 final results study). Lung Cancer. 2015;89:139-45
2. Zhang C, Ding XP, Zhao QN. et al. Role of alpha7-nicotinic acetylcholine receptor in nicotine-induced invasion and epithelial-to-mesenchymal transition in human non-small cell lung cancer cells. Oncotarget. 2016;7:59199-208
3. Zhou C, Wu YL, Chen G. et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735-42
4. Jotte RM, Spigel DR. Advances in molecular-based personalized non-small-cell lung cancer therapy: targeting epidermal growth factor receptor and mechanisms of resistance. Cancer Med. 2015;4:1621-32
5. Cortot AB, Janne PA. Molecular mechanisms of resistance in epidermal growth factor receptor-mutant lung adenocarcinomas. Eur Respir Rev. 2014;23:356-66
6. Yang J, Ramalingam SS, Janne PA. et al. LBA2_PR: Osimertinib (AZD9291) in pre-treated pts with T790M-positive advanced NSCLC: updated Phase 1 (P1) and pooled Phase 2 (P2) results. J Thorac Oncol. 2016;11:S152-3
7. Thress KS, Paweletz CP, Felip E. et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560-2
8. Wu YL, Kim DW, Felip E. et al. Phase (Ph) II safety and efficacy results of a single-arm ph ib/II study of capmatinib (INC280) + gefitinib in patients (pts) with EGFR-mutated (mut), cMET-positive (cMET+) non-small cell lung cancer (NSCLC). ASCO Annual Meeting: J Clin Oncol 34. 2016 (suppl; abstr 9020)
9. Zakowski MF, Ladanyi M, Kris MG. EGFR mutations in small-cell lung cancers in patients who have never smoked. N Engl J Med. 2006;355:213-5
10. Zhu J, Ma J, Wang X. et al. High Expression of PHGDH Predicts Poor Prognosis in Non-Small Cell Lung Cancer. Transl Oncol. 2016;9:592-9
11. Zhang B, Zheng A, Hydbring P. et al. PHGDH Defines a Metabolic Subtype in Lung Adenocarcinomas with Poor Prognosis. Cell Rep. 2017;19:2289-303
12. Mullarky E, Lucki NC, Beheshti Zavareh R. et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci U S A. 2016;113:1778-83
13. Pacold ME, Brimacombe KR, Chan SH. et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol. 2016;12:452-8
14. Wang Q, Liberti MV, Liu P. et al. Rational Design of Selective Allosteric Inhibitors of PHGDH and Serine Synthesis with Anti-tumor Activity. Cell Chem Biol. 2017;24:55-65
15. Ou Y, Wang SJ, Jiang L. et al. p53 Protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. J Biol Chem. 2015;290:457-66
16. Wu M, Neilson A, Swift AL. et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125-36
17. TeSlaa T, Teitell MA. Techniques to monitor glycolysis. Methods Enzymol. 2014;542:91-114
18. Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453-62
19. Zhang X, Bai W. Repression of phosphoglycerate dehydrogenase sensitizes triple-negative breast cancer to doxorubicin. Cancer Chemother Pharmacol. 2016;78:655-9
20. DeNicola GM, Chen PH, Mullarky E. et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nature genetics. 2015;47:1475-81
21. Morandi A, Indraccolo S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim Biophys Acta. 2017;1868:1-6
22. Ma P, Fu Y, Chen M. et al. Adaptive and Acquired Resistance to EGFR Inhibitors Converge on the MAPK Pathway. Theranostics. 2016;6:1232-43
23. Sun Y, Daemen A, Hatzivassiliou G. et al. Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells. Cancer Metab. 2014;2:20
24. Thiagarajan PS, Wu X, Zhang W. et al. Transcriptomic-metabolomic reprogramming in EGFR-mutant NSCLC early adaptive drug escape linking TGFbeta2-bioenergetics-mitochondrial priming. Oncotarget. 2016;7:82013-27
25. Locasale JW, Grassian AR, Melman T. et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869-74
26. Possemato R, Marks KM, Shaul YD. et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346-50
27. Samanta D, Park Y, Andrabi SA. et al. PHGDH Expression Is Required for Mitochondrial Redox Homeostasis, Breast Cancer Stem Cell Maintenance, and Lung Metastasis. Cancer Res. 2016;76:4430-42
28. Norberg E, Lako A, Chen PH. et al. Differential contribution of the mitochondrial translation pathway to the survival of diffuse large B-cell lymphoma subsets. Cell Death Differ. 2017;24:251-62
29. Fan J, Teng X, Liu L. et al. Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem Biol. 2015;10:510-6
30. Huang J, Liang X, Xuan Y. et al. A reference human genome dataset of the BGISEQ-500 sequencer. Gigascience. 2017;6:1-9
31. Cock PJ, Fields CJ, Goto N. et al. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38:1767-71
32. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357-60
33. Langmead B, Trapnell C, Pop M. et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25
34. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323
35. Tarazona S, Garcia-Alcalde F, Dopazo J. et al. Differential expression in RNA-seq: a matter of depth. Genome Res. 2011;21:2213-23
Author contact
![]() Corresponding authors: Ying Shen, Ph.D, Associate Research Fellow, Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Ph: 008621-63846590; Fax: 008621-54561329; Email: yshen0510edu.cn; Liang Zhu, Ph.D, Professor, Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Ph: 008621-63846590; Fax: 008621-54561329; Email: zhuliang17com; Hong-Zhuan Chen, Ph.D, Professor, Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Ph: 008621-63846590; Fax: 008621-54561329; Email: hongzhuan_chencom
Corresponding authors: Ying Shen, Ph.D, Associate Research Fellow, Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Ph: 008621-63846590; Fax: 008621-54561329; Email: yshen0510edu.cn; Liang Zhu, Ph.D, Professor, Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Ph: 008621-63846590; Fax: 008621-54561329; Email: zhuliang17com; Hong-Zhuan Chen, Ph.D, Professor, Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Ph: 008621-63846590; Fax: 008621-54561329; Email: hongzhuan_chencom