Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(6):1511-1526. doi:10.7150/thno.21986 This issue Cite
Research Paper
Elevated Expression of miR302-367 in Endothelial Cells Inhibits Developmental Angiogenesis via CDC42/CCND1 Mediated Signaling Pathways
Jingjiang Pi1,*, Jie Liu1,*, Tao Zhuang1,*, Lin Zhang1, Huimin Sun1, Xiaoli Chen1, Qian Zhao1, Yashu Kuang1, Sheng Peng1, Xiaohui Zhou1, Zuoren Yu1, Ting Tao2, Brian Tomlinson3, Paul Chan4, Ying Tian5, Huimin Fan1, Zhongmin Liu1, Xiangjian Zheng6, Edward Morrisey7, Yuzhen Zhang1, ![]()
1. Key Laboratory of Arrhythmias of the Ministry of Education of China, Research Center for Translational Medicine, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, 200120, China.
2. Department of Geriatrics, Ruijin Hospital, Jiaotong University, School of Medicine, Shanghai, 200025, China.
3. Department of Medicine and Therapeutics, The Chinese University of Hong Kong, Hong Kong SAR, China
4. Division of Cardiology, Department of Internal Medicine, Wan Fang Hospital, Taipei Medical University, Taipei, Taiwan
5. Department of Pharmacology, Center for Translational Medicine, Temple University School of Medicine, Philadelphia, PA 19140, USA.
6. Lab of Cardiovascular Signaling, Centenary Institute, Camperdown, NSW 2050, Australia.
7. Department of Cell and Developmental Biology (R.W., E.E.M.), Department of Medicine (E.E.M.), Penn Cardiovascular Institute (E.E.M.), and Penn Institute for Regenerative Medicine (E.E.M.), University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA.
* These authors contributed equally to this work
Received 2017-7-18; Accepted 2017-11-14; Published 2018-2-5
Abstract

Rationale: Angiogenesis is critical for embryonic development and microRNAs fine-tune this process, but the underlying mechanisms remain incompletely understood.
Methods: Endothelial cell (EC) specific miR302-367 line was used as gain-of-function and anti-miRs as loss-of-function models to investigate the effects of miR302-367 on developmental angiogenesis with embryonic hindbrain vasculature as an in vivo model and fibrin gel beads and tube formation assay as in vitro models. Cell migration was evaluated by Boyden chamber and scratch wound healing assay and cell proliferation by cell count, MTT assay, Ki67 immunostaining and PI cell cycle analysis. RNA high-throughput sequencing identified miR-target genes confirmed by chromatin immunoprecipitation and 3'-UTR luciferase reporter assay, and finally target site blocker determined the pathway contributing significantly to the phenotype observed upon microRNA expression.
Results: Elevated EC miR302-367 expression reduced developmental angiogenesis, whereas it was enhanced by inhibition of miR302-367, possibly due to the intrinsic inhibitory effects on EC migration and proliferation. We identified Cdc42 as a direct target gene and elevated EC miR302-367 decreased total and active Cdc42, and further inhibited F-actin formation via the WASP and Klf2/Grb2/Pak1/LIM-kinase/Cofilin pathways. MiR302-367-mediated-Klf2 regulation of Grb2 for fine-tuning Pak1 activation contributing to the inhibited F-actin formation, and then the attenuation of EC migration. Moreover, miR302-367 directly down-regulated EC Ccnd1 and impaired cell proliferation via the Rb/E2F pathway.
Conclusion: miR302-367 regulation of endothelial Cdc42 and Ccnd1 signal pathways for EC migration and proliferation advances our understanding of developmental angiogenesis, and meanwhile provides a rationale for future interventions of pathological angiogenesis that shares many common features of physiological angiogenesis.
Keywords: miR302-367, endothelial cells, developmental angiogenesis, cdc42, actin remodeling, cell cycle.
Introduction
Angiogenesis is critical both for physiological processes and in various diseases (1). The formation of blood vessels is a complex process and developmental and pathological angiogenesis share many common features. Therefore better understanding of the key regulatory mechanisms of developmental angiogenesis should help in the development of novel therapeutic strategies for pathological angiogenesis related diseases.
Angiogenesis is defined as the formation of new vessels by the sprouting of endothelial cells (ECs) of pre-existing vasculature. It commences in the embryo by E9.5 mediating the formation of the majority of embryonic blood vessels, which is responsible for the vascularization of organs including the brain (2). The hindbrain is vascularized early at E9.75, first sprouting from the perineural vascular plexus and growing rapidly towards the ventricular zone. From E10.0 the radial vessels extend sprouts laterally and meet to form a subventricular vascular plexus and perfuse vascular networks (3). Whole-mount immunolabelling of the embryonic hindbrain surface blood vessels and high-resolution imaging with easy quantification of the angiogenic sprouting network density permits the spatiotemporal analysis of its vascularization. Therefore, mouse embryonic hindbrain is a robust and adaptable model for studying in vivo sprouting angiogenesis and the underlying mechanisms (4).
It is now well understood that sprouting angiogenesis is a coordinated series of events centered on ECs involving migration, proliferation and remodeling (5). Cell migration involves actin remodelling for extension of filopodia and lamellipodia at the leading edge (6) and Rho-GTPase Cdc42 represents the central signal mechanism controlling this essential process during angiogenesis (7). The underlying mechanisms of cell proliferation have been extensively investigated and the cyclin D1 mediated retinoblastoma protein (Rb)/cyclin-dependent kinases (CDKs) pathway plays a key role in the transition of cells from G0 to S for cell proliferation in response to mitogenic signals (8, 9).
MicroRNAs, a new class of small RNA molecules, have emerged as key regulators of several cellular processes, including angiogenesis at a post-transcriptional level by targeting multiple pathways, therefore, microRNAs can represent a future therapeutic target for the treatment of pathological neovascularization-related diseases (10). MiR302-367 is expressed at high levels in embryonic stem cells (11, 12) and recent studies demonstrated that miR302-367 directed lung endoderm development, promoted mammalian cardiac repair and regeneration, and prevented tumor growth via restricting angiogenesis and improving vascular stability (13-15), but its role in embryonic developmental angiogenesis and the underlying mechanisms have not yet been fully elucidated.
Here we used a well-established mouse embryonic hindbrain angiogenesis model to look for the effects of gain- or loss-of-function of miR302-367 in ECs on embryonic developmental angiogenesis. We found that miR302-367 inhibited embryonic developmental angiogenesis through impaired cell migration and proliferation. Moreover, miR302-367 directly down-regulated Cdc42, leading to the reduction of F-actin formation via the Wasp pathways. In addition, miR302-367 mediated Klf2 upregulation inhibited Grb2 and fine-tuned Pak1 activation, and in turn the LIM-kinase/Cofilin pathway, together resulting in inhibition of EC migration. Finally, miR302-367 directly targeted Cyclin D1 (Ccnd1) leading to impaired cell proliferation via the Rb/E2F pathway. Taken together, EC-expressing miR302-367 intrinsically regulated EC migration and proliferation through multiple target genes, which are essential for embryonic developmental angiogenesis.
Results
Elevated expression of miR302-367 specifically in vascular endothelial cells reduces angiogenesis during embryonic development
Our previous investigation showed that elevated expression of miR302-367 restricted post-natal angiogenesis and tumor angiogenesis. However, miR302-367 expression levels were higher at E9.5-11.5, and their expression sharply decreased after E15.5 and were kept at low level after birth, suggesting that miR302-367 might display more important physiological effects in embryonic developmental angiogenesis. It is known that angiogenesis commences at E9.5 embryo and forms perfused vascular networks in the hindbrain from E10.5 (3), thus we proposed that miR302-367 might regulate hindbrain angiogenesis during embryonic development.
To investigate the cell lineage-specific mechanisms of miR302-367 on embryonic developing angiogenesis, EC specific R26R-miR302-367Tg/+; Cdh5(PAC)-CreERT2 (miR302-367ECTg) mice were generated by crossing the gain-of-function mouse miR302-367 (14) with the Cdh5 (PAC)-CreERT2 line (16).
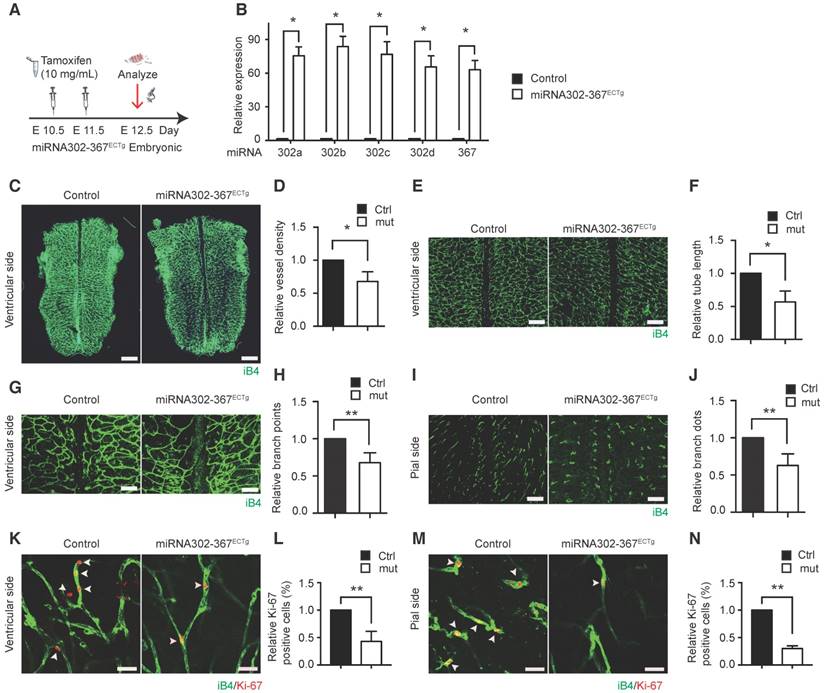
Tamoxifen administration for induction of elevated miR302-367 expression in mouse hindbrain ECs was shown in the schematic chart (Fig. 1A) and elevated miR302-367 expression was observed in miR302-367ECTg mutants compared to the littermate control mice (Fig. 1B). MiR302-367ECTg mutant mice exhibited a marked reduction of vessel density (Fig. 1C-D), tube length (Fig. 1E-F) and branching points (Fig. 1G-H) of the hindbrain ventricular side, and branching dots (Fig. 1I-J) of the pial side. This reduced developmental hindbrain angiogenesis correlated with a loss in EC proliferation, as shown by reduced Ki67/iB4 co-immunostaining of ventricular (Fig. 1K-L) and pial (Fig. 1M-N) sides at E12.5 embryonic hindbrain. The similar elevated expression of miR302-367 was found in retinal ECs of miR302-367ECTg mutant mice (Fig. S1A) and the mutant mice exhibited a significant reduction of retinal angiogenesis (Fig. S1B-D) as well as reduction of EC proliferation shown by reduced Ki67/iB4 co-immunostaining (Fig. S1E-F). In addition, elevated expression of miR302-367 in vascular smooth muscle cells (VSMCs) by a VSMC specific inducible Cre line SMMHC-CreER(T2) (17) was verified by qPCR (Fig. S2A) and the mutant mice miR302-367SMCTg exhibited no significant change of vessel density in the hindbrain and retina (Fig. S2B-G). These data indicate that elevated miR302-367 specifically in ECs reduces developmental angiogenesis through inhibition of EC proliferation.
Elevated expression of miR302-367 in vascular endothelial cells reduces sprouting angiogenesis and cell proliferation in embryonic hindbrain angiogenesis in vivo model. (A) The schematic chart of tamoxifen administration for induction of elevated miR302-367 expression in endothelial cells of E12.5 embryonic hindbrain. (B) miR302-367ECTg mutant mice exhibit significantly elevated miR302-367 expression in hindbrain endothelial cells compared to the littermate control mice. (C-J) Whole mount IB4 staining of E12.5 embryonic hindbrain in the WT and miR302-367ECTg mutant mice indicates significant reduction of hindbrain angiogenesis at ventricular side quantified by vessel density (C-D), tube length (E-F), branch points (G-H), and at pial side quantified by branch dots (I-J). (K-N) The reduced embryonic hindbrain angiogenesis correlates with loss of endothelial cell proliferation, as shown by reduced Ki67/IB4 co-immunostaining of both ventricular and pial sidesat E12.5 embryonic hindbrain. Data quantification is mean ± S.E.M (n = 6). All data were analyzed using Student's t-test unless otherwise noted. *, P<0.05; **, P<0.01. Scale bars: C, 2 mm; E, 500 μm; G, I, 250 μm; K-M, 10 μm.
Reduced expression of miR302-367 by anti-miRs enhances embryonic developmental angiogenesis
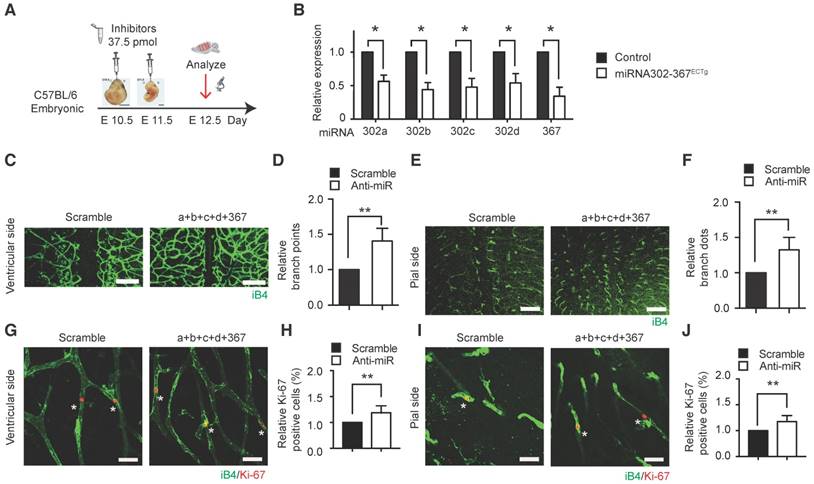
Inhibition of miR302-367 expression by anti-miRs was used to test miR302-367 loss-of-function in embryonic hindbrain developmental angiogenesis. Anti-miRs were administrated into mice via in utero electroporation to reduce miR302-367 expression as shown in the schematic chart (Fig. 2A), and a significant reduction of miR302-367 expression was confirmed in hindbrain ECs (Fig. 2B). The mice administrated with anti-miRs exhibited significantly increased branch points in both ventricular and pial sides of the hindbrain (Fig. 2C-F), as well as a significantly increased Ki67 staining in ECs (Fig. 2G-J) compared with the mice administrated with scramble miRNA, which was consistent with our gain-of-function study.
Inhibition of miR302-367 expression in hindbrain endothelial cells by anti-miRs enhances cell proliferation and embryonic hindbrain angiogenesis in vivo. (A) The schematic chart of anti-miRs administration for reduction of miR302-367 expression in endothelial cells of embryonic hindbrain. (B) Anti-miRs significantly reduce the miR302-367 expression of ECs in embryonic hindbrain compared to the scramble miRNA. (C-F) The mice administrated with anti-miRs exhibit significantly increased branch points in both ventricular and pial sides of embryonic hindbrain. (G-J) The increased embryonic hindbrain angiogenesis correlates with increase in endothelial cell proliferation, as shown by elevated Ki67/IB4 co-immunostaining of ventricular and pial sides at E12.5 embryonic hindbrain. Data quantification is mean ± S.E.M (n = 6). All data were analyzed using Student's t-test unless otherwise noted. *, P<0.05; **, P<0.01. Scale bars: C, E, 250 μm; G, I, 10 μm.
Elevated expression of miR302-367 attenuates in vitro sprouting angiogenesis through inhibition of migration and proliferation of endothelial cells
To better understand the molecular mechanisms of elevated EC miR302-367 expression responsible for the reduction of in vivo developmental angiogenic activity, we next analyzed the effects of miR302-367 on in vitro angiogenic activity of ECs using fibrin gel beads and tube formation assays, which mimic the multiple key steps of in vivo angiogenic processes including EC proliferation and migration.
Lentivirus carrying miR302-367 (Lentiviral-miR302-367) or nonfunctional control (Lentiviral-GFP) was generated to achieve elevated expression of miR302-367 in human umbilical vein endothelial cells (HUVECs). All members of miR302-367 cluster levels were significantly increased in HUVECs by infection of lentiviral-miR302-367 compared to lentiviral-GFP control (Fig. S2H). In the fibrin gel beads assay we observed markedly attenuated sprout angiogenic activity when miR302-367 expression was elevated in HUVECs, as shown by a significant reduction of sprouts (Fig. 3A, red points), the numbers of scattered cells (Fig.3A, blue points) and the quantification, respectively (Fig. 3B-C). The same markedly attenuated angiogenic tube formation with decreased branch points and total tube length was observed in HUVECs when miR302-367 expression was elevated (Fig. 3D-F). These data confirmed the reduction of in vitro sprout angiogenic activity upon elevated expression of miR302-367 in ECs, which is in agreement with the in vivo anti-angiogenic effect of elevated EC miR302-367 expression.
Elevated expression of miR302-367 in HUVECs reduces in vitro sprouting angiogenesis, cell migration and proliferation. (A-C) Using fibrin beads assay HUVECs with elevated miR302-367 expression display a profound attenuation of sprout angiogenic activity as shown by a significant reduction of sprouts (A, red points) and the numbers of scattered cells (A, blue points). (D-F) HUVECs with elevated miR302-367 expression have significant inhibition of tube formation as shown by a significant reduction of branch points and total tube length. (G-J) The reduced in vitro angiogenesis correlates with a significant reduction of endothelial cell migration as shown by decreased migrated cell numbers in Boyden chamber assay, and increased blank area in scratch wound healing assay upon elevated miR302-367 expression. (K-N) The reduced in vitro angiogenesis also correlates with loss of endothelial cell proliferation, as shown by reduced Ki67 staining of HUVECs and less cell growth measured by cell count and MTT assay upon elevated miR302-367 expression. (O-Q) Cell cycle analysis shows more cells stayed in G0/G1-phase and fewer cells in S-phase when miR302-367 expression was elevated in HUVECs. Data quantification is mean ± S.E.M (n = 3). All data were analyzed using Student's t-test unless otherwise noted. n.s., no significance; *, P<0.05; **, P<0.01.Scale bars: A, 100 μm; D, 100 μm; G, 100 μm; I, 200 μm; K, 50 μm.
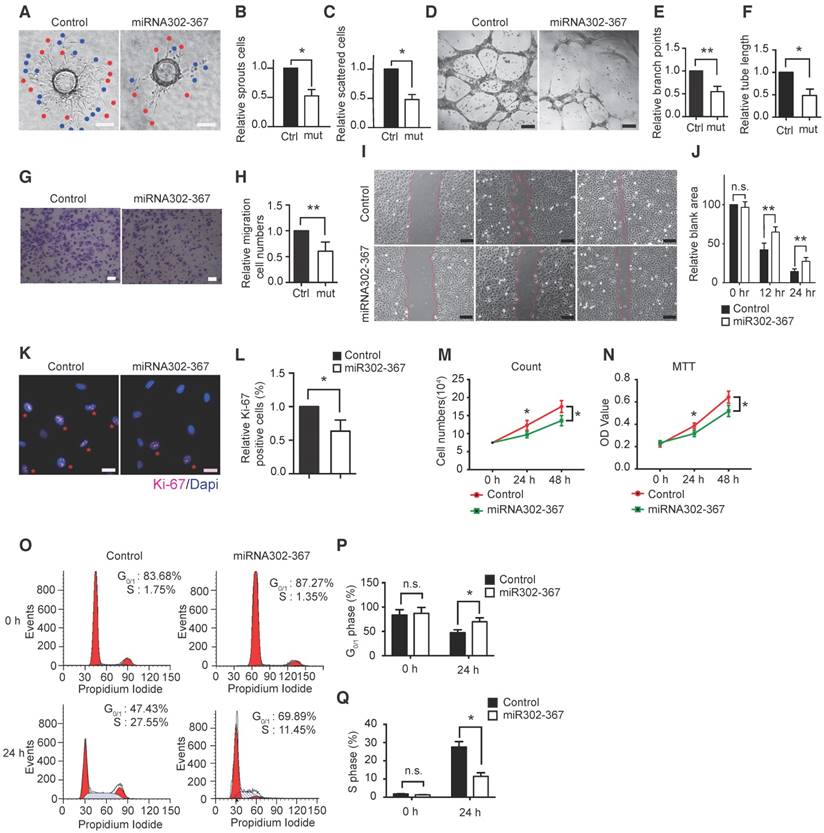
During vascular development, coordinated control of EC behavior at the levels of cell migration and proliferation is critical for functional blood vessel morphogenesis. To further explore the mechanisms of elevated miR302-367 expression on reduced angiogenic activity in vivo and in vitro, cell migration and cell proliferation studies were performed in HUVECs. Elevated expression of miR302-367 in HUVECs markedly decreased cell migration as shown by the Boyden chamber assay (Fig. 3G-H) and scratch wound healing assay (Fig. 3I-J), suggesting significantly less EC migration when miR302-367 expression was increased in HUVECs. Furthermore, we transferred Life-act-GFP plasmids into HUVECs to observe the actin dynamics and cell motility and the results showed significantly slower cell motility in the lentiviral-miR302-367 group compared with the lentiviral-GFP control (Fig. S3A-B, Video S1) with reduced cell center movement trajectory (Fig. S3C) and cell movement velocity (Fig. S3D). Moreover, elevated miR302-367 expression reduced the formation of filopodias and lamellipodias (Fig. S3E-F) which drived the cellular locomotion, and it also enhanced the formation of actin stress fibers which are essential for cell adhesion (Fig. S3G), while no significant difference of the cortex actin distribution was observed between the miRNA302-367 and control groups (Fig. S3H). These data suggest that miR302-367 control cell motility via an influence on actin dynamic to regulate EC behaviors including filopodias and lamellipodias formation.
Significantly less Ki67 positive staining cells (Fig. 3K-L) and reduced cell growth shown by fewer cell counts (Fig. 3M) and lower MTT OD value (Fig.3N) were also observed in HUVECs upon elevated expression of miR302-367, suggesting less EC proliferation occurred when miR302-367 expression was elevated in HUVECs. In addition, cell cycle analysis confirmed that more cells stayed in the G0/G1-phase and fewer cells were in S-phase when expression of miR302-367 was elevated in HUVECs (Fig. 3O-Q), while no significant changes in cleaved caspase-3 expression were observed in both basal and TNFα treated condition between the control and miRNA302-367 groups (Fig. S4A). Taken together, these results suggest that elevated expression of miR302-367 in ECs inhibits cell migration and proliferation, but not apoptosis, which contribute to the attenuated angiogenic activity.
Cdc42 is identified as the direct target of miR302-367 and the Cdc42-Wasp pathway regulates F-actin formation contributing to the miR302-367-mediated reduced cell migration and sprouting angiogenesis
MicroRNAs are paired with mRNAs to direct post-transcriptional repression by mRNA cleavage, or more commonly through mRNA direct translational repression, mRNA destabilization, or a combination of both (11). To identify the target genes of miR302-367 contributing to the reduced developmental angiogenesis, we performed RNA high-throughput sequencing of mouse lung eGFP-ECs obtained by FACS sorting the cells from R26R-miR302-367Tg/+;Cdh5(PAC)-CreERT2; R26R-tdTomato-EGFP mutant or corresponding WT control reporter mice and the results were shown in supplemental table 1. Together with analysis of microRNA database TargetScan, we identified Cdc42 as a possible target of miR302-367. Elevated expression of miR302-367 markedly reduced CDC42 expression in HUVECs (Fig. 4A) which was confirmed by reduced CDC42 total protein, especially the activated protein level and overall CDC42-GTPase activity in HUVECs (Fig.4B-C). We further separated ECs from hindbrain and found similar reduction in RNA and protein levels (Fig. S4B-C) without significant influence on RhoA and Rac1 protein expression (Fig. S4D). To confirm Cdc42 as a direct target gene of miR302-367, we performed sequence analysis and found miR302-367 binding sites in the 3'-UTR of CDC42 (Fig. 4D). Furthermore, we found that miR302-367 suppressed the luciferase activity, while mutation of the seed sequences of the miR302-367 binding site restored the luciferase activity (Fig. 4E). Similarly repressed luciferase activity was observed by individual miR302a/b/c/d mimics, miR367 mimic and most significantly by mimic mixture and miR302-367 vector (Fig. 4F). To clarify whether the inhibitory effect of miR302-367 on EC migration and sprouting angiogenesis is CDC42-dependent, elevated CDC42 expression via transfer of constitutively active CDC42 plasmids into HUVECs was performed. Constructive active CDC42 was able to reverse the reduced cell migration (Fig. 4G-H, Fig. S5A-B), as well as the attenuated angiogenic sprouts (Fig. 4I-J) and tube formation (Fig. S5C-D) in HUVECs upon elevated expression of miR302-367. Similar reversal of cell migration by constructive active CDC42 was observed under hypoxia conditions (Fig. S5E-F). Beyond regulation of cell migration, elevated CDC42 expression in HUVECs could reverse the reduced cell proliferation upon elevated miRNA302-367 expression, but the reversal degree was not as high as the elevated expression of CCND1 (Fig. S6A-C). Taken together, these results indicate that miR302-367 inhibits cell migration and proliferation contributing to the attenuated sprouting angiogenesis through the CDC42 signal pathway.
CDC42 is a direct target gene of miR302-367 and CDC42-WASP pathway contributes to actin remodeling and inhibition of cell migration in HUVECs. (A) qPCR results confirm the reduced CDC42 expression in HUVECs with elevated miR302-367 expression. (B-C) Elevated miR302-367 in HUVECs reduces total CDC42 protein level, and more significantly active CDC42 by western blot and by G-Lisa kit. (D) Sequence analysis shows miR302-367 binding sites in 3'-UTR of CDC42. (E) miR302-367 represses the luciferase reporter activity of CDC42 3'-UTR, while mutation of the seed sequence of miR302-367 binding site in 3'-UTR of CDC42 abolishes the luciferase repression. (F) Similar repression of the luciferase activity was observed by individual miR302a/b/c/d, miR367 mimic, and more significantly by miR302-367 mimic mixture or vector of miR302-367, and mutation of the binding site in 3'-UTR of CDC42 abolishes all individual miR302-367 mimic luciferase repression. (G-J) Elevated CDC42 expression by plasmid transfer in HUVECs is able to rescue the reduced cell migration (G-H), as well as angiogenic sprouts (I-J) when miR302-367 expression is elevated. (K) miR302-367ECTg mutant mice exhibit significantly decreased F-actin formation and increased free G-actin in endothelial cells of hindbrain and retina compared to the WT littermate control mice, as well as in HUVECs with elevated miR302-367 expression by transfer of the lentiviral-miR302-367 compared to the lentiviral-GFP control. (L-N) Constitutively active CDC42 reverses the reduction of total CDC42, CDC42-GTPase activity and phospho-WASP (L-M), further reverses the reduction of F-actin and elevation of G-actin (N) in HUVECs with elevated expression of miR302-367. Data quantification is mean ± S.E.M (n = 3). All data were analyzed using Student's t-test unless otherwise noted. *, P<0.05; **, P<0.01. Scale bars: G, 100 μm; I, 100 μm.
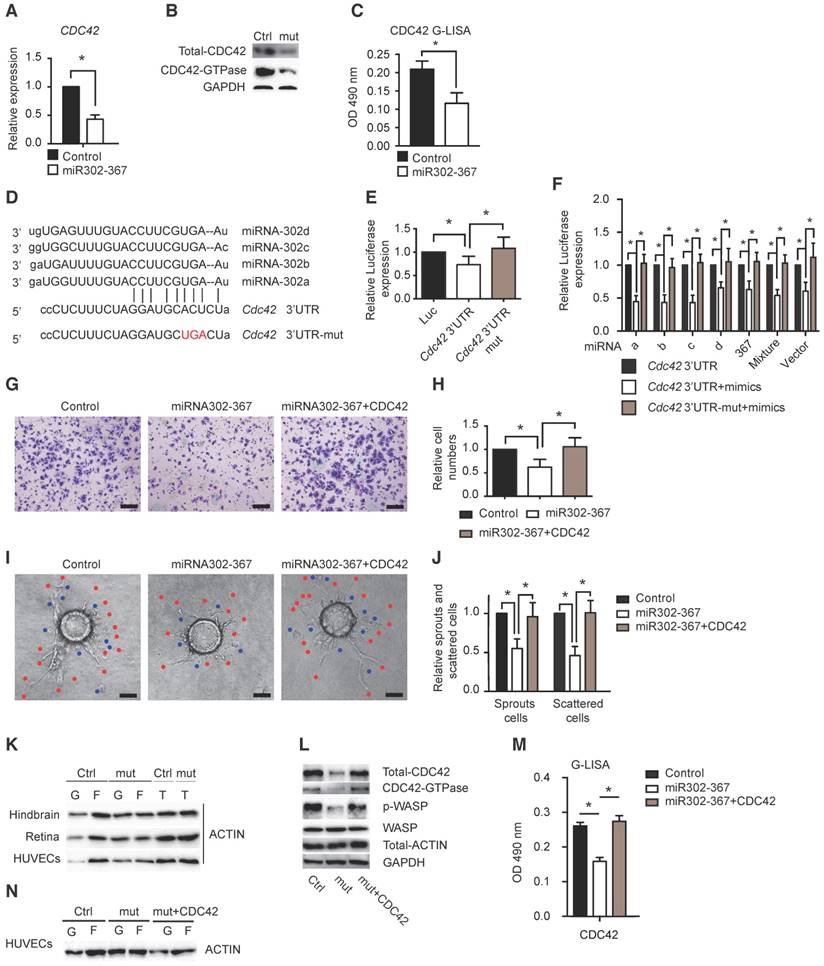
Actin is present in two forms within the cells, the monomeric globular molecule G-actin and the double-stranded filamentous polymer F-actin. In non-muscle cells, the actin cytoskeleton is a highly dynamic structure. G-actin has the ability to polymerize and form F-actin, and F-actin can depolymerize to G-actin. The ratio of G- and F-actin can alter rapidly and F-actin is primarily involved in crucial cellular processes, such as cell migration and division (18). In our study, we found that elevated expression of miR302-367 significantly increased G-actin but decreased F-actin in embryonic hindbrain ECs of miR302-367ECTg mutant mice compared to the WT littermate control mice. Similar results were observed in retinal ECs, as well as in HUVECs when miR302-367 expression was elevated by lentiviral-miR302-367 transfer compared to lentiviral-GFPcontrol (Fig. 4K). These data suggested that miR302-367-mediated reduced F-actin formation might be responsible for the attenuation of cell migration and sprouting angiogenesis.
Cdc42 has a conserved role in regulation of the actin cytoskeleton in many eukaryotic organisms and has been shown to have a role in migratory polarity. Constitutively active Cdc42 and dominant negative Cdc42 affect the formation of highly dynamic finger-like actin-rich protrusions known as filopodia, which are thought to be important for axon guidance and growth (19). Moreover Cdc42 was found to directly interact with and activate Wiskott-Aldrich syndrome protein (Wasp) family proteins, further activate the Arp2/3 complex and subsequently bind to the side of an actin filament and promote G-actin nucleation and F-actin formation for initiation of filopodia formation (20, 21), which is known to be important in axon guidance and growth, as well as cell migration and angiogenic sprouts. Our study found that elevated expression of miR302-367 in ECs increased G-actin but decreased F-actin (Fig. 4K), and CDC42 was identified as direct target of miR302-367 in ECs. Therefore, we hypothesized that miR302-367-mediated change of CDC42 and downstream pathways in ECs might have an influence on F-actin formation, and further on EC migration and angiogenic sprouts.
As expected, our findings showed that elevated miR302-367 expression in HUVECs decreased the expression of CDC42 and the downstream target gene WASP phosphorylation, whereas constitutively active CDC42 reversed the reduction of CDC42 GTPase activity and phospho-WASP (Fig.4L-M), and further reversed the reduction of F-actin (Fig.4N). Therefore, we conclude that miR302-367 attenuates EC migration possibly through the CDC42/WASP-mediated actin remodeling pathway.
MiR302-367 directly targets Cdc42 and regulates F-actin formation via the Pak1/LIM-kinase/Cofilin pathway with miR302-367-mediated Klf2 regulation of Grb2 for fine-tuning Pak1 activation
Another small GTP binding protein Cdc42 effector, p21-activated kinase family 1 (Pak1), is the first identified and most well-studied protein as an important positive regulator for actin dynamic function (22-24). Activated Pak1 could phosphorylate LIM-kinase, then actin-regulatory protein Cofilin leading to inactivation of F-actin depolymerization and increase of F-actin (25). Our study found that elevated miR302-367 expression in HUVECs significantly reduced active phospho-PAK1 (Fig. 5A) and LIM-kinase, producing more active Cofilin (Fig. 5L) to depolymerize actin without significant influence on other PAK isoforms, phospho-PAK2 and 4. This indicated that the CDC42/PAK1/LIM-kinase/Cofilin pathway might be involved in miR302-367-mediated F-actin reduction in ECs. To our surprise, constitutively active CDC42 was not able to fully reverse the reduction of PAK1 activation in miR302-367 highly expressing ECs (Fig. 5B), which indicating that other signaling molecule besides CDC42 might be involved in the disturbed PAK1/LIM-kinase/Cofilin pathway in ECs with elevated miR302-367 expression.
MiR302-367-mediated CDC42-PAK1-LIM kinase-Cofilin pathway regulates F-actin formation with miR302-367-ERK1/2-KLF2-GRB2 pathway fine-tuning the PAK1 activation. (A) Elevated miR302-367 expression in HUVECs significantly reduces phospho-PAK1 without significant change of phospho-PAK2 or PAK4. (B) Constitutively active CDC42 is not able to reverse the reduction of PAK1 activation in miR302-367 mutant HUVECs when high KLF2 expression is present. (C) Active CDC42 is not able to reverse the PAK1 activation in HUVECs with high KLF2 expression when the cells are stimulated by simvastatin. (D) KLF2 inhibition by siRNA increases GRB2 and PAK1 expression in HUVECs without change of PAK2 and PAK4. (E) KLF2 inhibition by siRNA reverses the reduced GRB2 expression, but cannot reverse the inhibition of PAK1 activation, in HUVECs with elevated miR302-367 expression. (F) Sequence analysis shows four KLF2 binding sites at GRB2 promoter region. (G) The proximal-1kb GRB2 promoter containing KLF2 binding sites is repressed by KLF2 in a dose-dependent manner. (H) ChIP assay shows association of KLF2 with GRB2 promoter in HUVECs by qPCR using GAPDH promoter as negative control. (I) Elevated miR302-367 expression in HUVECs significantly increases KLF2 binding to GRB2 when high KLF2 expression is present. (J) Co-IP with PAK1 antibody pulls down less GRB2 protein in HUVECs with elevated miR302-367 expression. (K) Co-IP with GRB2 antibody pulls down similar amount PAK1, and less PAK1 band due to decreased GRB2 in HUVECs with elevated miR302-367 expression. (L) In HUVECs when KLF2 is inhibited by siRNA, the constitutively active CDC42 reverses the miR302-367-mediated reduction of GRB2 and PAK1 activation, and in turn increases phospho-LIM-kinase, and phospho-Cofilin. (M) Phospho-PAK1 immunostaining shows significantly reduced phospho-PAK1 expression in the membrane of HUVECs with elevated miR302-367 expression, and constitutively active CDC42 is not able to reverse the reduced phospho-PAK1 staining in mutant HUVECs, but significantly increases the staining when higher KLF2 is inhibited by siRNA. Data quantification is mean ± S.E.M (n = 3). All data were analyzed using Student's t-test unless otherwise noted. n.s., no significance; *, P<0.05; **, P<0.01;***, P<0.005. Scale bars: M, 25 μm.
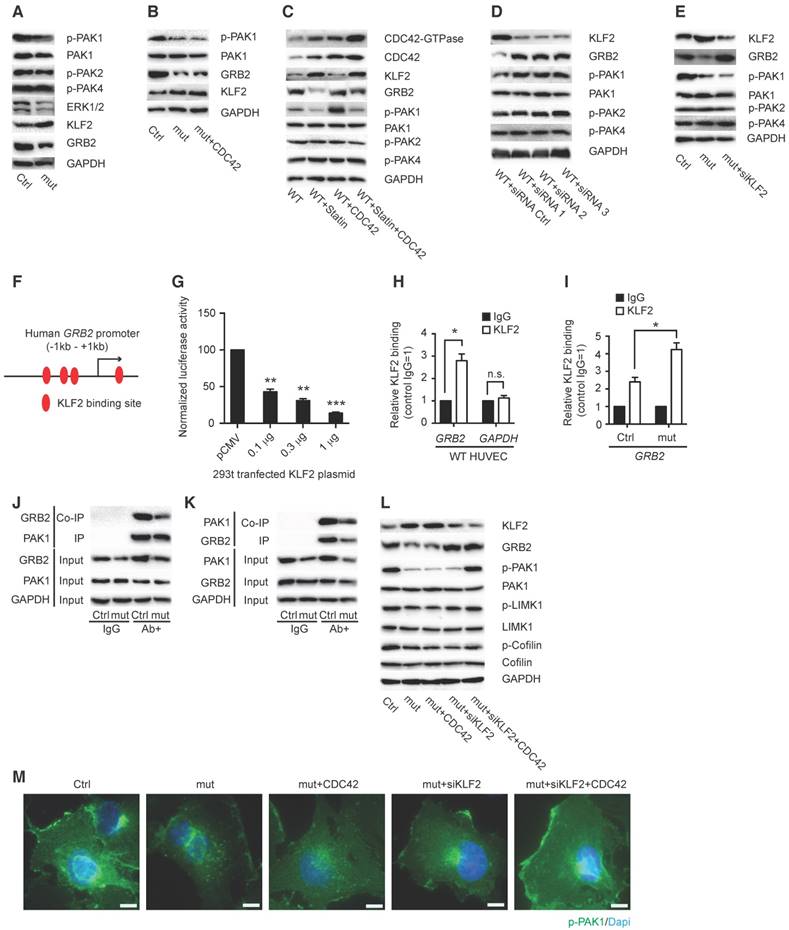
In resting cells, Pak1 is mainly localized in the cytosol and translocated to membrane ruffles and lamellipodia when cells are activated (21, 26). Adaptor protein Grb2 was reported to interact with Pak1 and bring it to the plasma membrane, where Cdc42 GTPases can activate it (27, 28), which further recruit and/or stimulate additional downstream signaling pathways leading to cytoskeletal rearrangement in immune cells (29). Therefore, we hypothesized that miR302-367 might regulate Grb2 and fine-tune Pak1 activation in ECs as well.
Our previous study showed that miR302-367 inhibited retinal and tumor angiogenesis via the Erk1/2-Klf2-S1pr1 pathway (15). In the present study we confirmed increased KLF2 expression and a profoundly reduced expression of CDC42, PAK1 and GRB2 in HUVECs (Fig.5A), and constitutively active CDC42 domain was not able to reverse the reduced PAK1 expression (Fig.5B). A similar non-reversal of the PAK1 reduction by CDC42 was also observed when KLF2 expression was increased by vasoprotective simvastatin in HUVECs (Fig. 5C). However, when KLF2 expression was inhibited by siRNA (Fig. 5D), significantly increased GRB2 expression was observed in HUVECs, with significant influence on PAK1, but not on PAK2 and PAK4 expression (Fig. 5D). KLF2 is a known flow-activated transcription factor which regulates gene expression to influence endothelial migration and angiogenesis (30). Elevated GRB2 expression was also observed in mutant HUVECs with elevated miR302-367 expression when high KLF2 expression was inhibited by siRNA (Fig. 5E). We then hypothesized that EC miR302-367-mediated KLF2 upregulation was involved in regulating GRB2 and in turn PAK1 activation for actin remodeling to have an influence on EC migration and sprouting angiogenesis.
Sequence analysis showed four KLF2 binding sites at the -1kb - +200 bp GRB2 promoter (Fig. 5F, Fig. S7A). To determine whether KLF2 regulates GRB2, we cloned the -820bp - +207bp GRB2 promoter into the pGL3 reporter vector (Fig. S7B-C). Forced expression of KLF2 could repress this -1kb proximal human GRB2 promoter luciferase reporter activity in a dose-dependent manner (Fig. 5G). Moreover, chromatin immunoprecipitation (ChIP) assays showed a direct association of KLF2 with the GRB2 promoter in HUVECs (Fig. 5H), and KLF2 binding to GRB2 was significantly elevated when KLF2 was upregulated in HUVECs with elevated miR302-367 expression (Fig. 5I). To determine the interaction of GRB2 and PAK1 in HUVECs, PAK1 and GRB2 antibodies were used to IP and Co-IP PAK1 and GRB2, and the house-keeping GAPDH antibody was used as loading control to confirm the same amount of total input protein. Elevated miR302-367 expression significantly reduced GRB2 expression without influence on PAK1 expression, and Co-IP results with PAK1 and GRB2 antibodies suggested significantly reduced GRB2-bound PAK1 when EC miR302-367 expression was elevated (Fig. 5J-K). Further study showed that the reduction of KLF2 expression in the mutant HUVECs by siRNA restored the reduced GRB2 expression, and the increased GRB2 can bring PAK1 to the membrane where it can be activated by constitutively active CDC42, which in turn phosphorylated and activated LIM-kinase, then phosphorylated and inactivated the actin-depolymerizing protein Cofilin, leading to increased F-actin formation and actin filament stabilization (Fig. 5L). Phospho-PAK1 immunostaining confirmed that constitutively active CDC42 is not able to reverse the reduced phospho-PAK1 staining in the membrane of HUVECs with elevated miR302-367 expression, but significantly increased the staining when high KLF2 level was inhibited by siRNA (Fig. 5M).
In summary these data suggest that miR302-367 directly targets CDC42, together with miR302-367-mediated-KLF2 regulation of GRB2, which fine-tunes the PAK1 activation, in turn influences on LIM-kinase and Cofilin, and finally leads to actin filament growth for cell migration and angiogenic sprouts.
CCND1 is the direct target of miR302-367 and exogenous CCND1 can reverse the significant reduction of cell proliferation and sprouting angiogenesis upon elevated expression of miR302-367 in endothelial cells
A single miRNA can regulate several, even hundreds, of gene transcripts. Using a similar approach as in cell migration we identified Ccnd1 as a possible target of miR302-367 contributing to cell proliferation for reduction of embryonic developmental angiogenesis. Elevated expression of miR302-367 markedly reduced CCND1 expression in HUVECs (Fig. 6A-B), and we further separated ECs from hindbrain and found similar reduced Ccnd1 expression in RNA and protein levels (Fig S4B-C) without significant influence on CDK4/6. Sequence analysis showed binding sites in the 3'-UTR of Ccnd1 (Fig. 6C), and miR302-367 suppressed the 3'-UTR luciferase reporter activity, whereas mutation of the seed sequences of the miR302-367 binding site restored the luciferase activity (Fig.6D). Similarly repressed luciferase activity was observed with individual miR302a/b/c/d mimics, miR367 mimic and most significantly with mimic mixture as miR302-367 vector (Fig. 6E). Elevated CCND1 expression produced by plasmid transfer was able to reverse the attenuated cell count (Fig.6F), MTT OD value (Fig.6G), reduced Ki67 staining (Fig. 6H-I), as well as reverse the decreased S-phase cells and increased G0/G1-phase cells (Fig. 6J-L) in HUVECs when miR302-367 expression was elevated.
MiR302-367 inhibits HUVEC proliferation through a CCND1 pathway. (A-B) qPCR and western blot show that elevated miR302-367 expression in HUVECs decreases CCND1 RNA (A) and protein (B) expression. (C) Sequence analysis shows miR302-367 binding sites in 3'-UTR of CCND1. (D) miR302-367 represses the luciferase activity of 3'-UTR of CCND1, and mutation of the binding site abolishes the luciferase repression. (E) Individual miR302a/b/c/d and miR367 mimic, miR302-367 mimic mixture as well as miR302-367 vector all repress the luciferase activity of CCND1. (F-I) Elevated CCND1 expression by plasmid transfer is able to reverse the attenuated cell count (F), MTT OD value (G) and Ki67 staining (H) in HUVECs with elevated miR302-367 expression. (J-M) Elevated CCND1 expression reverses the reduced CCND1 expression (J), reverses the increased G0/G1-phase cell proportion (K, L) and the decreased S-phase cell proportion (K, M) in HUVECs with elevated miR302-367 expression. (N) Elevated expression of miR302-367 in HUVECs dramatically reduces phospho-RB expression. (O) Co-IP with CDK4 antibody pulls down less CCND1 in HUVECs with elevated miR302-367 expression. (P) Co-IP with CCND1 antibody pulls down similar amount CDK4, and less CDK4 band due to decreased CCND1 in HUVECs with elevated miR302-367 expression.(Q) Elevated expression of CCND1 reverses the reduction of phospho-RB in HUVECs when elevated miR302-367 expression. Data quantification is mean ± S.E.M (n = 3). All data were analyzed using Student's t-test unless otherwise noted. n.s., no significance; *, P<0.05; **, P<0.01. Scale bars: H, 50 μm.
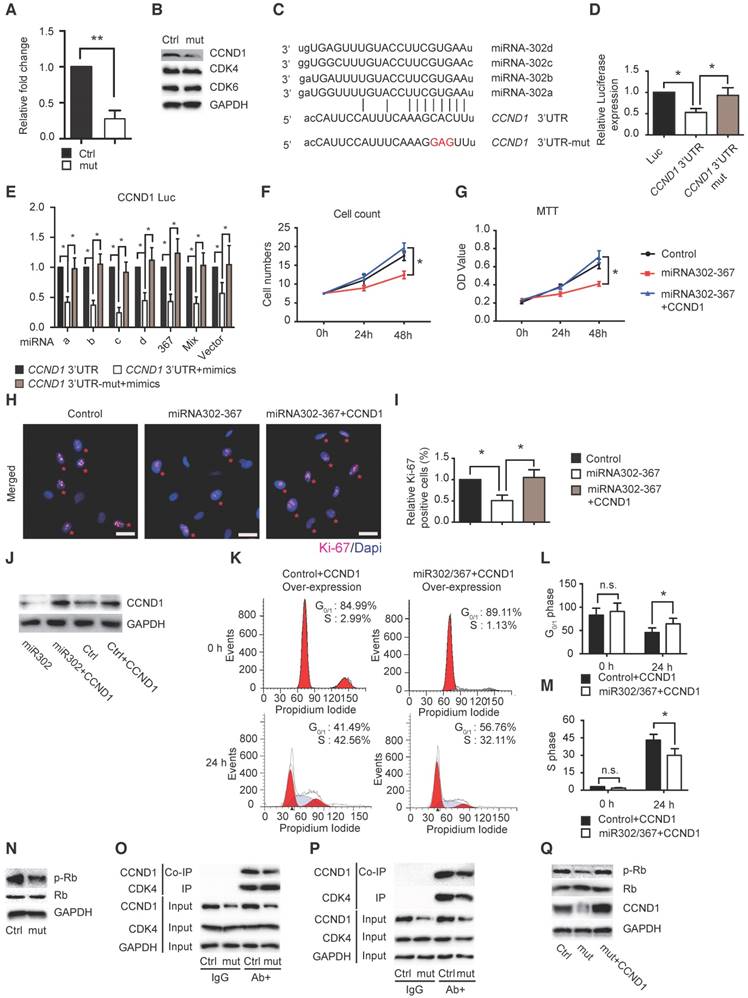
The mammalian cell cycle is tightly regulated, the key regulator of the G0 to S transition being the Ccnd1-Cdk4/6-Rb pathway (8). Our study showed that elevated expression of miR302-367 in HUVECs dramatically reduced the CCND1 expression (Fig. 6A-B) and its downstream target genes phospho-RB and E2F1 with no significant change of total RB and CDK4/6 expression (Fig. 6N). To determine the interaction of CCND1 and CDK4/6, we used the similar IP and Co-IP approach as in the study of GRB2 and PAK1 and found significantly reduced CCND1-bound CDK4 when expression of miR302-367 was elevated in ECs (Fig. 6O-P). Further study showed that elevated CCND1 expression restored the reduction of phospho-RB in HUVECs with elevated miR302-367 expression (Fig. 6Q). These results suggested that reduced interaction of CCND1 with CDK4/6 results in less RB phosphorylation, which further contributes to attenuation of EC proliferation when EC miR302-367 expression is elevated.
Target site blocker determined the Cdc42 and Ccnd1 pathways contributing significantly to the reduced developmental angiogenesis observed upon miR302-367 expression in endothelial cells
MicroRNAs usually regulate multiple target genes leading to the phenotypic changes, therefore Target Site Blockers (TSBs) were used to validate the importance of miR302 target genes in cell function phenotypes. The sequences of mouse or human immature stem-loop and mature miR302a-d were shown in Fig. S8A-D. Effects of TSBs against miR302 specific target genes Cdc42 or Ccnd1 contributing to EC migration and proliferation during developmental angiogenesis were shown in Fig. S8E-F. The predicted consequential pairing of miR-302 with the Cdc42/Ccnd1 target region was shown in Fig. S8G, and the custom-design TSB sequences to selectively impair miR302 cluster-mediated inhibition of Cdc42/CDC42 or Ccnd1/CCND1 shown in Fig. S8H.
The mouse TSBs studies were performed in the retinal developmental sprouting angiogenesis model and human TSBs studies in cultured HUVECs. The results showed that mouse TSB-miR302-Cdc42 enhanced the miR302-mediated reduction of Cdc42 RNA and protein levels in ECs without significant influence on Ccnd1 and Mapk1/3 compared to the scrambled sequence (Fig. S9A-B), and this resulted in significant reversal of the reduced angiogenic sprout filopodia (Fig. S9C-D) and EC proliferation (Fig. S9E-F) in the TSB-miR302-Cdc42 group as measured by quantification of angiogenic sprouts and Ki67/iB4 double positive cells. Similarly, in HUVECs the human TSB-miR302-CDC42 enhanced the miR302-mediated reduction of CDC42 expression compared to the scrambled sequence (Fig. S9G-H), and further reversed the reduced EC migration and proliferation with more influence on cell migration (1.96±0.23) than cell proliferation (1.40±0.19) (Fig. S9I-L). These results confirmed the important role of the miR302-Cdc42 pathway in EC migration and proliferation contributing to developmental angiogenesis.
Using a similar approach we found that TSB-miR302-Ccnd1/CCND1 specifically increased the reduction of Ccnd1/CCND1 expression upon elevated miR302 expression in ECs (Fig. S9M-N and S9Q-R), which reversed the reduced EC proliferation in in vivo developmental angiogenesis shown by Ki67/iB4 immunostaining (Fig. S9O-P), as well as in cultured HUVECs shown by Ki67 immunostaining, cell count and MTT assay (Fig. S9S-V), therefore confirming the important role of CCND1 in EC proliferation, and further influence on developmental angiogenesis when miR302 expression was elevated in ECs.
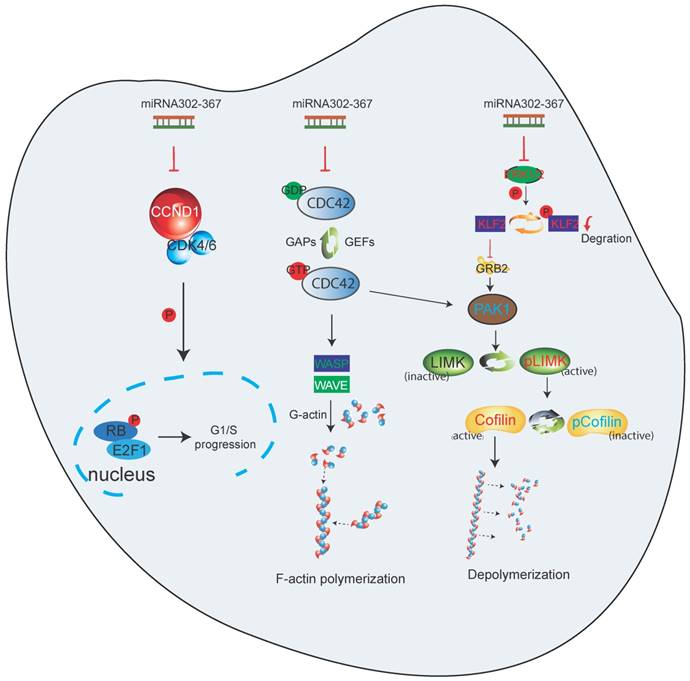
Taken together, these data indicate that miR302-367 targets multiple pathways of EC migration and proliferation, and thereby attenuates embryonic developmental angiogenesis. Elevated expression of miR302-367 in ECs inhibits cell proliferation through repression of Ccnd1, which influences the Cdk4/6-Rb-E2F1 pathway. The inhibition of cell motility by elevated EC miR302-367 is through repression of Cdc42, which regulates F-actin formation via Wasp, as well as Klf2/Grb2/Pak1/LIM-Kinase/Cofilin pathway, with Klf2 and adaptor Grb2, together with Cdc42, fine-tuning the Pak1 activation (Fig. 7).
Working model of endothelial specific elevated miRNA302-367 attenuating embryonic hindbrain developmental angiogenesis by targeting CDC42 and CCND1 multiple pathways. 1) Elevated expression of miR302-367 in endothelial cells inhibits cell proliferation through repression of CCND1 via the CCND1-CDK4/6-RB-E2F1 pathway; 2) Elevated expression of miR302-367 in endothelial cells reduces cell motility through repression of CDC42 and its effectors WASP and PAK1 activation, and moreover, PAK1 activation can be fine-tuned by KLF2 and the adaptor protein GRB2, which futher regulates LIM-kinase and Cofilin for F-actin formation and filament growth, and finally influences cell migration.
Discussion
We present here the evidence that EC specific miR302-367 targets diverse signal pathways, including the Rho GTPase Cdc42 pathway, which is essential for the control of cell motility, and Ccnd1, which regulates cell cycle and cell proliferation, and together tightly control angiogenic spouts and developmental angiogenesis in vivo. EC miR302-367 decreases the total and activated Cdc42 expression, and in turn inhibits F-actin formation via the Wasp pathway. Furthermore, EC miR302-367-mediated increased Klf2 expression regulates Grb2, which together with Cdc42, fine-tunes Pak1 activation, and this in turn reduces the phosphorylation of LIM-kinase and Cofilin for precise control of F-actin formation and filament growth for cell motility. We also reveal that miR302-367 directly targets Ccnd1 and results in impaired cell proliferation via the Rb/E2F pathway.
MicroRNAs represent evolutionary conserved small noncoding RNAs that can be transcribed individually or in cluster and are encoded by introns or intergenic regions. miR302-367, in the cluster of miR302b, c, d, a, and miR367 located in the intron 8 of gene Larp7, chromosome 3 (31), is highly expressed in early mouse development and plays important roles in diverse biological processes, such as maintaining pluripotency of human embryonic stem cells, tissue-specific progenitor identity, developing lung endoderm, promoting cardiomyocyte proliferation for regeneration, and restricting tumor angiogenesis (12-15). MicroRNAs modulate gene expression by either inhibiting mRNA translation or inducing mRNA degradation via imperfect sequence complementarities with the sites mostly located in the 3'UTR of mRNAs (11), and microRNAs may suppress multiple mRNA targets for precise control of cellular processes. Our results found that miR302-367 directly targeted 3'-UTR of Cdc42 and Ccnd1 and repressed their expression in ECs.
Embryonic organogenesis requires nutrient delivery, gas exchange and metabolic waste removal, and these processes depend upon the presence of a functional cardiovascular system. Angiogenesis refers to the formation of new blood vessels from preexisting ones, which is required in embryonic development. In this study we used a well-established in vivo angiogenesis hindbrain model and found that EC specific miR302-367 regulated the developmental angiogenesis.
EC proliferation is essential for angiogenesis and our results demonstrated that both in vivo miR302-367ECTg mutant mice and in vitro miR302-367-elevated HUVECs exhibited loss of EC proliferation. Cell cycle analysis showed fewer cells entering into S phase and we identified Ccnd1 as the direct target of miR302-367. Further studies showed less association of CCND1 with CDK4/6 forming less CCND1/CDK complex, less phosphor-RB in HUVECs when expression of miR302-367 was elevated, confirming that CCND1, together with CDKs, is crucial for phosphorylation of RB and release of E2F factors for progression from G0- into S-phase and cell proliferation, as reported previously (9).
During embryogenesis, formation of the primitive vascular network and subsequent vessel sprouting angiogenesis and remodeling require migration of ECs (32). Our results showed that elevated miR302-367 expression in ECs resulted in a significant reduction of EC migration contributing to the reduction of sprouting angiogenic activity in vitro and attenuation of embryonic developmental angiogenesis in vivo.
Actin, a major cytoskeleton component of ECs, is composed of 43-kDa G-actin that polymerizes into helical filaments of F-actin, and the constant remodeling of the actin cytoskeleton for formation of filopodia, lamellipodia, and stress fibers is essential for cell migration (32). Cdc42 plays a vital role in directional migration by modulating the actin cytoskeleton at the leading edge and regulating the formation of filopodia in motile cells (33). Cdc42 GAP-deficient mice showed elevated levels of activated Cdc42. HSC/P lacking Cdc42 GAP mice exhibited disorganized F-actin structures, reduced adhesion, impaired directional migration, defective short term- long term engraftment and impaired directional migration (34). Our study identified Cdc42 as the direct target of miR302-367 by high-throughput RNA sequencing and subsequent qPCR, 3'UTR luciferase reporter assay. The reduction of F-actin and cell migration by elevated EC miR302-367 expression can be restored by constitutively active Cdc42 domain.
Numerous effectors have been identified as Cdc42-mediated actin remodeling for cell migration. The activation of Wasp upon binding to Cdc42 induces the Arp2/3-mediated actin polymerization at the side of existing actin filaments and, thereby, generates the spike like projections called filopodia at the leading edge of a cell responsible for cell migration (20). Our study demonstrated that miR302-367ECTg mutant mice exhibited significantly decreased endothelial F-actin formation in vivo, as well as in vitro. Constitutively active CDC42 reversed the reduced CDC42 GTPase activity and phospho-WASP, restored the decreased F-actin formation and rescued the impaired cell migration as well as angiogenic sprouts in HUVECs with elevated miRNA302-367, suggesting the CDC42-WASP pathway involved in the actin filament remodel responsible for EC migration and in turn sprouting angiogenesis.
The Rho GTPase Cdc42 also regulates actin branching in the area of protrusion through the interaction of Pak (23, 35). Cdc42 activates Pak, which enhances the level of polymerized actin by activation of LIM-kinase leading to phosphorylation of Cofilin, and further impairment of actin depolymerization, thereby potentiating actin reorganization into stress fibers for EC migration (36). These signal pathways on the cytoskeleton reflect the fact that Cdc42 is an important regulator of cell motility (37). Our study found elevated miR302-367 expression in HUVECs significantly reduced phospho-PAK1 without significant change of phospho-PAK2 or PAK4, suggesting that PAK1 is the CDC42 downstream target in ECs, but constitutively active CDC42 was not able to reverse the reduction of PAK1 activation in miR302-367 mutant HUVECs.
The cytoskeleton remodeling for cell migration is a complex process, requiring a finely-tuned balance between numerous stimulatory and inhibitory signals, and Pak1 serves as an important mediator of Cdc42 GTPase for regulation of cytoskeleton dynamics. Grb2, a 25-kDa adapter protein, specifically interacted with Pak1 in vitro and in vivo, bringing Pak1 to the membrane edge for Cdc42 activation (28), while a flow-activated gene Klf2 was found as a novel Pak1 repressor in ECs via the Erk5-Klf2-Pak1 pathway (38). Our previous study found significantly increased expression of Klf2 in ECs when miR302-367 expression was elevated, thus we hypothesized that Klf2 upregulation by miR-302-367 might interact with Grb2 and regulate Pak1 activation.
Indeed, sequence analysis showed four Klf2 binding sites at the -1kb Grb2 promoter region, and forced Klf2 expression repressed this proximal -1kb Grb2 promoter in a dose-dependent manner, and Ch-IP analysis confirmed the direct association of Grb2 with the flow-activated transcription factor Klf2. Co-IP assay indicated interaction of Grb2 with Pak1, suggesting that Klf2-Grb2 signaling can fine-tune Pak1 activation. Further study showed that inhibition of KLF2 upregulation in miR302-367 mutant HUVECs by siRNA abolished the GRB2 blockage, resulting in more GRB2 bringing PAK1 to the cell membrane where active CDC42 led to PAK1 activation, and in turn LIM-kinase Cofilin to prevent F-actin degradation, which might be responsible for the reversal of impaired cell motility with elevated miR302-367 expression in ECs.
In conclusion, this study demonstrates that miR302-367 regulates multiple pathways including Ccnd1, Cdc42 and effector Wasp/Pak1, and Erk1/2-Klf2-Grb2 fine-tuning the Pak1 activation in the endothelium to inhibit cell proliferation and migration, and further restricts developmental angiogenesis. As pathological angiogenesis shares many common features with the physiological pathway, the clarification of the underlying mechanisms in developmental angiogenesis would provide opportunities for future therapeutic intervention of pathological angiogenesis related diseases.
Materials and Methods
miR302-367 gain- and loss-of-function in mouse hindbrain developmental angiogenesis model
Mouse developmental embryonic hindbrain angiogenesis in vivo model (4) was used and tamoxifen (50 mg/kg) was given by i.p. injection to pregnant mice at E10.5 for induction of elevated EC miR302-367 expression in miR302-367ECTg mice (39). The embryonic hindbrain was dissected out at E 12.5, fixed with cold 4% (wt/vol) formaldehyde and stained with iB4 antibody (Vector, B-1205) for vasculature analysis. MiR302-367 anti-miRs were administrated by in utero electroporation as previously described (40). Briefly mice at E10.5 and E11.5 were anesthetized with an i.p. injection of ketamine (120 mg/kg) and xylazine (10 mg/kg). The uterine horns were exposed and 37.5 pmol of each anti-miR mixture or control scramble microRNA injected through the uterus into the lateral ventricles of embryos. Electroporation was accomplished by square electric pulses (5 pulses, duration 50 ms each, interval 950 ms) delivering to the embryos using electroporator CUY-21 (Protech International). The electroporated mice were killed at E12.5 and embryonic hindbrains were collected and analyzed. The mouse studies were approved by Tongji University Institutional Animal Care and Use Committee, which is the appropriate animal care committee in accordance with the Institutional Animal Care and Use of Laboratory Animals.
Statistical Analysis
Data are presented as mean ± SEM of at least three independent assays unless otherwise noted. The standard two-tailed Student's t-test was used for statistical analysis, and p < 0.05 was considered significant.
Abbreviations
ECs: Endothelial Cells; Erk1/2: Extracellular Regulated Protein Kinases; 3′-UTR: 3′-UnTranslated Region; miRNAs: MicroRNAs; Cdc42: Cell Division Control Protein 42 homolog; Pak1: P21-Activated Kinase; Klf2: Krüppel-Like Factor 2; qPCR: Quantitative Polymerase Chain Reaction; Grb2: Growth Factor Receptor-bound Protein 2; Wasp: Wiskott-Aldrich Syndrome protein; Ccnd1: Cyclin-D1.
Acknowledgements
The study was supported by funds from National Natural Science Foundation of China (81703074, 91639112, 81770259, 81470472, 81670234, 81370433, 81470393 and 81370434), Program Project Grant for Important Diseases of Shanghai Municipal Health and Family Planning Commission (2014ZYJB0502) and Pudong Health Bureau of Shanghai (PW2013E-1). China Scholarship Council supported Jingjiang Pi for one-year study at the University of Pennsylvania.
Author contributions
J.J. Pi, J. Liu and T. Zhuang participated in the study design and performed the experiments; L. Zhang T. Tao and X.H. Zhou helped the experiments and data analysis; H.M. Sun, X.L. Chen, Q. Zhao, Y.S. Kuang, S. Peng performed the experiments; L. Zhang, Z.R. Yu, B. Tomlinson, P. Chan, Y. Tian, H.M. Fan, Z.M. Liu, X.J. Zheng and E. Morrisey helped design the study, gave comments and revised the manuscript; Y.Z. Zhang designed the study, analyzed the data and wrote the manuscript.
Supplementary Material
Supplementary methods, figures and tables.
Supplemental video 1.
Competing interests
The authors declare no conflict of interest.
References
1. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298-307
2. Patel-Hett S, D'Amore PA. Signal transduction in vasculogenesis and developmental angiogenesis. Int J Dev Biol. 2011;55:353-63
3. Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, Prykhozhij S. et al. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood. 2010;116:829-40
4. Fantin A, Vieira JM, Plein A, Maden CH, Ruhrberg C. The embryonic mouse hindbrain as a qualitative and quantitative model for studying the molecular and cellular mechanisms of angiogenesis. Nat Protoc. 2013;8:418-29
5. Karamysheva AF. Mechanisms of angiogenesis. Biochemistry (Mosc). 2008;73:751-62
6. Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359-69
7. Nayak RC, Chang KH, Vaitinadin NS, Cancelas JA. Rho GTPases control specific cytoskeleton-dependent functions of hematopoietic stem cells. Immunol Rev. 2013;256:255-68
8. Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220-7
9. Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859-69
10. Anand S, Cheresh DA. MicroRNA-mediated regulation of the angiogenic switch. Curr Opin Hematol. 2011;18:171-6
11. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215-33
12. Card DA, Hebbar PB, Li L, Trotter KW, Komatsu Y, Mishina Y. et al. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol Cell Biol. 2008;28:6426-38
13. Anokye-Danso F, Trivedi CM, Juhr D, Gupta M, Cui Z, Tian Y. et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8:376-88
14. Tian Y, Liu Y, Wang T, Zhou N, Kong J, Chen L. et al. A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci Transl Med. 2015;7:279ra38
15. Pi J, Tao T, Zhuang T, Sun H, Chen X, Liu J. et al. A MicroRNA302-367-Erk1/2-Klf2-S1pr1 Pathway Prevents Tumor Growth via Restricting Angiogenesis and Improving Vascular Stability. Circ Res. 2017;120:85-98
16. Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A. et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483-6
17. Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S. et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64-8
18. dos Remedios CG, Chhabra D, Kekic M, Dedova IV, Tsubakihara M, Berry DA. et al. Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev. 2003;83:433-73
19. Gupton SL, Gertler FB. Filopodia: the fingers that do the walking. Sci STKE. 2007;2007:re5
20. Machesky LM, Insall RH. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr Biol. 1998;8:1347-56
21. Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T. et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221-31
22. Delorme-Walker VD, Peterson JR, Chernoff J, Waterman CM, Danuser G, DerMardirossian C. et al. Pak1 regulates focal adhesion strength, myosin IIA distribution, and actin dynamics to optimize cell migration. J Cell Biol. 2011;193:1289-303
23. Kiosses WB, Daniels RH, Otey C, Bokoch GM, Schwartz MA. A role for p21-activated kinase in endothelial cell migration. J Cell Biol. 1999;147:831-44
24. Kiosses WB, Hood J, Yang S, Gerritsen ME, Cheresh DA, Alderson N. et al. A dominant-negative p65 PAK peptide inhibits angiogenesis. Circ Res. 2002;90:697-702
25. Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253-9
26. Sells MA, Pfaff A, Chernoff J. Temporal and spatial distribution of activated Pak1 in fibroblasts. J Cell Biol. 2000;151:1449-58
27. Parrini MC, Matsuda M, de Gunzburg J. Spatiotemporal regulation of the Pak1 kinase. Biochem Soc Trans. 2005;33:646-8
28. Puto LA, Pestonjamasp K, King CC, Bokoch GM. p21-activated kinase 1 (PAK1) interacts with the Grb2 adapter protein to couple to growth factor signaling. J Biol Chem. 2003;278:9388-93
29. Rudd CE. Adaptors and molecular scaffolds in immune cell signaling. Cell. 1999;96:5-8
30. Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW. et al. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. 2006;107:4354-63
31. Vidigal JA, Ventura A. Embryonic stem cell miRNAs and their roles in development and disease. Semin Cancer Biol. 2012;22:428-36
32. Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100:782-94
33. Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247-69
34. Yang L, Wang L, Geiger H, Cancelas JA, Mo J, Zheng Y. Rho GTPase Cdc42 coordinates hematopoietic stem cell quiescence and niche interaction in the bone marrow. Proc Natl Acad Sci U S A. 2007;104:5091-6
35. Vadlamudi RK, Li F, Adam L, Nguyen D, Ohta Y, Stossel TP. et al. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat Cell Biol. 2002;4:681-90
36. Gong C, Stoletov KV, Terman BI. VEGF treatment induces signaling pathways that regulate both actin polymerization and depolymerization. Angiogenesis. 2004;7:313-21
37. Allen WE, Zicha D, Ridley AJ, Jones GE. A role for Cdc42 in macrophage chemotaxis. J Cell Biol. 1998;141:1147-57
38. Komaravolu RK, Adam C, Moonen JR, Harmsen MC, Goebeler M, Schmidt M. Erk5 inhibits endothelial migration via KLF2-dependent down-regulation of PAK1. Cardiovasc Res. 2015;105:86-95
39. Sun Y, Liang X, Najafi N, Cass M, Lin L, Cai CL. et al. Islet 1 is expressed in distinct cardiovascular lineages, including pacemaker and coronary vascular cells. Dev Biol. 2007;304:286-96
40. Sun G, Ye P, Murai K, Lang MF, Li S, Zhang H. et al. miR-137 forms a regulatory loop with nuclear receptor TLX and LSD1 in neural stem cells. Nat Commun. 2011;2:529
Author contact
![]() Corresponding author: Yuzhen Zhang, MD, PhD, Key Laboratory of Arrhythmias of the Ministry of Education of China Research Center for Translational Medicine, Shanghai East Hospital, Tongji University School of Medicine, 150 Jimo Rd, Pudong New District, Shanghai, 200120, China. Tel: 86-21-61569673, e-mail: yzzhang-tjedu.cn
Corresponding author: Yuzhen Zhang, MD, PhD, Key Laboratory of Arrhythmias of the Ministry of Education of China Research Center for Translational Medicine, Shanghai East Hospital, Tongji University School of Medicine, 150 Jimo Rd, Pudong New District, Shanghai, 200120, China. Tel: 86-21-61569673, e-mail: yzzhang-tjedu.cn