Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(6):1540-1557. doi:10.7150/thno.22952 This issue Cite
Research Paper
The role of PTRF/Cavin1 as a biomarker in both glioma and serum exosomes
Kai Huang1,*, Chuan Fang2,3,*, Kaikai Yi1, Xing Liu4, Hongzhao Qi5, Yanli Tan6, Junhu Zhou1, Ying Li1, Mingyang Liu7, Yuqing Zhang7, Jingxuan Yang7, Jianning Zhang2 ![]() , Min Li7
, Min Li7 ![]() , Chunsheng Kang1,
, Chunsheng Kang1, ![]()
1. Tianjin Neurological Institute, Key Laboratory of Post-neurotrauma Neuro-repair and Regeneration in Central Nervous System, Ministry of Education and Tianjin City, Tianjin 300052, China;
2. Department of Neurosurgery, Tianjin Medical University General Hospital, Tianjin 300052, China;
3. Department of Neurosurgery, Hebei University Affiliated Hospital, Baoding 071000, China;
4. Beijing Neurosurgical Institute, Capital Medical University,Beijing,100050,China;
5. Tianjin Key Laboratory of Composite and Functional Materials, School of Materials Science and Engineering, Tianjin University , Tianjin 300072, China;
6. College of Fundamental Medicine, Hebei University, Baoding 071000, China;
7. Department of Medicine, Department of Surgery, the University of Oklahoma Health Sciences Center, Oklahoma City, OK, 73104, USA.
* These authors contributed equally to this work
Received 2017-9-22; Accepted 2017-12-5; Published 2018-2-7
Abstract

Exosomes play critical roles in intercellular communication in both nearby and distant cells in individuals and organs. Polymerase I and transcript release factor (PTRF), also known as Cavin1, has previously been described as a critical factor in caveola formation, and aberrant PTRF expression has been reported in various malignancies. However, the function of PTRF in tumor progression remains controversial, and its role in glioma is poorly understood. In this study, we report that PTRF is associated with malignancy grade and poor prognosis in glioma patients. Our previous study using two proteomics methods, tandem mass tag (TMT) and data-independent acquisition (DIA), showed that EGFRvIII overexpression increased PTRF expression at the protein level. In contrast, blocking PI3K and AKT using LY294002 and MK-2206, respectively, decreased PTRF expression, showing that PTRF is regulated in the EGFR/PI3K/AKT pathway. ChIP-PCR analysis showed that PTRF is transcriptionally regulated by the H3K4me3 and H3K27me3 modifications. Furthermore, PTRF overexpression increased exosome secretion and induced cell growth in vitro. More importantly, overexpressing PTRF induced the malignancy of nearby cells in vivo, suggesting that PTRF alters the microenvironment through intercellular communication via exosomes. Furthermore, analysis of clinical samples showed a positive correlation between tumor grade and PTRF expression in both tumor tissues and exosomes isolated from blood harvested from glioma patients, and PTRF expression in exosomes isolated from the sera of GBM patients was decreased after surgery. In conclusion, PTRF serves as a promising biomarker in both tumor samples and serum exosomes, thus facilitating the detection of glioma and potentially serving as a therapeutic target for glioblastoma multiforme.
Keywords: PTRF/Cavin1, Extracellular vesicle, GBM, exosome
Introduction
Glioblastoma multiforme (GBM) is the most common type of intracranial malignant tumor and is associated with a dismal prognosis. Despite advanced therapeutic methods, the median survival time of GBM is merely 14 months. The RTK/PI3K signaling pathway was proven to be altered in ~90% of GBMs, and epidermal growth factor receptor (EGFR) amplification and mutations occur in 40-60% of GBMs [1-3]. EGFR variant III (EGFRvIII), the most common EGFR mutation, occurs in ~25% of GBM patients [2] and is caused by an in-frame deletion of EGFR gene exons 2-7 [4], leading to a constitutively active tyrosine kinase [5]. Furthermore, EGFRvIII enhances tumorigenicity [6] and confers radioresistance to tumor cells [7].
In GBM cells, EGFR and EGFRvIII function as key regulators of the formation of cell membrane infoldings called caveolae by increasing the expression of caveola-associated protein caveolin-1 (Cav-1) [8]. Caveolae are flask-shaped invaginations of the plasma membrane 50-100 nm in diameter that are implicated in various physiological processes [9-11], and Cav-1 was first characterized as a caveola marker [12]. Recently, another family of proteins called cavins was shown to be required together with caveolins for caveola formation and function. Polymerase I and transcript release factor (PTRF), also known as Cavin1, was originally defined as a regulator of RNA polymerase I (Pol I) in transcription [13] and plays a critical role in caveola biogenesis [14]. PTRF was shown to colocalize with Cav-1 at the plasma membrane but not in the Golgi apparatus [14, 15]. . However, its role in glioma microenvironment alteration is poorly understood.
Exosomes are 30-100 nm membrane vesicles secreted into the extracellular microenvironment by almost all cell types and participate in various biological processes, including intercellular communication [16, 17]. Exosomes can attach to discrepant receptors on the surfaces of target cells and excrete their components into recipient cells after fusing with their membranes [18]. Exosomes are natural transporters of RNA and proteins, and delivery of this molecular information can change the physiology of recipient cells at transcriptional, posttranscriptional, and epigenetic levels. In contrast to microvesicles, which are generated by budding from the plasma membrane, exosomes are derived from membrane invagination and released because of fusion between vesicle-laden endosomes, or multivesicular bodies (MVBs), and the plasma membrane [19-21].
Researchers are searching for novel biomarkers for cancer diagnoses and treatment, and compared with biomarkers in tumor tissues, circulating biomarkers are more easily available and less invasive, facilitating early screening and assisting diagnosis for suspected cases. Thus far, numerous proteins have been successfully established as biomarkers for various cancers, and a number of potential protein biomarkers, such as MGMT [22], EGFR [23] and IDH1/2 mutant [24], have shown to be promising therapeutic targets for glioma. However, few proteins have been proven as biomarkers in both tumor tissues and exosomes and simultaneously used for treatment. Therefore, a reliable biomarker for both exosomes and tumor tissues is urgently needed.
In this study, we first proved that PTRF is regulated by the EGFRvIII/PI3K/AKT pathway via histone modification (H3K4me3 and H3K27me3) in GBMs. Enriched in the mesenchymal GBM subtype, PTRF overexpression is associated with poor prongosis. Furthermore, PTRF overexpression increases exosome secretion, and exosomes induced by PTRF enhance the proliferation of recipient cells in vitro and in vivo. More importantly, PTRF expression is detectable in both tumor tissues and serum exosomes from clinical glioma samples of different grades, thus making PTRF an ideal candidate for diagnostic and prognostic indicators. Furthermore, PTRF expression in exosomes isolated from the blood of GBM patients was decreased after surgery. Together, these data indicate that PTRF is a promising marker that can be detected in both tumor samples and serum exosomes. Silencing PTRF reversed the malignancy of GBM in vivo and in vitro, showing that PTRF may also be a promising therapeutic target of GBM.
Materials and methods
Cell culture and lentiviruses
The human glioma cell lines LN229, U87 and U251 were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA) and cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS, HyClone, Logan, UT, USA). Primary cell lines (TBD0207, TBD0224, TBD0313 and TBD0314) were derived directly from GBM patients who underwent surgery at Hebei University Affiliated Hospital according to a protocol reported by Dong et al [25]. EGFRvIII cell lines were derived via lentiviral transduction of the cell lines with GV341 constructs containing EGFRvIII cDNA (Shanghai Genechem Co. LTD., Shanghai, China) followed by puromycin selection for 7 days. The cells were maintained in a humidified 5% CO2 atmosphere at 37℃. Lentiviruses encoding the PTRF-EGFP fusion protein and PTRF siRNA-1, PTRF siRNA-2 or NC siRNA were obtained from Genechem. The PTRF siRNA-1, PTRF siRNA-2 and NC siRNA sequences are presented in supplementary table 1.
Clinical specimens
Glioma samples (n=4) for primary cell line culture were resected and freshly retained from patients at Hebei University Affiliated Hospital, and blood samples collected from the patients one day prior and seven days after surgery were stored at -80℃. Another 36 glioma samples and their relative donated blood were obtained from Tiantan Hospital and stored at -80℃. Complete clinical data were collected, and the collection and processing of primary human GBM tumor samples was in accordance with ethical standards of the 2008 Helsinki Declaration. All patients provided written consent for the use of their samples for biomedical research. Tumor grades were determined according to the 2007 World Health Organization (WHO) classification of nervous system tumors.
Standard protocol for Extracellular vesicle isolation
Exosomes and microvesicles were isolated and purified according to an isolation protocol established by Thery C, et al. [26]. Supernatants were collected from equivalent amounts of culture medium and normalized by equivalent amounts of cells. Briefly, glioma cells were grown to 80% confluence and incubated in serum-free medium for 24 h, and the cell supernatants were pooled and centrifuged at 300×g for 10 min and 2,000×g for 20 min (4℃) to remove cell debris. The supernatants were then centrifuged at 10,000×g for 30 min (4℃, Beckman Coulter Avanti J-26 XP, Pasadena, CA, USA) to collect microvesicles. Subsequently, the supernatants were transferred to ultracentrifuge tubes (Beckman Coulter) for the first ultracentrifugation step at 100,000×g for 70 min (4℃). The supernatants were discarded, and the remaining pellet was washed with phosphate-buffered saline (PBS) and centrifuged again at 100,000×g for 70 min (4℃). The final pellets enriched in exosomes were re-suspended in 50 µL of PBS. Microvesicles and exosomes were frozen at -80℃ for further research. Exosome size and size distribution were measured by dynamic light scattering (BI-90Plus, Brookhaven Instruments Ltd., USA), and exosome morphology and composition were visualized using a high-resolution transmission electron microscope (JEM-2100F, JEOL Ltd., Japan) [27].
To isolate exosomes from blood donated from glioma patients, serum was first collected by centrifugation at 2000×g for 20 min after blood coagulation. Thereafter, exosomes were isolated from the serum according to the same protocol as that used for supernatant isolation.
Western blot analysis
Western blot analysis of cells, samples and exosome lysates was performed according to the manufacturer's instructions and a previously described protocol [28]. Briefly, cells, samples and exosomes were lysed with RIPA buffer (Solarbio, Beijing, China) supplemented with phenylmethylsulfonyl fluoride (PMSF) and a phosphatase inhibitor (Solarbio), and lysates were incubated on ice for 30 min to ensure membrane disruption. Protein concentrations were determined using a bicinchoninic acid (BCA) kit (Beyotime, Shanghai, China) following the manufacturer's recommendations, and electrophoresis was performed using 10% acrylamide gels. The following primary antibodies were used: anti-EGFRvIII, anti-EGFR, and anti-p-AKT Ser473 (Cell Signaling Technology (CST), Boston, MA, USA, 1:1,000 dilution), anti-PTRF (Abcam, Cambridge, MA, USA, 1:1,000 dilution), anti-CD63 (Santa Cruz Biotechnology, CA, USA, 1:500 dilution) and anti-GAPDH (Millipore, Billerica, MA, USA, 1:2,000 dilution). Anti-mouse and anti-rabbit horseradish peroxidase-conjugated secondary antibodies were purchased from Promega (Madison, WI, USA, 1:10,000 dilution). Immunoblots were developed using G:BOX F3 (Syngene, Cambridge, UK).
Quantitative RT-PCR
Total RNA was isolated from cells and exosomes using TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Total RNA (2 μg) was used as the template for reverse transcription. cDNA synthesis was performed for one cycle at 42℃ for 1 hour and 70℃ for 15 minutes. qRT-PCR was performed using SYBR Green Master Mix (Applied Biosystems/Thermo Fisher, Austin, TX, USA), and normalization was conducted using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6 as endogenous controls for mRNA and microRNA, respectively. qRT-PCR reactions were performed on a CFX96™ PCR cycler (Bio-Rad, Hercules, CA, USA) for 40 cycles. The PCR thermal profile was as follows: 95℃ for 5 min, 95℃ for 15 s, 53-58℃ for 1 min, and 72℃ for 10 min. The primer sequences used are presented in supplementary table 2. The specificity of the qRT-PCR reaction was confirmed using melting curve analysis and electrophoresis on 1.2% agarose gels stained with GelRed fluorescent dye (Biotium, Hayward, CA, USA). Relative gene expression was calculated as the 2-ΔΔCt fold change.
Cell proliferation assay
Cell proliferation assays were performed using the CellTiter 96® AQueous One Solution Cell Proliferation Assay Kit (COS, Promega) according to the manufacturer's protocol. Briefly, cells were plated in 96-well plates in triplicate at approximately 2,000 cells per well and cultured in growth medium from 0 to 96 h. At the indicated time, 10 μl of COS was added to each well, and the cells were cultured for 1 h. The absorbance of the plate was measured at 490 nm using a microplate reader (Synergy2, BioTek, Winooski, VT, USA).
Immunohistochemistry
For immunohistochemistry, a primary PTRF antibody (Abcam, 1:100 dilution) was used according to protocol, and slides were analyzed using National Institutes of Health (NIH) Image J software.
Confocal imaging
Immunofluorescence assays were performed according to a previously described protocol [29]. Briefly, cells were seeded on poly-L-lysine-coated glass coverslips, fixed with ice-cold polyformaldehyde at 4℃ overnight, blocked with 1% bovine serum albumin, and incubated with antibodies against PTRF (Abcam, 1:200 dilution) and anti-EGFRvIII (CST, 1:200 dilution). Primary antibody labeling was detected by Alexa Fluor 488- or 546-conjugated secondary antibodies (Alexa Fluor, Molecular Probes/Life Technologies, Eugene, Oregon, USA, 1:100 dilution). Images were captured using an Olympus FluoView 1200 confocal microscope (Olympus, Tokyo, Japan). To objectively compare differences in immunofluorescence due to various treatments, all confocal scanning parameters were kept constant, and images were minimally processed to maintain data integrity.
Patients and samples for microarray data
The Cancer Genome Atlas (TCGA) data were downloaded from the University of California Santa Cruz (UCSC) Cancer Genome Browser (https://genome-cancer.ucsc.edu), and only patients with full datasets were included. Copy number data were based on TCGA GBM segmented delete germline cnv (n=595), RNAseq data were based on TCGA illuminaHiSeq (n=561) and GBM gene expression data were based on the AffyU133a array (n=539) and AgilentG4502A (n=483). Data were normalized and log2 transformed.
The RNAseq database from the Chinese Glioma Genome Atlas database (CGGA, http://www.cgcg.org.cn/, n=325).
Chromatin immunoprecipitation (ChIP) sequencing data for histone modification
ChIP-sequencing datasets of glioma cell line histone modifications were obtained from the Gene Expression Omnibus website (http://www.ncbi.nlm.nih.gov/geo/, accession no. GSE 46016). Density maps were generated with read extensions to 200 bp with IGVtools based on the hg19 human reference genome. Histone methylation data for other cell lines were obtained as interactive tracks from the UCSC Genome Browser (http://genome.ucsc.edu/).
ChIP and ChIP-qPCR assays
ChIP assays were performed using a ChIP Assay Kit (Millipore) [29]. Briefly, after crosslinking chromatin with 1% formaldehyde for 10 min and neutralizing with glycine for 5 min at room temperature, cells were washed with cold PBS, scraped, collected and stored on ice. Cells were then lysed and sonicated 90 times for 3 s at 9 s intervals in ice water using a Sonics Vibra-Cell processor (Sonics & Materials Inc., USA). An equal amount of chromatin was immunoprecipitated at 4℃ overnight with at least 1.5 μg of the following antibodies: H3K27me3 (CST), H3K4me3 (CST) and normal mouse IgG (Millipore). Immunoprecipitated products were collected after incubation with Protein G agarose beads. The beads were washed, and bound chromatin was eluted in ChIP Elution Buffer. RNA was then digested with RNase A for 30 min at 37℃, and proteins were digested with Proteinase K for 2 h at 45℃. After DNA purification, the PTRF promoter binding site was evaluated using qPCR and normalized by total chromatin (input). Normal mouse IgG was used as the negative control, and the primers are described in supplementary table 3. The qPCR conditions were based on a three-step method that consisted of 1 cycle at 95℃ for 3 min followed by 40 cycles at 95℃ for 15 s, 53℃ for 30 s, and 72℃ for 30s, and a final incubation at 72℃ for 5 min with fluorescence acquisition. The PCR products were then electrophoresed on 2% agarose gels stained with GelRed.
Scanning ion conductance microscopy (SICM)
LN229 cells were seeded at 50% confluence and washed with L15 medium three times before being observed by SICM (Ionscope Ltd., UK). A pipette filled with L15 medium was immersed into the bath solution (L15 medium). After immersion, ion current flow through the pipette was pushed by a 200 mV DC voltage and measured by an external Axon MultiClamp 700B amplifier and a Digidata 1440A analog-to-digital converter (Molecular Devices, USA). The position of the z-dimensional piezo was recorded as the height of the cell at this imaging point, providing a 3D spatial map of the cell morphology. The time required to scan a 100*100 µm area at a typical resolution of 128*128 pixels was approximately 10 min. All SICM experiments were carried out at room temperature, and 3D image reconstruction and topographical feature analysis with the obtained images were performed using SICMImage Viewer 2010.
Intracranial mouse model
5-week-old female nude mice (Chinese Academy of Medical Science Cancer Institute) were used to establish intracranial GBM xenografts, and LN229 and U87EGFRvIII cells were used as the cell models. Previously, LN229 cells were transduced with lentivirus expressing RFP (LN229-RFP) and luciferase, and U87EGFRvIII cells were transduced with the negative control (NC) vector or PTRF-EGFP (U87EGFRvIII-PTRF-EGFP). Orthotopic glioma models were classified into three groups according to the intracranial injection of LN229-RFP cells (G1) and LN229-RFP cells mixed with U87EGFRvIII cells transduced with NC lentivirus (1:1, G2) or PTRF-EGFP lentivirus (1:1, G3), respectively. A total of five hundred thousand cells were injected to each mouse under the guidance of a stereotactic instrument. Bioluminescence imaging was used to detect intracranial tumor growth on days 7, 14, 21 and 28. For the PTRF-silenced orthotopic glioma model, U87 and U87EGFRvIII cells were transduced with the PTRF siNC or PTRF siRNA-1 lentiviruses, respectively. Five hundred thousand cells were injected into each mouse as previously described. Bioluminescence imaging was used to detect intracranial tumor growth on days 10 and 20. The data were normalized to bioluminescence detected at the initiation of treatment for each animal. The error bars shown in the figures indicate standard deviation (SD). Kaplan-Meier survival curves were plotted to show survival time.
Statistical analysis
Statistical analysis was performed using the statistical package SPSS (version 16.0, SPSS Inc., Chicago, IL, USA). Data are presented as the mean ± SD of three independent experiments. One-way analysis of variance (ANOVA) was used for comparisons between the different groups. When ANOVA was significant, post-hoc testing of the differences between groups was performed using a least significant difference (LSD) test. Kaplan-Meier survival curves were analyzed by log-rank tests using GraphPad Prism software. Differences were considered to be significant at P < 0.05.
Results
PTRF expression confers a worse prognosis for glioma patients of the TCGA cohort
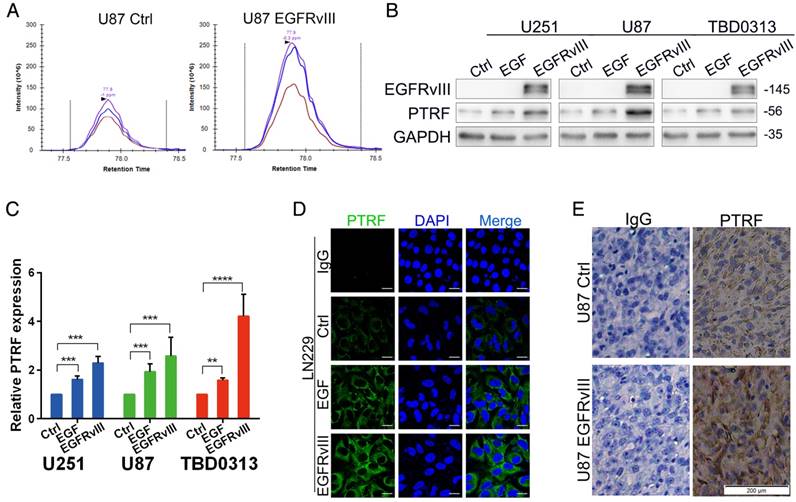
Although seldom studied in glioma, PTRF is a proven cofactor in caveola formation. Our previous study showed that PTRF expression is increased by EGFRvIII overexpression using two important proteomic methods, data-independent acquisition (DIA) (Fig. 1A) and tandem mass tags (TMT) (Fig. S1A)[30], indicating that PTRF is a downstream gene of EGFRvIII. To further confirm this result, GBM cell lines (U251, U87 and TBD0313) were either stimulated by recombinant EGF protein (Proteintech, final concentration of 100 pg/ml) or transduced by EGFRvIII for 48 hours, PTRF expression was increased at both the protein (Fig. 1B) and mRNA level (Fig. 1C), and EGFRvIII showed a higher efficiency to increase PTRF expression. A confocal assay also showed higher PTRF expression after EGF stimulation and EGFRvIII transduction (Fig. 1D and Fig. S1B). Furthermore, an orthotopic xenograft experiment involving the injection of U87 NC or U87EGFRvIII cells into nude mice showed that EGFRvIII regulated PTRF expression in vivo (Fig. 1E). EGFR amplification and the EGFRvIII mutation were proven to be major factors driving glioma development. In the copy number variation (CNV) analysis of 595 patient samples from TCGA data, the CNV of genes in the PI3K/AKT pathway were not well-correlated with EGFR gene alteration (Fig. S1C), whereas AKT1 mRNA expression was positively associated with EGFR mRNA expression (Fig. S1E), showing that the EGFR/PI3K/AKT pathway plays a critical role in gliomagenesis. The CNV of the PTRF gene was also not well-correlated with EGFR (Fig. S1D), but the mRNA level was positively correlated with EGFR mRNA expression (Fig. S1F). Together, the abovementioned data indicated that PTRF is regulated by EGFR/EGFRvIII but not by copy number variation.
PTRF expression is positively associated with EGFR and EGFRvIII. (A) After stably overexpressing EGFRvIII, DIA proteomic methods were used to evaluate total protein expression. Among these proteins, PTRF expression was increased. (B) GBM cell lines were either stimulated by EGF or transduced with EGFRvIII for 48 hours, and the expression of EGFRvIII, PTRF and GAPDH was evaluated by western blot. GAPDH served as the negative control. (C) RT-qPCR experiments were performed to detect PTRF mRNA expression. After EGF stimulation and EGFRvIII transduction, PTRF mRNA expression was gently increased. Error bars in the RT-qPCR results indicate the standard error of the mean. Significant differences in the experimental groups were compared with those of the control group (**P < 0.01, ***P<0.001, ****P<0.0001). (D) Confocal microscopy detected increased PTRF expression in LN229 cells stimulated by EGF or transduced with EGFRvIII. Scale bar: 20 μm. (E) Representative immunostaining results of PTRF in tumors from each group of nude mice. EGFRvIII increased PTRF expression in vivo. Scale bar: 200 μm.
PTRF expression is correlated with glioma grade and confers a worse prognosis in glioma patients
We employed the RNAseq data from TCGA and CGGA cohorts to show whether PTRF is correlated with glioma grade. According to the WHO classification criteria, these gliomas were classified as grade II, III or IV. As shown in Fig. 2A and D, PTRF expression was significantly associated with tumor grade (P <0.0001). PTRF is an important gene for distinguishing the mesenchymal subtype in TCGA tumors [31]. Hence, the PTRF expression levels in the classical and mesenchymal subtypes were higher than those in the proneural subtypes (Fig. 2B and E, P<0.0001). Next, we further evaluated the prognostic values of PTRF using Kaplan-Meier survival curve analysis with a log-rank comparison. Regardless of low-grade gliomas (LGG), high-grade gliomas (HGG) and all grade glioma, PTRF showed an association with decreased survival in the TCGA cohort compared with that in patients in the low PTRF expression group, and those in the high PTRF expression group had worse prognoses (Fig. 2C and F, P<0.01).
PTRF is an independent biomarker in glioma diagnosis. (A and D) RNAseq data from the CGGA (A) and TCGA (D) cohorts were used to show PTRF expression levels in WHO II-IV gliomas. PTRF is positively associated with glioma WHO grades. (B and E) PTRF is enriched in patients with classical and mesenchymal GBM. (C and F) Kaplan-Meier curve showing that PTRF expression is associated with poor prognosis in glioma patients. (G) Cox proportional hazards regression analyses of PTRF expression and other characteristics in relation to overall survival in GBM from CGGA cohort.
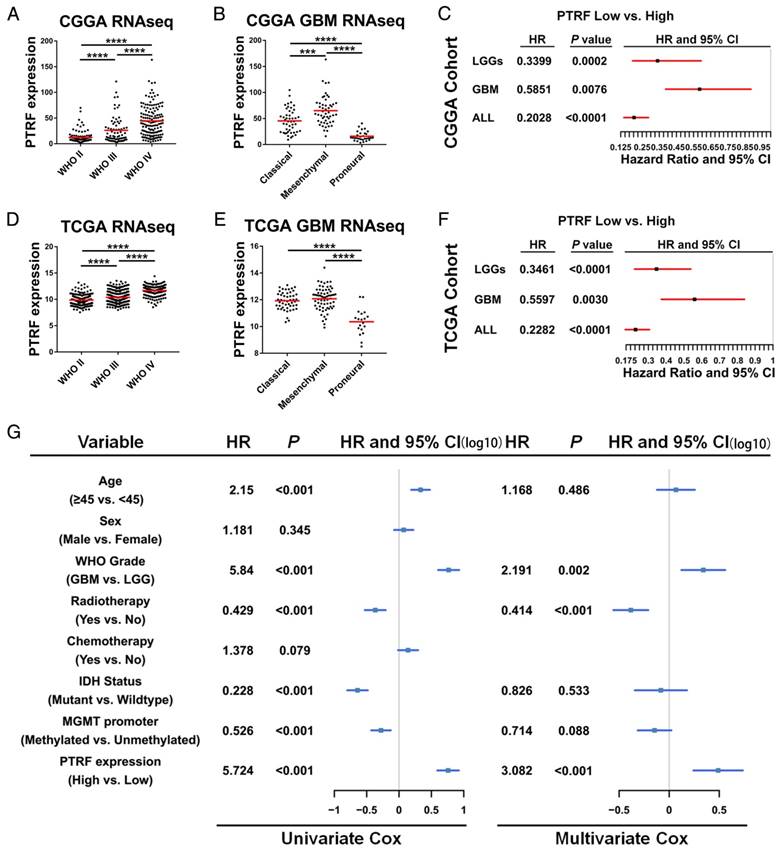
We next explored cDNA microarray data from the TCGA cohort for more evidence. two datasets showed that PTRF mRNA levels were higher in Classical and Mesenchymal subtypes than Proneural subtypes (Fig. S2A and C), and GBM patients with high PTRF expression had worse prognoses than those with low PTRF expression groups (Fig. S2B and D). Immunohistochemistry analysis of a tissue microarray containing normal brain, LGG and HGG tissue sections showed that PTRF was expressed higher at the protein level in HGG (http://www.proteinatlas.org) (Fig. S2E). Thereafter, univariate Cox regression analysis was firstly used for GBM patients from the CGGA and TCGA cohorts and found that high expression of PTRF, high age, IDH1 mutation and MGMT promoter methylation were associated with overall survival (Fig. 2G and Fig. S2F). Further analysis using a multivariate Cox proportional hazards model revealed that PTRF expression correlated independently with overall survival (Fig. 2G and Fig. S2F , hazard ratio[HR] = 3.082, P <0.001). These findings showed that PTRF expression is positively associated with glioma WHO grades and that PTRF overexpression correlates to significantly worse survival outcome, indicating that PTRF is a promising independent biomarker in glioma diagnoses.
PTRF is regulated by H3K4me3 and H3K27me3 histone modifications in the EGFR/PI3K/AKT pathway
To identify whether PTRF is regulated in the EGFR/PI3K/AKT pathway, we used LY294002 (Selleck, USA, 20 μM) and MK-2206 (Selleck, USA, 10 μM) to inhibit the activity of PI3K and AKT, respectively. After treatment with LY294002 and MK-2206 for 48 h, total protein and mRNA were collected to detect PTRF expression by western blot and qRT-PCR. PTRF was attenuated in U251, U87 and TBD0313 EGFRwt/vIII cells at the protein (Fig. 3A) and mRNA levels (Fig. 3B), indicating that PTRF is regulated in the EGFR/PI3K/AKT pathway.
PTRF is regulated by the H3K4me3 and H3K27me3 histone modifications in the EGFR/PI3K/AKT pathway. (A) Western blot showing that p-AKT and PTRF expression decreased with LY294002 and MK-2206 treatment. GAPDH served as the negative control. (B) qRT-PCR showing that the PTRF mRNA levels were decreased after treatment with LY294002 and MK-2206. (C) Using IGV, the PTRF promoter was enriched with H3K4me3 and H3K27me3 in glioma cell lines, but H3K4me3 binding was more important in the GSE46016 dataset. (D) ChIP-PCR assays showing that H3K4me3 exhibited increased binding to the PTRF promoter, while H3K27me3 exhibited decreased binding to the PTRF promoter after stimulation by EGF or transduction with EGFRvIII. (E) Chip-PCR assays showing that H3K4me3 exhibited decreased binding to the PTRF promoter, while H3K27me3 exhibited increased binding to the PTRF promoter after LY294002 and MK-2206 treatment. (F) Schematic illustration of the mechanism underlying PTRF regulation by the EGFRvIII/wt/PI3K/AKT pathway. Stimulation by EGF or transduction with EGFRvIII increased PTRF expression, while blocking PI3K and AKT decreased PTRF expression via the H3K4me3 and H3K27me3 modifications.
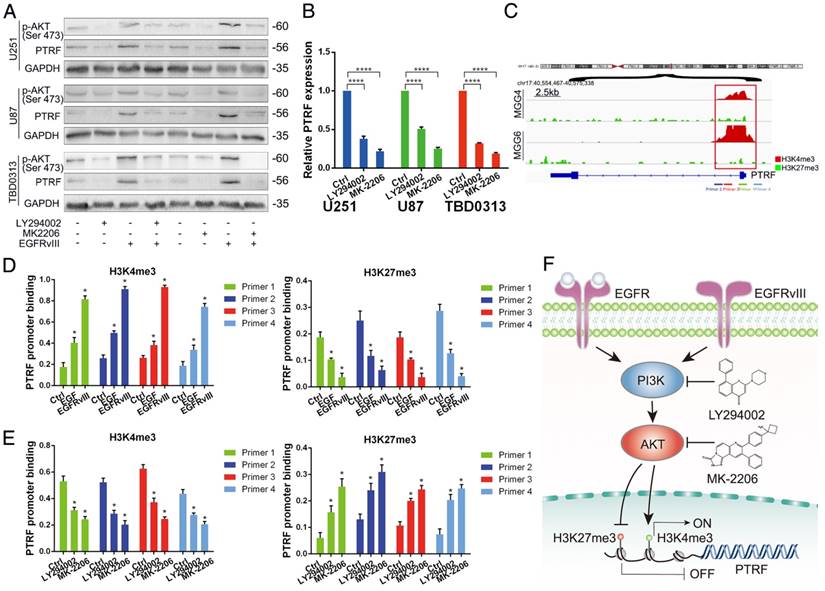
To explore how PTRF is regulated by PI3K and AKT, we gathered information regarding the regulation of PTRF promoter region histone modifications from the UCSC Genome Browser. As shown in Fig. S3A, PTRF promoters in human cancers are enriched with numerous H3K4me3 modifications, while H3K27me3 modifications are few. Similarly, in MGG cells (GBM cell stem cells), the PTRF promoter is characterized mostly by H3K4me3 enrichment, while H3K27me3 enrichment is poor (Fig. 3C) [32]. To evaluate the chromatin state of the PTRF promoter regulated by recombinant EGF protein and EGFRvIII in GBM cell lines, we performed ChIP-PCR analysis using antibodies against H3K4me3 and H3K27me3 and 4 genomic PCR primers specific for the PTRF promoter. The amount of H3K4me3 binding the promoter region increased, and the amount of H3K27me3 binding the promoter region decreased (Fig. 3D and Fig. S3B). By contrast, blocking PI3K and AKT using LY294002 and MK-2206, respectively, yielded a totally different result (Fig. 3E and Fig. S3C). These studies revealed that PTRF transcription is regulated by H3K4me3 and H3K27me3 histone modifications in the EGFRvIII/wt/PI3K/AKT pathway (Fig. 3F).
PTRF is a novel biomarker of glioma exosomes and enhances exosome secretion
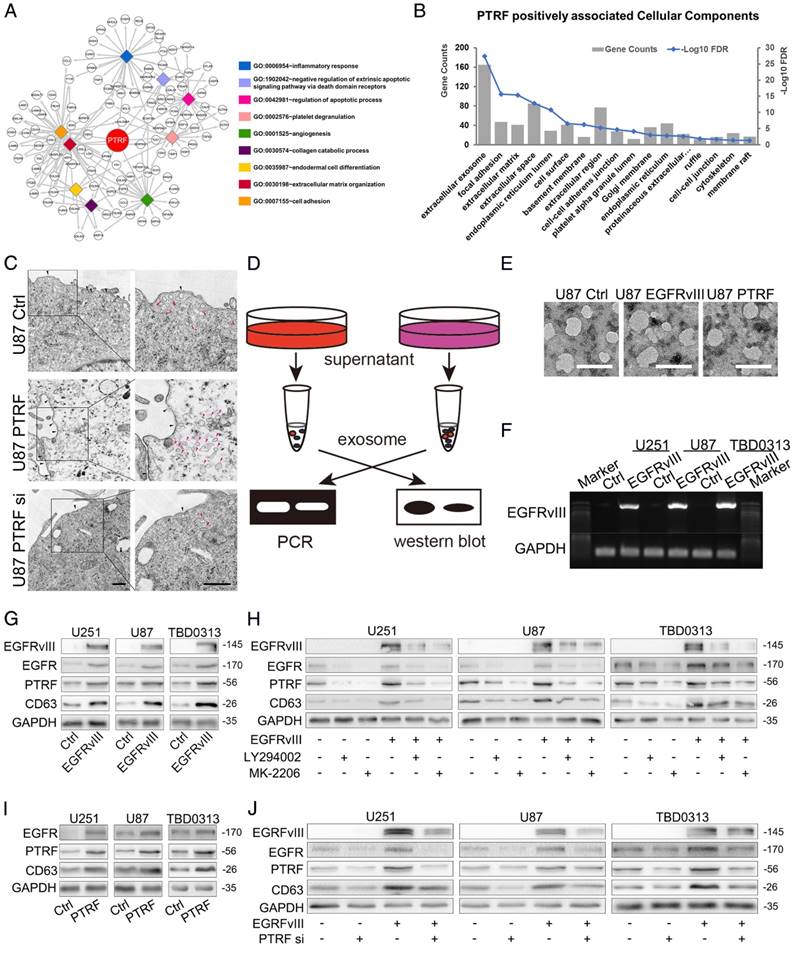
PTRF was previously proven to be a cofactor of caveola formation, and the fact that exosomes are formed by endosomes inspired our hypothesis that PTRF may be an exosome component. From TCGA and CGGA cohorts, we selected genes positively correlated with PTRF expression to subject to gene ontology (GO) analysis. One of the most important biological processes (BP) of these genes was extracellular matrix organization (Fig. 4A). Further analysis by cellular component (CC) displayed that these genes were mainly enriched in extracellular exosome (Fig. 4B), giving us more confidence that PTRF may enhance exosome secretion. We observed the caveolea and endocytic vesicles with transmission electron microscopy (TEM) by overexpression or knockdown of PTRF and found that the numbers of caveolea and endocytic vesicles were both positively associated with PTRF expression (Fig. 4C). To validate our hypothesis, we analyzed components after the isolation and purification of exosomes using a modified protocol (Fig. 4D). TEM showed that the isolated exosomes exhibited the typical spherical structure and were of the correct size (Fig. 4E). Size distribution analysis of exosomes transduced with EGFRvIII or PTRF as measured by dynamic light scattering (BI-90Plus, Brookhaven Instruments Ltd., USA) showed that those exosomes were smaller than the controls (Fig. S4A). EGFRvIII mRNA and miR-21 were first discovered in exosomes isolated from the supernatants of GBM cells and the sera of GBM patients by Skog et al [33]. After transduction of EGFRvIII lentivirus, EGFRvIII mRNA was found in exosomes isolated from culture medium (Fig. 4F), and more miR-21 was also found embedded in these exosomes (Fig. S4B). Western blot showed that in addition to that of CD63, EGFR, EGFRvIII and PTRF expression was also increased (Fig. 4G). Further, EGFR and CD63 protein expression in exosomes was decreased by inhibiting PI3K and AKT (Fig. 4H), indicating that PI3K and AKT play pivotal roles in exosome secretion. PTRF was transduced into GBM cells to evaluate whether exosome secretion could be altered by PTRF. The increased miR-21 (Fig. S4C), EGFR and CD63 protein (Fig. 4I) expression indicated that the number of exosomes was increased by PTRF. To further evaluate whether PTRF depletion influences GBM exosome secretion, we first transduced GBM cells with two pairs of siRNAs (siRNA-1 and siRNA-2) to knock down PTRF expression. Of these siRNAs, siRNA-1 was more efficient at suppressing PTRF expression (Fig. S4D). After persistently silencing PTRF, the number of exosomes was significantly decreased (Fig. 4J). Taken together, we confirmed that EGFR, PI3K, AKT, and PTRF enhance exosome secretion.
PTRF promotes exosome formation. (A) Genes from TCGA and CGGA cohorts positively correlated with PTRF expression were subjected to gene ontology (GO) analysis. The biological processes (BP) of these genes were mainly enriched in inflammatory response, regulation of apoptotic process, and most importantly, extracellular matrix organization. (B) The cellular components (CC) of these genes were mainly enriched in extracellular exosome. (C) Cultured cells were analyzed by transmission electron microscopy (TEM) scanning. U87 cells were treated by overexpression or knockdown of PTRF. Compare with control group, the numbers of caveolea and endocytic vesicles were both positively associated with PTRF expression. Black arrow head: caveolea, pink arrow head: endocytic vesicles, scale bar: 500nm. (D) Schematic overview of exosome detection. Exosomes were isolated from supernatants and subjected to PCR and western blot analysis. (E) Transmission electron microscopy was used to detect exosome products. Scale bar: 100 nm. (F) Exosomes were isolated from the supernatants of U87, U251 and TBD0313 cells transduced with vector or EGFRvIII. mRNA was then extracted, and PCR was performed. Electrophoresis was used to show that EGFRvIII mRNA can be detected in exosomes. (G) Western blot showing that exosome components, such as PTRF, EGFRvIII and EGFR, were increased after the transduction of EGFRvIII. (H) Western blot showing that PTRF, EGFRvIII and EGFR expression in exosomes was decreased after LY294002 and MK-2206 treatment. (I) Exosome components, such as PTRF and EGFR, were increased after the overexpression of PTRF, as determined by western blot. (J) Western blot showing that PTRF, EGFRvIII and EGFR expression in exosomes was decreased after PTRF silencing. For (G), (H), (I) and (J), GAPDH served as the negative control of the relative cell lysate levels.
PTRF enhances intercellular communication by exosomes
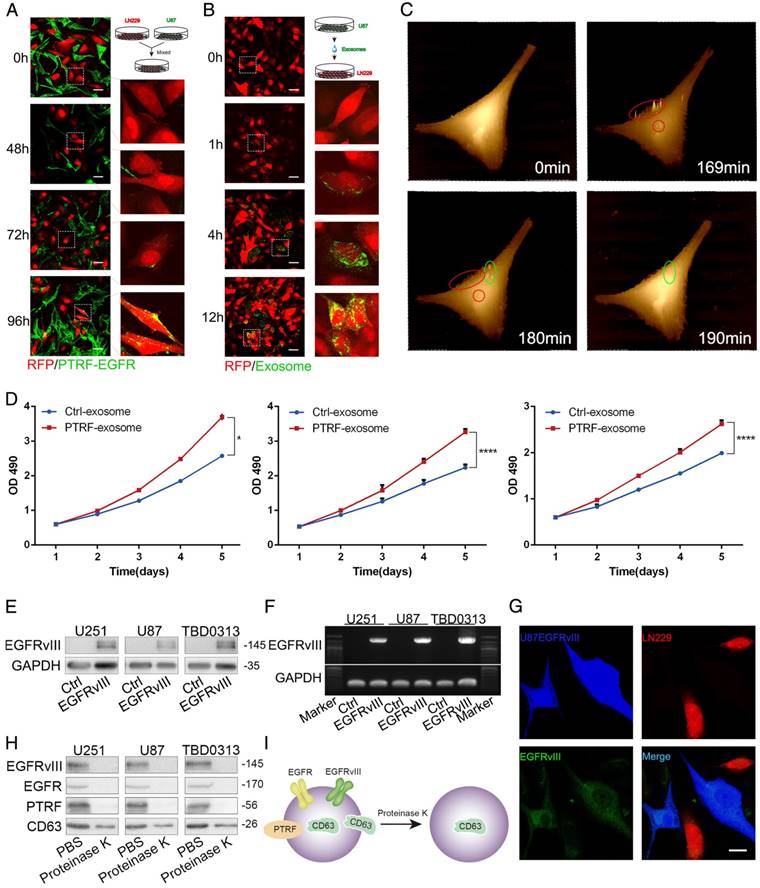
To determine whether exosomes secreted by PTRF-overexpressing cells are related to tumor malignancy, we transduced GBM cell lines with lentivirus expressing the PTRF-EGFP fusion protein. By mixing LN229-RFP and U87 cells transduced with the PTRF-EGFP lentivirus (U87-PTRF-EGFP), PTRF-EGFP was transported into LN229 cells for 72 h (Fig. 5A). To further confirm that exosomes indeed transported PTRF-EGFP into nearby cells, we isolated exosomes from the supernatants of U87-PTRF-EGFP cells. Thereafter, exosomes were added to LN229-RFP cells to show that they were absorbed by LN229 cells in a time-dependent manner (Fig. 5B). Furthermore, microvesicles isolated from the supernatants of U87PTRF-EGFP cells showed little fluorescence (Fig. S5A), suggesting that PTRF does not exist in microvesicles. SICM showed that exosomes were absorbed by the cells within ten minutes (Fig. 5C). After adding 100 µg of exosomes carrying PTRF to each well containing LN229 cells every day, their proliferation rate was significantly increased (Fig. 5D). To determine whether exosomes transport EGFRvIII and its mRNA to recipient cells, exosomes from GBM EGFRvIII supernatants were isolated and added to LN229 cells, and EGFRvIII mRNA and protein levels in the recipient cells were detected 24 h later (Fig. 5E and 5F). After mixing U87EGFRvIII and LN229-RFP together, EGFRvIII was detectable in LN229-RFP cells (Fig. 5G), whereas LN229-RFP mixed with U87 didn't show any EGFRvIII (Fig. S5B). To explore whether PTRF exists on exosome membranes, we treated exosomes with proteinase K and found that EGFRvIII, EGFR and PTRF were all degraded, while some CD63 remained (Fig. 5H). Thus, EGFRvIII, EGFR and PTRF exist on exosome membranes, and CD63 exists both on exosome membranes and inside exosomes (Fig. 5I). In conclusion, our results showed that exosomes induced by PTRF regulate intercellular communication and exert a malignant effect.
PTRF induces intercellular communication via exosomes. (A) LN229 cells transduced with lenti-RFP and U87 cells transduced with PTRF-EGFP were mixed together for 0, 48, 72 or 96 hours. Confocal microscopy showed that PTRF-EGFP could be detected in LN229 cells. (B) Exosomes isolated from the supernatants of U87 cells transduced with PTRF-EGFP were added to LN229-RFP cells for 0, 1, 4 or 12 hours, and confocal microscopy showed that PTRF-EGFP was transferred to LN229 cells. The scale bars for (A) and (B) correspond to 50 μm. (C) LN229 cells were seeded in a 35-mm Petri dish and washed three times with L15 medium. Next, 2 ml of L15 medium was added to the Petri dish, and the cells were observed with a scanning ion conductive microscope (SICM). One photo was taken as the control, and 5 μg of exosomes isolated from U87 PTRF cell supernatants was then added. After continuously scanning for 4 hours, exosomes were determined to be absorbed by the cells within ten minutes (within the red and green circles). (D) LN229 cells were treated with equal amounts of exosomes isolated from the supernatants of U87, U251 and TBD0313 cells transduced with vector or PTRF. Compared with that of the control group, the proliferation rate of the PTRF group was increased. (E and F) Exosomes isolated from the supernatants of U87, U251 and TBD0313 cells transduced with vector or EGFRvIII were added to LN229 cells for 24 hours. Thereafter, proteins were extracted from the cells, and EGFRvIII was analyzed by western blot. Detection of the EGFRvIII protein showed its transfer to recipient cells (E). mRNA was extracted from the cells, and PCR was performed. Electrophoresis was used to show that EGFRvIII mRNA could be transferred to recipient cells (F). (G) LN229-RFP cells and U87EGFRvIII cells expressing EGFP were mixed together for 96 hours. A confocal assay staining for EGFRvIII showed that EGFRvIII was transferred to LN229 cells. Blue represents U87EGFRvIII cells, red represents LN229 cells and green represents stained EGFRvIII. The staining of U87 and LN229 mixing model was served as negative control which was seen in Fig. S5B. The scale bar corresponds to 20 μm. (H) Ten micrograms of exosomes isolated from the supernatants of U87, U251 and TBD0313 cells transduced with EGFRvIII were treated with 10 μl of proteinase K or PBS for 10 min. Western blot analysis showed that EGFRvIII, EGFR and PTRF were totally degraded, while CD63 was partly intact. (I) Cartoon showing EGFRvIII, EGFR and PTRF expression on exosome membranes and CD63 expression on both exosome membranes and inside exosomes.
PTRF promotes exosome secretion and enhances recipient cell proliferation in vivo
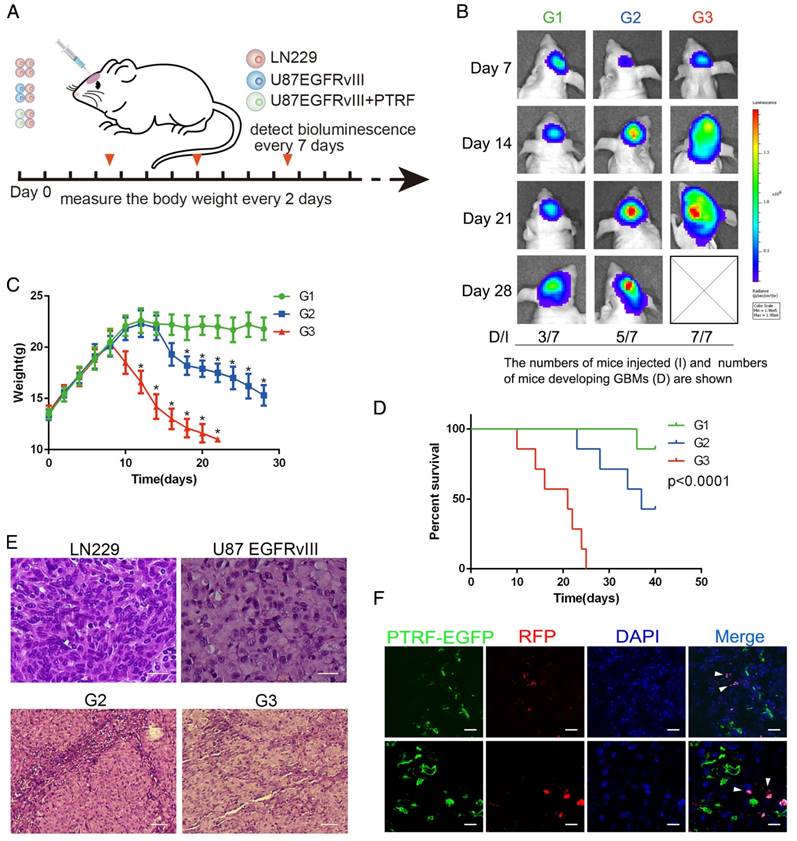
To determine whether PTRF plays a role in exocytosis in vivo, we used the LN229 and U87EGFRvIII cell models. We first transduced LN229 cells with RFP and luciferase and U87EGFRvIII cells with only the vector or PTRF-EGFP lentivirus. Orthotopic glioma models were classified into three groups according to whether they were injected with LN229 cells (G1 group), LN229 cells mixed with U87EGFRvIII (1:1, G2 group), or LN229 cells mixed with U87EGFRvIII-PTRF-EGFP (1:1, G3 group) (Fig. 6A). Bioluminescence imaging showed a significantly faster tumor growth rate and higher percentage of LN229 tumor formation in the G3 group (7/7) than in the G1 (3/7) and G2 (5/7) groups (Fig. 6B). The body weights of mice in G3 decreased faster than those in G1 and G2 during their overall survival time (Fig. 6C). Thereafter, we employed Kaplan-Meier survival curve analysis to show that mice transduced with tumors with PTRF had poorer prognoses (Fig. 6D). Hematoxylin and eosin (H&E) staining also showed that LN229 cells mixed with U87EGFRvIII-PTRF-EGFP exhibited a more aggressive growth phenotype by day 21 (Fig. 6E). Importantly, confocal imaging showed that PTRF-EGFP was transferred from U87 EGFRvIII cells to LN229 cells (Fig. 6F). All the evidence described above showed that PTRF promotes not only GBM itself but also peritumoral cells, leading to devastating results.
PTRF overexpression increases exosome secretion in vivo. (A) Schematic representation of the mixed cell in vivo experiment. Mice were orthotopically injected with LN229 (G1), LN229+U87 EGFRvIII (G2) or LN229+EGFRvIII/PTRF (G3), and only LN229 cells were transduced with luciferase and RFP. Bioluminescence was detected every 7 days, and body weight was measured every other day. (B) Representative pseudocolor bioluminescence images of mice implanted with intracranial tumors on days 7, 14, 21 and 28. Tumors comprising LN229 cells were detected in 3/7, 5/7 and 7/7 mice of the G1, G2 and G3 groups, respectively. (C) Body weights of nude mice as measured every 2 days. (D) Kaplan-Meier curve showing that G3 mice had a shorter survival time than G1 and G2 mice. (E) H&E staining showed that LN229 cells grew more aggressively after being mixed with U87 EGFRvIII/PTRF. The scale bar corresponds to 50 μm (upper) and 20 μm (lower).(F) U87EGFRvIII/PTRF (green) and LN229 (red) cells were mixed before being orthotopically injected into mice, and tumor sections observed by confocal microscopy showed that PTRF-EGFP was transferred to LN229 cells (white arrow). The scale bars correspond to 50 μm (upper) and 20 μm (lower).
PTRF is a promising therapeutic target in vitro and in vivo
PTRF was proven to be enriched in mesenchymal GBMs and associated with poor prognosis in GBM patients. To confirm these results, lentiviruses expressing PTRF and PTRF siRNA-1 were used to overexpress PTRF and knock down PTRF, respectively. PTRF overexpression increased the proliferation of the GBM cell lines (Fig. 7A), while PTRF depletion suppressed their proliferation (Fig. 7B), implying that PTRF is a tumor promoter during gliomagenesis.
PTRF expression positively influences GBM cell proliferation in vivo and in vitro. (A) PTRF overexpression increased the proliferation rate of GBM cell lines in vitro. (B) Proliferation of GBM cell lines was inhibited after silencing PTRF. (C) Schematic representation of PTRF silencing in vivo. Bioluminescence was detected every 10 days, and mouse body weights were measured every other day. Mice were injected with U87, U87 PTRF RNAsi-1, U87EGFRvIII or U87EGFRvIII+PTRF siRNA-1. (D) Representative pseudocolor bioluminescence images of mice implanted with intracranial tumors on days 10 and 20. (E) Body weights of orthotopic nude mice were measured every 2 days. (F) Kaplan-Meier curve showing that mice with tumors transduced with PTRF siRNA-1 had better prognoses.
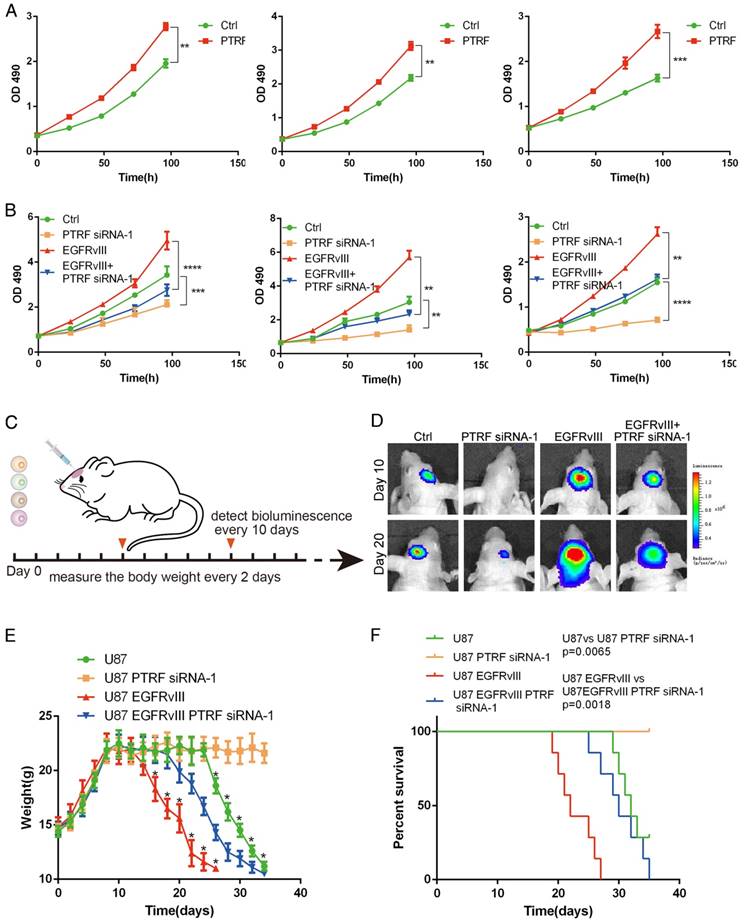
To further confirm that PTRF silencing suppresses GBM proliferation in vivo, we investigated the effects in an orthotopic mouse model by transducing siPTRF-1 lentivirus into U87 and U87EGFRvIII cells (Fig. 7C). Bioluminescence imaging showed a significantly slower growth rate after PTRF knockdown (Fig. 7D). Additionally, mice of the PTRF-suppressing groups lost their body weights at much slower rates during their overall survival time (Fig. 7E). Furthermore, Kaplan-Meier survival curve analysis showed that mice with PTRF-silenced tumors had a better prognosis (Fig. 7F). In conclusion, these data showed that PTRF is a biomarker positively related to GBM progression and that PTRF depletion may be a promising therapeutic strategy for GBM patients.
PTRF as a biomarker of glioma tumor diagnosis
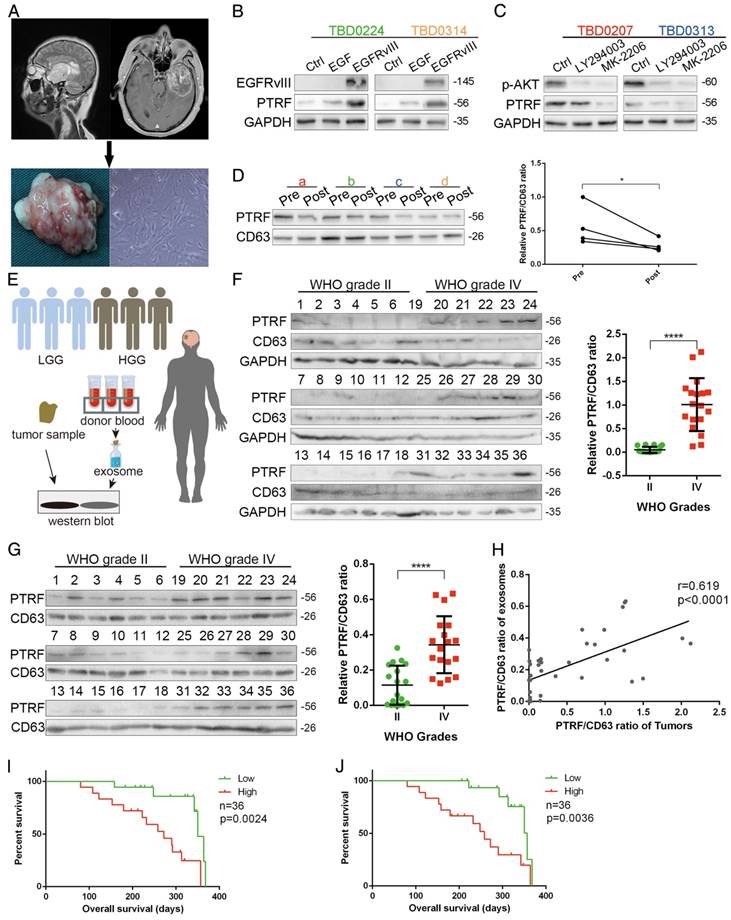
To explore whether PTRF could be a biomarker for glioma tumor diagnosis, we first propagated primary cell lines from glioma samples obtained from 4 different patients (Fig. 8A). PTRF and CD63 expression was detected, showing that the TBD0207 and TBD0313 cell lines had higher levels of endogenous PTRF expression, while the TBD0224 and TBD0314 cell lines had lower levels of endogenous PTRF expression (Fig. S6A). Stimulation by EGF and EGFRvIII in TBD0224 and TBD0314 cells increased PTRF expression (Fig. 8B), and treatment with LY294002 and MK-2206 in TBD0207 and TBD0313 cells decreased PTRF expression (Fig. 8C), showing that PTRF expression is regulated by the EGFRvIII/PI3K/AKT pathway in primary glioma cells. Thereafter, exosomes were isolated from blood samples collected from the four glioma patients one day before and one week after surgery. PTRF and CD63 expression was detected by western blot, and the PTRF/CD63 ratio was significantly decreased in the post-surgery blood samples (Fig. 8D), implying that the PTRF/CD63 ratio can be used as a marker to judge surgical effects or tumor volume. In the TCGA cohort, a higher PTRF/CD63 ratio also indicated poor prognosis (Fig. S6B). To further prove that the PTRF/CD63 ratio can serve as a marker in tumor tissues and exosomes, we analyzed tumor samples and relative donated blood from 18 WHO grade II samples (patients 1-18) and 18 WHO grade IV samples (patients 19-36) (Fig. 8E). A higher PTRF/CD63 ratio at the protein level was detected in WHO grade IV samples in both tumor tissues and serum exosomes (Fig. 8F and G), and PTRF/CD63 ratios in tumor samples and exosomes were positively related (Fig. 8H). According to the clinical data, higher PTRF/CD63 ratios in tumor tissues and serum exosomes also indicated poor prognosis (Fig. 8I and J). Taken together, the above evidence suggested that the PTRF/CD63 ratio can be a biomarker for GBM diagnosis based on the examination of tumor tissues and exosomes. Furthermore, this ratio can also be a surgical effect criterion based on exosome examination.
PTRF down-regulation is detectable after surgery. (A) Primary cell lines were isolated from four glioma patients and named TBD0207, TBD0224, TBD0313 and TBD0314. (B) TBD0224 and TBD0314 stimulated by recombinant EGF and transduced with EGFRvIII exhibited gradually increased PTRF expression, as determined by western blot. (C) p-AKT and PTRF expression was decreased significantly by PI3K and AKT inhibition by LY294002 and MK-2206, respectively, in the TBD0207 and TBD0313 cell lines. (D) Exosomes were isolated from sera of GBM patients before and after surgery. PTRF expression was down-regulated after surgery, as detected by western blot. CD63 was used as the negative control (left panel). Paired t-tests were used to analyze alterations of the PTRF/CD63 ratio (right panel, p<0.05, paired t-test) (E) Schematic illustration of clinical tumor samples and serum exosome detection. In total, 18 grade II and 18 grade IV glioma samples and their relative donated blood samples were analyzed. Total proteins from the tumor samples and exosomes extracted from the sera were lysed. Western blot was used to detect PTRF and CD63 expression. (F) PTRF and CD63 protein expression in 18 grade II and 18 grade IV glioma samples were detected by western blot (left panel). The PTRF/CD63 ratio was significantly higher in WHO grade IV glioma samples than in grade II glioma samples (right panel, p<0.0001, t-test). (G) Exosomes were isolated from sera of 18 grade II and 18 grade IV gliomas, and PTRF and CD63 protein expression was detected by western blot (left panel). The PTRF/CD63 ratio was significantly higher in WHO grade IV glioma samples than in grade II glioma samples (right panel, p<0.0001, t-test). For (B), (C), (D), (G) and (I), GAPDH served as the negative control. (H) The correlation between PTRF/CD63 ratios in tumor tissues and exosomes was analyzed. Each point denotes a tumor sample. Both ratios were positively related (r=0.619, p<0.0001). (I) The overall survival of 36 patients was evaluated according to their PTRF/CD63 ratio. The Kaplan-Meier survival curve showed that the PTRF/CD63 ratio was positively related to poor prognosis (p=0.0024). (J) The overall survival of 36 patients was evaluated according to their PTRF/CD63 ratio. The Kaplan-Meier survival curve showed that the PTRF/CD63 ratio was positively related to poor prognosis (p=0.0036).
Discussion
In the past decade, PTRF/Cavin1 was proven to work in conjunction with Cav family proteins to form caveolae [14], which play critical roles in cellular signaling, endocytosis, lipid and cholesterol homeostasis and cell migration. PTRF is necessary for multidrug resistance (MDR) in breast cancer cells via the fortification of lipid rafts [34] and was shown to promote GBM chemoresistance to imatinib [35]. However, some studies have indicated that PTRF is a tumor suppressor in numerous cancers, including breast [36], colorectal [37], prostate [38] and lung cancer [39]. In this study, we proved that PTRF promotes the proliferation of glioma cells, thus acting as a tumor promoter. PTRF was enriched in classical and mesenchymal GBM subtypes, and its expression was associated with tumor grade, demonstrating that high PTRF expression is correlated with poor prognosis. To date, an increasing number of studies have proven that PTRF plays dual roles in cancer [40].
Although the biological functions of PTRF are well studied, the mechanism underlying PTRF transcriptional regulation is largely unknown. Using TMT and DIA, two important proteomic methods, PTRF was determined to be expressed in U87 EGFRvIII cells at high levels. When these cells were treated with LY294002 and MK-2206, PTRF expression decreased, indicating that PTRF is regulated in the EGFR/PI3K/AKT pathway. AKT has been proven to regulate histone modification in different ways [41, 42], and we also found PTRF to be regulated by AKT via the alteration of H3K4me3 and H3K27me3 modifications. Our gain- or loss-of-function studies showed that H3K4me3 and H3K27me3 enrichment at the promoter region of PTRF was dampened. These results support that histone methylation of the PTRF promoter region plays a major role in the transcriptional regulation of PTRF expression.
The fact that endosomes are formed by caveolae inspired the hypothesis that PTRF may promote the secretion of exosomes, which are small membrane vesicles of endocytic origin secreted by most cell types that are released into the extracellular microenvironment upon the exocytic fusion of MVBs with the plasma membrane [43, 44]. Exosomes play important roles in intercellular communication as natural carriers by delivering functional cargo, including proteins, mRNAs, and miRNAs. Exosomes carrying different RNA and protein components released by diseased patients can be measured as biomarkers [45]. However, the mechanisms involved in the promotion of exosome secretion by PTRF are unclear. In this study, we first demonstrated that PTRF exists on exosome membranes in cell medium supernatants and that EGFRvIII and PTRF increased exosome secretion. Then, exosomes from PTRF-overexpressing cells were transferred to recipient cells, leading to increased rates of tumor formation and proliferation of the recipients in vitro and in vivo.
Cav1, a close partner of PTRF to form caveolea on the plasma membrane, was shown negatively expressed in breast cancer [46], prostate cancer [47] and colon cancer [48]. On the contrary, the high expression of Cav1 in GBM is associated with high EGFR expression [8]. Consistently, our study proved that the expression of Cav1 was positively associated with PTRF, indicating that the caveolea induced by PTRF and Cav1 promotes tumorigenesis. More and more studies have proved that PTRF and Cav1 play the dual role in cancers [40].
Noninvasive diagnosis is an emergent need for glioma patients, and recent genome-wide studies on LGG and HGG have substantially driven biomarker clinical research. The most widely accepted molecular biomarkers are MGMT promoter methylation, co-deletion of 1p and 19q, IDH1/2 mutations and select pathway-associated mutations [49]. However, the MGMT, IDH1/2 mutant and EGFR proteins can currently be detected by only immunohistochemistry [22-24, 50]. EGFRvIII was found to be detectable in serum exosomes with high diagnostic sensitivity [33]. However, EGFRvIII only exists in ~25% of glioma patients [2], and distinguishing low- and high-grade gliomas based on their EGFRvIII levels is difficult because most gliomas with EGFRvIII are HGG. Here, we demonstrated that PTRF can serve as a diagnostic biomarker in not only glioma tissues but also in serum exosomes. The fact that the PTRF/CD63 ratio could predict exosome secretion demonstrates its good criterion ability for glioma diagnosis, prognosis and surgical effects. More importantly, glioma diagnosis based on the PTRF level in serum exosomes is minimally invasive, as it only requires a blood draw.
The overexpression or silencing of PTRF in vitro and in vivo indicated that PTRF plays a critical role in tumor progression. Glioma progression was suppressed by silencing PTRF in an orthotopic mouse model, suggesting that PTRF may be a promising therapeutic target in glioma treatment, although more clinical investigations will be needed for better therapy.
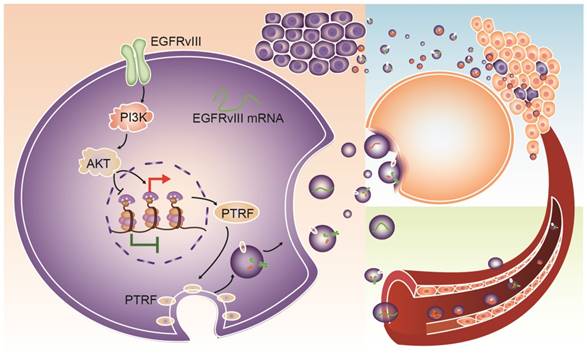
In conclusion, PTRF is a downstream effector of the EGFR/PI3K/AKT pathway via H3K4me3 and H3K27me3 modifications. PTRF enhanced GBM cell proliferation and generated exosomes to transport functional cargo to peripheral and distant cells (Fig. 9), leading to worse prognosis. Importantly, PTRF was detected in glioma tissue and exosomes, suggesting its potential use as a biomarker and as a promising therapeutic target of GBM.
Schematic mechanism for intercellular communication via exosomes. PTRF is a downstream effector of the EGFR/PI3K/AKT pathway via the H3K4me3 and H3K27me3 modifications. Exosomes from GBM cells delivered biological components, such as EGFRvIII, PTRF and EGFRvIII mRNA, to nearby cells and induced their malignancy. Exosomes could also be released into the blood and serve as a detectable biomarker.
Acknowledgements
This work was supported by the National Key Research and Development Program (NO. 2016YFC0902502), the National Natural Science Foundation of China (81772667), the Tianjin Science and Technology Committee (15ZXJZSY00040) and the Training of Clinical Medicine Talents and Basic Research Project in 2015 of Hebei Province (NO. 361007).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061-8
2. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR. et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462-77
3. Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS. et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A. 1992;89:2965-9
4. Ekstrand AJ, James CD, Cavenee WK, Seliger B, Pettersson RF, Collins VP. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 1991;51:2164-72
5. Chu CT, Everiss KD, Wikstrand CJ, Batra SK, Kung HJ, Bigner DD. Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (EGFRvIII). The Biochemical journal. 1997;324( Pt 3):855-61
6. Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD. et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. The Journal of biological chemistry. 1997;272:2927-35
7. Lammering G, Valerie K, Lin PS, Hewit TH, Schmidt-Ullrich RK. Radiation-induced activation of a common variant of EGFR confers enhanced radioresistance. Radiotherapy and oncology: journal of the European Society for Therapeutic Radiology and Oncology. 2004;72:267-73
8. Abulrob A, Giuseppin S, Andrade MF, McDermid A, Moreno M, Stanimirovic D. Interactions of EGFR and caveolin-1 in human glioblastoma cells: evidence that tyrosine phosphorylation regulates EGFR association with caveolae. Oncogene. 2004;23:6967-79
9. Fielding CJ, Fielding PE. Membrane cholesterol and the regulation of signal transduction. Biochemical Society transactions. 2004;32:65-9
10. Pelkmans L, Helenius A. Endocytosis via caveolae. Traffic. 2002;3:311-20
11. Meng F, Joshi B, Nabi IR. Galectin-3 Overrides PTRF/Cavin-1 Reduction of PC3 Prostate Cancer Cell Migration. PloS one. 2015;10:e0126056
12. Glenney JR Jr, Soppet D. Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in Rous sarcoma virus-transformed fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:10517-21
13. Jansa P, Mason SW, Hoffmann-Rohrer U, Grummt I. Cloning and functional characterization of PTRF, a novel protein which induces dissociation of paused ternary transcription complexes. The EMBO journal. 1998;17:2855-64
14. Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ. et al. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell. 2008;132:113-24
15. Liu L, Pilch PF. A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. The Journal of biological chemistry. 2008;283:4314-22
16. Losche W, Scholz T, Temmler U, Oberle V, Claus RA. Platelet-derived microvesicles transfer tissue factor to monocytes but not to neutrophils. Platelets. 2004;15:109-15
17. Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A. et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nature cell biology. 2008;10:619-24
18. Parolini I, Federici C, Raggi C, Lugini L, Palleschi S, De Milito A. et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. The Journal of biological chemistry. 2009;284:34211-22
19. Kowal J, Tkach M, Thery C. Biogenesis and secretion of exosomes. Current opinion in cell biology. 2014;29:116-25
20. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annual review of cell and developmental biology. 2014;30:255-89
21. Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). The Journal of biological chemistry. 1987;262:9412-20
22. Dahlrot RH, Dowsett J, Fosmark S, Malmstrom A, Henriksson R, Boldt H. et al. Prognostic value of O-6-methylguanine-DNA methyltransferase (MGMT) protein expression in glioblastoma excluding nontumour cells from the analysis. Neuropathology and applied neurobiology. 2017
23. Montgomery RM, Queiroz Lde S, Rogerio F. EGFR, p53, IDH-1 and MDM2 immunohistochemical analysis in glioblastoma: therapeutic and prognostic correlation. Arquivos de neuro-psiquiatria. 2015;73:561-8
24. Takano S, Ishikawa E, Sakamoto N, Matsuda M, Akutsu H, Noguchi M. et al. Immunohistochemistry on IDH 1/2, ATRX, p53 and Ki-67 substitute molecular genetic testing and predict patient prognosis in grade III adult diffuse gliomas. Brain tumor pathology. 2016;33:107-16
25. Dong F, Eibach M, Bartsch JW, Dolga AM, Schlomann U, Conrad C. et al. The metalloprotease-disintegrin ADAM8 contributes to temozolomide chemoresistance and enhanced invasiveness of human glioblastoma cells. Neuro-oncology. 2015;17:1474-85
26. Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current protocols in cell biology. 2006 Chapter 3: Unit 3 22
27. Qi H, Liu C, Long L, Ren Y, Zhang S, Chang X. et al. Blood Exosomes Endowed with Magnetic and Targeting Properties for Cancer Therapy. ACS nano. 2016;10:3323-33
28. Qian X, Ren Y, Shi Z, Long L, Pu P, Sheng J. et al. Sequence-dependent synergistic inhibition of human glioma cell lines by combined temozolomide and miR-21 inhibitor gene therapy. Molecular pharmaceutics. 2012;9:2636-45
29. Huang K, Yang C, Wang QX, Li YS, Fang C, Tan YL. et al. The CRISPR/Cas9 system targeting EGFR exon 17 abrogates NF-kappaB activation via epigenetic modulation of UBXN1 in EGFRwt/vIII glioma cells. Cancer letters. 2017;388:269-80
30. Huang K, Yang C, Wang QX, Li YS, Fang C, Tan YL. et al. The CRISPR/Cas9 system targeting EGFR exon 17 abrogates NF-kappaB activation via epigenetic modulation of UBXN1 in EGFRwt/vIII glioma cells. Cancer letters. 2016;388:269-80
31. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010;17:98-110
32. Rheinbay E, Suva ML, Gillespie SM, Wakimoto H, Patel AP, Shahid M. et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell reports. 2013;3:1567-79
33. Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M. et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nature cell biology. 2008;10:1470-6
34. Yi JS, Mun DG, Lee H, Park JS, Lee JW, Lee JS. et al. PTRF/cavin-1 is essential for multidrug resistance in cancer cells. Journal of proteome research. 2013;12:605-14
35. Wang X, Liu T, Bai Y, Liao H, Qiu S, Chang Z. et al. Polymerase I and transcript release factor acts as an essential modulator of glioblastoma chemoresistance. PloS one. 2014;9:e93439
36. Bai L, Deng X, Li Q, Wang M, An W, Deli A. et al. Down-regulation of the cavin family proteins in breast cancer. Journal of cellular biochemistry. 2012;113:322-8
37. Wang F, Zheng Y, Orange M, Yang C, Yang B, Liu J. et al. PTRF suppresses the progression of colorectal cancers. Oncotarget. 2016
38. Aung CS, Hill MM, Bastiani M, Parton RG, Parat MO. PTRF-cavin-1 expression decreases the migration of PC3 prostate cancer cells: role of matrix metalloprotease 9. European journal of cell biology. 2011;90:136-42
39. Gamez-Pozo A, Sanchez-Navarro I, Calvo E, Agullo-Ortuno MT, Lopez-Vacas R, Diaz E. et al. PTRF/cavin-1 and MIF proteins are identified as non-small cell lung cancer biomarkers by label-free proteomics. PloS one. 2012;7:e33752
40. Gupta R, Toufaily C, Annabi B. Caveolin and cavin family members: dual roles in cancer. Biochimie. 2014;107(Pt B):188-202
41. Gherardi S, Ripoche D, Mikaelian I, Chanal M, Teinturier R, Goehrig D. et al. Menin regulates Inhbb expression through an Akt/Ezh2-mediated H3K27 histone modification. Biochimica et biophysica acta. 2017;1860:427-37
42. Liu X, Li Z, Song Y, Wang R, Han L, Wang Q. et al. AURKA induces EMT by regulating histone modification through Wnt/beta-catenin and PI3K/Akt signaling pathway in gastric cancer. Oncotarget. 2016;7:33152-64
43. Harding C, Heuser J, Stahl P. Endocytosis and intracellular processing of transferrin and colloidal gold-transferrin in rat reticulocytes: demonstration of a pathway for receptor shedding. European journal of cell biology. 1984;35:256-63
44. Pan BT, Teng K, Wu C, Adam M, Johnstone RM. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. The Journal of cell biology. 1985;101:942-8
45. Barile L, Vassalli G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacology & therapeutics. 2017;174:63-78
46. Chiu WT, Lee HT, Huang FJ, Aldape KD, Yao J, Steeg PS. et al. Caveolin-1 upregulation mediates suppression of primary breast tumor growth and brain metastases by stat3 inhibition. Cancer research. 2011;71:4932-43
47. Ayala G, Morello M, Frolov A, You S, Li R, Rosati F. et al. Loss of caveolin-1 in prostate cancer stroma correlates with reduced relapse-free survival and is functionally relevant to tumour progression. The Journal of pathology. 2013;231:77-87
48. Bender FC, Reymond MA, Bron C, Quest AF. Caveolin-1 levels are down-regulated in human colon tumors, and ectopic expression of caveolin-1 in colon carcinoma cell lines reduces cell tumorigenicity. Cancer research. 2000;60:5870-8
49. Staedtke V, Dzaye ODA, Holdhoff M. Actionable Molecular Biomarkers in Primary Brain Tumors. Trends in cancer. 2016;2:338-49
50. Chaurasia A, Park SH, Seo JW, Park CK. Immunohistochemical Analysis of ATRX, IDH1 and p53 in Glioblastoma and Their Correlations with Patient Survival. Journal of Korean medical science. 2016;31:1208-14
Author contact
![]() Corresponding authors: Chunsheng Kang, PhD, Tianjin Neurological Institute, Key Laboratory of Post-neurotrauma Neuro-repair and Regeneration in Central Nervous System, Ministry of Education and Tianjin City, Tianjin 300052, China. Tel: +86-022-6081-7499, Fax: +86-022-2781-3550, E-mail: kang97061edu.cn. and Min Li, PhD, Department of Medicine, Department of Surgery, the University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104. Tel: (405) 271-1796, Fax: (405) 271-1476, Email: Min-Liedu. and Jianning Zhang, M.D, Department of Neurosurgery, Tianjin Medical University General Hospital, Tianjin 300052, Tianjin China. Tel: +86-022-8333-6717, E-mail: jianningzhangcom.
Corresponding authors: Chunsheng Kang, PhD, Tianjin Neurological Institute, Key Laboratory of Post-neurotrauma Neuro-repair and Regeneration in Central Nervous System, Ministry of Education and Tianjin City, Tianjin 300052, China. Tel: +86-022-6081-7499, Fax: +86-022-2781-3550, E-mail: kang97061edu.cn. and Min Li, PhD, Department of Medicine, Department of Surgery, the University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104. Tel: (405) 271-1796, Fax: (405) 271-1476, Email: Min-Liedu. and Jianning Zhang, M.D, Department of Neurosurgery, Tianjin Medical University General Hospital, Tianjin 300052, Tianjin China. Tel: +86-022-8333-6717, E-mail: jianningzhangcom.