Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2018; 8(6):1575-1590. doi:10.7150/thno.23085 This issue Cite
Research Paper
Receptor-targeted aptamer-siRNA conjugate-directed transcriptional regulation of HIV-1
Jiehua Zhou1 ![]() , Daniel Lazar2, Haitang Li1, Xin Xia1, Sangeetha Satheesan1,4, Paige Charlins3, Denis O'Mealy2, Ramesh Akkina3, Sheena Saayman5, Marc S. Weinberg5, John J. Rossi1,4, Kevin V. Morris2
, Daniel Lazar2, Haitang Li1, Xin Xia1, Sangeetha Satheesan1,4, Paige Charlins3, Denis O'Mealy2, Ramesh Akkina3, Sheena Saayman5, Marc S. Weinberg5, John J. Rossi1,4, Kevin V. Morris2 ![]()
1. Department of Molecular and Cellular Biology, Beckman Research Institute of City of Hope, Duarte, CA.
2. Center for Gene Therapy, Beckman Research Institute of City of Hope, Duarte, CA.
3. Department of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, CO
4. Irell and Manella Graduate School of Biological Sciences, Beckman Research Institute of City of Hope, CA, USA
5. HIV Pathogenesis Research Unit; Department of Molecular Medicine and Hematology, School of Pathology; University of the Witwatersrand, South Africa
Received 2017-9-28; Accepted 2017-12-9; Published 2018-2-7
Abstract

Gene-based therapies represent a promising therapeutic paradigm for the treatment of HIV-1, as they have the potential to maintain sustained viral inhibition with reduced treatment interventions. Such an option may represent a long-term treatment alternative to highly active antiretroviral therapy.
Methods: We previously described a therapeutic approach, referred to as transcriptional gene silencing (TGS), whereby small noncoding RNAs directly inhibit the transcriptional activity of HIV-1 by targeting sites within the viral promoter, specifically the 5' long terminal repeat (LTR). TGS differs from traditional RNA interference (RNAi) in that it is characterized by concomitant silent-state epigenetic marks on histones and DNA. To deliver TGS-inducing RNAs, we developed functional RNA conjugates based on the previously reported dual function of the gp120 (A-1) aptamer conjugated to 27-mer Dicer-substrate anti-HIV-1 siRNA (dsiRNA), LTR-362.
Results: We demonstrate here that high levels of processed guide RNAs localize to the nucleus in infected T lymphoblastoid CEM cell line and primary human CD4+ T-cells. Treatment of the aptamer-siRNA conjugates induced TGS with an ~10-fold suppression of viral p24 levels as measured at day 12 post infection. To explore the silencing efficacy of aptamer-siRNA conjugates in vivo, HIV-1-infected humanized NOD/SCID/IL2 rγnull mice (hu-NSG) were treated with the aptamer-siRNA conjugates. Systemic delivery of the A-1-stick-LTR-362 27-mer siRNA conjugates suppressed HIV-1 infection and protected CD4+ T cell levels in viremia hu-NSG mice.
Principle conclusions: Collectively these data suggest that the gp120 aptamer-dsiRNA conjugate design is suitable for systemic delivery of small RNAs that can be used to suppress HIV-1.
Keywords: Aptamer, HIV-1, gp120, transcriptional gene silencing, RNAi
Introduction
Nucleic acid aptamers are emerging as bona fide therapeutics, functionally comparable to antibodies, with several distinct advantages such as small physical size, enhanced stability and immunological inertness as well as flexible structure and modular design characteristics (reviewed in [1]). Many RNA aptamers have been designed to target specific cell receptors and to also deliver small interfering RNA (siRNA) payloads (reviewed in [2, 3]). One highly effective receptor-targeted aptamer, the anti-HIV-1 gp120 aptamer A-1, directed to the HIV-1 gp120 receptor on the surface of infected cells, was shown to deliver anti-HIV-1 siRNAs selectively to HIV-1-infected cells as well as inhibit virus entry by blocking HIV-1 envelope interactions with the CD4 receptor [4-6]. These previous studies of gp120 aptamer-directed siRNA targeting of HIV-1 relied on siRNAs that were directed against the viral Tat/Rev transcripts and silenced viral expression via post-transcriptional gene silencing (PTGS). An alternative form of siRNA targeting, that is long-lasting and stable, can be established when siRNAs are directed to a gene's promoter (reviewed in [7]). This form of small RNA targeting lasts longer because the effector antisense strand of the siRNA guides epigenetic silencing complexes to the targeted promoter, inducing histone and DNA methylation and chromatin remodeling [8, 9], ultimately resulting in transcriptional gene silencing (TGS) (reviewed in [10]).
One small RNA, LTR-362, which is directed to the HIV-1 LTR specifically at the conserved NF-kB doublet and is unique to HIV-1, has been found to potently target HIV-1 and modulate TGS both in vitro [8, 11-13] and in vivo [14, 15]. We sought here to determine to what extent the LTR-362-directed siRNA, LTR-362, could be delivered to HIV-1-infected cells by the action of the previously reported gp120 aptamer A-1 and induce stable TGS of HIV-1. We report here that the gp120 aptamer can deliver functional LTR-362 siRNA and induce TGS of HIV-1 both in vitro and in vivo. This is the first example of an aptamer delivering a promoter-targeted small RNA capable of inducing transcriptional gene silencing (TGS) and has implications not only in targeting stable silencing of HIV-1 but also for the delivery of therapeutic RNAs to epigenetically silence other genes involved in disease.
Materials and Methods
Unless otherwise noted, all chemicals were purchased from Sigma-Aldrich, all restriction enzymes were obtained from New England BioLabs (NEB) and all cell culture products were purchased from GIBOC (Gibco BRL/Life Technologies, a division of Invitrogen). Sources for the other reagents were: DuraScribe T7 transcription Kit (Lucigen Corp.); Silencer siRNA Labeling Kit (ThermoFisher); Hoechst 33342 (nuclear dye for live cells) (ThermoFisher); M-MLV Reverse transcriptase, Superscript III reverse transcriptase and Random primers (ThermoFisher); Bio-Spin 30 Columns (Bio-Rad); Lipofectamine 2000 (ThermoFisher). CCRF-CEM cells were purchased from ATCC. CHO-Env Transfectants (CHO-WT gp160 cells and CHO-EE cells), HIV-1 LAV infected Jurkat E6 Cells (J1.1), HIV-1 viruses (IIIB, NL4-3 and Bal) were obtained from the AIDS Research and Reference Reagent Program.
DNA oligos, primers and siRNAs were purchased from Integrated DNA Technologies (IDT, Coralville, IA, USA).
Synthesis and formulation of aptamer-siRNA conjugates
Synthetic RNA materials (A-1-stick, stick-sense and stick-antisense RNAs) were purchased from TriLink (San Diego, CA, USA) for the in vitro validation assay and small group animal pilot test. For the big group animal test, synthetic RNA materials were chemically synthesized by the Synthetic and Biopolymer Chemistry Core in the City of Hope as described previously [4]. The synthetic A-1-stick RNA was refolded in HBS buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 2.7 mM KCl), heated to 95 °C for 3 min and then slowly cooled to 37°C. The incubation was continued at 37°C for 10 min. The Sense-stick or Antisense-stick strands were annealed to their complementary partner using the same molar amounts as the corresponding partner strand to form the stick-siRNAs. The same amount of the refolded A-1-stick was added and incubated at 37°C for 15 min in HBS buffer to form the A-1-stick-siRNA conjugates.
- A-1-stick: 5'-GGG AGG ACG AUG CGG AAU UGA GGG ACC ACG CGC UGC UUG UUG UGA UAA GCA GUU UGU CGU GAU GGC AGA CGA CUC GCC CGA XXXXXXX GUA CAU UCU AGA UAG CC-3'
- Tat/rev N-1: Antisense-Stick: 5'-UGA UGA GCU CUU CGU CGC UGU CUC CGC XXXXX GGC UAU CUA GAA UGU AC-3'; Sense strand: 5'-GCG GAG ACA GCG ACG AAG AGC UCA UCA UU-3'
- LTR362 N-1: Antisense-stick: 5'-GAA AGU CCC CAG CGG AAA GUC CCU U XXXXX GGC UAU CUA GAA UGU AC-3'; Sense strand: 5'-AAG GGA CUU UCC GCU GGG GAC UUU CUU-3'
- Scrambled N-1: Antisense-stick: 5'-UUC ACC GAA CGU CUC ACG UAG CGU U XXXXX GGC UAU CUA GAA UGU AC-3'; Sense strand: 5'-AAC GCU ACG UGA GAC GUU CGG UGA AUU-3'
The bold nucleotides indicate 2'-Flourine-modified sugars. The stick portion is underlined. This contains 2'-OMe-modified A and G and 2'-F-modified U and C. The italic X indicates the 3-carbon linker (C3) between the aptamer/siRNA and stick sequences.
Generation of aptamer and aptamer-siRNA chimeras by in vitro transcription
The design, synthesis and in vitro efficacies of the aptamer A-1 and aptamer-siRNA conjugates have been described previously [4]. Double-stranded DNA templates were directly generated by PCR, and the resulting PCR products were recovered using a QIAquick Gel Purification Kit (Qiagen, Valencia, CA). Chimera sense strands were transcribed from their PCR-generated DNA templates using the DuraScription T7 Kit (Lucigen Corp., Middleton, WI) in accordance with the manufacturer's instruction. In the transcription reaction mixture, the canonical cytidine triphosphate and uridine triphosphate were replaced with 2'-fluoro-cytidine triphosphate and 2'-fluoro-uridine triphosphate to produce RNA that is resistant to RNase degradation. The reactions were incubated at 37 °C for 6 h, and subsequently purified using Bio-Spin 30 Columns (Bio-Rad, Hercules, CA) after phenol extraction and ethanol precipitation.
Fluorescent dye-labeled RNAs were generated using the Silencer siRNA Labeling Kit (ThermoFisher, Waltham, MA) in accordance with the manufacturer's instructions.
The bold nucleotides indicate 2'-Flourine modified sugars. The sense strands of the chimeras are underlined. The italic UU is the linker between the aptamer and siRNA portions. The assembly of these chimeric constructs was described previously.
- A-1 aptamer: 5'-GGG AGG ACG AUG CGG AAU UGA GGG ACC ACG CGC UGC UUG UUG UGA UAA GCA GUU UGU CGU GAU GGC AGA CGA CUC GCC CGA-3';
- A-1-LTR362 DsiRNA chimera (N-1) sense strand: 5'-GGG AGG ACG AUG CGG AAU UGA GGG ACC ACG CGC UGC UUG UUG UGA UAA GCA GUU UGU CGU GAU GGC AGA CGA CUC GCC CGA UU AAG GGA CUU UCC GCU GGG GAC UUU CUU-3'; Antisense strand: 5'-GAA AGU CCC CAG CGG AAA GUC CCU UUU-3';
- A-1-Tat/rev DsiRNA chimera sense strand: 5'-GGG AGG ACG AUG CGG AAU UGA GGG ACC ACG CGC UGC UUG UUG UGA UAA GCA GUU UGU CGU GAU GGC AGA CGA CUC GCC CGA UU GCG GAG ACA GCG ACG AAG AGC UCA UCA-3'; Antisense strand: 5'-UGA UGA GCU CUU CGU CGC UGU CUC CGC UU-3';
- A-1-scrambed siRNA chimera sense strand: 5'-GGG AGG ACG AUG CGG AAU UGA GGG ACC ACG CGC UGC UUG UUG UGA UAA GCA GUU UGU CGU GAU GGC AGA CGA CUC GCC CGA UU ACG UGA GAC GUU CGG UGA AUU-3'; Antisense strand: 5'-UUC ACC GAA CGU CUC ACG UdTdT-3';
- BAFF-R aptamer R-1-LTR-362 DsiRNA chimera sense strand: 5'-GGG AGG ACG AUG CGG GAG GCU CAA CAA UGA UAG AGC CCG CAA UGU UGA UAG UUG UGC CCA GUC UGC AGA CGA CUC GCC CGA UU AAG GGA CUU UCC GCU GGG GAC UUU CUU-3'; Antisense strand: 5'-GAA AGU CCC CAG CGG AAA GUC CCU UUU-3';
Cell lines and cell culture
CCRF-CEM cells
Cells were purchased from ATCC. CCRF-CEM cell lines were suspension cell lines and split 1:10 once per week upon reaching confluence. They were cultured in RPMI-1640 supplemented with 10% FBS and PenStrep (100 U/mL penicillin, and 100 µg/mL streptomycin). Cells were cultured in a humidified 5% CO2 incubator at 37°C.
CHO-WT and CHO-EE cells
Cells were obtained through the AIDS Research and Reference Reagent Program and were grown in GMEM-S medium as instructed. Cells were cultured in a humidified 5% CO2 incubator at 37 °C.
HIV-1 LAV-infected Jurkat E6 Cells (J1.1)
Cells were obtained from the AIDS Research and Reference Reagent Program. J1.1 cell lines were suspension cell lines and split 1:10 once per week upon reaching confluence. They were cultured in RPMI-1640 supplemented with 2 mM L-glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin and 10% fetal bovine serum. Cells were cultured in a humidified 5% CO2 incubator at 37°C.
Ethics statement
Human fetal liver tissue was obtained from Advance Bioscience Resources (Alameda, CA), a nonprofit organization, in accordance with federal and state regulations. The vendor has its own Institutional Review Board (IRB) and is compliant with human subject protection requirements. Because these human specimens will be obtained through a third party with their own IRB, there is an overwhelming likelihood that the City of Hope IRB will grant exempt status.
The research involves blood specimens from anonymous human subjects with no identifiers to age, race, ethnicity, or gender. All human tissue specimens will be obtained from healthy, anonymous donors from third party sources. We use discarded peripheral blood from anonymous, healthy adult donors from the City of Hope Blood Donor Center (Duarte, CA), for isolation of peripheral blood mononuclear cells (PBMCs) and subsequent primary CD4+ T cell cultures. The information provided for the above was evaluated and determined to not involve human subjects research (45 CFR 46.102 (d)(f)). Therefore, it does not need to be approved nor does it need to undergo continuing review by the Institutional Review Board (IRB) in the City of Hope. IRB#/REF#: 97071 / 075546.
PBMCs
Peripheral blood mononuclear samples were obtained from healthy donors from the City of Hope National Medical Center. PBMCs were isolated from whole blood by centrifugation through a Ficoll-Hypaque solution (Histopaque-1077, Sigma). CD8+ T cells (T-cytotoxic / suppressor cells) were depleted from the PBMCs by CD8 Dynabeads (Invitrogen, CA) according to the manufacturer's instructions. CD8+ T cell-depleted PBMCs were washed twice in PBS and resuspended in culture media (RPMI 1640 with 10% FBS, PenStrep and 100 U/mL interleukin-2). Cells were cultured in a humidified 5% CO2 incubator at 37°C.
Internalization and intracellular localization studies (live-cell confocal microscopy analyses
The CHO-WT gp160 and CHO-EE cells were grown in 35 mm plates (Glass Bottom Dish, MatTek, Ashland, MA) with seeding at 0.3×106 in GMEM-S medium to allow about 70% confluence in 24 h. On the day of the experiments, cells were washed with 1 mL of prewarmed PBS and incubated with 1 mL of pre-warmed complete growth medium for 30 min at 37°C. The cells were first stained by treatment with 0.15 mg/mL Hoechst 33342 (nuclear dye for live cells, ThermoFisher) according to the manufacturer's instructions. Subsequently, Cy3-labeled RNAs at a 100 nM final concentration were added into the media and incubated for live-cell confocal microscopy in a 5% CO2 microscopy incubator at 37 °C. The images were collected every 15 min using a Zeiss LSM 510 Meta inverted 2 photon confocal microscope system under water immersion at 40× magnification.
HIV-1 challenge and p24 antigen assays
CCRF-CEM cells or human PBMCs-CD4+ cells were infected with HIV-1 IIIB or NL4-3 for 5 days (MOI 0.001). HIV-1 LAV-infected Jurkat E6 Cells (J1.1) were gently washed with 1 mL PBS three times to remove free viruses and then cultured one day for HIV-1 induction. Prior to RNA treatments the infected cells were gently washed with PBS three times to remove free virus. Next 1×106 infected cells and 1×106 uninfected cells were incubated with formulated RNA conjugates at 800 nM final concentration in 24-well plates at 37°C. The formulated RNA conjugates were added at days 1, 3 and 5. The culture supernatants were collected at different times (7 d and 10 d). The p24 antigen analyses were performed using a HIV-1 p24 Antigen Assay (Alliance HIV-1 p24 Antigen ELISA kit, PerkinElmer, CA) according to the manufacturer's instructions.
Detection of LTR-362 siRNA in nuclear and cytoplasm of HIV-1 infected cells
At 7 and 10 days after treatment of the formulated RNA conjugates, HIV-1 infected cells (CCRF-CEM or PBMCs) were collected and total RNAs were isolated with TriZol agent (ThermoFisher) according to the manufacturer's instructions. The total RNAs from nuclear and cytoplasmic fractions were isolated according to the protocol developed by David Corey's lab [16]. The siRNA quantification was performed with TaqMan MicroRNA Assay according to manufacturer's recommended protocol (Applied Biosystems). Fifty nanograms of small RNA, 0.2 µM stem-loop RT primer, RT buffer, 250 µM dNTPs, 3.33 units/mL MultiScribe reverse transcriptase (RT) and 0.25 units/mL RNase inhibitor were used in 15 µL RT reactions for 30 min at 16°C, 30 min at 42°C, and 5 min at 85°C, using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems). For real-time PCR, 1.33 µL of cDNA, 0.2 mM TaqMan Probe, 1.5 mM forward primer, 0.7 mM reverse primer, and TaqMan Universal PCR Master Mix were added in 20 µL reactions for 10 min at 95°C and 40 cycles of 15 s at 95°C and 1 min at 60°C. All real-time PCR experiments were done with an iCycler iQ system (Bio-Rad). Primers for Tat/Rev siRNA were as follows: Site I Looped RT primer: 5'-GTC GTA TCC AGT GCA GGG TCC GAG GTA TTC GCA CTG GAT ACG ACA CAG CG-3'; Site I Forward Primer: 5'-GCT GAT GAG CTC TTC GTC G-3'; Site I Reverse Primer: 5'-GTG CAG GGT CCG AGG T-3'; Site I probe primer: 5'-6-FAM-TCG CAC TGG ATA CGA CAC AGC GAC GA-BHQ1-3'. Custom Taqman small RNA assay (Assay ID: CSX0Z2N) for LTR362 siRNA detection was ordered from ThermoFisher. In this case, a synthetic 27-mer duplex RNA was used as positive control. U6 snRNA (expressed in the nucleus) and tubulin-alpha (expression in the cytoplasm) were used to monitor the fraction purity.
HIV-1 challenge with/without epigenetic repressive chemicals (Trichostatin A or 5′-Aza-2'-deoxycytidine) and p24 antigen assay by ELISA
CCRF-CEM cells were infected with HIV-1 IIIB for 5 days (MOI 0.001). Prior to RNA treatments the infected cells were gently washed with PBS three times to remove free virus. On the day of the experiments (day 1), 2.5×105 infected cells and 2.5×105 uninfected cells were mixed and then incubated with refolded experimental RNAs at 800 nM final concentration in 12-well plates at 37°C at day 1 and day 3 (twice treatment), which were accompanied by addition of the buffer (mock) or Trichostatin A (TSA) at 1 µM final concentration or 5-Aza-2'-deoxycytidine (5' AzaC) at 4 µM final concentration. The culture supernatants were collected at day 7. As control, irrelative aptamer-siRNA chimera (BAFF-R aptamer R-1-LTR-362 DsiRNA chimera) and A-1 aptamer-scrambled siRNA chimera were used here. The HIV-1 p24 antigen analyses were performed using an Alliance HIV-1 p24 Antigen ELISA kit (PerkinElmer, CA) according to the manufacturer's instructions.
Gene knockdown experiment by qRT-PCR
As previously described, CCRF-CEM cells were infected with HIV-1 IIIB for 5 days (MOI 0.001) and then treated twice with experimental RNAs at 800 nM final concentration in 12-well plates at day 1 and day 3. After 7 days of incubation, total RNA was isolated with STAT60 (TEL-TEST, Friendswood, TX, USA) according to the manufacturer's instructions. Residual DNA was digested using the DNA-free kit per the manufacturer's instructions (Ambion, CA, USA). cDNA was produced using 2 µg of total RNA. Reverse transcription was carried out using Moloney murine leukaemia virus reverse transcriptase (MMLV-RT) and random primers in a 15 µL reaction according to the manufacturer's instructions (Invitrogen, CA, USA). Expression of the TNPO3 coding RNAs was analyzed by quantitative RT-PCR using 2× iQ SyberGreen Mastermix (Bio-Rad) and specific primer sets at a final concentration of 400 nM. Gapdh expression was used for normalization of the qPCR data. Primers were as follows: HIV-1 IIIB tat/rev forward primer: 5'-GGC GTT ACT CGA CAG AGG AG-3'; HIV-1 IIIB tat/rev reverse primer: 5'-TGC TTT GAT AGA GAA GCT TGA TG-3'. HIV-1 Pol p128 forward primer: 5'-AGG GAT GGA AAG GAT CAC CAG CAA-3'; HIV-1 Pol p129 HIV reverse primer: 5'-CCC ACC TCA ACA GAT GTT GTC TCA-3'. GAPDH forward primer: 5'-CAT TGA CCT CAA CTA CAT G-3'; GAPDH reverse primer: 5'-TCT CCA TGG TGG TGA AGA C-3'.
Nuclear run-on assay with/without epigenetic repressive chemicals (Trichostatin A or 5'-Aza-2'-deoxycytidine)
As described above, CCRF-CEM cells were infected with HIV-1 IIIB for 5 days (MOI 0.001) and then treated twice with experimental RNAs at 800 nM final concentration in 12-well plates at day 1 and day 3 (twice treatment), which were accompanied by addition of the buffer (mock) or Trichostatin A (TSA) at 1 µM final concentration or 5-Aza-2'-deoxycytidine (5' AzaC) at 4 µM final concentration as described above. Nuclear run-on was performed as described in [17], although subsequent analysis was slightly modified to use biotin-targeted transcripts [18]. After 7 days of incubation, the cell pellets were collected, washed with cold PBS and re-suspended into 1.5 mL NP-40 lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40) twice for 5 min on ice. After lysis, the pellets were collected by immediate centrifugation at 231 crf for 5 min at 4 °C. The nuclear pellets were re-suspended in 200 µL glycerol storage buffer (50 mM Tris-HCl, pH 8.3, 0.1 mM EDTA, 40% glycerol, 5 mM MgCl2) and were then ready for in vitro transcription.
50 µL of nuclear pellet sample was incubated with 60 µL 2×transcription buffer (20 mM Tris-HCl, pH 8, 5 mM MgCl2, 300 mM KCl, 4 mM DTT) and 10 µL dNTP mixture (2 mM ATP, 2 mM CTP, 2 mM GTP, 1 mM biotin-16-AA-5'-UTP) at 30°C for 45 min. After DNAse and Proteinase K (10 mg/mL) treatment, the reaction was extracted by acid phenol/chloroform. The resultant biotinylated RNA transcripts were pulled down using Dynabeads (Invitrogen) according to the manufacturer's instructions. The biotinylated RNA expression was analyzed by quantitative RT-PCR. Primers were as follows: HIV-1 Pol p128 forward primer: 5'-AGG GAT GGA AAG GAT CAC CAG CAA-3'; HIV-1 Pol p129 HIV reverse primer: 5'-CCC ACC TCA ACA GAT GTT GTC TCA-3'.
Generation and characterization of humanized NOD/ SCID/IL2rγnull mice (hu-NSG) [19]
Animal care
All animal care and procedures have been performed according to protocols reviewed and approved by the City of Hope Institutional Animal Care and Use Committee (IACUC) held by the PI of this application (Dr. John Rossi, IACUC #12034). The animal care facilities at the City of Hope Animal Resources Center (ARC) provide high quality animal care consistent with the Public Health Service Policy on Humane Care and Use of Laboratory Animals, Guide for the Care and Use of Laboratory Animals, Animal Welfare Act, and other applicable state and local regulations. The vivarium is fully accredited and animal protocols are rigorously monitored for compliance. The ARC will provide all animal veterinary care, including husbandry, veterinary medical care, training, surgical facilities, and breeding. We performed all experimental procedures, according to protocols reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) and held by the lead investigator of this study.
Isolation of hematopoietic stem/progenitor cells
Human CD34+ HSCs were isolated from fetal liver tissue obtained from Advanced Bioscience Resources following regulatory guidelines. The fetal liver tissue at 16-24 weeks of gestation was digested with collagenase then filtered through a sterile 70 μm nylon mesh filter. Immunomagnetic enrichment for CD34+ cells were performed using the magnetic-activated cell sorting (MACS) system (Miltenyi Biotec), per the manufacturer's instructions, with the modification that the initial purified CD34+ population was put through a second column and washed three times with 3 mL of the supplied buffer per wash before the final elution. This additional step gave a >99% pure CD34+ population, as measured by FACS analysis using the anti-CD34 antibody.
Injection of human CD34+ HSCs in NSG mice
NOD.Cg-Prkdc scid Il2rg tm1Wj/SzJ (NOD/ SCID/IL2rγnull, NSG) mice were obtained from Jackson Laboratories. A modified intrahepatic injection technique was used for engraftment of the neonatal pups within 48 h of birth. A custom-made Hamilton 80508 syringe/needle set-up was used for the injections. The needle specifications are: 30 gauge, 51 mm long needle with a beveled edge attached to a 50 μL glass syringe. The maximum volume used for injection with this needle/ syringe was 25 μL. Animals were pre-irradiated with 100 cGy and then transplanted with 0.5-1×106 CD34+/CD90+ HSC each. 12-14 weeks after transplantation, blood was collected and the engraftment was verified using multi parameter flow cytometry analysis.
Mouse blood and tissue collection
Peripheral blood samples were collected at ~10-12 weeks of age, using retro-orbital sampling. The red blood cells were lysed using Red Cell Lysis Buffer Solution (Sigma Aldrich) and cells were washed with PBS. This was followed by blocking in FBS and further staining for FACS analyses. Tissue samples were collected at necropsy and processed immediately for cell isolation and FACS analysis. Tissue samples were manually agitated in PBS before filtering through a sterile 70 μm nylon mesh screen (Fisher Scientific) and suspension cell preparations were produced as previously described [20, 21]. Intestinal samples were processed as per the published protocol, with the modification that the mononuclear cell population was isolated after incubation in citrate buffer and collagenase enzyme for 2 h, followed by nylon mesh filtration and ficoll-hypaque gradient isolation (GE Healthcare).
Flow cytometric analysis of human leukocytes in PB and tissue
FACS analysis of human cells was performed using a FACSCalibur instrument (BD Biosciences), BD Fortessa (BD Biosciences) and FlowJo software version 8.8.6. The gating strategy performed was an initial forward scatter versus side scatter (FSC/SSC) gate to exclude debris, followed by a human CD45 gate. For analysis of lymphocyte populations in peripheral blood, a further lymphoid gate (low side scatter) was also applied to exclude cells of monocytic origin. All antibodies used were fluorochrome conjugated and human specific, and obtained from BD Biosciences: phycoerythrin (PE)-Texas Red-conjugated CD45 (clone 2D1), phycoerythrin (PE)-conjugated CD19 (clone HIB19), allophycocyanin (APC)-Alexa Flour 750-conjugated CD14 (clone MϕP9), peridinin-chlorophyll protein (PerCP)-Cy7-conjugated CD3 (clone SK7), pacific blue-conjugated CD4 (clone SK3), fluorescein isothiocyanate (FITC)-conjugated CD8 (clone HIT8a) and brilliant violet-conjugated CCR5 (2D7). Gates were set using fluorescence minus one controls, where cells were stained with all antibodies except the one of interest. Specificity was also confirmed using isotype-matched nonspecific antibodies (BD Biosciences) and with tissues from animals that had not been engrafted with human cells.
In vivo HIV-1 challenge with hu-NSG mice
A cell-free virus stock of CCR5-tropic HIV-1 clone Bal was obtained from the AIDS Research and Reference Reagent Program (ARRRP), Division of AIDS, NIAID, NIH. HIV-1 Bal virus was propagated in human peripheral blood mononuclear cells and harvested 10 days' post-infection. Virus was titrated using the Alliance HIV-1 p24 ELISA kit (PerkinElmer, Waltham, MA).
Hu-NSG mice with stable human leukocyte reconstitution (with a 20% or higher percentage of human CD45+ T cells) were infected with the HIV-1 Bal (200 ng p24/mouse) by intraperitoneal injection (i.p.) while under inhalant general anesthesia. Plasma viremia was assayed using one-step reverse transcriptase real-time PCR [22] with automated CFX96 TouchTM Real Time PCR Detection System (Bio-Rad). HIV-1 levels in peripheral blood were determined by extracting RNA from 5×105 cells using the QIAamp Viral RNa mini kit (Qiagen) and performing Taqman qPCR using a primer and probe set targeting the HIV-1 LTR region, using the TaqMan Fast Virus 1-Step Master Mix as per the manufacturer's recommendations. The limit of detection in this assay is 40 copies/mL or less, but due to dilution, the limit of detection of our assays was 500-800 copies/mL, depending on the available sample volume.
Oral administration of cART in HIV-1-infected hu-NSG mice
The infected mice with demonstrable viral infection was treated for two weeks with combinatorial antiretroviral therapy composed of drugs that block new infections, but not drugs that inhibit the viral production of infected cells. The ART regimen consisting of tenofovir disoproxil fumarate (TDF; 300 mg/capsule), emtricitabine (FTC; 200 mg/capsule) and raltegravir (RAL; 400 mg/capsule), scaled down to the equivalent mouse dosage using the appropriate conversion factor was administered in a drinking water formulation (sweetened water gel, Medidrop® Sucralose). We calculated human equivalent doses of ∼0.4 mg total dose of raltegravir, 0.1 mg total dose of emtricitabine and 2.14 mg total dose of tenofovir based on Km values of 37 and 3 for humans and mice, respectively [23]. The medication tablets were powdered thoroughly and re-suspended in the sweetened water gel formulation.
In vivo evaluation of formulated RNA conjugates in the HIV-1-infected hu-NSG mice
Hu-NSG mice with detectable HIV-1 infection were treated for 2 weeks with cART administered daily via the drinking water formulation described above. After 2 weeks of oral cART, treatment of RNA conjugates was done by intraperitoneal injection (i.p.) on the last day of week 2 with 1.0 nmol experimental RNAs (1.65-2.15 mg/kg aptamer-stick-siRNA conjugates) in a 200 µL volume, followed by another the next week. The injections were continued a weekly basis for 2 weeks. Mice were bled by retroorbital route, and the plasma viral loads and CD4+ T cell populations were assayed periodically (every two weeks).
Measurement of viral load in plasma
To quantify cell-free HIV-1 by qRT-PCR, RNA was extracted from 50-140 µL of EDTA-treated plasma with the QIAamp Viral RNA Kit (QIAGEN). cDNAs were produced with Superscript III Reverse Transcriptase (ThermoFisher) with a primer set specific for the HIV-1 LTR sequence, and qPCR was performed with the same primer set and a LTR-specific probe using Supermix UDG (Invitrogen) as described [24]. If there was no detectable viral RNA we established this as a value of 1 (100) to allow for the use of logarithmic values on the Y-axis.
Detection of CD4 level by Flow cytometry
Whole blood was collected and red blood cells were lysed as reported. FACS analysis of human cells was performed as described above using a FACSCalibur instrument (BD Biosciences) with either BD Fortessa (BD Biosciences) or FlowJo software version 8.8.6. CD4+ T-cell levels were calculated as a ratio of the entire CD3 population (CD4+CD3+: CD4-CD3+). To establish baseline CD4+ T-cell ratios, blood samples of all mice were analyzed prior to infection. Each individual mouse was bled two times prior to HIV-1 infection and averaged within treatment groups to establish a baseline CD4:CD3 level.
Detection of LTR-362 siRNA by Taqman qPCR assa
At 6 weeks after RNA conjugates treatment, blood samples were collected and total RNAs were isolated with RiboPure-Blood Kit (ThermoFisher) according to the manufacturer's instructions. The siRNA quantification was performed with TaqMan MicroRNA Assay according to manufacturer's recommended protocol (Applied Biosystems) (Assay ID: CSX0Z2N for LTR-362 siRNA) as described above.
DNA methylation analysis by ChIP and bisulfite sequencing
In order to assess the methylated state of cytosines in the HIV-1 LTR, mouse spleens were harvested and genomic DNA was isolated using the Qiagen Mini Kit. Aliquots of the isolated genomic DNA were utilized and ChIP carried out using anti-5-methylcytosine (5-mC) antibody (33D3, ab10805) for methylated CpGs (protocol described in detail in [18]) with 5' LTR primer 5'-TTTCCGCTGGGGACTTTCCAG-3' and 3' LTR primer 5'-ACTCAAGGCAAGCTTTATTGAGGC-3'. For bisulfite sequencing, ~600 ng of genomic DNA was bisulfite converted using the Qiagen EpiTect Bisulfite Kit (Qiagen - 59104), converting the genomic DNA. The resultant bisulfite-converted genomic DNA was then amplified using primers ltrR 5'-AAA TCT AAA AAA TCT CTA ATT ACC AAA ATC-3' and ltrF 5'-NNN NNN TGG TAG AAT TAT ATA TTA GGG TTA GGG ATT-3' incorporating a 6 bp sample barcode (indicated by Ns) and PCR amplicons were purified using the Qiagen PCR Kit. Sequencing libraries were prepared using the KAPA Hyper Prep Kit (Kapa Biosystems, Wilmington, Massachusetts) and sequenced on an Illumina MiSeq by the City of Hope Integrative Genomics Core, a fee-for-service facility. We demultiplexed reads using the seal package from BBDuk (BBMap - Bushnell B.; http://sourceforge.net/projects/bbmap/ (accessed 26 Sep 17)), used TrimGalore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed 26 Sep 17)) with the --rrbs option to trim paired reads, aligned them using the default parameters of Bismark [25] with no deduplication and used cgmaptools [26] to visualize the results.
Results
Design of gp120 aptamer-siRNA chimeras that suppress HIV-1 infection in vitro
In our previous studies, we demonstrated that the 2'-fluoropyrimidine-modified gp120 aptamer functions in a twofold manner: it can both (1) bind to free virus and block the interaction of gp120 and the CD4 receptor; and, (2) it can deliver a siRNA targeted to HIV-1. Two different aptamer-siRNA designs have been developed including a covalent aptamer-siRNA chimera and a noncovalent aptamer-stick-siRNA conjugate [4, 27]. In the covalent aptamer-siRNA chimera design, the aptamer is covalently fused with the sense strand of siRNA via a two-nucleotide linker (i.e., UU in the present study) through an in vitro T7 RNA polymerase-mediated transcription. The aptamer-sense strand is subsequently annealed with the antisense strand of siRNA to make a chimeric RNA conjugate. In the noncovalent aptamer-stick-siRNA design, the chemically synthetic aptamer and siRNA portions are hybridized using a GC-rich sticky bridge sequence, which provides a facile approach to conjugating various aptamers or siRNAs for combination treatment. Systemic administration of both aptamer-siRNA conjugates in vivo suppressed HIV-1 replication by several orders of magnitude and prevented the depletion of CD4 T+ cell which is the hallmark of the acute HIV-1 infection [5, 6].
Building on this technology, we therefore set out to determine to what extent the gp120 aptamer-siRNA conjugates (A-1-LTR-362 conjugates) could direct TGS of HIV-1 (shown schematically in Figure 1A). Due to the time-consuming and expensive nature of RNA chemical synthesis, the covalent aptamer-siRNA chimeras provide a cost-efficient alternative for quick synthesis and functional screening of aptamer-siRNA conjugates. In the present study, several covalent gp120 aptamer-siRNA chimeras were firstly designed and tested, including A-1-LTR-362 21 and A-1-LTR-362 27, which both target the LTR-362 site with varying length siRNAs, e.g., 21 or 27 bp (Figure 1B). Control gp120 aptamer conjugates were also developed including the A-1-Tat/rev, the gp120 aptamer conjugate, which was shown previously to repress HIV-1 in a PTGS manner in vitro and in vivo [4-6], A-1-Scr 21, a scrambled siRNA control, and R-1-LTR-362 27, an unrelated aptamer control (Figure 1B).
Aptamer-directed TGS. (A) A schematic is shown depicting gp120 aptamer conjugates containing the LTR-362 siRNA directed to transcriptionally silence HIV-1 LTR activity and inhibit HIV-1 expression. (B) The candidate gp120-aptamer siRNA chimeras and controls assessed are shown. (C) The effects of the various gp120-aptamer-siRNA chimeras and controls on HIV-1 expression in HIV-1-infected CCRF-CEM cells following one 800 nM treatment. A schematic depicting the experimental procedure is shown along with p24 values from days 3-10 post-aptamer treatment.
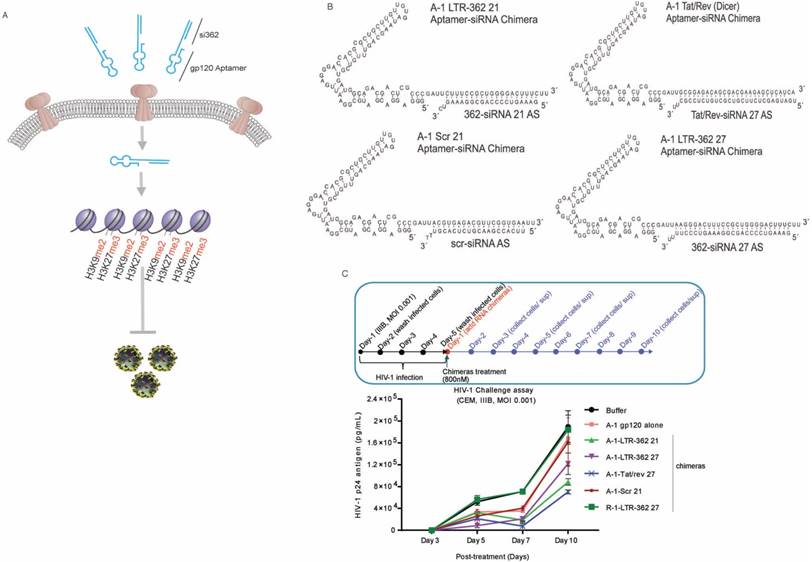
To determine the efficacy of the various gp120 aptamer-siRNA conjugates to repress HIV-1, we screened the various RNA conjugates in an in vitro HIV-1 challenge assay. In the assay, the experimental RNA chimeras were incubated with HIV-1-infected CCRF-CEM cells four days after the cells were challenged with the HIV-1 IIIB virus. At different days' post-incubation, cell-free supernatants were collected for viral p24 antigen testing. Both the A-1-Tat/rev 27 and A-1-LTR-362 27 chimeras demonstrated an ability to repress HIV-1 expression 10 days' post-treatment (Figure 1C). Notably, the A-1-Scr 21 and A-1 aptamer alone also repressed HIV-1 expression, presumably through direct interactions with free virus, an observation that has been previously reported [4]. Consistent with previous observations, these A-1-anti-HIV-1 siRNA chimeras showed stronger suppression of HIV-1 than A-1 alone or A-1-Scr control, suggesting the siRNA components were functioning along with the aptamer in the infected cells.
Gp120 aptamer directed PTGS vs. TGS in HIV-1 infected CCRF-CEM cells
To interrogate to what extent multiple dosing may have on the level of aptamer conjugate suppression, CCRF-CEM cells were infected and treated twice with the respective aptamer conjugates (Figure 2A). Following these conditions, the A-1-LTR-362 27 aptamer conjugate demonstrated the most potent repression of HIV-1 (Figure 2B).
Gp120 aptamer-siRNA 362 represses HIV-1 by transcriptional gene silencing. (A) A schematic depicting the experimental plan to test the various aptamer conjugates. (B) The effects of the various aptamer siRNA conjugates on HIV-1 expression in HIV-1-IIIB-infected CCRF-CEM cells (MOI=0.001). (C-E) Nuclear run-on analysis of LTR transcription in differentially treated aptamer-siRNA conjugates in (C) the absence of any drug and (D) the presence of TSA (1 µM) or (E) 5' AzaC (4 µM). (F-H) expression of HIV p24 in (F) untreated, TSA- or 5' AzaC-treated HIV-1-infected CCRF-CEM cultures. (G) Nuclear localization of the antisense strand of LTR-362-directed siRNAs following gp120 aptamer delivery in gp120 positive CHO-WT cells. (H) The cytoplasmic vs. nuclear distribution of siRNA Tat/rev, LTR-362 siRNA, and Scr siRNA in aptamer conjugate-treated HIV-1-infected CCRF-CEM cells. For B-F triplicate treated cells are shown with the standard deviations and (*) representing p <0.05 from a paired T-test.
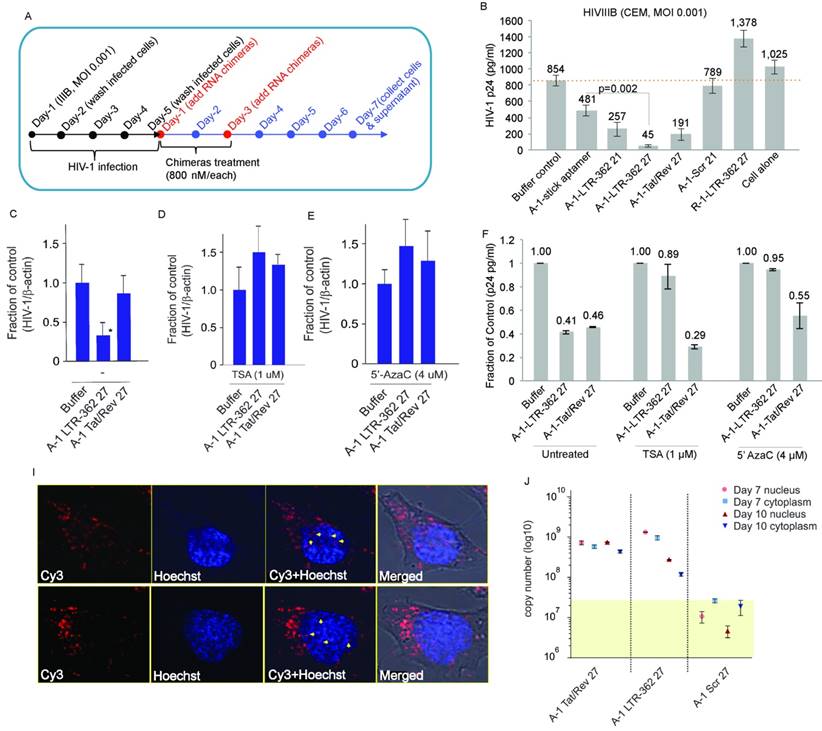
The A-1-LTR-362 27 aptamer conjugate should repress HIV-1 in a transcriptional manner, as the LTR-362 site is one of the most well studied sites for non-coding RNA directed transcriptional gene silencing (TGS) [11, 12, 28-30] (reviewed in [7]). To determine to what extent the A-1-LTR-362 27 aptamer conjugate is driving TGS, a nuclear run-on experiment was carried out in the differentially treated cells in the presence and absence of known epigenetic repressive chemicals, Trichostatin A (TSA) and 5'-Aza-2'-deoxycytidine (5' AzaC). While transcription was repressed by the action of A-1-LTR-362 27 (Figure 2C), it was lost by the addition of TSA (Figure 2D and 2F) and 5' AzaC (Figure 2E and 2F) to the treated cultures. We found that the presence of TSA or 5' AzaC compromised the ability of A-1-LTR-362 27 (aptamer-directed TGS) rather than A-1-Tat/rev 27 (aptamer-directed PTGS) to repress HIV-1 expression. These data suggest that the aptamer-siRNA conjugate, A-1-LTR-362 27 is targeting HIV-1-infected cells and is able to direct TGS, similar to previous observations with LTR-362 siRNA directly transfected [8, 12] or lentiviral vector transduced [11, 30] into HIV-1-infected cells. Additionally, supporting the mechanistic notion that A-1-LTR-362 27 is functional through RNA-directed TGS are observations that A-1-Tat/rev 27 repression is refractory to both TSA and 5' AzaC treatment (Figure 2F), as the Tat/rev siRNA is directed to the HIV-1 mRNA and not the 5' LTR promoter region.
Interestingly, the aptamer-siRNA chimera, A-1-LTR-362 27, was found to associate with the nucleus (Figure 2G) and both the A-1-LTR-362 27 and A-1-Tat/rev 27 chimeras were capable of delivering their siRNA payloads to both the cytoplasm and nucleus (Figure 2H). Collectively, these data support the notion that the gp120 aptamer can deliver siRNAs targeted to HIV-1 via gp120 receptor, with one siRNA, LTR-362 siRNA, shown to functionally inhibit viral transcription in the nucleus. This is the first experimental observation of an aptamer delivering an RNA that negatively controls gene transcription in the nucleus.
Design of gp120 aptamer-stick-siRNA conjugates that suppress HIV-1 infection in primary CD4+ cells and in vivo
The A-1-LTR-362 27 chimera demonstrated the most potent repression of HIV-1 in HIV-1-infected CCRF-CEM cells. Next, to determine the ability of the gp120 aptamer-directed TGS to repress HIV-1 in primary peripheral blood mononuclear cells (PBMCs) in vivo, noncovalent aptamer-stick-siRNA conjugates (A-1-stick-LTR-362 27) were designed and chemically synthesized in milligram scales (Figure 3A). Human PBMC-CD4+ T cells were isolated and challenged with HIV-1 virus for four days as described previously (Figure 3B). HIV-1-infected PBMC-CD4+ cells were subsequently treated three times with the chemically synthetic aptamer conjugates. Similar to previous observations in the CCRF-CEM cells culture, both the chemically synthetic A-1-stick-LTR-362 27 and A-1-stick-Tat/rev 27 aptamer conjugates were capable of repressing HIV-1 expression in primary cells infected with HIV-1 (Figures 3C).
Aptamer-stick-siRNA conjugate suppression of HIV-1 in PBMC-CD4+ T cells. (A) A schematic depicting the aptamer-stick-siRNA scheme for those aptamers developed and tested. (B) A schematic depicting the infection and treatment scheme used for PBMC-CD4+ T cells with the various aptamer-stick-siRNA conjugates. (C) Relative expression of HIV-1 in aptamer-siRNA conjugate-treated cultures at days 7 and 10. The average of triplicate treated cells is shown with the standard deviations. (D) A schematic depicting the treatment scheme for HIV-1-infected ART-treated hu-NSG mice (n = 3 per group). ART drug was removed from the drinking water of the HIV-1-infected mice at day 0 when the mice were treated with the various aptamer-stick-siRNAs. (E) The viral loads in aptamer scrambled siRNA 362 (A-1-stick-Scr)- and aptamer siRNA 362 (A-1-stick LTR-362 27)-treated mice (n = 3) at weeks 0 and 4. (F) Percentage of CD4 to CD3 in blood from the aptamer-siRNA conjugate-treated mice at weeks 0 and 4. (G) The copy number of the antisense strand for LTR-362 siRNA in A-1-stick-Scr and A-1-stick-LTR362 27 conjugates-treated mice at week 4.
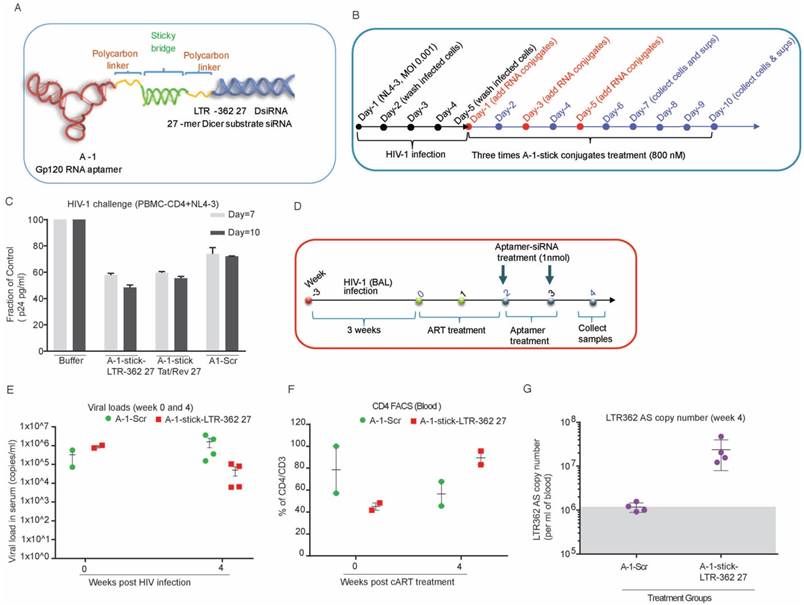
To quickly evaluate the potential efficacy of the aptamer-stick-siRNA conjugates in vivo, we tested their anti-HIV-1 ability in the antiretroviral (ART)-treated humanized NOD/ SCID/IL2rγnull (hu-NSG) mouse model in a short time scheme (Figure 3D). As shown in Figure 3E, the hu-NSG mice injected intraperitoneally with HIV-1 Bal virus (200 ng of p24 units) became viremic by 3 weeks after HIV-1 infection, with plasma viral loads averaging 106 per mL, indicating an established infection (at week 0, box and cycle as shown in Figure 3E). At 3 weeks' post-infection, these viremic hu-NSG mice were administrated daily with combinatorial antiretroviral therapy (cART) that blocks new infections, but does not inhibit the viral production of infected cells. After two weeks of treatment, cART was discontinued and was then replaced with the aptamer-stick-siRNA conjugates treatment weekly for an additional 2 weeks (at weeks 2 and 3 as shown in Figure 3D). During this treatment, viral loads and CD4+ T cell counts were monitored by quantitative RT-PCR (qRT-PCR) and flow cytometry, respectively. A sufficient repression was observed in the cART-treated viremic hu-mice within two weeks of treatment (at week 2, box and cycle as shown in Figure 3D). These mice treated with the A-1-stick-LTR-362 27 conjugate demonstrated notable suppression relative to A-1-stick-scr siRNA controls (at week 4 as shown in Figure 3E). Moreover, suppression of viremia following the A-1-stick-LTR-362 27 conjugates resulted in recovery of human CD4+ T cells in the peripheral blood (week 2 vs. week 4, box as shown in Figure 3F), indicating that HIV-1 killing of CD4+ T-cells was largely halted. As expected, a general pattern of decreased CD4+ T cell count was seen after infection in the control A-1-stick-Scr conjugate group blood (week 2 vs. week 4, cycle as shown in Figure 3F).
To validate that the A-1-stick-LTR-362 RNA conjugate delivered the LTR-362 siRNA to infected T lymphocytes, we collected PBMCs after 2 weeks of RNA treatment (at week 4 as shown in Figure 3G). Real-time TaqMan quantitative RT-PCR assay was conducted to measure small RNAs for the presence of the LTR-362 siRNA. Notably, the A-1-stick-LTR-362 27 conjugates were detectable in PBMCs from all the A-1-stick-LTR-362 conjugates at week 4 (Figure 3G), suggesting that LTR-362 siRNA is present to be internalized in HIV-1-infected T cells in vivo.
Gp120 aptamer directed PTGS vs. TGS in HIV-1-infected Hu-NSG mice
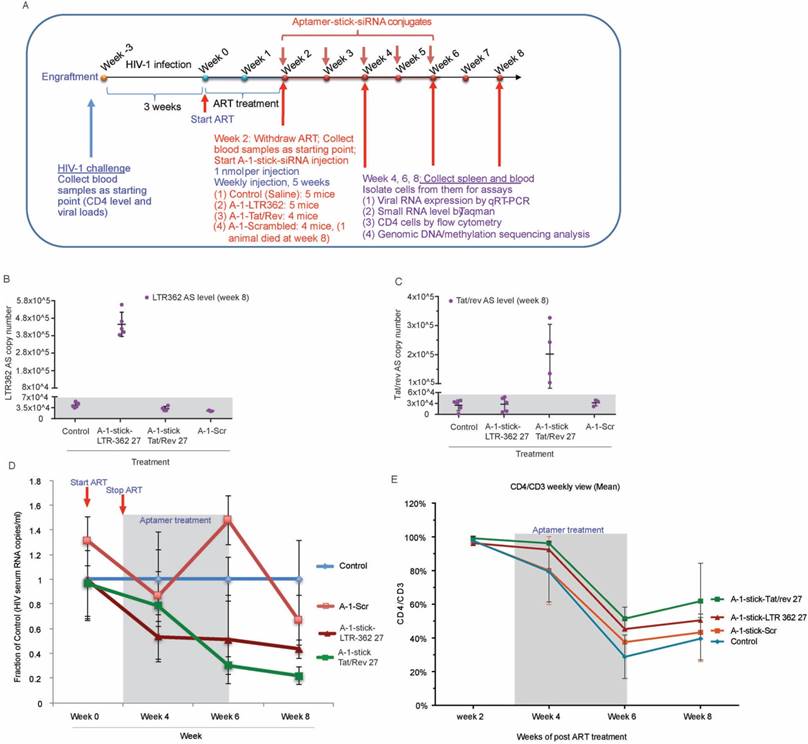
In a short duration scheme, the A-1-stick-LTR-362 27 conjugates demonstrated potent suppression of HIV-1 in vivo (Figures 3B-G). To determine the relative potency and duration of suppression for both the A-1-stick-LTR-362 27 and A-1-stick-Tat/rev 27 RNA conjugates in vivo, an extended time scheme with multiple dosing was performed with cART-treated hu-NSG mice as described above.
As shown in Figure 4A, at 3 weeks' post HIV-1 infection, the viremic hu-NSG mice were treated with oral cART regimen for 2 weeks, then ART was removed and replaced with A-1-stick-LTR-362 27 and A-1-stick-Tat/rev 27 RNA conjugate treatment, which was done by intraperitoneal injection on a weekly basis for a total of 5 weeks. Blood samples were drawn routinely at two week intervals, and the cellular and plasma fractions were separated for analysis. Consistent with our previous observation, both the LTR-362 27 mer and Tat/rev 27 mer antisense RNAs were detectable by TaqMan qRT-PCR in the PBMCs at week 8, two weeks after the last RNA conjugate injection (Figure 4B-C). Notably, both the A-1-stick-LTR-362 27 and A-1-stick-Tat/rev 27 RNA conjugates suppressed viral loads (Figure 4D). A general pattern of reduced viral loads was observed after treatment in the A-1-stick-LTR-362 27 and A-1-stick-Tat/rev 27 RNA conjugates-treated mice and this suppression persisted throughout the treatment period (weeks 4 and 6) and up to 2 weeks following the last RNA conjugates injection (week 8). Moreover, we measured CD4+ T cells in peripheral blood collected during and after RNA conjugate treatment. Compared to the untreated control group, a modest protection in CD4 T-cell depletion was observed throughout the treatment period (Figure 4E). Collectively, these data along with those presented in Figure 3, suggest that A-1-stick-LTR-362 27 can effectively repress HIV-1 in vivo. Notably, A-1-stick-LTR-362 27 did not appear to be as potent as A-1-stick-Tat/rev 27 at repressing HIV-1.
Aptamer-stick-siRNA conjugate suppression of HIV-1 in vivo. (A) A schematic depicting the treatment scheme for HIV-1-infected cART-treated hu-NSG mice. cART drug was removed from the drinking water of the HIV-1-infected mice at week 2 when the mice were treated with the various chimeric aptamer-siRNAs. (B-C) Detection of (B) LTR-362 antisense RNA and (C) Tat/rev antisense RNA in Control, A-1-stick-Scr, A-1-stick-LTR-362 27 and A-1-stick-Tat/rev 27 conjugates-treated mice at week 8. (D) The viral loads in aptamer-stick-siRNA conjugates- (A-1-stick-Scr, A-1-stick-LTR-362 27, and A-1-stick-Tat/rev 27) treated mice at weeks 0-8 as determined by qRT-PCR from plasma. (E) Percentage of CD4 to CD3 in blood from the aptamer-siRNA conjugate-treated mice at weeks 2, 4, 6 and 8.
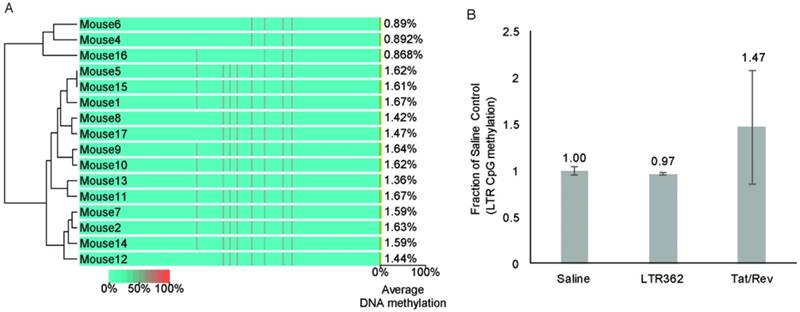
One inherent difference between A-1-stick-LTR-362 27 and A-1-stick-Tat/rev 27 is that A-1-stick-LTR-362 27 appears to direct TGS (reviewed in [7]). To determine to what extent A-1-stick-LTR-362 27 was actively modulating the epigenetic state of HIV-1 in the hu-NSG mice, genomic DNA from spleens of the differentially treated mice were assessed for CpG methylation. No apparent CpG methylation was observed in the spleens of the cART-aptamer-siRNA-treated hu-NSG mice at week 8 by either bisulfite sequencing (Figure 5A) or chromatin immunoprecipitation analysis (ChIP) (Figure 5B). Collectively, these data suggest that the observed suppression of HIV-1 in the A-1-stick-LTR-362 27 and A-1-stick-Tat/rev 27 in the cART-hu-NSG mice treated with A-1-stick-LTR-362 27 was predominantly the result of siRNA directed post-transcriptional gene silencing.
CpG methylation at the HIV-1 LTR from mouse spleens. (A) Cytosine methylation was determined at the LTR by bisulfite sequencing. Genomic DNA from mouse spleens. The groups Saline (mice 1-2, 9, 16-17), A-1-Tat/rev (mice 3-5, 15), A-1-Scrambled (mice 6-8) and A-1-LTR362 (mice 10-14) were assessed by bisulfite sequencing of the LTR. (B) Cytosine methylation was also assessed by CpG methyl ChIP. The averages of the pooled samples are shown with the standard deviations.
Discussion
RNA aptamers are promising molecules for cell receptor-specific targeting. The ability to utilize RNA aptamers as a delivery vehicle to bring siRNAs to target cells presents a unique opportunity to develop cell-targeted siRNA-regulated therapeutics [1, 31]. Previous studies have shown that the gp120 aptamer [4, 27] can functionally deliver siRNAs targeted to the Tat/Rev transcript and impair HIV-1 expression [5, 6]. This previous body of work was carried out in the Rag-hu mouse model challenged with HIV-1 CXCR4-tropic NL4-3 virus using the A-1-stick-multiplexed anti-HIV-1 siRNA conjugates targeting HIV-1 Tat/Rev or the HIV-1 dependency factors (CD4 and transportin-3 (TNPO3)), whereas the data presented here were generated in the Hu-NSG mouse model infected with HIV-1 CCR5-tropic Bal virus with cART treatment contrasting both the A-1-stick-Tat/rev 27 and A-1-stick-LTR362 27 mer. The use of a 2-week cART treatment allowed for delivery of the aptamer siRNA conjugates to those cells that were infected, as free-floating virus could be expected to also engage aptamers and “soak” aptamer conjugates away from intended cellular targets. A related higher virema level was also observed in our Hu-NSG model after 3 weeks post HIV-1 infection, indicating plasma viral loads averaging 106 per mL in hu-NSG mice vs. 105 per mL in Rag-hu mice [6], ultimately suggesting that the hu-NSG mouse model may be more susceptible to HIV-1 infection. Therefore, variation in the degree of suppression between the two studies is somewhat expected; however, our aptamer-siRNA conjugates have proven their anti-HIV-1 potency in different in vivo systems. Notably, the suppression observed in the Hu-NSG mouse model with the A-1-stick-Tat/rev 27 was not as potent as previous observations with similar aptamer-cocktailed siRNA conjugates in the Rag-hu mouse model [5, 6], suggesting that a combinatorial formulation of aptamer-siRNA conjugates may be more effective for the control of HIV-1.
To date, all previous aptamer conjugate studies utilized the A-1 aptamer conjugated to the Tat/rev siRNA to target HIV-1, a method that predominately targets the Tat/rev transcript of HIV-1 through post-transcriptional silencing. The approach outlined here utilizing aptamer conjugates with the LTR-362 siRNA targets the LTR and functionally represses HIV-1 transcription via transcriptional gene silencing [12, 14], as well as post-transcriptionally, like the Tat/rev siRNAs, by targeting those LTRs embedded in the viral mRNA. Our observation in Figure 2C-F that known epigenetic repressive chemicals (e.g., TSA and 5'AzaC) only partially attenuated the anti-HIV-1 activity of A-1-LTR-362 27 provide additional evidence that LTR-362-siRNA represses HIV-1 expression transcriptionally.
One advantage of transcriptional gene silencing is that the repression is established at the level of chromatin through epigenetic changes (reviewed in [7]). Previous studies have demonstrated that siRNA-directed transcriptional gene silencing of HIV-1 can be established in mouse models in which the effector RNAs are expressed as short hairpin RNAs targeting HIV-1 promoter regions (shPromA-JRFL) delivered by lentiviral transduction of human PBMCs [14]. The experiments shown here suggest that LTR-targeted silencing can be established by direct targeting with a cell-type specific aptamer via the cell surface receptor. However, compared to previous in vitro studies [11, 12, 29, 30, 32], the observed silencing in vivo from gp120 aptamer-directed delivery appeared to lack robust epigenetic silencing marks such as CpG methylation at the siRNA-targeted LTR. Collectively, these data suggest that aptamer conjugates can deliver promoter-targeted siRNAs to instill transcriptional gene silencing in vitro, but in vivo the observed silencing appears to function in the absence of CpG methylation and presumably a robust epigenetic effect, suggesting ultimately that a lentiviral vector-mediated delivery approach may be a more efficient means to instilling transcriptional gene silencing of the HIV-1 LTR [14]. Although one can't rule out the potential that the observed silencing is via histone methylation; indeed, histone methylation appears to occur first in TGS followed only later by DNA methylation and stable epigenetic silencing [33, 34]. Based on the observations presented here it's clear that this approach may benefit from further optimizations, such as a prolonged time scheme with additional dosing, altered cessation of cART, rational chemical modification for efficient Dicer processing, and proper conjugation for improved circulation time. Alternatively, the combination of two different targets, e.g., Tat/rev and the LTR-362, with one gp120 receptor-targeted aptamer, may prove to be a promising adjunctive therapy to treat cART refractory HIV-1-infected individuals [35].
The HIV-1-susceptible hematopoietic stem cells (HSC) engrafted-NSG (hu-NSG) mouse model was used to demonstrate proof-of-principle of the potential in vivo efficacy of aptamer-directed TGS, recapitulating the key aspects of HIV-1 infection and dynamic response to therapeutic agents in vivo. Using the cART-suppressive HSC-reconstituted hu-NSG mouse model, we attempt here to simulate conditions in human HIV-1 treatment and determine whether the replacement of aptamer-directed transcriptional gene silencing can control chronic infection upon withdrawal of antiretroviral therapy. It was noted that, as in patients taking cART, during treatment interruption there was a rapid rebound of plasma viremia to levels comparable to those detected prior to initiation of therapy. In this study, although discontinuation of cART therapy resulted in rapid viral rebound and loss of peripheral CD4+ T cells in the control group, the substitutive treatment with aptamer-siRNA conjugates substantially suppressed viral rebound and protected CD4+ T cell depletion, as shown in Figures 3E, 3F, 4D and 4E. Collectively, the observations presented here suggest that receptor-directed aptamers can deliver small RNAs that can functionally modulate HIV-1 infection as well as viral transcription, opening up an avenue to utilize nucleic acid-delivered therapeutics to target and control gene expression.
Abbreviations
HIV: human immunodeficiency virus; cART: combinatorial anti-retroviral therapy; TGS: transcripttional gene silencing; PTGS: post transcriptional gene silencing; LTR: long terminal repeat; RNAi: RNA interference; siRNA: small interfering RNA; PBMC: Peripheral blood mononuclear cell; hu-NSG mice: humanized NOD/SCID/IL2 rγnull mice; HSC: hematopoietic stem cell; TSA: Trichostatin A; 5' AzaC: 5'-Aza-2'-deoxycytidine; TDF: tenofovir disoproxil fumarate; FTC: emtricitabine; RAL: raltegravir; TNPO3: transportin-3; MOI: multiplicity of infection.
Acknowledgements
Funding & Support
This project was supported by National Institute of Health NIAID P01 AI099783-01, NIH DK104681-02 and ARC Future Fellow FT130100572 to K.V.M, NIH AI29329, HL07470 to J.J.R, and National Cancer Institute of the National Institutes of Health P30CA033572 to support City of Hope Integrative Genomics and Analytical Cytometry Core. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: CHO-WT, CHO-EE, J1.1 cells, HIV-1 IIIB, NL4-3, Bal viruses.
We would like to thank City of Hope Animal Resources Center (ARC) for the support of the animal use and care, Analytical Cytometry Core for cell sorting and technical assistance for flow cytometry analysis, and Pathology Core for the postmortem analysis of blood and tissue specimens.
Author contributions statement
J.Z., D.L., W.S.M., J.J.R., and K.V.M. conceived and designed the experiments; J.Z. conducted in vitro evaluation assays and prepared the animal IACUC protocol; J.Z. and K.V.M. wrote the main manuscript and prepared the tables/figures; D.L. performed deep sequencing analysis; H.L. conducted in vitro HIV-1 challenge assay; H.L., X.X. and S.S. performed human cell isolation, engraftment, establishment and characterization of hu-NSG mice and HIV-1-infected hu-NSG mice; P.C. and R.K., established the Rag-hu model and pre-test of HIV-1-infection; S.S. performed nuclear-run on assay; J. J. R and K.V.M. provided funding. All authors reviewed the manuscript.
Conflict of Interest
J. J. R. and J. Z. have an issued patent entitled “Cell-type specific aptamer-siRNA delivery system for HIV-1 therapy”. USPTO, No. US 8, 222, 226 B2, issued date: July 17, 2012. J.J.R., J. Z., M.S.W and K.V.M. have an issued patent entitled “Cell-specific internalizing RNA aptamers against human CCR5 and used therefore”, USPTO, No. US 9,605,266, issued date: March 28, 2017.
References
1. Zhou J, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov. 2017;16:181-202
2. Shum KT, Zhou J, Rossi JJ. Aptamer-based therapeutics: new approaches to combat human viral diseases. Pharmaceuticals (Basel). 2013;6:1507-42
3. Zhou J, Rossi JJ. Cell-type-specific, Aptamer-functionalized Agents for Targeted Disease Therapy. Mol Ther Nucleic Acids. 2014;3:e169
4. Zhou J, Swiderski P, Li H, Zhang J, Neff CP, Akkina R. et al. Selection, characterization and application of new RNA HIV gp 120 aptamers for facile delivery of Dicer substrate siRNAs into HIV infected cells. Nucleic Acids Res. 2009;37:3094-109
5. Neff CP, Zhou J, Remling L, Kuruvilla J, Zhang J, Li H. et al. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4(+) T cell decline in humanized mice. Sci Transl Med. 2011;3:66ra6
6. Zhou J, Neff CP, Swiderski P, Li H, Smith DD, Aboellail T. et al. Functional in vivo delivery of multiplexed anti-HIV-1 siRNAs via a chemically synthesized aptamer with a sticky bridge. Mol Ther. 2013;21:192-200
7. Weinberg MS, Morris KV. Transcriptional gene silencing in humans. Nucleic Acids Res. 2016(14):6505-17
8. Weinberg MS, Villeneuve LM, Ehsani A, Amarzguioui M, Aagaard L, Chen ZX. et al. The antisense strand of small interfering RNAs directs histone methylation and transcriptional gene silencing in human cells. RNA. 2006;12:256-62
9. Brooks DG, Hamer DH, Arlen PA, Gao L, Bristol G, Kitchen CM. et al. Molecular characterization, reactivation, and depletion of latent HIV. Immunity. 2003;19:413-23
10. McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, Weissman IL. The SCID-hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science. 1988;241:1632-9
11. Turner AM, Ackley AM, Matrone MA, Morris KV. Characterization of an HIV-targeted transcriptional gene-silencing RNA in primary cells. Hum Gene Ther. 2012;23:473-83
12. Suzuki KT, Shijuuku T, Fukamachi J. et al. Kelleher. Prolonged transcriptional silencing and CpG methylation induced by siRNAs targeted to the HIV-1 promoter region. J RNAi Gene Silencing. 2005;1:66-78
13. Brechtl JR, Breitbart W, Galietta M, Krivo S, Rosenfeld B. The use of highly active antiretroviral therapy (HAART) in patients with advanced HIV infection: impact on medical, palliative care, and quality of life outcomes. J Pain Symptom Manage. 2001;21:41-51
14. Suzuki K, Hattori S, Marks K, Ahlenstiel C, Maeda Y, Ishida T. et al. Promoter Targeting shRNA Suppresses HIV-1 Infection In vivo Through Transcriptional Gene Silencing. Mol Ther Nucleic Acids. 2013;2:e137
15. Re MC, Vitone F, Biagetti C, Schiavone P, Alessandrini F, Bon I. et al. HIV-1 DNA proviral load in treated and untreated HIV-1 seropositive patients. Eur J Clin Microbiol. 2010;16:640-6
16. Gagnon KT, Li L, Janowski BA, Corey DR. Analysis of nuclear RNA interference in human cells by subcellular fractionation and Argonaute loading. Nat Protoc. 2014;9:2045-60
17. Morris KV, Chan SW, Jacobsen SE, Looney DJ. Small interfering RNA-induced transcriptional gene silencing in human cells. Science. 2004;305:1289-92
18. Zhang MX, Ou H, Shen YH, Wang J, Wang J, Coselli J. et al. Regulation of endothelial nitric oxide synthase by small RNA. Proc Natl Acad Sci U S A. 2005;102:16967-72
19. Satheesan S, Li H, Burnett JC, Takahashi M, Li S, Wu SX, Synold TW, Rossi JJ, Zhou J. HIV replication and latency in a humanized NSG mouse model during suppressive oral combinational antiretroviral therapy. J Virol. 2018;92:e02118-17
20. Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA. et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med. 2006;12:1316-22
21. Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V. et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839-47
22. Jiang J, Lee EJ, Gusev Y, Schmittgen TD. Real-time expression profiling of microRNA precursors in human cancer cell lines. Nucleic Acids Res. 2005;33:5394-403
23. Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659-61
24. Berges BK, Akkina SR, Folkvord JM, Connick E, Akkina R. Mucosal transmission of R5 and X4 tropic HIV-1 via vaginal and rectal routes in humanized Rag2-/- gammac -/- (RAG-hu) mice. Virology. 2008;373:342-51
25. Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571-2
26. Guo W. et al. CGmapTools improves the precision of heterozygous SNV calls and supports allele-specific methylation detection and visualization in bisulfite-sequencing data. Bioinformatics. 2017 Sep 18. doi: 10.1093/bioinformatics/btx595
27. Zhou J, Li H, Li S, Zaia J, Rossi JJ. Novel Dual Inhibitory Function Aptamer-siRNA Delivery System for HIV-1 Therapy. Mol Ther. 2008;16:1481-9
28. Ahlenstiel C, Mendez C, Lim ST, Marks K, Turville S, Cooper DA. et al. Novel RNA Duplex Locks HIV-1 in a Latent State via Chromatin-mediated Transcriptional Silencing. Mol Ther Nucleic Acids. 2015;4:e261
29. Suzuki K, Ishida T, Yamagishi M, Ahlenstiel C, Swaminathan S, Marks K. et al. Transcriptional gene silencing of HIV-1 through promoter targeted RNA is highly specific. RNA Biol. 2011;8:1035-46
30. Turner AM, De La Cruz J, Morris KV. Mobilization-competent Lentiviral Vector-mediated Sustained Transcriptional Modulation of HIV-1 Expression. Mol Ther. 2009;17:360-8
31. Zhou J, Rossi JJ. Aptamer-targeted cell-specific RNA interference. Silence. 2010;1:4
32. Suzuki K, Juelich T, Lim H, Ishida T, Watanebe T, Cooper DA. et al. Closed chromatin architecture is induced by an RNA duplex targeting the HIV-1 promoter region. J Biol Chem. 2008(34):23353-63
33. Hawkins PG, Santoso S, Adams C, Anest V, Morris KV. Promoter targeted small RNAs induce long-term transcriptional gene silencing in human cells. Nucleic Acids Res. 2009(9):2984-95
34. Kim DH, Villeneuve LM, Morris KV, Rossi JJ. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat Struct Mol Biol. 2006;13:793-7
35. Nanfack AJ, Redd AD, Bimela JS, Ncham G, Achem E, Banin AN. et al. Multimethod Longitudinal HIV Drug Resistance Analysis in Antiretroviral-Therapy-Naive Patients. J Clin Microbiol. 2017;55:2785-800
Author contact
![]() Corresponding authors: Jzhouorg and Kmorrisorg
Corresponding authors: Jzhouorg and Kmorrisorg