Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviation
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(6):1494-1510. doi:10.7150/thno.21717 This issue Cite
Research Paper
XIAP Limits Autophagic Degradation of Sox2 and Is A Therapeutic Target in Nasopharyngeal Carcinoma Stem Cells
Jiao Ji1,2#, Yan Yu1#, Zhi-Ling Li1#, Ming-Yuan Chen3, Rong Deng1, Xiang Huang4, Guang-Feng Wang2, Meng-Xia Zhang5, Qi Yang1, Senthilkumar Ravichandran1, Gong-Kan Feng1, Xue-Lian Xu1, Chen-Lu Yang6, Miao-Zhen Qiu7, Lin Jiao1, Dajun Yang1,2 ![]() , Xiao-Feng Zhu1
, Xiao-Feng Zhu1 ![]()
1. State Key Laboratory of Oncology in South China; Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University Cancer Center, Guangzhou, China
2. Ascentage Pharma Group Corp. Limited, Jiangsu, China
3. Department of Nasopharyngeal Carcinoma, Sun Yat-sen University Cancer Center, Guangzhou, China
4. Department of Biochemistry, the School of Medicine, Jinan University, Guangzhou, China
5. Department of Radiotherapy, Fujian Medical University Union Hospital, China
6. Department of Gynecology, Women and children hospital of Guangdong Province, Guangzhou, China
7. Department of Medical Oncology, Sun Yat-sen University Cancer Center, Guangzhou, China
# Jiao Ji, Yan Yu and Zhi-Ling Li are equally contributed to this article.
Received 2017-7-1; Accepted 2017-11-14; Published 2018-2-5
Abstract

Rationale: Nasopharyngeal carcinoma (NPC) is the most frequent head and neck tumor in South China. The presence of cancer stem cells (CSCs) in NPC contributes to tumor maintenance and therapeutic resistance, while the ability of CSCs to escape from the apoptosis pathway may render them the resistant property to the therapies. Inhibitor of apoptosis proteins family proteins (IAPs), which are overexpressed in nasopharyngeal carcinoma stem cells, may play an important role in maintaining nasopharyngeal cancer stem cell properties. Here, we develop a novel CSC-targeting strategy to treat NPC through inhibiting IAPs.
Methods: Human NPC S-18 and S-26 cell lines were used as the model system in vitro and in vivo. Fluorescence activated cell sorting (FACS) assay was used to detect nasopharyngeal SP cells and CD44+ cells. The characteristics of CSCs were defined by sphere suspension culture, colony formation assay and cell migration. The role of XIAP on the regulation of Sox2 protein stability and ERK1-mediated phosphorylation of Sox2 signaling pathway were analyzed using immunoblotting, immunoprecipitation, immunofluorescence, phosphorylation mass spectrometry, siRNA silencing and plasmid overexpression. The correlation between XIAP and Sox2 in NPC biopsies and their role in prognosis was performed by immunohistochemistry. APG-1387 or chemotherapies-induced cell death and apoptosis in S-18 and S-26 were determined by WST, immunoblotting and flow cytometry assay.
Results: IAPs, especially X chromosome-linked IAP (XIAP), were elevated in CSCs of NPC, and these proteins were critically involved in the maintenance of CSCs properties by enhancing the stability of Sox2. Mechanistically, ERK1 kinase promoted autophagic degradation of Sox2 via phosphorylation of Sox2 at Ser251 and further SUMOylation of Sox2 at Lys245 in non-CSCs. However, XIAP blocked autophagic degradation of Sox2 by inhibiting ERK1 activation in CSCs. Additionally, XIAP was positively correlated with Sox2 expression in NPC tissues, which were associated with NPC progression. Finally, we discovered that a novel antagonist of IAPs, APG-1387, exerted antitumor effect on CSCs. Also, the combination of APG-1387 with CDDP /5-FU has a synergistic effect on NPC.
Conclusion: Our study highlights the importance of IAPs in the maintenance of CSCs in NPC. Thus, XIAP is a promising therapeutic target in CSCs and suggests that NPC patients may benefit from a combination treatment of APG-1387 with conventional chemotherapy.
Keywords: XIAP, nasopharyngeal cancer, cancer stem-cells, Sox2, SMAC mimetics
Introduction
Nasopharyngeal carcinoma (NPC) is an epithelial malignancy that is highly prevalent in Southern China and Southeast Asia [1]. Significant progress in its treatment has been made in recent years, yet the 5-year survival rate is only 10% to 40% in advanced NPC because of the high incidence of treatment resistance, recurrence and lymph node metastasis [2, 3]. In fact, there is no standard treatment for platinum-resistant patients [4]. Therefore, identifying the cellular and molecular mechanisms of treatment resistance and metastasis has become a prime target for novel drug discovery and design.
Accumulating evidence has shown that cancer stem cells (CSCs) are critical for tumor initiation, maintenance, regeneration, recurrence and metastasis in NPC [5-8]. Escaping apoptosis is one of the important properties of CSCs and a key mechanism of therapeutic resistance [9-11].The IAPs, including XIAP, cellular IAP-1 (cIAP-1), cellular IAP-2 (cIAP-2) and melanoma-linked IAP (ML-IAP), are often upregulated in cancers and are believed to underlie the resistance of many tumors to chemotherapeutics [12]. In addition, IAPs regulate nuclear factor κB (NF-κB) activation and the mitogen-activated protein kinase (MAPK) pathway through RING domain E3 ligase activity, thus controlling inflammation [13], immunity [14], cell migration [15-17], and cell survival. However, little is known about the role of IAPs in CSCs.
Sex-determining region Y (SRY)-box 2 (Sox2) is an important regulator of embryonic stem cell fate and is aberrantly expressed in a great variety of cancer stem cells owing to DNA amplification, transcriptional activation and post-transcriptional modifications, including in CSCs from head and neck [18], lung and oesophagus [19] cancers. Increasing evidence has shown that Sox2 may have important roles in the 'stemness' and progression of head and neck squamous cell carcinomas (HNSCC) including NPC. Sox2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma, and behaves as a bystander stemness marker. Constitutive expression of Sox2 in HNSCC cells generates a CSC-like population [20-22].
Inhibition of IAPs suppresses proliferation and induces apoptosis in various tumor cells [23-25]. Second mitochondria-derived activator of caspases (SMAC) mimetics, which are designed based on the IAP-Binding Motif (IBM) of the potent endogenous antagonist of the IAPs, have been demonstrated to induce death in a subset of cancer cells in a caspase-8- and TNFα-dependent manner. In the last decade, a large number of small molecule SMAC mimetics have been designed and developed for cancer treatment. Both monovalent and bivalent SMAC mimetics have been developed to clinical trial phases [26, 27]. We previously reported that monovalent SMAC mimetics AT-406 and SM-164 have potent antitumor effects on NPC stem-like cells [28]. APG-1387, a potent bivalent SMAC mimetic, was developed as a therapy for cancer patients by Ascentage Pharma Group Inc. APG-1387 is currently being evaluated in a phase I clinical trial in patients with solid tumor in China and Australia. Our findings suggest that patients with NPC may benefit from treatment with a combination of conventional chemotherapy and APG-1387 by inhibiting the CSCs of NPC.
In this study, we showed that XIAP maintains the stemness of cancer cells in NPC via enhancing the stability of stem cell transcription factor Sox2 by inhibiting ERK1-mediated autophagic degradation. Consistently, strong correlation between XIAP and Sox2 expression in NPC tumor tissue was found in patients. Antagonizing XIAP by APG-1387 in addition to chemotherapy showed a better antitumor effect to conventional chemotherapy alone both in vitro and in vivo. Thus, targeting CSCs by IAP inhibitor APG-1387 combined with chemotherapy may be a novel therapeutic strategy for the treatment of NPC.
Materials and Methods
Cell culture
The clones of the human nasopharyngeal carcinoma cell line CNE2, S18 and S26 were kind gifts from Dr. Chaonan Qian (Sun Yat-sen University Cancer Center, China). HEK293T was obtained from the American Type Culture Collection in 2010. All cells were cultivated in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (Gibco) at 37°C in 5% CO2.
Smac mimetic APG-1387
APG-1387 was provided by Ascentage Pharma Group Inc. For in vitro studies, APG-1387 was dissolved in DMSO at a concentration of 10 μM and stored at -20°C. For in vivo studies, APG-1387 was dissolved in 10% PEG400/5% EL/85% PBS.
SP cells analysis
The S18 or S26 cells were untreated or treated with the compounds to test for 24 h, then harvested and resuspended at 106 cells/mL density in ice-cold DMEM (supplemented with 2% fetal bovine serum). The DNA binding dye hoechst 33342 (Sigma-Aldrich) was added at a final concentration of 5 μg/mL and incubated for 90 min at 37°C in the dark with interval mixing. As a negative control, a subset of the cells were incubated with 5 μM fumitremorgin C (FTC, an inhibitor of ABCG2 that could block the pumping out of hoechst 33342 in CSCs, Merck) for 5 min prior to hoechst 33342 dyeing. After hoechst staining, cells were washed twice then pelleted and maintained at 4°C before FACS analysis. FACS analysis was performed on COULTER EPICS ALTRA™ Flow Cytometer (Beckman Coulter). The hoechst dye was excited with UV laser at 350 nm and its fluorescence was measured at two wavelengths using a 450/40 BP filter (hoechst Blue) and a 675 long pass filter (hoechst Red). Flow cytometry data were analyzed using FlowJo software. At least three independent experiments were performed.
CD44 cells analysis
1×106 S18 or S26 cells were suspended in 100 µL PBS for CD44-PE or IgG-PE antibody labeling. CD44-PE antibody (clone: DB105) and negative control IgG-PE antibody were obtained from Miltenyi Biotec GmbH (Germany) and used to label cells following the manufacturer's instructions. Flow cytometric analysis was performed using a Beckman Coulter filtered with a 488 nm laser. At least three independent experiments were performed.
Quantitative real-time PCR
Total RNA of S18 or S26 cell was extracted using trizol reagent (Invitrogen) according to the manufacturer's instructions. cDNA was synthesized using High Capacity RNA-to-cDNA Kit (Applied BiosystemsTM) according to the manufacturer's instructions. Real-time PCR amplification was performed by SYBR® Green PCR Master Mix (Applied BiosystemsTM) on a Hard-Shell PCR Plates (Bio-Rad). Relative quantification of each target gene was normalized by using an endogenous control (GAPDH). qPCR and analyses were performed using a CFX Connect Real-Time PCR Detection System (Bio-Rad).
Cell proliferation and cytotoxicity assay
Cell proliferation and cytotoxicity was measured using Cell Counting Kit-8 (Dojindo). S18 and S26 cells were counted and plated in triplicate at 2000-3000 cells per well (200 µL) in 96-well plates (Falcon), and allowed to adhere overnight. For individual groups, compounds (cisplatin, 5-fluorouracil, APG-1387) were added to the wells in concentration gradients. Cell viability was measured 72 h later by adding 10 µL CCK8 per well and incubated 1-4h. The observation value was detected at 450 nm, Prism software was used to calculate the IC50. All experiments were performed in 6 replicates per trial, with three independent trials in total and the average percentages of cell viability are shown.
Colony formation assay
S18 or S26 cells were plated in triplicate at 100 cells per well in 6-well plates (Falcon), and cultured in DMEM (supplemented with 10% fetal bovine serum) for 7-10 days. Then, the cells were washed twice with PBS and fixed in methanol for 10 min. After washing with PBS twice, the cells were dyed with crystal violet for 30 min. Then, the crystal violet was washed out and the number of colonies was counted. Images are shown as representatives of three independent experiments.
Sphere formation assay
S18 or S26 cells were plated in triplicate at 1000 cells per well in ultra-low attachment 6-well plates (Corning), and cultured in DMEM/F12 medium (Invitrogen) with 20 ng/mL recombinant human basic fibroblast growth factor (Invitrogen), 20 ng/mL recombinant human epidermal growth factor (BD Biosciences), B-27 supplement (Invitrogen) and compounds to be tested for ~2 weeks. The spheres were counted under a light microscope. Images are shown as representatives of three independent experiments.
In vitro migration assay
S18 or S26 cells were suspended in serum-free DMEM at a density of 1×106 cells/ml. 300 µL cell suspended with the compounds to be tested was placed in the upper chamber of an 8 μm 24-well transwell plate (Corning), and 700 µL DMEM supplemented with 10% fetal bovine serum was placed in the lower chamber. The plate was cultivated at 37°C in 5% CO2 for 16-18 h. The cells were fixed in 75% ethanol for 20 min and dyed with crystal violet for 30 min. The cells on the upper surface of the chamber were wiped away. Then, the crystal violet was washed out and the number of colonies was counted by microscopy. Ten fields were randomly taken to count the number of cells. Images are shown as representatives of three independent experiments.
Wound healing assay
Cells were seeded into 6-well tissue culture plates at an appropriate density (reaching ~70-80% confluence of the well as a monolayer after 24 h of growth). Cells were cultured in DMEM medium supplemented with 10% FBS for 24 h. A straight-line-wound was made by scraping a 20 µL pipette tip across the cell monolayer. After scratching, detached cells were washed away by gently the rinsing the well twice with medium and cultured with DMEM medium for 48 h. The wound was imaged at 0, 24 and 72 h under microscopy. Five random fields were snapped. Images are shown as representatives of three independent experiments.
Plasmids and Transfection
XIAP shRNA and control shRNA plasmids were purchased from Shanghai Genechem Co.Ltd. Plasmid PLVX-DsRED-XIAP was constructed by subcloning pcDNA3-XIAP (addgene, Plasmid #11833) constructs into PLVX-DsRED (Clontech) Lentivirus vector. The plasmid pMSCV-Flag-hSox2 was obtained from addgene (Plasmid #20073). Plasmids transient transfection was performed using Lipofectamine 2,000 (Invitrogen) according to the manufacturer's instructions. To generate retroviruses, HEK293T cells were transfected with Lentivirus packaging plasmids and PLVX-DsRED-XIAP or Vector using Lipofectamine 2,000. Medium was changed 24 h after transfection, and the cell supernatant was collected on the third and fourth day. Target cells were infected by retrovirus supernatant in the presence of 8 μg/mL polybrene every 12 h for three rounds. After infection, the cells were selected using 1 μg/ mL puromycin for 3 days.
Western blotting and Immunoprecipitation
Whole-cell extracts were generated by direct lysis with 1× Cell Lysis Buffer (Cell Signaling Technology, #9873) with 1 mM phenylmethylsulphonyl fluoride (PMSF) added immediately before use. Samples with added 6× SDS sample buffer were heated for 10 min at 100 °C and resolved using SDS-PAGE. For immunoprecipitation, cells were lysed by E1A lysis buffer (25 mM NaCl, 50 mM HEPES (pH 7.5), 0.1% NP-40, 5 mM EDTA, protease inhibitor cocktail (Roche)). Immunoprecipitation was carried out either by incubating Flag beads (Sigma) at 4 °C with lysate overnight or by incubating appropriate antibody with cell lysate for 2-3 h, followed by incubating the Protein-A/G beads overnight (Roche). Immunoprecipitates were washed three times with cold lysis buffer and eluted with SDS loading buffer by heating the samples at 95-100 °C for 10 min. Densitometric analyses were performed on at least two different expositions to assure the linearity of each acquisition using ImageJ software (v1.46r). Three replicates were performed.
Antibodies and Reagents
Anti-cIAP1 (#7065), Anti-cIAP2 (#3130), Anti-survivin (#2808) , Anti-Oct4 (#2750), Anti-klf4 (#12173), Anti-SUMO1 (#4930), Anti- HA-Tag (#3724), Phospho-p44/42 MAPK (Erk1/2) ( #4377), Cleaved-PARP (#5625) antibodies were purchased from Cell Signaling Technology; Anti-Naong ( ab21624), Anti-Sox2 ((ab92494) were obtained from Abcam; Anti-phospho-Sox2(S251) (AP3742a) antibody was purchased from Abgent; Anti-LC3 (NB100-2220) antibody was obtained from Novus; Anti-Flag (F3165) and Anti-α-Tubulin (T9026) antibodies were obtained from Sigma; Anti-XIAP(sc-55551), Anti- ERK1/2(sc-292838), Anti-p62/SQSTM1 (sc-28359), Anti-caspase-3(sc-56046), goat anti-mouse IgG-HRP (sc-2005), goat anti-rabbit IgG-HRP (sc-2030) antibodies were purchased from Santa Cruz Biotechnology.
Bafilomycin A1, Chloroquine, MG-132, CHX were purchased from Sigma.
siRNA transfection
All siRNAs were produced from GenePharma Inc. and transfected using lipofectamine RNAimax transfection reagent (life Technologies) according to the manufacturer's protocol. Target sequences for siRNAs were as follows:
P62 #1 (5'-GAGGAAUUGACAAUGGCCAUGUCCU-3'),
P62 #2 (5'-ACAGAUGCCAGAAUCGGAA-3'),
Lamp2 #1 (5'-CCAUCAGAAUUCCAUUGAATT-3'),
Lamp2 #2 (5'-GUUGCUUCAGUUAUUAACATT-3').
Immunofluorescence
S-18/XIAPsh and S-18/CTLsh cells were seeded in 24-well plates containing NUNC Thermanox™ coverslips. Upon treatment with rapamycin, CQ or APG-1387 for indicated time points and concentrations, cells were fixed with 4% paraformaldehyde fixative solution at room temperature for 20 min and washed three times in PBS. Cells were permeabilized with 0.25% Triton X-100 (Sigma-Aldrich) in PBS at 4°C for 10 min and blocked with 4% BSA (MP Biomedicals) blocking solution at 37°C for 30 min. The cells were incubated with the primary antibody (1:300) overnight at 4°C and the fluorophore-conjugated secondary antibody (1:200) at room temperature for 1 h. DAPI was added for 5 min to visualize nuclei. After using an antifluorescence quenching reagent, the coverslips were stored at 4°C. Cells were observed using a laser scanning confocal microscope (Olympus, FV-1000). Three replicates were performed.
LC3 fluorescence microscopy
S-18/XIAPsh or S-18/CTLsh cells transfected with pCMV-C Vector, pCMV-C-GFP orpCMV-C-GFP-LC3 plamids were cultured on glass coverslips in 24-well plates at 37 °C. GFP-LC3 punctuation/aggregation was observed with live cell imaging under a confocal microscope (Olympus, FV-1000). pCMV-C Vector or pCMV-C-GFP transfected cells served as negative or positive controls, respectively. Samples were imaged with a 488 nm excitation laser. Cells representing intense GFP-LC3 aggregates in the cytosol but not in the nucleus were classified as autophagic LC3 puncta, whereas cells presenting a mostly diffuse distribution of GFP-LC3 in the cytoplasm and nucleus were considered as non-autophagic. The GFP-LC3 puncta cells were counted in 10 random areas. Data represent the mean ± SE from 10 areas from 5 experiments. Images are shown as representatives of three independent experiments.
GST-Sox2 fusion protein purification
The bacterial expression constructs (PGEX-6P-1-Sox2) containing the indicated genes were transformed into BL21-competent cells (Agilent Technologies). Cells were induced to overexpress protein under 0.1 mM isopropyl-b-D-thiogalactoside (Sigma-Aldrich) at 16 °C for 16-18 h. Cells were resuspended in PBS containing Protease Inhibitor Cocktail (Roche) and Phosphatase Inhibitor Cocktail (Roche), followed by ultrasonication. The proteins were purified using GST agarose beads according to the manufacturer's protocol (Thermo Fisher). Three replicates were performed.
GST-pulldown
GST-Sox2 fusion protein was incubated with ERK1-untagged active kinase (sigma) at 4°C for 4-6 h, then samples were added to 6× SDS sample buffer and heated for 10 min at 100 °C and resolved using SDS-PAGE. Three replicates were performed.
In vitro kinase assays and Mass spectrometry
GST-Sox2 fusion protein and ERK1-untagged active kinase (sigma) were co-incubated using in vitro kinase assay kit according to the manufacturer's protocol (Cell Signaling Technology). The samples were added to 6× SDS sample buffer and heated for 10 min at 100 °C and resolved using SDS-PAGE. The Sox2 bands were excised from the gel and subjected to trypsin digestion and mass spectrometry. Protein and modification identification was performed with a database search, and peptide identifications were validated with Peptide Prophet. Three replicates were performed.
Cell apoptosis analysis
Drug-induced apoptosis was evaluated by Annexin V and Propidium Iodide (PI) staining using an Annexin-V-FLUOS Staining kit (Roche). The S18 or S26 cells were untreated or treated with compounds to be tested for 48 h, and then cells were harvested and stained with Annexin-V-FLUOS solution and PI solution for 15 min in the dark at room temperature. The stained cells were analyzed on a Cytomics FC500 flow cytometer (BeckmanCoulter) using 488 nm excitation and 515 nm band pass filter for fluorescein detection and a 600 nm longpass filter for PI detection. Flow cytometric analysis clearly differentiated living cells with low Annexin and low PI staining, apoptotic cells with high Annexin and low PI staining, and necrotic cells with high Annexin and high PI staining. Each experiment was analyzed in triplicate and at least three independent experiments were performed.
Immunohistochemistry
108 paraffin-embedded tissue specimens of NPC patients who were diagnosed and treated from 2008 to 2009 at Sun Yet-sen University Cancer Center were dewaxed and rehydrated in xylene and graded ethanol solutions for antigen retrieval, then blocked with 3% H2O2, goat serum, avidin solution, and biotin solution. Primary XIAP and Sox2 were added and then probed with biotinylated rabbit secondary antibody (Vector Laboratories) and high-sensitivity streptavidin-HRP conjugate. To visualize staining, slides were incubated in 3,3'-diaminobenzidine in 0.1% H2O2 in Tris-HCl buffer, and subsequently counterstained with Hematoxylin QS (Vector Laboratories). XIAP and Sox2 positive samples were defined as those showing cytoplasmic staining pattern of tumor tissue. XIAP and Sox2 scores used were the sum of the percentage of cytoplasm positive cells (0-100%) and the staining intensity (0, negative; 1, weak; 2, moderate and 3, strong). Samples with scores 0 to 1.92 were considered low expression for Sox2 protein and those with score 1.92 to 3 were considered high expression for Sox2 protein; samples with scores 0 to 1.95 were considered low expression for XIAP protein and those with scores 1.95 to 3 were considered high expression for XIAP protein. The immunostained slide was evaluated under a microscope followed by correlation with clinicopathological parameters.
Animal experiments
For in vivo antitumor activity assay, 4- to 6-week-old BALB/c nude mice were obtained from Hunan SlacJingda Laboratory Animal Co., Ltd (Shanghai, China). Primary tumor xenografts were established by subcutaneously injecting 5×106 S-18 cells into the mice on each side of the axillary area. When the xenograft tumors developed to ~100 mm3, the mice were randomly divided into 6 groups with each group containing 6 mice. The treatments, including control (solvent control), APG-1387 (3 mg/kg, 5/w, iv), CDDP (3 mg/kg, 1/w, ip), 5-FU (25 mg/kg, 2/w, ip), APG-1387 (3 mg/kg) + CDDP, APG-1387(3 mg/kg) + 5-FU. Tumor volume and body weights were measured twice per week. For the secondary xenograft experiments, fresh cells from the dissociated primary tumors were suspended and counted. All of the 6 secondary nude mice were inoculated with 1×106 cells from each group of primary mouse tumors. The growth of the tumors was monitored, and the tumor volumes were measured twice weekly. The tumor volume (V) was determined according to the equation V= 1/2×a×b2, where L is the longest axis and W is the axis perpendicular to L, and H is the axis perpendicular to the L-W plane. Endpoint criteria were defined as when the longest axis of the tumor exceeded 1.5 cm or there was active ulceration of the tumor. Survival curves were determined by the Kaplan-Meier method using the tumor implant day as day 0. Mice weights were recorded throughout the survival period. All animal studies were approved by the Sun Yat-sen Univiersity Cancer Center Animal Care and Ethics Committee.
Statistical analysis
Results were obtained from at least three independent experiments and are expressed as the mean ± SD. The log survival probabilities of the expression of Sox2 and XIAP were analyzed using the Kaplan-Meier method. The correlation between Sox2 and XIAP was analyzed with COX regression analysis and evaluated using a chi-square or Student's t-test (SPSS, IBM, USA, version 21.0). Differences among the treatment groups were assessed using ANOVA and the appropriate statistical software. (GraphPad Prism, version 7.00). P values <0.05 (*), <0.01 (**) or <0.001 (***) were considered significant for all analyses.
Results
XIAP maintains the cancer stem cell (CSC) properties of NPC
S-18 and S-26 cell lines present different abilities for migration and invasion, although they are two clones derived from the same NPC cell line CNE-2 [29]. To find an appropriate model to explore how NPC cells survive from therapeutics, we conducted a series of experiments to evaluate the stem cell properties of the two clones. We confirmed that S-18 cells exhibit a higher proportion of side population cells (SP cells) (Fig. S1A), CD44-positive cells (Fig. S1B), and higher ABCG2 gene expression level (Fig. S1C) than S-26 cells. Furthermore, compared to S-26 cells, S-18 cells demonstrated stronger sphere-forming, clonogenic and migratory abilities (Fig. S1D), and elevated resistance to cisplatin (CDDP) or 5-fluorouracil (5-FU) treatment (Fig. S1E). Therefore, S-18 cells had stronger cancer stem-like properties and could be a fine model for NPC stem cells.
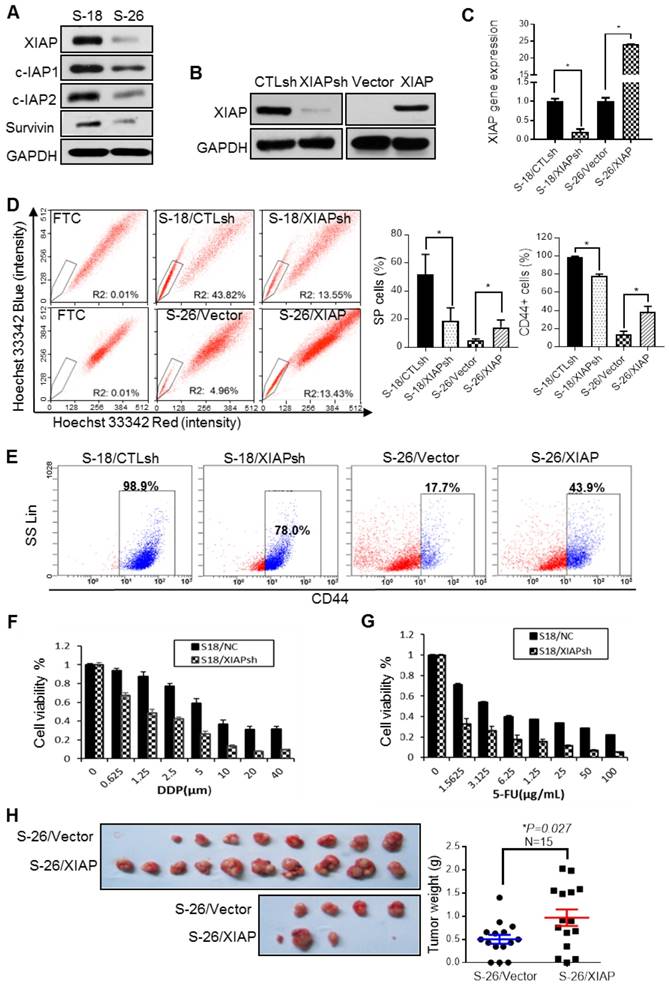
The IAP family proteins, especially XIAP, were highly expressed in S-18 cells (Fig. 1A). To validate the hypothesis that XIAP may play a role in maintaining CSC characteristics, we established an XIAP stable knockdown (KD) cell line in S-18 cells (S-18/XIAPsh) and an XIAP-overexpressing cell line in S-26 cells (S-26/XIAP) (Fig. 1B-C). In the XIAP KD cell line, the SP and CD44-positive populations were reduced (Fig. 1D-E), accompanied by a decrease in sphere formation and cell migratory ability (Fig. S2A-C). Converse results were observed in the XIAP-overexpressing cell line. Moreover, XIAP KD cells were more sensitive to CDDP or 5-FU treatment (Fig. 1F-G). In in vivo tumorigenesis experiments, overexpression of XIAP led to a dramatic increase in tumor growth (Fig. 1H). These data indicated that XIAP played a key role in maintaining the stemness properties of NPC.
The correlation between XIAP expression level and the cancer cell properties of NPC. (A) Western blotting for IAPs family proteins in S-18 and S-26 cells. (B, C) S-18 cells stably transfected with control short hairpin RNA (CTLsh) or XIAP short hairpin RNA (XIAPsh) and S-26 cells stably transfected with Vector or PLVX -XIAP. The effect of XIAP interference and overexpression was verified by western blot analysis and qPCR. Depicted results represent one of three independent experiments performed (A and B). (D) S-18/CTLsh, S-18/XIAPsh and S-26/Vector, S-26/XIAP cells were stained with hoechst 33342 and analyzed by flow cytometry with or without treatment with Fumitremorgin C (FTC). Quantification of SP cells proportion is shown on the right. The data are represented as mean ± SD of three independent experiments normalized to FTC. (E) S-18/CTLsh, S-18/XIAPsh, and S-26/Vector, S-26/XIAP cells were stained with CD44-PE and analyzed by flow cytometry. Quantification of CD44-positive cells proportion is shown on the right. The data are represented as mean ± SD of three independent experiments. (F, G) S-18/CTLsh, S-18/XIAPsh and S-26/Vector, S-26/XIAP cells were treated with a range of concentrations of cisplatin or 5-fluorouracil for 72 h, and cell proliferation and cytotoxicity was measured by using the Cell Counting Kit-8. (H) S-26/Vector or S-26/XIAP cells (2 x 104 cells) were injected into the left or right upper back of each nude mouse, respectively. The tumors were isolated and tumor weights were measured after three weeks of implantation.
XIAP enhances protein stability of Sox2 via suppressing autophagy
To explore the molecular mechanism by which XIAP maintains stem the cell properties of NPC cells, we measured the expression of the 4 main stem cell transcription factors: octamer-binding transcription factor 4 (Oct4), SRY (sex determining region Y)-box 2 (Sox2), c-Myc and Kruppel-like factor 4 (Klf4). Surprisingly, XIAP knockdown led to a dramatic decrease in Sox2 protein level, whereas XIAP overexpression showed the opposite effect (Fig. 2A). Meanwhile, the mRNA level of Sox2 was unaffected (data not shown). Increasing evidence has shown that Sox2 does not merely behave as a bystander stemness marker, but controls tumor initiation and cancer stem-cell functions in squamous-cell carcinoma and. Constitutive expression of Sox2 in head and neck squamous cell carcinoma (HNSCC) generates a CSC-like population [20-22].
XIAP regulates Sox2 stability by modulating autophagy. (A) Western blotting for stem cell factors Sox2, Oct4, C-myc, Klf4, and Nanog in S-18/CTLsh, S-18/XIAPsh and S-26/Vector, S-26/XIAP cells. (B) S-18/CTLsh and S-18/XIAPsh cells were treated with cyclohexane (CHX, 20 µM) for 6, 12, 24, 36, and 48 h. The half-life of Sox2 was determined by western blotting. (C) S-18/CTLsh and S-18/XIAPsh cells were treated with proteasome inhibitor MG132 (10 μM, 6 h) or autophagy inhibitors bafilomycin A1 (100 nM, 6 h) and chloroquine (CQ, 20 μM, 6 h). The protein level of Sox2 was then measured using western blotting. Blots shown are representative of three independent experiments (A) (B) and (C). Error bars present the SEM from three independent experiments **, P<0.01 (B) (C). (D) Representative images of GFP-LC3 staining inS-18/CTLsh and S-18/XIAPsh cells with stable expression of GFP-LC3. Quantification of LC3 punctate cells is shown on the right. The data are represented as mean ± SD of three independent experiments. (E) S-18/CTLsh and S-18/XIAPsh cells were treated with Bafilomycin A1 (100 nM, 6 h) or cultivated with EBSS for 6 h, then protein level of Sox2, p62 and LC3 were measured by western blotting. (F, G) SiRNA was used to knockdown LAMP2a or p62 in S-18 cells. The expression of Sox2 was measured by western blotting. (H) The interaction between Sox2 and p62 or LAMP2a was analysed by immunoprecipitation in S-18/CTLsh and S-18/XIAPsh cells. Blots shown are representative of three independent experiments (E) (F) (G) and (H).
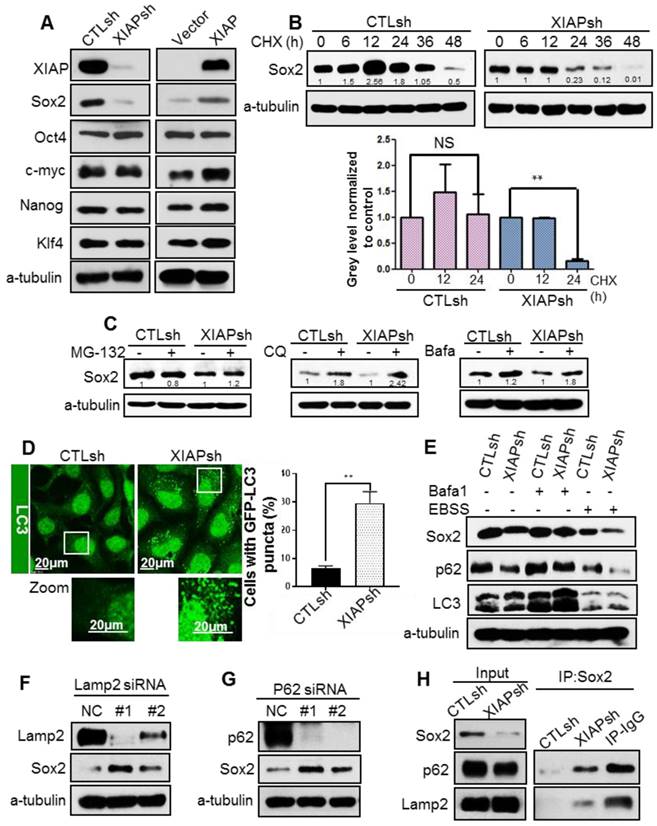
Therefore, we assumed that XIAP may regulate stemness in NPC by affecting the protein stability of Sox2. To determine whether XIAP affects Sox2 protein stability, the half-life of Sox2 was examined. XIAP knockdown resulted in a dramatic reduction in the half-life of Sox2 (Fig. 2B). We further explored the degradation of Sox2 and found that degradation of Sox2 can be reversed by autophagy inhibitors bafilomycinA1 and chloroquine (CQ), but not proteasome inhibitor MG132 (Fig.2C). Thus, we speculated that XIAP may limit the autophagic degradation. XIAP was reported to inhibit autophagy via the mouse double minute 2 homolog (Mdm2)-p53 pathway. We first assessed the basal autophagy level in S-18/CTLsh and S-18/XIAPsh cells by tracking the formation of GFP-LC3 puncta using confocal microscopy. We found that the percentage of cells with GFP-LC3 puncta was much higher in cells S-18/XIAPsh than in control cells (Fig. 2D). The subsequent western blot analysis also showed that LC3-II was increased when XIAP was KD in S-18 cells. In addition, the basal p62 level was lower in XIAP KD cells, and recovered when autophagy was blocked with bafilomycin A1 treatment, and then reduced when autophagy was activated by Earle's balanced salt solution (EBSS) treatment. The expression levels of Sox2 changed along with p62 (Fig. 2E).
To confirm that Sox2 was degraded by autophagy, autophagic receptor protein p62 or lysosomal-associated membrane protein (LAMP2) were knocked down by using small interfering RNA (siRNA), and the protein level of Sox2 was significantly increased (Fig. 2F-G), suggesting that the stability of Sox2 was regulated by the autophagy-lysosomal pathway. To further confirm that XIAP inhibits Sox2 degradation via autophagy, immuneprecipitation assays were performed and showed that the interaction between Sox2 and p62 or LAMP2 was enhanced in XIAP KD S-18 cells (Fig. 2H).
Since Sox2 has been shown to localize in the nucleus mostly, we determined if Sox2 was translocated into the cytoplasm, and further captured by p62, and finally degraded by the lysosome. Immunofluorescence staining under a confocal microscope showed that the nuclear expression of Sox2 was normal, however, the cytoplasmic distribution of Sox2 and its co-localization with p62 and LAMP2 were increased in XIAPsh cells (Fig. S3A-B). Moreover, the co-localization of Sox2 with p62 and LAMP2 was higher after treatment with CQ or bafilomycin A1 (Fig. S3A-B). These data demonstrated that Sox2 was degraded via the autophagy lysosome pathway, and XIAP limited Sox2 degradation by inhibiting autophagy.
XIAP inhibits ERK1-mediated phosphorylation of Sox2 at Ser251
Next, we wanted to know how Sox2 was translocated from the nucleus to the cytoplasm and recognized by p62. Sox2 can be post-translationally modified by multiple mechanisms, including SUMOylation [30], phosphorylation [31, 32], and ubiquitination [33, 34], which are crucial in the regulation of the activity and stability of Sox2. We hypothesized that post-translational modification of Sox2 resulted in its translocation to the cytoplasm and recognition by p62. Then, we analysed whether XIAP affected the post-translational modification of Sox2. The Group-based Phosphorylation Predication System (GPS V3.0) computer program was employed to analyse the Sox2 protein sequence. The results showed that S251 of Sox2 was a predicted phosphorylation site of Sox2 and could be phosphorylated by ERK1/2 (Fig. 3A). Moreover, S251 of Sox2 flanks the upstream K245 SUMOylation site of Sox2. The K245 SUMOylation of Sox2 has been proven to inhibit the DNA binding of Sox2 and affect its transcriptional activity and protein stability [30]. This motif resembles the classic phosphorylation-dependent SUMOylation motif (PDSM), JKxExxSP, which has been highly conserved throughout evolution (Fig. 3B). The phosphorylation of Sox2 at S249-S251 by an unknown kinase has been confirmed to promote Sox2 SUMOylation [31]. To determine if ERK1/2 could bind to and phosphorylate Sox2 at S251, the Sox2-glutathione-S-transferase (GST) fusion protein was first purified. Then, an in vitro GST pull-down assay showed that ERK1 bound directly to Sox2 (Fig. 3C). Furthermore, an ex vivo immunoprecipitation assay detecting the binding of Sox2 and ERK1 indicated that ERK activation enhanced binding and ERK inactivation inhibited binding (Fig. 3D). Mass spectrum analysis was employed after an in vitro kinase assay in which cells were incubated with or without ERK1-active kinase, and the results demonstrated that Sox2 was phosphorylated by ERK1 directly at S251 (Fig. 3E), which was further verified by western blotting for the specific antibody targeting S251-phosphorylated Sox2 after the in vitro kinase assay (Fig. 3F). To further examine this in vivo, we analyzed total and phosphorylated protein levels of Sox2-S251 in EGF or U0126 treated cells. We found that phosphorylation of Sox2-S251 was increased after ERK was activated without altering the total Sox2 protein level (Fig. 3G).
XIAP regulates ERK activation and phosphorylation of Sox2 at Ser251. (A) Based on the Group-based Phosphorylation Predication System (GPS V3.0), the Ser251 of Sox2 was strongly predicted to be an ERK phosphorylation site. (B) Consensus sequence of phosphorylation-dependent SUMOylation (left). Ser251 flanks the upstream SUMOylation site of Sox2. This motif resembles the phosphorylation-dependent SUMOylation motif (PDSM). (C) Sox2 and ERK1 binding assessed by an in vitro GST pull-down assay. (D) ERK activation stimulated by EGF (20 ng/mL, 30 min) or inhibited by U0126 (20 μM, 30 min) in S-18 cells. The levels of Sox2 binding with ERK were tested by immunoprecipitation. (E, F) The Sox2-GST fusion protein was analysed using an in vitro kinase assay with ATP and active ERK1 kinase. Sox2 Ser251 phosphorylation was tested by Mass spectrum analysis or Western blotting. (G) ERK activation stimulated by EGF (20 ng/mL, 30 min) or inhibited by U0126 (20 μM, 30 min) in S-18 cells. The expression of Sox2 p-S251 and total Sox2 were tested by Western blot. (H) XIAP knockdown in S-18 cells induced constitutive ERK1/2 activation. (I) XIAP knockdown in S-18 cells induced elevated ERK1/2 activation following treatment with EGF (20 ng/mL). Blots and images shown are representative of three independent experiments (C-I).
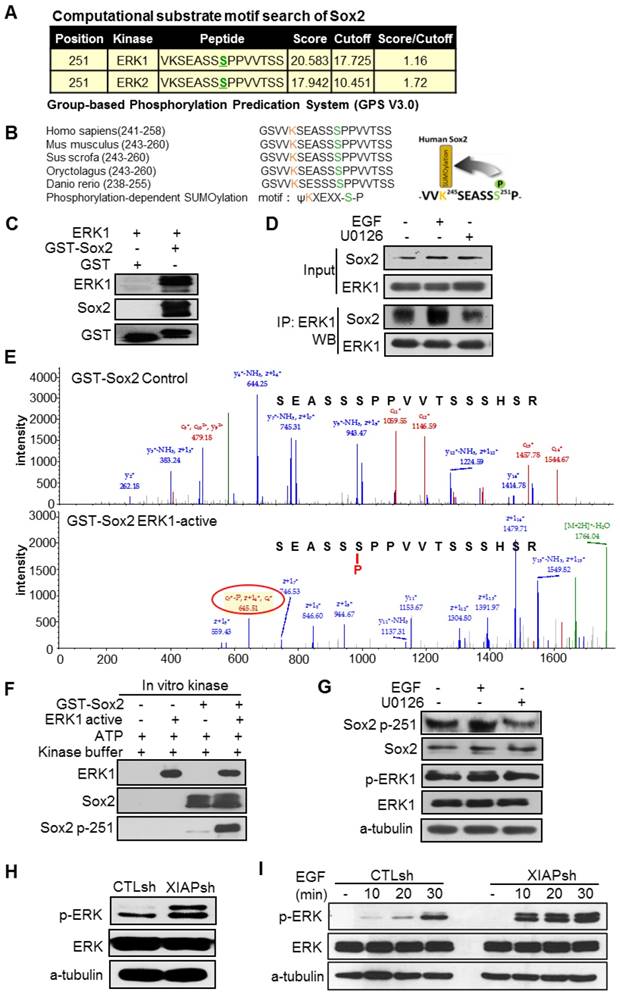
While XIAP has been reported to limit activation of the MAPK/ERK pathway through its E3 ligase activity, we presumed that XIAP might inhibit the SUMOylation of Sox2, thus affecting the protein stability of Sox2 through limiting the MAPK/ERK pathway. First, we examined the ERK1/2 phosphorylation level in XIAP KD cells and found that the ERK/MAPK pathway was more activated in XIAP KD cells than in control cells (Fig. 3H). Moreover, upon EGF stimulation, prolonged and elevated MAPK/ERK pathway activation were observed when XIAP was down regulated (Fig. 3I).
XIAP suppresses ERK1-mediated SUMOylation of Sox2
Next, we explored whether Sox2 phosphorylation by ERK1 promoted Sox2 SUMOylation. We found that the SUMOylation level of Sox2 was higher when ERK1/2 was activated by EGF stimulation, while the opposite result was observed when ERK1/2 was inhibited by U0126 (Fig. 4A), suggesting that Sox2 phosphorylation by ERK1/2 could promote its SUMOylation. Furthermore, immunoprecipitation showed that the Sox2 SUMOylation level was higher in XIAP KD cells (Fig. 4B). Substitution of the target lysine to arginine (K245R), or 251 serine to alanine (S251A), decreased the SUMO conjugation of Sox2 (Fig. 4C), but substitution of S249A or S250A had no such effect (Fig. 4D). The co-binding of Sox2 with LAMP2 increased upon treatment with XIAP inhibitor APG-1387 or CQ, while the binding of the K245R or S251A mutant displayed little alteration (Fig. 4E). Furthermore, XIAP small-molecule inhibitor APG-1387 significantly induced the translocation of wild-type Sox2 to the cytoplasm, but K245R or S251A mutant Sox2 had no such effect (Fig. 4F-G).
The regulation of Sox2 SUMOylation and degradation by XIAP. (A) S-18 cells co-transfected with HA-SUMO-1 and Flag-Sox2 with ERK activation stimulated by EGF (20 ng/mL, 30 min) or inhibited by U0126 (20 μM, 30 min). The levels of Sox2 SUMOylation were measured by immunoprecipitation. (B) S-18/CTLsh or S-18/XIAPsh cells co-transfected with HA-SUMO-1 and Flag-Sox2. The levels of Sox2 SUMOylation were measured by immunoprecipitation. (C) S-18 cells co-transfected with HA-SUMO-1 and mCherry-Flag-Sox2 or its mutants K245R or S251A. The levels of Sox2 SUMOylation were measured by immunoprecipitation. (D) S-18 cells co-transfected with HA-SUMO-1 and mCherry-Flag-Sox2 or its mutants S249A, S250A or S251A. The levels of Sox2 SUMOylation were measured by immunoprecipitation. (E) S-18 cells co-transfected with HA-SUMO-1 and mCherry-Flag-Sox2 or its mutants K245R or S251A, were treated with CQ (20 µM, 6 h) and with or without APG-1387 (100 nM, 12 h). The co-binding level of Sox2 and Lamp2 was tested by immunoprecipitation. (F) S-18 cells co-transfected with HA-SUMO-1 and mCherry-Flag-Sox2 or its mutants K245R or S251A, were treated with CQ (20 µM) and with or without APG-1387 (100 nM, 12 h). The nucleus was then isolated from the cytoplasm. The levels of Sox2 in each fraction were measured by western blotting. Blots shown are representative of three independent experiments (A-F). (G) S-18 cells co-transfected with HA-SUMO-1 and mCherry-Flag-Sox2 or its mutants K245R or S251A, treated with CQ (20 µM) and with or without APG-1387 (100 nM, 12 h). The subcellular localization of Sox2 was observed by confocal microscopy, and the number of cells with Sox2 puncta in the cytoplasm per 100 cells was counted in three different experiments. **, P<0.01.
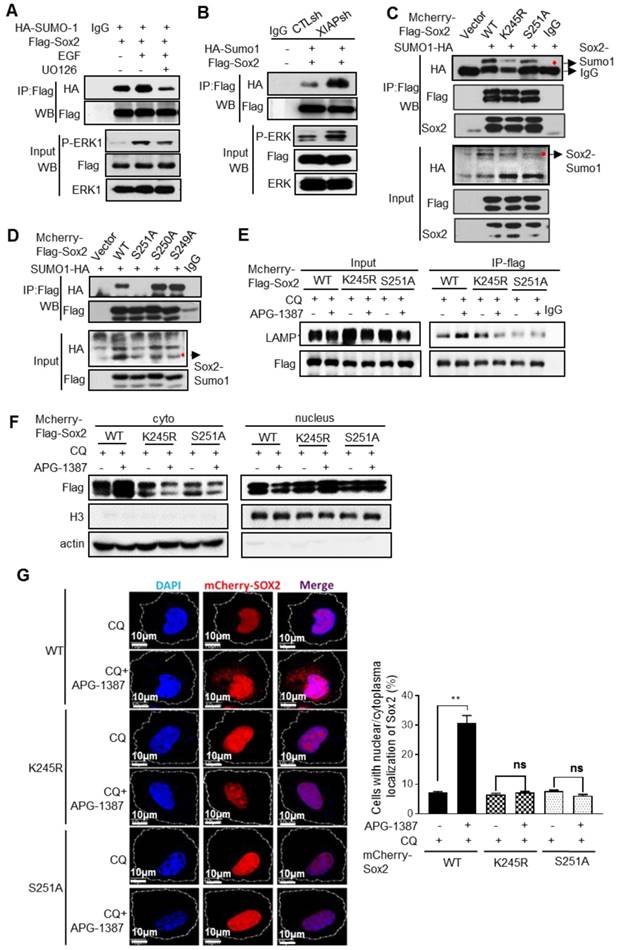
These results suggested that Sox2 phosphorylation by ERK1/2 at S251 could promote Sox2 SUMOylation. XIAP suppressed the SUMOylation of Sox2 via limiting ERK1/2 activation, thereby enhancing the protein stability of Sox2.
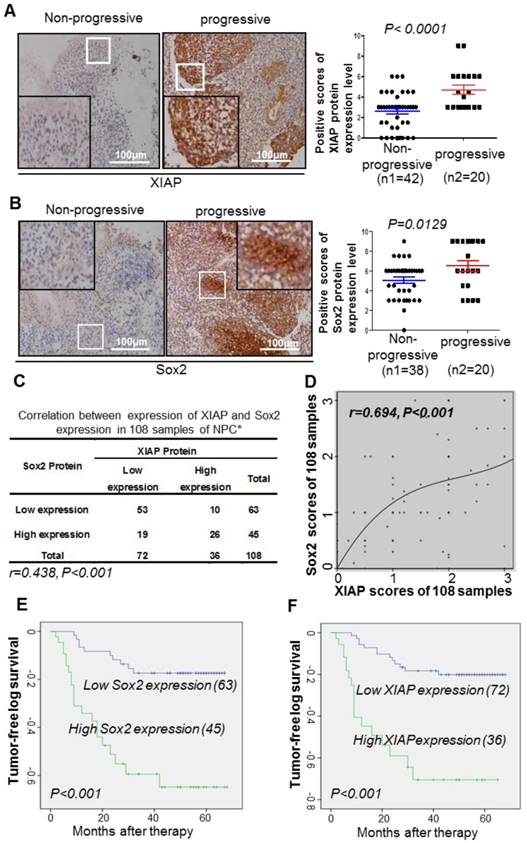
XIAP positively correlates with Sox2 expression in NPC tissues
We collected 108 clinical samples of paraffin-embedded tissue specimens from NPC patients who received radiotherapy or radiotherapy-based comprehensive treatment, and performed immunohistochemistry to detect the expression of XIAP and Sox2. Patients with a high expression level of XIAP or Sox2 displayed a high risk of tumor progression, namely, recurrence or metastasis (Fig. 5A-B). Among the 108 cases of NPC, 53 cases (49%) showed low expression level of Sox2 and XIAP, and 26 cases (24%) showed high expression level of XIAP and Sox2. Spearman correlation analysis revealed that the expressions of XIAP and Sox2 were positively correlated (r = 0.348, p <0.001) in the 108 cases of NPC (Fig. 5C), while the cubic curve of Cox regression analysis of XIAP and Sox2 protein showed that correlation between these two proteins was very strong (r = 0.694, p <0.001, Fig. 5D). Multivariable analysis indicated that XIAP and Sox2 were two independent prognostic factors in the whole population. Low expression of XIAP was positively correlated with a longer tumor-free interval (P<0.001); the median tumor-free interval in patients with low XIAP expression was 59.26 months, while that in patients with high XIAP expression was only 38.15 months (Fig. 5E). Low expression of Sox2 was also positively correlated with a longer tumor-free interval (P<0.001); the median tumor-free interval in patients with low expression of Sox2 was 58.36 months, while that in patients with high expression of Sox2 was only 42.17 months (Fig. 5F). Together, our findings defined a central role of XIAP as a protector to maintain the stability of Sox2 and cell stemness in NPC, suggesting its potential in novel strategies to inhibit therapeutic-resistant CSCs and reduce tumor recurrence or metastasis.
Correlation between XIAP and Sox2 expression in 108 cases of nasopharyngeal carcinoma. (A, B) Representative immunohistochemistry photos of XIAP or Sox2 protein level in progressive and non-progressive specimens with immunostaining in the same NPC tissues from the same first-visit patients (200x). The XIAP and Sox2 scores are shown. Student's t-test was used for statistical analysis. (C, D) The Spearman correlation test or Cox regression analysis was used to validate the correlation between XIAP and Sox2 expression in NPC samples. (E) Kaplan-Meier analysis of XIAP expression with the tumor-free survival interval of NPC. The log-rank test was used to perform the statistical analysis. *, P<0.05; **, P<0.01. (F) Kaplan-Meier analysis of Sox2 expression with the tumor-free survival interval of NPC. The log-rank test was used to perform the statistical analysis. *, P<0.05; **, P<0.01.
IAP inhibitor APG-1387 exerts an antitumor effect on nasopharyngeal carcinoma cancer stem cells
The results above suggested that XIAP may be a promising target in the treatment of NPC stem cells. APG-1387 is a novel SMAC mimetic and has strong binding affinity to XIAP and cIAP1/2. APG-1387 potently induced cIAP1/2 and XIAP degradation in a time- and dose-dependent manner in S-18 and S-26 cells (Fig. 6A-B).
The effect of XIAP small-molecule inhibitor APG-1387 on NPC cancer stem cells in vitro. (A, B) S-18 or S-26 cells were treated with multiple doses of APG-1387 (1-104 nM) for 24 h or APG-1387 (1 μM) for the indicated time (1-12 h). Total isolated protein was analysed by immunoblotting with the indicated antibodies. (C) S-18 cells were treated with multiple doses of APG-1387 (1-104 nM) for 24 h. The expression levels of Sox2, Notch1, Oct4 and c-myc were determined by western blotting. Blots shown are representative of three independent experiments (A) (B) and (C). (D) S-18 and S-26 cells were treated with APG-1387 (10-100 nM) for 24 h, labelled with hoechst 33342 dye, and then analysed by flow cytometry. A set of representative flow cytometry dot plots are shown. Quantification of SP cells proportion is shown below. The data are represented as mean ± SD of three independent experiments.
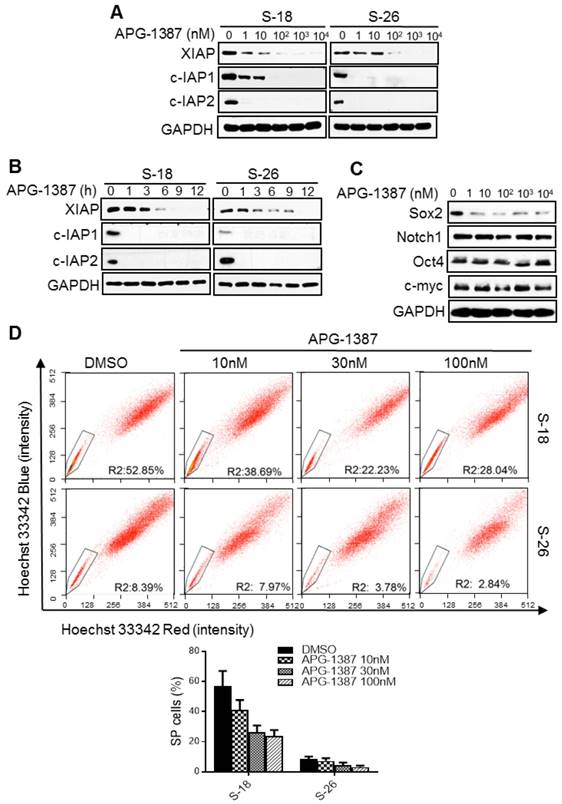
Next, we examined whether APG-1387 could inhibit the NPC cancer stem-like cells in vitro. Firstly, we found that 10 nM APG-1387 decreased the protein level of Sox2 (Fig. 6C) and the percentage of SP cells (Fig. 6D), and the reduction in SP cells was much greater in S-18 cells than in S-26 cells (Fig. 6D). Additionally, APG-1387 decreased cell migration ability in a dose-dependent manner (Fig. S2D). Lastly, a low concentration of APG-1387 (1 nM) treatment significantly inhibited sphere formation in S-18 cells, while CDDP single treatment had no effect (Fig. S2E). These data suggested that APG-1387 effectively inhibited the NPC stemness properties in vitro.
APG-1387 effectively sensitizes NPC to conventional chemotherapy in vitro and in vivo
CDDP and 5-FU are clinically active agents against NPC. However, drug resistance occurs owing to the presence of CSCs and apoptosis evasion. Here, we tested whether the combination of APG-1387 with conventional cancer therapy could completely induce cancer eradication and avoid recurrence. We combined APG-1387 with CDDP or 5-FU for treatment of NPC to study their synergistic antitumor effect in vivo and in vitro.
In the present study, CDDP or 5-FU alone increased the SP cell proportion in S-18 and S-26 cells, while APG-1387 treatment decreased the SP cell proportion (Fig. 7A-B). To determine if APG-1387 could enhance the chemo-sensitivity of NPC cells, either S-18 or S-26 cells were treated with a concentration gradient of CDDP or 5-FU alone or in combination with APG-1387 (3, 10, 30 nM) for 72 h. APG-1387 significantly enhanced the chemo-sensitivity of S-18 or S-26 cells, but the effect was more significant in S-18 cells (Fig. S4A). To investigate whether XIAP regulated drug resistance of other NPC cells lines displaying stem-like properties, we measured XIAP expression level and drug sensitivity of CDDP or 5-FU upon XIAP inhibition by APG-1387. APG-1387 significantly enhanced the chemo-sensitivity of HONE1, CNE1 and SUNE1 cells (Fig. S5). Then, we tested whether APG-1387 increased the chemo-sensitivity of NPC cells by re-enabling apoptosis. Annexin V-FITC/propidium iodide double staining assay showed that APG-1387 (30 nM) with CDDP (2.5 μM) or 5-FU (2.5 μM) treatment induced apoptosis in up to 62.8% and 38.4% of S-18 cells, respectively, and in 57.5% and 32% of S-26 cells, respectively (Fig. S4B, Fig. 7C). APG-1387 alone or combined with CDDP or 5-FU resulted in significant cleavage of poly (ADP-ribose) polymerase (PARP; Fig. 7D). Results above indicated that treatment with APG-1387 in combination with CDDP or 5-FU selectively targeted NPC CSCs in vitro.
The combinatorial effect of APG-1387 with CDDP or 5-FU on NPC cancer stem cells in vitro and in vivo. (A, B) S-18 or S-26 cells were treated with CDDP (2.5 μM), 5-FU (2.5 μg/mL), APG-1387 (100 nM) or a combination of APG-1387 with CDDP or 5-FU for 24 h, and were labelled with hoechst 33342 dye and analysed by flow cytometry. (C) S-18 or S-26 cells were treated with CDDP (2.5 μM), 5-FU (2.5 μg/mL), APG-1387 (30 nM) or a combination of APG-1387 with CDDP or 5-FU for 2 days, and stained with annexin V / propidium iodide, and measured by flow cytometry. Data shown are representative of three independent experiments. (D) (E) Average tumor growth for mice treated with single drug APG-1387, CDDP or 5-FU, or combined therapeutic of APG-1387 with CDDP or 5-FU. The tumor volume and body weights were monitored twice per week. *, P<0.05; **, P<0.01. Error bars represent the SEM from three independent experiments. (F, G) Survival curves were determined by Kaplan-Meier method using the days after CDDP or 5-FU treatment. Endpoint criteria were defined as when the longest axis of tumor exceeded 1.5 cm or active ulceration of the tumor. (H) Mice were implanted with cells derived from each group of primary tumor xenografts as shown in (E), and then tumor growth was measured twice per week. Error bars represent the SEM from three independent experiments. *, P<0.05.
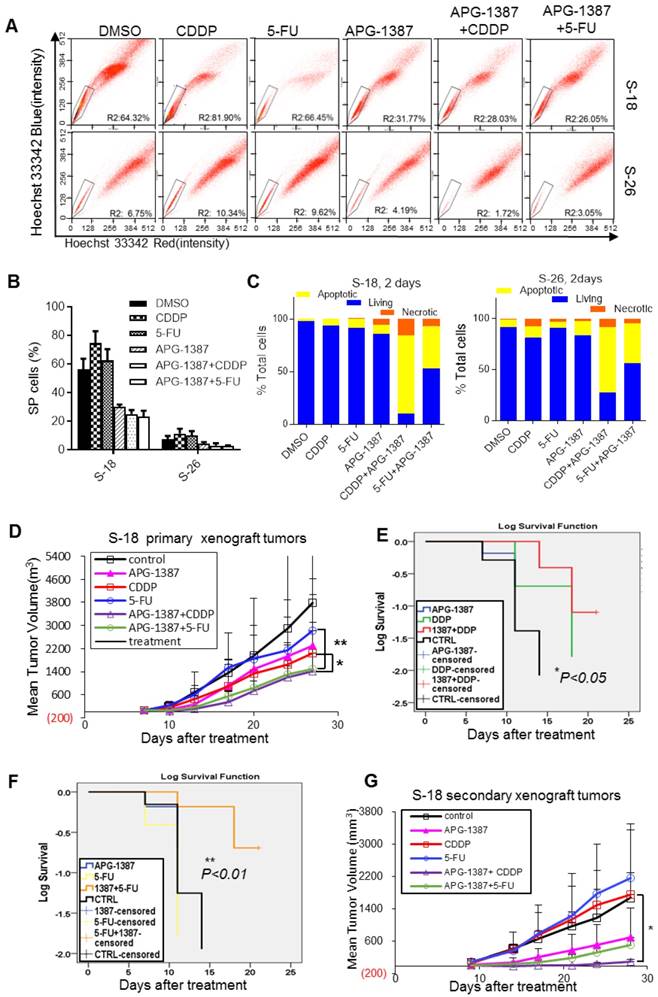
A tumor xenograft model was employed to test whether APG-1387 could affect tumor growth when combined with CDDP or 5-FU in vivo. S-18 cells were subcutaneously injected into mice, and when the resulting palpable tumors reached a size of 100 mm3, the mice were treated with normal saline, APG-1387 (3 mg/kg, 5 days per week, intravenous (iv)), CDDP (3 mg/kg, one day per week, intraperitoneal (ip)), 5-FU (25 mg/kg, two days per week, ip) or in combination (APG-1387, five days per week with CDDP or 5-FU) for three weeks. The tumor volumes and body weights were measured twice per week. We found that the tumor volumes of mice treated with APG-1387(10 mg/kg) in combination with CDDP or 5-FU were much smaller than those of mice treated with the single agent; the inhibition of tumor volume was 92.5% and 91.1% in the APG-1387 combined with CDDP or 5-FU groups, respectively (Fig. 7E-G). And notably, the average interval between endpoints (tumor reaching 1.5 cm on the longest axis and/or tumor ulceration) and starting point (first dose or 7 days after implantation) of the APG-1387 combined 5-FU group (18.33 days) was much longer than the APG-1387 (11.83 days) or 5-FU (10.16 days) single treatment groups (Fig. 7G). Importantly, no obvious toxicity was observed in mice receiving the treatments, and the body weights of the mice were unaffected (Fig. S6A-B).
To define stem-cell property of S-18 cells after combined treatment with APG-1387 and conventional therapy in vivo, we implanted nonobese diabetic/severe combined immunodeficient (NOD-SCID) mice with cells derived from the primary xenograft in each group. The cancer cells obtained from the APG-1387-treated mice resulted in a slower tumor growth, reaching a final tumor size ranging from 500 mm3 to 600 mm3 in the secondary mouse xenograft experiment (Fig. 7H). However, the cancer cells from the CDDP- or 5-Fu singly treated mice exhibited rapid tumor regrowth, reaching a final tumor size ranging from 1400 mm3 to 1600 mm3 in the secondary mouse xenograft experiment. Above all, the tumor regrowth of cells from mice with APG-1387 combined with CDDP treatment was the slowest among the 6 groups (Fig. 7H). These data illustrated that APG-1387 in combination with traditional chemotherapeutics showed a significant synergistic effect on the inhibition of NPC cells in vivo.
Discussion
In this study, we have shown that a member of the IAPs family, XIAP, is critical to CSCs maintenance and self-renewal in NPC (Fig. 1). We have demonstrated that XIAP regulates the stability of Sox2, a CSCs marker that is important for NPC CSCs maintenance and self-renewal [35, 36], through regulating the activation of ERK1 (Fig. S7). We identified ERK1 as a serine kinase that promotes the phosphorylation of Sox2 at S251, permitting the SUMOylation of Sox2 at K245, and thus leading it to autophagic degradation. The SUMOylation of Sox2, which is conserved in human Sox2 at K245, negatively regulates its transcriptional role by impairing DNA binding [30]. In addition, SUMOylation is important for cancer stem cell maintenance and self-renewal [33, 37]. The clinical significance of this finding is suggested by the analysis of clinical samples and xenograft tumors. Importantly, NPC patients with a high level of XIAP or Sox2 expression in tumor tissues exhibited a higher risk of relapse or metastasis after years of chemotherapy. Indeed, we reconstructed a Sox2-GST fusion protein and purified it from Escherichia coli. Using this fusion protein as a substrate, we found that ERK1 and Sox2 could interact with one another, and ERK1 phosphorylated Sox2 in vivo (Fig. 3). As Sox2 activity has been identified as a marker for CSCs in many cancer types [19, 34, 38-40], our findings are likely applicable to a broad range of cancers.
We also uncovered a unique mechanism of SUMOylation-dependent regulation of protein stability. SUMOylation regulates Sox2 stability through autophagy, which is dependent on the fusion of autophagosome and lysosome. The SUMO-dependent control of Sox2 degradation is distinct from the well-established paradigm of SUMO-dependent ubiquitin E3 ligase-induced ubiquitination and proteasome degradation; for example, RNF4, a prototype of such ubiquitin E3 ligases, targets SUMOylated proteins for ubiquitination and proteasome degradation through recognition of poly-SUMO chains [41-43]. Our results revealed a unique mechanism of SUMO-dependent autophagic degradation of Sox2 through binding with autophagic receptor p62.
We recently reported that SMAC mimetics AT-406 and SM-164 exerted an antitumor effect on NPC CSCs with TNF-related apoptosis inducing ligand (TRAIL) [28]. We now provide evidence that XIAP, a representative IAPs, predominantly affects the stability of Sox2 and maintains CSCs. However, a novel SMAC mimetic, APG-1387, can induce the apoptosis of CSCs and sensitize chemotherapy-resistant S-18 cells to CDDP or 5-FU treatment. These findings imply that the other members of the IAPs family may play a role in CSCs maintenance, including cIAP1, cIAP2, and survivin, although we focused on XIAP, mainly because it had the lowest expression in S26 cells compared to the S18 cell line. The important role of IAPs in CSCs as shown in this study is consistent with a previous study demonstrating that the combination of the IAPs inhibitors birinapant and carboplatin significantly improved disease-free survival in vivo compared with either therapy alone in tumor-bearing mice; thus, the combination overcame platinum resistance in a tumor-initiating subpopulation of ovarian cancer, indicating that IAPs may play a crucial role in cancer therapeutic resistance and recurrence [44]. This study provides a novel approach for using SMAC mimetics combined with chemotherapeutics for the treatment of NPC.
Abbreviation
CSCs: cancer stem cells; IAPs: inhibitors of apoptosis protein family; XIAP: X-linked IAP; cIAP1: cellular IAP1; cIAP2: cellular IAP2; Smac/DIABLO: second mitochondria-derived activator of caspases/direct IAP binding protein with low isoelectric point; SP: side population; NPC: nasopharyngeal carcinoma; FTC: fumitremorgin C; SUMO: small ubiquitin-like modifier.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81630079, 81572605, 81572732, 81772587, 81602066), the Natural Science Foundation of Guangdong Province (2014A030313010, 2017A030313481), the Science and Technology project of Guangdong Province (2014B050504004, 2015B050501005), the Science and Technology Project of Guangzhou (201504010038), the Fundamental Research Funds for the Central Universities (17ykjc25), the Major Science and Technology Project of National Basic Research Program (973 Program) of China (2012CB967004), and the Instrumental Analysis & Research Center, Sun Yat-sen University.
Supplementary Material
Supplementary figures 1-7.
Conflict of Interest
Dr. Dajun Yang has ownership interest (including patents) in Ascentage Pharma Group Inc. Jiao Ji and Guang-Feng Wang are employees of Ascentage Pharma Group Inc. No potential conflicts of interest were disclosed by the other authors.
References
1. Lo KW, To KF, Huang DP. Focus on nasopharyngeal carcinoma. Cancer Cell. 2004;5:423-8
2. Bensouda Y, Kaikani W, Ahbeddou N, Rahhali R, Jabri M, Mrabti H. et al. Treatment for metastatic nasopharyngeal carcinoma. Eur Ann Otorhinolaryngol Head Neck Dis. 2011;128:79-85
3. Kamran SC, Riaz N, Lee N. Nasopharyngeal carcinoma. Surg Oncol Clin N Am. 2015;24:547-61
4. Zhang L, Huang Y, Hong S, Yang Y, Yu G, Jia J. et al. Gemcitabine plus cisplatin versus fluorouracil plus cisplatin in recurrent or metastatic nasopharyngeal carcinoma: a multicentre, randomised, open-label, phase 3 trial. Lancet. 2016;388:1883-92
5. Wang J, Guo LP, Chen LZ, Zeng YX, Lu SH. Identification of cancer stem cell-like side population cells in human nasopharyngeal carcinoma cell line. Cancer Res. 2007;67:3716-24
6. Zhang HB, Ren CP, Yang XY, Wang L, Li H, Zhao M. et al. Identification of label-retaining cells in nasopharyngeal epithelia and nasopharyngeal carcinoma tissues. Histochem Cell Biol. 2007;127:347-54
7. Su J, Xu XH, Huang Q, Lu MQ, Li DJ, Xue F. et al. Identification of cancer stem-like CD44+ cells in human nasopharyngeal carcinoma cell line. Arch Med Res. 2011;42:15-21
8. Janisiewicz AM, Shin JH, Murillo-Sauca O, Kwok S, Le QT, Kong C. et al. CD44(+) cells have cancer stem cell-like properties in nasopharyngeal carcinoma. Int Forum Allergy Rhinol. 2012;2:465-70
9. Dragu DL, Necula LG, Bleotu C, Diaconu CC, Chivu-Economescu M. Therapies targeting cancer stem cells: Current trends and future challenges. World J Stem Cells. 2015;7:1185-201
10. Kakarala M, Wicha MS. Cancer stem cells: implications for cancer treatment and prevention. Cancer J. 2007;13:271-5
11. Clayton S, Mousa SA. Therapeutics formulated to target cancer stem cells: Is it in our future? Cancer Cell Int. 2011;11:7
12. Hong X, Lei L, Glas R. Tumors acquire inhibitor of apoptosis protein (IAP)-mediated apoptosis resistance through altered specificity of cytosolic proteolysis. J Exp Med. 2003;197:1731-43
13. Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer. 2010;10:561-74
14. Beug ST, Cheung HH, LaCasse EC, Korneluk RG. Modulation of immune signalling by inhibitors of apoptosis. Trends Immunol. 2012;33:535-45
15. Mehrotra S, Languino LR, Raskett CM, Mercurio AM, Dohi T, Altieri DC. IAP regulation of metastasis. Cancer Cell. 2010;17:53-64
16. Oberoi TK, Dogan T, Hocking JC, Scholz RP, Mooz J, Anderson CL. et al. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. EMBO J. 2012;31:14-28
17. Dogan T, Harms GS, Hekman M, Karreman C, Oberoi TK, Alnemri ES. et al. X-linked and cellular IAPs modulate the stability of C-RAF kinase and cell motility. Nat Cell Biol. 2008;10:1447-55
18. Lee SH, Oh SY, Do SI, Lee HJ, Kang HJ, Rho YS. et al. SOX2 regulates self-renewal and tumorigenicity of stem-like cells of head and neck squamous cell carcinoma. Br J Cancer. 2014;111:2122-30
19. Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG. et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41:1238-42
20. Tam WL, Ng HH. Sox2: masterminding the root of cancer. Cancer Cell. 2014;26:3-5
21. Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M. et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;511:246-50
22. Keysar SB, Le PN, Miller B, Jackson BC, Eagles JR, Nieto C. et al. Regulation of Head and Neck Squamous Cancer Stem Cells by PI3K and SOX2. J Natl Cancer Inst. 2017:109
23. Yang C, Wang H, Zhang B, Chen Y, Zhang Y, Sun X. et al. LCL161 increases paclitaxel-induced apoptosis by degrading cIAP1 and cIAP2 in NSCLC. J Exp Clin Cancer Res. 2016;35:158
24. Moon JH, Shin JS, Hong SW, Jung SA, Hwang IY, Kim JH. et al. A novel small-molecule IAP antagonist, AZD5582, draws Mcl-1 down-regulation for induction of apoptosis through targeting of cIAP1 and XIAP in human pancreatic cancer. Oncotarget. 2015;6:26895-908
25. Zappavigna S, Scuotto M, Cossu AM, Ingrosso D, De Rosa M, Schiraldi C. et al. The 1,4 benzoquinone-featured 5-lipoxygenase inhibitor RF-Id induces apoptotic death through downregulation of IAPs in human glioblastoma cells. J Exp Clin Cancer Res. 2016;35:167
26. Bai L, Smith DC, Wang S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacol Ther. 2014;144:82-95
27. Finlay D, Teriete P, Vamos M, Cosford NDP, Vuori K. Inducing death in tumor cells: roles of the inhibitor of apoptosis proteins. F1000Res. 2017;6:587
28. Wu MS, Wang GF, Zhao ZQ, Liang Y, Wang HB, Wu MY. et al. Smac mimetics in combination with TRAIL selectively target cancer stem cells in nasopharyngeal carcinoma. Mol Cancer Ther. 2013;12:1728-37
29. Qian CN, Berghuis B, Tsarfaty G, Bruch M, Kort EJ, Ditlev J. et al. Preparing the "soil": the primary tumor induces vasculature reorganization in the sentinel lymph node before the arrival of metastatic cancer cells. Cancer Res. 2006;66:10365-76
30. Tsuruzoe S, Ishihara K, Uchimura Y, Watanabe S, Sekita Y, Aoto T. et al. Inhibition of DNA binding of Sox2 by the SUMO conjugation. Biochem Biophys Res Commun. 2006;351:920-6
31. Van Hoof D, Munoz J, Braam SR, Pinkse MW, Linding R, Heck AJ. et al. Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell Stem Cell. 2009;5:214-26
32. Jeong CH, Cho YY, Kim MO, Kim SH, Cho EJ, Lee SY. et al. Phosphorylation of Sox2 cooperates in reprogramming to pluripotent stem cells. Stem Cells. 2010;28:2141-50
33. Suresh B, Lee J, Kim KS, Ramakrishna S. The Importance of Ubiquitination and Deubiquitination in Cellular Reprogramming. Stem Cells Int. 2016;2016:6705927
34. Weina K, Utikal J. SOX2 and cancer: current research and its implications in the clinic. Clin Transl Med. 2014;3:19
35. Lun SW, Cheung ST, Cheung PF, To KF, Woo JK, Choy KW. et al. CD44+ cancer stem-like cells in EBV-associated nasopharyngeal carcinoma. PLoS One. 2012;7:e52426
36. Zhang Y, Peng J, Zhang H, Zhu Y, Wan L, Chen J. et al. Notch1 signaling is activated in cells expressing embryonic stem cell proteins in human primary nasopharyngeal carcinoma. J Otolaryngol Head Neck Surg. 2010;39:157-66
37. Du L, Li YJ, Fakih M, Wiatrek RL, Duldulao M, Chen Z. et al. Role of SUMO activating enzyme in cancer stem cell maintenance and self-renewal. Nat Commun. 2016;7:12326
38. Favaro R, Appolloni I, Pellegatta S, Sanga AB, Pagella P, Gambini E. et al. Sox2 is required to maintain cancer stem cells in a mouse model of high-grade oligodendroglioma. Cancer Res. 2014;74:1833-44
39. Wen Y, Hou Y, Huang Z, Cai J, Wang Z. SOX2 is required to maintain cancer stem cells in ovarian cancer. Cancer Sci. 2017;108:719-31
40. Garros-Regulez L, Garcia I, Carrasco-Garcia E, Lantero A, Aldaz P, Moreno-Cugnon L. et al. Targeting SOX2 as a Therapeutic Strategy in Glioblastoma. Front Oncol. 2016;6:222
41. Sun H, Leverson JD, Hunter T. Conserved function of RNF4 family proteins in eukaryotes: targeting a ubiquitin ligase to SUMOylated proteins. EMBO J. 2007;26:4102-12
42. Kosoy A, Calonge TM, Outwin EA, O'Connell MJ. Fission yeast Rnf4 homologs are required for DNA repair. J Biol Chem. 2007;282:20388-94
43. Xie Y, Kerscher O, Kroetz MB, McConchie HF, Sung P, Hochstrasser M. The yeast Hex3.Slx8 heterodimer is a ubiquitin ligase stimulated by substrate sumoylation. J Biol Chem. 2007;282:34176-84
44. Janzen DM, Tiourin E, Salehi JA, Paik DY, Lu J, Pellegrini M. et al. An apoptosis-enhancing drug overcomes platinum resistance in a tumour-initiating subpopulation of ovarian cancer. Nat Commun. 2015;6:7956
Author contact
![]() Corresponding author: Xiao-Feng Zhu M.D., Ph.D., State Key Laboratory of Oncology in South China, Sun Yat-sen University Cancer Center, 651 Dongfeng Road East, Guangzhou, 510060, China. Tel: 8620-87343149; Fax: 8620-87343170; E-mail: zhuxfengsysu.edu.cn or Dajun Yang M.D., Ph.D., State Key Laboratory of Oncology in South China, Sun Yat-sen University Cancer Center, 651 Dongfeng Road East, Guangzhou, 510060, China. Tel: 8620-8732285; Fax: 8620-87343171; E-mail: yangdjorg.cn
Corresponding author: Xiao-Feng Zhu M.D., Ph.D., State Key Laboratory of Oncology in South China, Sun Yat-sen University Cancer Center, 651 Dongfeng Road East, Guangzhou, 510060, China. Tel: 8620-87343149; Fax: 8620-87343170; E-mail: zhuxfengsysu.edu.cn or Dajun Yang M.D., Ph.D., State Key Laboratory of Oncology in South China, Sun Yat-sen University Cancer Center, 651 Dongfeng Road East, Guangzhou, 510060, China. Tel: 8620-8732285; Fax: 8620-87343171; E-mail: yangdjorg.cn