Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(5):1350-1360. doi:10.7150/thno.22736 This issue Cite
Research Paper
Targeting Tumor Hypoxia Using Nanoparticle-engineered CXCR4-overexpressing Adipose-derived Stem Cells
Xinyi Jiang1, Christine Wang2, Sergio Fitch2, Fan Yang1,2, ![]()
1. Department of Orthopaedic Surgery, Stanford University, Stanford, CA 94305, USA
2. Department of Bioengineering, Stanford University, Stanford, CA 94305, USA
Received 2017-9-7; Accepted 2017-11-28; Published 2018-2-2
Abstract

Hypoxia, a hallmark of malignant tumors, often correlates with increasing tumor aggressiveness and poor treatment outcomes. Due to a lack of vasculature, effective drug delivery to hypoxic tumor regions remains challenging. Signaling through the chemokine SDF-1α and its receptor CXCR4 plays a critical role in the homing of stem cells to ischemia for potential use as drug-delivery vehicles. To harness this mechanism for targeting tumor hypoxia, we developed polymeric nanoparticle-induced CXCR4-overexpressing human adipose-derived stem cells (hADSCs). Using glioblastoma multiforme (GBM) as a model tumor, we evaluated the ability of CXCR4-overexpressing hADSCs to target tumor hypoxia in vitro using a 2D migration assay and a 3D collagen hydrogel model. Compared to untransfected hADSCs, CXCR4-overexpressing hADSCs showed enhanced migration in response to hypoxia and penetrated the hypoxic core within tumor spheres. When injected in the contralateral brain in a mouse intracranial GBM xenograft, CXCR4-overexpressing hADSCs exhibited long-range migration toward GBM and preferentially penetrated the hypoxic tumor core. Intravenous injection also led to effective targeting of tumor hypoxia in a subcutaneous tumor model. Together, these results validate polymeric nanoparticle-induced CXCR4-overexpressing hADSCs as a potent cellular vehicle for targeting tumor hypoxia, which may be broadly useful for enhancing drug delivery to various cancer types.
Keywords: malignant tumors, hypoxia, nanoparticles, adipose-derived stem cells, C-X-C chemokine receptor type 4 (CXCR4), glioblastoma.
Introduction
Hypoxia is a hallmark of malignant tumors and often correlates with increasing tumor aggressiveness and poor clinical outcomes.[1, 2] Most malignant tumors have been linked with tumor hypoxia, including glioblastoma multiforme (GBM),[3, 4] breast cancer,[5] prostate cancer,[6] and melanoma.[7] Malignant tumors contain a vascularized circumference, hypoxic intermediate layers, and a necrotic core. The increasing hypoxia within tumor core leads to upregulation of stromal cell-derived factor-1α (SDF-1α), also known as C-X-C motif chemokine 12 (CXCL12), and SDF-1α signaling has been observed in multiple cancers. [8, 9] Cancer stem cells and drug-resistant cancer cells are typically sequestered in the tumor's hypoxic and necrotic regions,[10] and the hypoxic microniche facilitates tumor initiation, progression, angiogenesis, and invasion.[10, 11]
Given the central role of hypoxia in tumor progression and resistance to chemotherapy and radiation, targeting hypoxic tumor cells is critical for enhancing therapeutic outcomes. Previous attempts to target hypoxia sought to develop prodrugs that could be activated via enzymatic reduction in hypoxic tissue, or small-molecule inhibitors against molecular targets involved in the survival of tumor cells in hypoxic tissues.[12, 13] However, these prodrugs and molecular inhibitors require efficient transport to target tissues, which is often greatly hindered in hypoxic tissues due to irregular blood flow and increased interstitial pressure.[14] As such, there remains a critical need to develop effective strategies for targeting tumor hypoxia for drug delivery.
Hypoxia-inducible factor 1α (HIF-1α), a central mediator of tissue hypoxia, strongly induces expression of SDF-1α in cancer cells, and SDF-1α is correlated with increasing tumor cell motility and invasiveness.[15] Multiple growth factors and receptors modulate the homing capacity of stem cells toward ischemia,[16-18] including SDF-1α and its receptor CXCR4,[16, 19] suggesting that stem cells could serve as drug-delivery vehicles for targeting cancer. HIF-1-induced SDF-1α upregulation has been shown to increase the homing of circulating CXCR4-positive endothelial progenitor cells to ischemic tissue in a skin wound healing model and in a hind-limb ischemia model.[20, 21] In addition to their ability to home to ischemia, stem cells can migrate toward tumor tissues in vivo.[22, 23] Although the SDF-1α/CXCR4 signaling axis is also believed to play an important role in this homing to cancer,[24], [34-37] harnessing stem cells for targeting tumor hypoxia remain largely unexplored.
Neural stem cells,[25-27] mesenchymal stem cells, [27, 28] embryonic stem cells[29] and induced pluripotent stem cells[30, 31] possess a remarkable capacity for migrating toward GBM cells and extensively investigated for targeted therapy and imaging. Although CXCR4 is highly expressed in mesenchymal stem cells in vivo, this expression markedly decreases during ex vivo expansion,[32, 33] which compromises the homing efficacy of these cells toward GBM. Given that in vitro expansion is often necessary to obtain clinically relevant numbers of cells for transplantation, it is critical to restore high CXCR4 expression in stem cells if they are to be used to target cancer in vivo. Viral vectors are the current gold standard for achieving high gene-delivery efficiency into various human stem cells,[28, 34, 35] but broad clinical applications are limited by the possibility of immune responses and unintended genomic integration.[36, 37] To overcome these safety concerns, biodegradable polymeric nanoparticles (NPs) have been recently reported to enable efficient gene delivery to human stem cells.[24, 38]
Here we have used our optimized polymeric NPs to restore high expression of CXCR4 in human adipose-derived stem cells (hADSCs) for targeting tumor hypoxia in vivo. We determined that polymeric NP-mediated transfection led to robust upregulation of CXCR4 in hADSCs with high cell viability, as characterized by flow cytometry, immunofluorescence, and western blotting. The hypoxia-targeting efficiency of CXCR4-overexpressing hADSCs was first evaluated in vitro using a transwell model and a 3D tumor sphere model. The ability of CXCR4-ADSCs to migrate toward tumors in vivo was subsequently examined using a mouse model of GBM. By comparing local intracranial delivery with systemic intravenous delivery, we characterized the effect of delivery route on the efficiency of targeting hypoxia. The results of this study validate the potential of nanoparticle-engineered hADSCs as a novel strategy for targeting tumor hypoxia in vivo, which may be broadly useful for enhancing drug delivery to various cancer cell types.
Materials and Methods
Cell Isolation and Cell Lines
hADSCs were isolated from the abdominal fat of a patient, as previously described [39]. All procedures were approved (APLAC 28120) and performed in accordance with the Stanford Institutional Review Board protocol. Cells were expanded in growth medium containing Dulbecco's Modified Eagle Medium (DMEM), 10% fetal bovine serum, 1% penicillin/streptomycin, and 10 ng/mL FGF-2. For brain tumor cells, we used cell line U87MG from the American Type Culture Collection. U87MG cells were transfected with GFP to facilitate in vivo cell tracking. All experiments were performed on cells in the logarithmic phase of growth.
Synthesis of PBAE and Self-assembly of NPs
Amino group-terminated PBAE was synthesized in a two-step reaction as described previously [38, 40]. Briefly, 1,4 butanediol diacrylate (C) (Sigma Aldrich) was reacted with 5-amino-pentanol (32) (Alfa Aesar) through a Michael addition reaction at 90 °C, and acrylate-terminated PBAE C32 was subsequently modified with an amino end group using tetraethyleneglycoldiamine (122) (Molecular Biosciences). After an overnight reaction at room temperature, amino-terminated PBAE was precipitated and purified in anhydrous diethyl ether (Fisher Scientific) and dissolved in 100 mg/mL dimethyl sulfoxide. Polymer stock solutions were stored in small aliquots at -20 °C until use.
To self-assemble PBAE-plasmid DNA NPs, PBAE and plasmid DNA were each dissolved in 25 mM sodium acetate buffer solution [pH 5.2]. Appropriate amounts of PBAE solution were mixed with diluted plasmid DNA solution in a 1.5 mL Eppendorf tube and vortexed for 10 s. PBAE/pDNA NPs were incubated at room temperature for 10 min to allow self-assembly via electrostatic interactions.
The size and zeta potential of PBAE-DNA NPs were determined via dynamic light scattering with a Zetasizer Nano ZS (Malvern Instruments, Ltd.). The shape and morphology of NPs were observed using transmission electron microscopy (JEOL-JEM 1400). For dynamic light scattering, PBAE-DNA NPs were suspended in fully supplemented DMEM containing 10% fetal bovine serum to reach a final DNA concentration of 10 μg/mL. For transmission electron microscopy, the NP sample was negatively stained with sodium phosphotungstatic solution (2%, w/v) and air-dried.
Optimizing NP-mediated Transfection
DNA encoding EGFP was used as a reporter to titrate NP-mediated transfection in hADSCs. The day before transfection, 6 x 104 hADSCs were seeded in each well of 24-well plates pre-coated with 0.1% gelatin, allowed to adhere overnight, and treated with complete DMEM containing 10% fetal bovine serum and PBAE-pEGFP NPs with various w/w ratios of PBAE to plasmid DNA. The optimum DNA dose (3.3 µg per 6 x 104 cells ) was used for transfection, as we previously reported [41] . After 4 h of incubation, NPs were removed and replaced with fresh culture medium. After another 44 h of incubation at 37℃ and 5% CO2, EGFP expression in cells was detected via fluorescence microscopy (Carl Zeiss, ApoTome).
Transfection efficiency was also quantified via flow cytometry. Briefly, culture medium was removed and cells were trypsinized, washed, and re-suspended in phosphate-buffered saline (PBS). Fluorescence-activated cell sorting of EGFP-positive cells was performed on a Scanford analyzer (Beckman Center, Stanford University). Cells transfected with pcDNA, a control vector, were used as a negative control and untransfected hADSCs were used to set the background. As a positive control, hADSCs were transfected with Lipofectamine 2000 and a DNA transgene in accordance with the manufacturer's protocol (Invitrogen). Data were analyzed with Flowjo 1.50i software (Tree Star).
Immunocytochemistry
Immunostaining was performed to detect CXCR4 protein expression in hADSCs after transient transfection with PBAE-CXCR4 NPs. Briefly, hADSCs were placed on 8-well Millicell EZ slides (Merck Millipore) at a density of 3×104 cells/cm2 in growth medium. Cells were transfected with PBAE/pCXCR4 NPs as described above. After 48 h of incubation at 37℃ and 5% CO2, spent medium was removed; cells were rinsed once with PBS and fixed with 2% paraformaldehyde in PBS for 10 min at room temperature. Cells were rinsed 3 times with PBS and permeabilized in 0.1% Triton X-100 in PBS that contained 1% bovine serum albumin (PBS-BSA) and 100 μg/mL RNase (Invitrogen) for 10 min at room temperature. After washing, cells were incubated with a blocking/permeabilization solution containing 10% normal goat serum (Dako) for 1 h at room temperature. After washing three times with PBS-BSA, cells were treated with rhodamine-phalloidin (1:100, Molecular Probes) in PBS-BSA for 20 min in the dark. Cells were then incubated with rabbit polyclonal anti-human CXCR4 (Abcam) diluted 1:100 in 0.5% Triton X-100 overnight at 4 °C. Secondary antibodies (Alexa Fluor 488 goat anti-rabbit secondary antibody; Invitrogen Life Technologies) diluted 1:500 in 0.5% Triton X-100 were subsequently added and incubated for another 2 hours at room temperature. After three washes, cell nuclei were counterstained with 10 mM Hoechst 33342 (Sigma-Aldrich) for 10 min at room temperature. Cells were imaged via fluorescence microscopy (Carl Zeiss, ApoTome).
Quantitative Reverse-transcription PCR (qRT-PCR)
Total RNA was isolated by homogenizing cells in buffer RLT and purifying the RNA via the Qiagen RNA extraction kit. Complementary DNA was synthesized using the SuperScript III First-Strand Synthesis kit (Life Technologies) in accordance with the manufacturer's instructions. Cycling was performed on an Applied Biosystems 7900 Real-Time PCR system (Applied Biosystems, Life Technologies) using Power SYBR Green PCR Master Mix (Applied Biosystems, Life Technologies) in accordance with the manufacturer's protocol. Primers were: GAPDH, 5′-ACAGTCAGCCCGCATCTTCTT-3′ and 5′-CGACCAAATCCGTTGACTC-3′; hCXCR4, 5′- TTCTACCCCAATGACTTGTG-3′ and 5′- ATGTAGTAAGGCAGCCAACA-3′.
Western Blotting
Cells were lysed with RIPA buffer plus proteinase inhibitors. Protein concentrations were measured via the Pierce BCA Protein Assay (Thermo Scientific) and equal amounts of cell extracts were loaded for western blotting. The primary antibodies used in this study were anti-human CXCR4 (Abcam, ab124824), anti-β-actin (Abcam, ab8227), and anti-PARP (Abcam, ab32138).
Cytotoxicity Assay
Cell viability was quantified using a colorimetric assay. The reduction of MTS tetrazolium by viable cells yields a colored formazan product that is soluble in cell culture medium. Passage 3 hADSCs were seeded into 96-well plates (1 x 104 cells/well), grown overnight at 37 ℃ and 5% CO2, and transfected with PBAE-pcDNA, PBAE-pEGFP, or PBAE-pCXCR4 NPs as described above. hADSCs were transfected with positive controls Lipofectamine 2000/pEGFP and Lipofectamine 2000/pCXCR4 in accordance with the manufacturer's instructions. Forty-four hours after transfection, the CellTiter 96® AQueous One Solution Cell Proliferation Assay kit (Promega) was used to determine cell viability in accordance with the manufacturer's instructions.
Conditioned Medium
To examine the effects of hypoxia on SDF-1α release, 2 x 106 U87MG cells were suspended in 2.5 mL DMEM and cultured under hypoxic (1% O2) or normoxic (20% O2) conditions for 72 h. Conditioned media were collected, centrifuged for 10 min at 600 × g, and stored at -80 °C until use.
hADSC Migration Toward Tumor-derived Conditioned Medium Using a Transwell Assay
The effects of tumor-derived conditioned medium on stem-cell migration were assessed using a transwell migration assay (ChemoTx® System, Neuro Probe Inc.). Passage 3 hADSCs (1 x 104 cells in 33 μL medium) pre-labeled with PKH26 (Sigma-Aldrich) were loaded into each transwell. Tumor-derived conditioned medium (hypoxia or normoxia) and base medium (55 μL/well) were loaded into a 96-well plate, into which the transwell was inserted. After 4 h of incubation, cells in the upper chamber were removed via aspiration and the porous membrane was carefully removed. Non-migrating cells on the origin side of the membrane were removed by gently wiping with a cotton swab. The nuclei of the cells on the membrane were counterstained with 10 mM Hoechst 33342 (Sigma-Aldrich) for 10 min at room temperature. At least five random fields of view on each membrane were selected, and cells were imaged with a fluorescence microscope (Carl Zeiss, ApoTome). Automated counting of nuclei was performed with ImageJ 1.5i (NIH). To determine whether the CXCR4 antagonist AMD3100 hindered hADSC migration, hADSCs were pre-incubated with 100 ng/mL AMD3100 (Sigma-Aldrich) for 1 h before they were placed in the upper compartment of the transwell.
Polarization and Penetration of hADSCs into GBM Spheroids
To determine whether CXCR4 overexpression in hADSCs enhanced their sensing of tumors after transfection with biodegradable NPs, we also performed in vitro 3D co-culture using a collagen gel model that mimics a GBM spheroid grown in an extracellular matrix. Spheroids were formed via the “hanging drop” technique. Briefly, 15 μL of culture medium containing 1.5 x 103 U87MG cells were dropped onto the inside cover of a 15-cm petri dish, which was filled with 15 mL PBS to maintain humidity. The dish was inverted and incubated at 37 °C with 5% CO2. After 4 days, spheroids were gently washed twice with DMEM. Each spheroid plus 600 hADSCs were re-suspended in 200 μL 1 mg/mL collagen I (Rat tail-derived, Thermo Fisher Scientific) in DMEM. The solution was transferred to one well of a 48-well plate pre-coated with 1% agarose. The plate was incubated at 37 °C with 5% CO2 and imaged at various time points.
Forty-eight hours after incubation, the collagen gel containing GBM spheroids and hADSCs was transferred to a tissue-culture plate, fixed with formaldehyde (10% w/v in PBS) for 30 min, and placed in a cavity microscope slide (Merck Millipore, Billerica MA). Z-stack images were obtained starting at the top of the spheroid in 20-μm intervals for a total of 100 μm into the spheroid using confocal laser scanning microscopy (Leica TCS SP5).
Tropism and In Vivo Targeting of Hypoxia by hADSCs
All animal procedures were performed with approval from the Stanford University Administrative Panel on Laboratory Animal Care and Use Committee. All procedures adhered to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals.
For mouse subcutaneous xenograft models, 5 x 106 U87MG cells in 100 μL PBS were injected into the subcutaneous tissue of the right hind legs of 8-week-old male athymic nude mice (Taconic, Nude NCr). When the tumors reached 0.7-0.9 cm in diameter, 2.5 x 106 untransfected hADSCs or 2.5 x 106 CXCR4-overexpressing hADSCs were injected into the tail vein of tumor-bearing mice (3 mice per group). After an additional 5 days, the hypoxic marker hypoxyprobe-1 (pimonidazole) was injected intraperitoneally (60 mg/kg, 20 min before euthanasia); the mice were then perfused intracardially with 4% paraformaldehyde in PBS. Tumor tissues were harvested, sectioned for hypoxia staining with the HypoxyprobeTM-1 kit (Chemicon) in accordance with the manufacturer's instructions, and mounted with Vectashield mounting medium with DAPI (VECTOR Lab. Inc.). Imaging was performed with a fluorescence microscope (Carl Zeiss, ApoTome).
For orthotopic human GBM xenografts, 4 x 106 U87MG cells (in 6 μL PBS) were inoculated into the right striatum of anesthetized male NCr nude mice (4-5 weeks of age; Taconic) through a 29 G burr hole (stereotactic coordinates: 1.8 mm lateral, 0.6 mm anterior to the bregma, and 3 mm of depth). Twelve days after tumor inoculation, 2 x 106 PKH26-labeled hADSCs were implanted into the hemisphere contralateral to the tumor using the same coordinates (3 mice per group). Five days later, the hypoxic probe hypoxyprobe-1 was injected intraperitoneally (60 mg/kg, 20 min before euthanasia); then the mice were perfused intracardially with 4% paraformaldehyde in PBS. Brains were harvested and sectioned for hypoxia staining using the procedure described above.
Statistical Analyses
All variables were expressed as mean ± standard deviation. GraphPad Prism 6.0 (GraphPad Software) was used to perform statistical analysis. Statistical differences between test conditions were determined via Student's t test. P < 0.05 was considered significant.
Results
Polymeric NPs Led to Robust Upregulation of CXCR4 in hADSCs with High Cell Viability
The optimal transfection formulation for hADSCs using NPs was first determined using enhanced green fluorescent protein (EGFP) encoded on plasmid DNA as a reporter. Flow cytometry indicated that compared with Lipofectamine 2000, a commercially available transfection reagent, the leading NP formulation using (poly (β-amino ester) (PBAE)s (w/w/ ratio of polymer:pEGFP = 20:1) enhanced the transfection efficiency by ~5.48 fold (Figure S3A, B). Importantly, NP-mediated transfection led to high cell viability that was comparable to that of untransfected cells (Figure S3C).
The biophysical properties of polymeric NPs that yielded the highest transfection efficiency were characterized using dynamic light scattering and transmission electron microscopy. The leading PBAE NPs had an average diameter of ~180 nm (Figure 1A), were spherical (Figure 1B), and had a zeta potential of 43.22 ± 2.33 mV (Figure 1C). We also determined that assembling NPs in deionized water led to smaller NPs with positive surface changes, while serum-containing medium led to larger NPs with negative surface charge (Table S1). Varying the sequence of the plasmid had minimal effect on NP size and surface charge (Table S1).
Polymeric NPs containing human CXCR4 DNA led to robust upregulation of CXCR4 in hADSCs with high cell viability. Physical characterization of NPs (20:1 PBAE/pCXCR4) using dynamic light scattering revealed the size distribution (A); transmission electron microscopy image (B) and surface charge (C) of NPs. CXCR4 expression was verified via (D) immunofluorescence, (E) flow cytometry (**p<0.01), and (F) western blotting. Scale bars = 20 μm. P, passage. (G) The MTS assay showed that cell viability did not significantly differ between untransfected hADSCs (defined as 100% viability) and cells transfected with NPs/pCXCR4 (48 h after transfection). N.S., no significant difference.
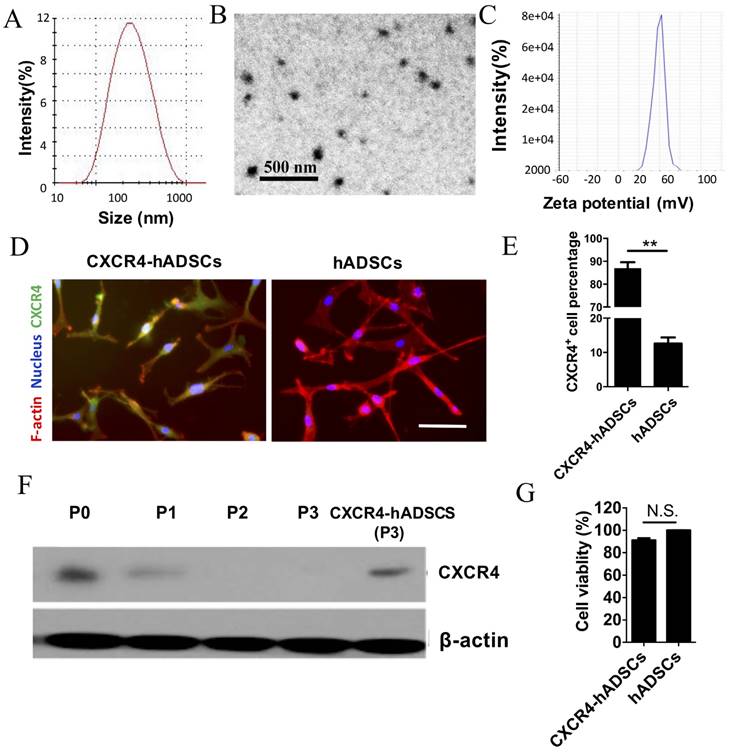
Using the leading PBAE NP formulation, hADSCs were transfected with a plasmid encoding human CXCR4. Immunofluorescence confirmed robust CXCR4 expression in hADSCs 48 h after transfection, while minimal signal was detected in untransfected passage 3 hADSCs (Figure 1D). Flow cytometry showed that 86.64 ± 8.32% of NP-transfected hADSCs expressed CXCR4 (Figure 1E). While CXCR4 was expressed by untransfected hADSCs (passage 0) on western blotting, expression was rapidly lost after passage 1, and became undetectable by passage 3 (Figure 1F). Transfection with NPs carrying CXCR4 plasmid DNA successfully restored CXCR4 expression in passage 3 hADSCs to a level comparable to that of untransfected hADSCs (Figure 1F) without compromising cell viability (Figure 1G).
CXCR4 Overexpression Enhances the Migration of hADSCs toward Tumor-derived Conditioned Medium Under Hypoxia In Vitro
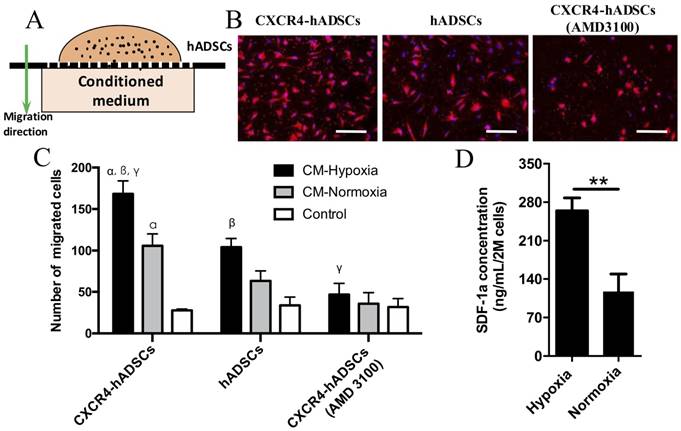
We next evaluated the behavior of CXCR4-overexpressing hADSCs toward conditioned medium collected from GBM cells cultured under hypoxic or normoxic conditions in vitro (Figure 2A). Data from our transwell migration assay indicated that conditioned medium from GBM cells cultured under hypoxia significantly enhanced the migration of hADSCs versus conditioned medium from cells cultured under normoxia; CXCR4-overexpressing hADSCs displayed the greatest migration (Figure 2B, C, Figure S5). Moreover, pre-incubation with AMD3100, an antagonist of CXCR4, reduced the migration of hADSCs (Figure 2B, C, Figure S5), confirming that CXCR4 plays an important role in targeting tumor hypoxia. Our enzyme-linked immunosorbent assay (ELISA) data revealed that hypoxia significantly upregulated the level of SDF-1α secreted by GBM cells by 2.52-fold (Figure 2D). Together, our results demonstrate that CXCR4 overexpression enhances the migration of hADSCs toward tumor-derived soluble factors; in vitro, this migration is both induced and enhanced by the upregulation of SDF-1α secreted by GBM cells under the hypoxic conditions found in vivo.
GBM-derived conditioned medium (CM) cultured under hypoxia enhanced hADSCs migration, and CXCR4-overexpression in hADSCs showed further enhanced migration. (A) Schematic of a transwell-mediated chemotactic migration assay of hADSCs toward tumor-derived conditioned medium cultured under hypoxia or normoxia. Tumor growth medium alone was included as a control. (B) Compared with untransfected hADSCs, overexpression of CXCR4 in hADSCs increased cell migration toward tumor-derived CM under normoxia. CM derived under hypoxia (O2, 1%) further enhanced migration of both hADSCs and CXCR4-hADSCs. Pre-incubation of hADSCs with the CXCR4 antagonist AMD3100 abolished such migration of both hADSCs and CXCR4-overexpressing hADSCs to a minimal level as the control. hADSCs were stained red with the fluorescent dye PKH26; nuclei were labeled blue with Hoechst 33342. Scale bar = 200 μm. (C) Quantification of cell migration. CM, conditioned medium. αp<0.05 vs. hADSC migrated towards conditioned medium from normoxia (O2, 20%); βp<0.05 vs. hADSCs migrated towards CM-Hypoxia, γp<0.05 vs. CXCR4-hADSCs migrated towards CM-Normoxia. (D) Hypoxia significantly up-regulates SDF-1α secreted in U87MG glioma cells. The amount of SDF-1α in conditioned media from U87MG under hypoxia (O2, 1%) or normoxia (O2, 20%) was quantified using ELISA. **P<0.01.
CXCR4 Overexpression Enhances hADSC Migration Toward and Penetration Into the Hypoxic Core of 3D Tumor Spheroids
Large multicellular spheroids are 3D in vitro tissue models with an inherent gradient of oxygen: a central hypoxic core surrounded by quiescent viable cells and an outer layer of cells actively proliferating under normoxic conditions. To mimic tumor hypoxia in vivo, here we fabricated large 3D spheroids ~500-600 µm in diameter with a hypoxic core (Figure 3A). To evaluate the ability of hADSCs to target tumor hypoxia in 3D, next we co-cultured tumor spheroids and PKH26-labeled hADSCs in a 3D collagen hydrogel (Figure 3B). CXCR4 overexpression enhanced hADSC migration toward tumor spheroids. CXCR4-overexpressing hADSCs responded most quickly by migrating toward GBM spheroids as early as 8 h, with migration increasing over time (Figure 3C, D). By 48 h, CXCR4-overexpressing cells appeared on the surface of the spheroids (Figure 3C, D). In contrast, unmodified hADSCs responded more slowly and did not migrate as far toward the spheroid.
hADSCs respond to hypoxia in 3D tumor spheroids and penetrate into solid tumor spheroids. (A) Tumor spheroids comprise a normoxic outer shell, surrounding a hypoxic core. Hypoxia immuno-staining of 3D multicellular glioma spheroids using Hypoxyprobe-1 kit. Blue: Nucleus stained by DAPI. Red: Hypoxic region stained by hypoxyprobe-1 kit (pimonidazole). Scale bar=100 μm. (B) The schematic drawing shows a spheroid and hADSCs 3D co-culture model that mimicks tumor extracellular matrix (ECM). (C) CXCR4-hADSCs showed the faster response and migrated towards tumor spheroids over 48 h, while hADSCs showed a moderate level of tropism towards tumor spheroid. Scale bar=600 μm. (D) Quantification of hADSCs polarized to tumor spheroids. **p<0.01; N.S., non-significant. (E) Penetration of hADSCs into solid tumor spheroids. Confocal images of glioma spheroids after 48 h migration showed some of the red cells penetrated inside the glioma spheroid and accessed the hypoxic cells inside the spheroids. Z-stack images were obtained starting at the top of the spheroid in 25 μm intervals for a total of 100 μm into the spheroid. Red: PKH 26-labeled migrating cells. Green: GFP-positive tumor cells (U87MG). Gray: Bright field of collagen matrix. Yellow arrow indicates the migrated cells. Scale bar = 300 μm. (F) Quantification of hADSCs penetrated into tumor spheroids. *p<0.05; **p<0.01; N.S., no significant difference; N.A., not available.
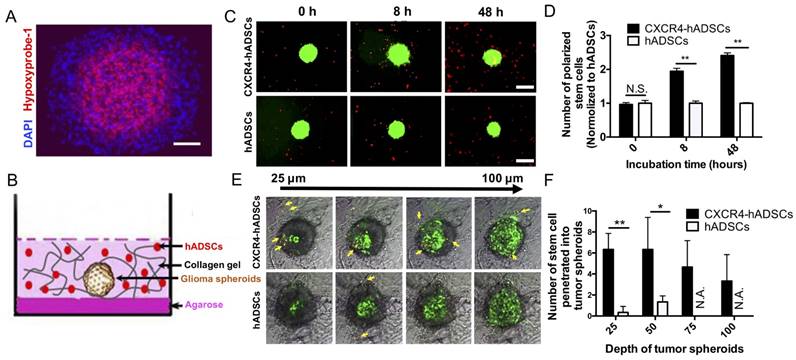
To test the capability of hADSCs to penetrate tumor spheroids, we performed confocal scanning of GBM spheroids after 48 h of co-culture in a 3D collagen gel. CXCR4-overexpressing hADSCs penetrated the spheroid and accessed the hypoxic core; untransfected ADSCs did not (Figure 3E, F), suggesting that CXCR4 overexpression is required for effective targeting of the hypoxic tumor core in 3D.
Intracranial Delivery of CXCR4-overexpressing hADSCs Targets Tumor Hypoxia in an In Vivo Mouse Intracranial Brain Tumor Xenograft Model
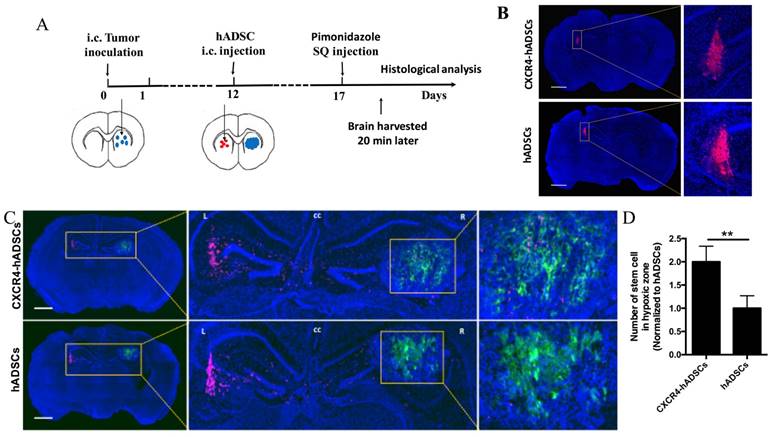
To examine the ability of hADSCs to target brain tumor hypoxia in vivo, we utilized a mouse intracranial brain tumor xenograft model [39]. hADSCs were injected into the contralateral brain hemisphere of the tumor site, and brain tissues were harvested 5 days later for histological analyses of migration (Figure 4A). To facilitate the visualization of brain-tumor hypoxia, animals were injected with pimonidazole, which labels hypoxic cells green (Figure 4A). In the absence of tumor, hADSCs remained at the injection site; CXCR4 overexpression did not induce migration (Figure 4B). In contrast, in tumor-bearing mice, both CXCR4-overexpressing hADSCs and untransfected hADSCs migrated long distances from the initial injection site toward tumor hypoxia (Figure 4C, D). Only 5 days after contralateral injection, CXCR4-overexpressing hADSCs reached the tumor and co-localized with the hypoxic tumor cells (Figure 4C, D).
CXCR4-overexpressing hADSCs enhance targeting of the hypoxic core of brain tumors in a mouse intracranial GBM xenograft model 5 days after intracranial delivery. (A) Schematic of experimental design. i.c., intracranial. hADSCs was injected in the contralateral side of the brain from the intracranial GBM tumor. (B and C) Representative brain sections illustrate the migratory behavior of CXCR4-overexpressing hADSCs and untransfected hADSCs 5 days after injection in the mouse brain tissue without or with GBM xenografts. (B) In the absence of tumor, both untransfected hADSCs and CXCR4-overexpressing hADSCs did not show any migration and remained at the site of injection. (C) In the presence of brain tumor, hADSCs (red) demonstrated long-range tropism across the brain toward tumor hypoxia (green). CXCR4 overexpression led to further enhanced co-localization of hADSCs and hypoxic tumor cells. Blue, cell nuclei stained with DAPI. Scale bars = 1mm. (D) Quantification of stem cells localized in hypoxia zone in tumor tissue. **p<0.01.
Since the blood-brain barrier constitutes a major obstacle in the treatment of brain tumors, we sought to determine whether hADSCs could cross this barrier and migrate toward intracranial brain tumors. We therefore performed systemic delivery of hADSCs via intravenous injection through the tail vein (Figure S6A). Neither CXCR4-overexpressing hADSCs engineered with NPs nor untransfected hADSCs traversed the blood-brain barrier, and no hADSCs were detected in the brain (Figure S6B). Taken together, these results show that intracranial delivery is required for targeting brain tumor hypoxia using CXCR4-overexpressing hADSCs.
Systemic Intravenous Delivery of CXCR4-overexpressing hADSCs Targets Tumor Hypoxia in a Subcutaneous Cancer Model
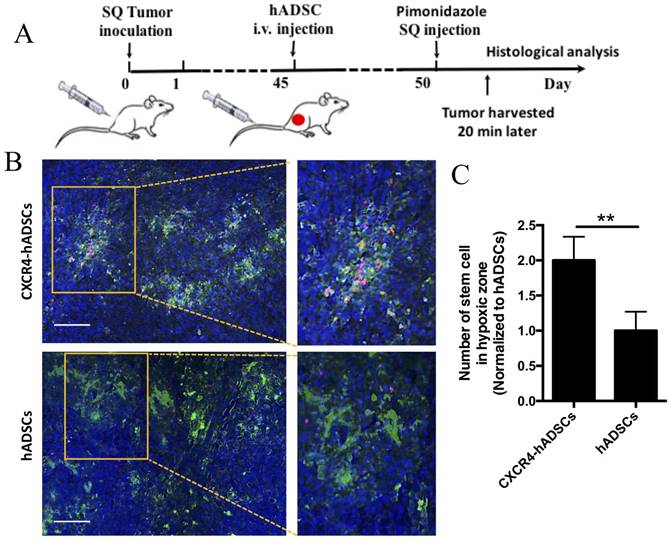
We next investigated whether intravenous injection of NP-assembled, CXCR4-overexpressing hADSCs targeted tumor hypoxia outside the brain using a mouse subcutaneous GBM xenograft model (Figure 5A). Five days after intravenous delivery, hADSCs were found in solid GBM tumors, and CXCR4-expressed hADSCs displayed enhanced tumor hypoxia targeting versus unmodified hADSCs (Figure 5B, C). These results suggest that systemic delivery of hADSCs overexpressing CXCR4 via NPs enables these stem cells to penetrate solid tumors outside the brain, leading to the enrichment of hADSCs in the hypoxic zones of malignant tumors.
CXCR4 overexpression increased the enrichment of hADSCs in the tumor hypoxic region of a mouse subcutaneous GBM xenograft model 5 days after tail-vein injection. (A) Schematic of experimental design. i.v., intravenous; SQ, subcutaneous. (B) Representative sections of the hypoxia targeting of CXCR4-overexpressing hADSCs (C) and untransfected hADSCs in subcutaneous tumor tissue. Blue, cell nuclei stained with DAPI; green, tumor hypoxic regions stained with pimonidazole; red, hADSCs labeled with PKH26. Scale bars = 100 μm. (C) Quantification of stem cells localized in hypoxia zone in tumor tissue. **p<0.01.
Discussion
The cores of solid malignant tumors are hypoxic core due to structural abnormalities in tumor microvessels and damaged microcirculation [42, 43]. This hypoxic core promotes more aggressive cancer phenotypes and leads to the resistance to conventional chemotherapy of cancer stem cells and progenitor cells within tumor tissues [10, 44]. Effective strategies for targeting tumor hypoxia remain lacking, posing a critical bottleneck for enhancing treatment outcomes for different types of malignant tumors. To overcome this obstacle, here we established a novel strategy for targeting tumor hypoxia by employing NP-engineered stem cells as cellular vehicles for potential drug delivery and harnessing CXCR4/SDF-1α signaling to further enhance the enrichment of stem cells within the hypoxic region of tumors. We specifically chose hADSCs due to their relative abundance, ease of isolation, and promise as an autologous cell source from individual patients. Using GBM as a model tumor, we demonstrated that hADSCs migrate toward tumor hypoxia in vitro (Figure 2) and in vivo (Figures 4, 5). NP-induced CXCR4 overexpression further enhanced hADSC penetration into the hypoxic core of solid tumors (Figure 3).
Hypoxia leads to upregulation of HIF-1α, which interacts with HIF-1β and translocates to the nucleus, resulting in the transcription of downstream target genes including SDF-1α [10] [11]. Consistent with previous findings, here hypoxia led to upregulation and secretion of SDF-1α in tumor cells (Figure 2D). SDF-1α/CXCR4 signaling is one of the major pathways responsible for stem-cell homing to tumor tissues.[24, 45-48] CXCR7 is another chemokine receptor with high binding affinity to SDF-1α, however, the activation of CXCR7 by SDF-1α does not contribute to cell chemotaxis [49]. Here, CXCR4 overexpression significantly enhanced the migration of hADSCs toward tumor-derived conditioned medium under hypoxic conditions (Figure 2B, C). Further, when co-cultured in 3D hydrogels with tumor spheroids, CXCR4-overexpressing hADSCs migrated more quickly toward tumor spheroids than untransfected hADSCs (Figure 3C, D), and penetrated the hypoxic core of tumor spheroids more robustly than untransfected hADSCs (Figure 3E, F).
To target tumor hypoxia in vivo, enabling the use of cells as drug-delivery vehicles, we sought to determine the optimal route for cell delivery. Intracranial injection of hADSCs led to long-range migration across the brain hemisphere toward GBM (Figure 4C). It is worth noting that both hADSCs and CXCR4-hADSCs can target both normoxic tumor and hypoxic regions, and CXCR4 overexpression enhanced the speed of migration of hADSCs towards tumor, and also increased the co-localization within the tumor hypoxic region (Figure 4C, D). The biodistribution of MSCs has been widely investigated in both animals and in human patients [2, 3]. Intravenous injection of MSCs resulted in the majority of the cells trapped initially in the lung, which was mostly cleared out by day 10. In contrast, more MSCs would be entrapped in spleen tissues by day 10 after transplant. Similar bio-distribution of MSCs have been observed in humans and animal models [3]. Given the similarities of MSCs isolated from bone marrow and adipose tissues [50], we expect a similar bio-distribution of hADSCs in tumor-bearing mice. When injected intravenously through the tail vein, neither CXCR4-overexpressing hADSCs nor hADSCs were detected in the brain (Figure S5B), suggesting that stem cells cannot overcome the blood-brain barrier when delivered systemically. Consistent with our findings, previous studies using neural stem cells to target brain tumors also reported that intracranial delivery is required for targeting intracranial brain tumors.[25, 27, 51-53] If the tumor is located outside the brain, then systemic delivery of NP-engineered hADSCs still effectively targets tumor hypoxia (Figure 5B, C). Thus, our data indicate that intracranial delivery is required for tumors within the brain, but systemic delivery is sufficient for malignant tumors located outside the brain.
For clinical translation of stem cell-based therapies for tumor targeting, biosafety is an important aspect to consider. In a recent preclinical study on treating patients with mammary carcinoma using MSCs, Koç et al. demonstrated that intravenous injection MSCs in patients at a dose of 1 million cells per kg body weight resulted no noticeable side effects [2]. Safety of intravenous delivery of human adipose-derived MSCs was also confirmed in animal models and patients, with no detectable chromosomal abnormalities or tumor formation 8 months after injection [6]. These studies suggest human ADSCs can serve as a safe cellular vehicle for targeting tumor hypoxia when used at appropriate doses. Several biological drugs have been delivered by stem cells, including chemotherapeutics,[54-56] prodrugs,[57] and genetic signals.[58] Future studies may employ NP-engineered hADSCs as vehicles to deliver various therapeutic agents into hypoxic tumor cores, helping overcome the challenges of effectively targeting tumor hypoxia and enhancing therapeutic outcomes for drug-resistant malignant tumors. While this study employed GBM as a model tumor, the concept of harnessing NP-engineered hADSCs for targeting tumor hypoxia and enhancing drug delivery can be broadly applied to the treatment of other cancer types.
Acknowledgements
This work was supported by an Alliance for Cancer Gene Therapy Young Investigator award grant (F.Y.). X.Y.J. acknowledges support from a Stanford School of Medicine Dean's Postdoctoral fellowship and a Child Health Research Institute Grant and Postdoctoral fellowship. This work was also supported by NIH R01DE024772 (F.Y.), NSF CAREER award CBET-1351289 (F.Y.), California Institute for Regenerative Medicine Tools and Technologies Award RT3-07804 (F.Y.), a Stanford Chem-H Institute Biomaterials Seed grant (F.Y.), the Stanford Bio-X Interdisciplinary Initiative Program (F.Y.), and a Stanford Child Health Research Institute Faculty Scholar Award (F.Y.).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nature Reviews Cancer. 2004;4:437-47
2. Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nature Reviews Cancer. 2002;2:38-47
3. Ferrarelli LK. Hypoxia micromanages glioma. Sci Signal. 2016;9:ec11-ec
4. Evans SM, Judy KD, Dunphy I, Jenkins WT, Hwang W-T, Nelson PT. et al. Hypoxia is important in the biology and aggression of human glial brain tumors. Clinical Cancer Research. 2004;10:8177-84
5. Schindl M, Schoppmann SF, Samonigg H, Hausmaninger H, Kwasny W, Gnant M. et al. Overexpression of hypoxia-inducible factor 1α is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clinical Cancer Research. 2002;8:1831-7
6. Zhong H, Semenza GL, Simons JW, De Marzo AM. Up-regulation of hypoxia-inducible factor 1α is an early event in prostate carcinogenesis. Cancer detection and prevention. 2004;28:88-93
7. Hartmann A, Kunz M, Köstlin S, Gillitzer R, Toksoy A, Bröcker E-B. et al. Hypoxia-induced up-regulation of angiogenin in human malignant melanoma. Cancer research. 1999;59:1578-83
8. Sorrentino C, Miele L, Porta A, Pinto A, Morello S. Activation of the A2B adenosine receptor in B16 melanomas induces CXCL12 expression in FAP-positive tumor stromal cells, enhancing tumor progression. Oncotarget. 2016;7:64274
9. Guo J-C, Li J, Zhou L, Yang J-Y, Zhang Z-G, Liang Z-Y. et al. CXCL12-CXCR7 axis contributes to the invasive phenotype of pancreatic cancer. Oncotarget. 2016;7:62006
10. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nature Reviews Cancer. 2005;5:275-84
11. Yang L, Lin C, Wang L, Guo H, Wang X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Experimental cell research. 2012;318:2417-26
12. Mohindra JK, Rauth AM. Increased cell killing by metronidazole and nitrofurazone of hypoxic compared to aerobic mammalian cells. Cancer research. 1976;36:930-6
13. Mason RP, Holtzman JL. The role of catalytic superoxide formation in the O 2 inhibition of nitroreductase. Biochemical and biophysical research communications. 1975;67:1267-74
14. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nature Reviews Cancer. 2006;6:583-92
15. Zhao D, Najbauer J, Garcia E, Metz MZ, Gutova M, Glackin CA. et al. Neural stem cell tropism to glioma: critical role of tumor hypoxia. Molecular Cancer Research. 2008;6:1819-29
16. Liu H, Liu S, Li Y, Wang X, Xue W, Ge G. et al. The role of SDF-1-CXCR4/CXCR7 axis in the therapeutic effects of hypoxia-preconditioned mesenchymal stem cells for renal ischemia/reperfusion injury. PloS one. 2012;7:e34608
17. Chavakis E, Urbich C, Dimmeler S. Homing and engraftment of progenitor cells: a prerequisite for cell therapy. Journal of molecular and cellular cardiology. 2008;45:514-22
18. Chavakis E, Dimmeler S. Homing of progenitor cells to ischemic tissues. Antioxidants & redox signaling. 2011;15:967-80
19. Marquez-Curtis LA, Janowska-Wieczorek A. Enhancing the migration ability of mesenchymal stromal cells by targeting the SDF-1/CXCR4 axis. BioMed research international. 2013. 2013
20. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME. et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nature medicine. 2004;10:858-64
21. Deveza L, Choi J, Lee J, Huang N, Cooke J, Yang F. Polymer-DNA Nanoparticle-Induced CXCR4 Overexpression Improves Stem Cell Engraftment and Tissue Regeneration in a Mouse Hindlimb Ischemia Model. Theranostics. 2016;6:1176
22. Sasportas LS, Kasmieh R, Wakimoto H, Hingtgen S, van de Water JA, Mohapatra G. et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proceedings of the National Academy of Sciences. 2009;106:4822-7
23. Kosaka H, Ichikawa T, Kurozumi K, Kambara H, Inoue S, Maruo T. et al. Therapeutic effect of suicide gene-transferred mesenchymal stem cells in a rat model of glioma. Cancer gene therapy. 2012;19:572-8
24. Kim D-S, Kim JH, Kwon Lee J, Choi SJ, Kim J-S, Jeun S-S. et al. Overexpression of CXC chemokine receptors is required for the superior glioma-tracking property of umbilical cord blood-derived mesenchymal stem cells. Stem Cells and Development. 2009;18:511-20
25. Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W. et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proceedings of the National Academy of Sciences. 2000;97:12846-51
26. Benedetti S, Pirola B, Pollo B, Magrassi L, Bruzzone MG, Rigamonti D. et al. Gene therapy of experimental brain tumors using neural progenitor cells. Nature medicine. 2000;6:447-50
27. Kauer TM, Figueiredo J-L, Hingtgen S, Shah K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nature neuroscience. 2012;15:197-204
28. Loebinger MR, Eddaoudi A, Davies D, Janes SM. Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer. Cancer research. 2009;69:4134-42
29. Ruan J, Song H, Li C, Bao C, Fu H, Wang K. et al. DiR-labeled embryonic stem cells for targeted imaging of in vivo gastric cancer cells. Theranostics. 2012;2:618
30. Liu Y, Yang M, Zhang J, Zhi X, Li C, Zhang C. et al. Human induced pluripotent stem cells for tumor targeted delivery of gold nanorods and enhanced photothermal therapy. ACS nano. 2016;10:2375-85
31. Yang M, Liu Y, Hou W, Zhi X, Zhang C, Jiang X. et al. Mitomycin C-treated human-induced pluripotent stem cells as a safe delivery system of gold nanorods for targeted photothermal therapy of gastric cancer. Nanoscale. 2017;9:334-40
32. Son BR, Marquez-Curtis LA, Kucia M, Wysoczynski M, Turner AR, Ratajczak J. et al. Migration of bone marrow and cord blood mesenchymal stem cells in vitro is regulated by stromal-derived factor-1-CXCR4 and hepatocyte growth factor-c-met axes and involves matrix metalloproteinases. Stem cells. 2006;24:1254-64
33. Honczarenko M, Le Y, Swierkowski M, Ghiran I, Glodek AM, Silberstein LE. Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem cells. 2006;24:1030-41
34. Ehtesham M, Kabos P, Gutierrez MA, Chung NH, Griffith TS, Black KL. et al. Induction of glioblastoma apoptosis using neural stem cell-mediated delivery of tumor necrosis factor-related apoptosis-inducing ligand. Cancer research. 2002;62:7170-4
35. Grisendi G, Bussolari R, Cafarelli L, Petak I, Rasini V, Veronesi E. et al. Adipose-derived mesenchymal stem cells as stable source of tumor necrosis factor-related apoptosis-inducing ligand delivery for cancer therapy. Cancer research. 2010;70:3718-29
36. Brown BD, Sitia G, Annoni A, Hauben E, Sergi LS, Zingale A. et al. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood. 2007;109:2797-805
37. Nayak S, Herzog RW. Progress and prospects: immune responses to viral vectors. Gene therapy. 2010;17:295-304
38. Yang F, Cho S-W, Son SM, Bogatyrev SR, Singh D, Green JJ. et al. Genetic engineering of human stem cells for enhanced angiogenesis using biodegradable polymeric nanoparticles. Proceedings of the National Academy of Sciences. 2010;107:3317-22
39. Jiang X, Fitch S, Wang C, Wilson C, Li J, Grant GA. et al. Nanoparticle engineered TRAIL-overexpressing adipose-derived stem cells target and eradicate glioblastoma via intracranial delivery. Proceedings of the National Academy of Sciences. 2016. 2016 15396
40. Keeney M, Ong S-G, Padilla A, Yao Z, Goodman S, Wu JC. et al. Development of Poly (β-amino esters)-Based Biodegradable Nanoparticles for Non-Viral Delivery of Minicircle DNA. Acs Nano. 2013;7:7241
41. Deveza L, Choi J, Imanbayev G, Yang F. Paracrine release from nonviral engineered adipose-derived stem cells promotes endothelial cell survival and migration in vitro. Stem cells and development. 2012;22:483-91
42. Secomb TW, Dewhirst MW, Pries AR. Structural adaptation of normal and tumour vascular networks. Basic & clinical pharmacology & toxicology. 2012;110:63-9
43. Pries AR, Höpfner M, Le Noble F, Dewhirst MW, Secomb TW. The shunt problem: control of functional shunting in normal and tumour vasculature. Nature Reviews Cancer. 2010;10:587-93
44. Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. Journal of the National Cancer Institute. 2007;99:1441-54
45. Ehtesham M, Yuan X, Kabos P, Chung NH, Liu G, Akasaki Y. et al. Glioma tropic neural stem cells consist of astrocytic precursors and their migratory capacity is mediated by CXCR4. Neoplasia. 2004;6:287-93
46. Van Der Meulen A, Biber K, Lukovac S, Balasubramaniyan V, Den Dunnen W, Boddeke H. et al. The role of CXC chemokine ligand (CXCL) 12-CXC chemokine receptor (CXCR) 4 signalling in the migration of neural stem cells towards a brain tumour. Neuropathology and applied neurobiology. 2009;35:579-91
47. Xu Q, Yuan X, Xu M, McLafferty F, Hu J, Lee BS. et al. Chemokine CXC receptor 4-mediated glioma tumor tracking by bone marrow-derived neural progenitor/stem cells. Molecular cancer therapeutics. 2009;8:2746-53
48. Park SA, Ryu CH, Kim SM, Lim JY, Park SI, Jeong CH. et al. CXCR4-transfected human umbilical cord blood-derived mesenchymal stem cells exhibit enhanced migratory capacity toward gliomas. International journal of oncology. 2011;38:97-103
49. Carbajal KS, Schaumburg C, Strieter R, Kane J, Lane TE. Migration of engrafted neural stem cells is mediated by CXCL12 signaling through CXCR4 in a viral model of multiple sclerosis. Proceedings of the National Academy of Sciences. 2010;107:11068-73
50. Li C-y, Wu X-y, Tong J-b, Yang X-x, Zhao J-l, Zheng Q-f. et al. Comparative analysis of human mesenchymal stem cells from bone marrow and adipose tissue under xeno-free conditions for cell therapy. Stem cell research & therapy. 2015;6:55
51. Aboody KS, Najbauer J, Metz MZ, D'Apuzzo M, Gutova M, Annala AJ. et al. Neural stem cell-mediated enzyme/prodrug therapy for glioma: preclinical studies. Science translational medicine. 2013;5:184ra59-ra59
52. Bagci-Onder T, Du W, Figueiredo J-L, Martinez-Quintanilla J, Shah K. Targeting breast to brain metastatic tumours with death receptor ligand expressing therapeutic stem cells. Brain. 2015 awv094
53. Redjal N, Zhu Y, Shah K. Combination of Systemic Chemotherapy with Local Stem Cell Delivered S-TRAIL in Resected Brain Tumors. Stem Cells. 2015;33:101-10
54. Bonomi A, Silini A, Vertua E, Signoroni PB, Coccè V, Cavicchini L. et al. Human amniotic mesenchymal stromal cells (hAMSCs) as potential vehicles for drug delivery in cancer therapy: an in vitro study. Stem cell research & therapy. 2015;6:1
55. Pacioni S, D'Alessandris QG, Giannetti S, Morgante L, De Pascalis I, Coccè V. et al. Mesenchymal stromal cells loaded with paclitaxel induce cytotoxic damage in glioblastoma brain xenografts. Stem cell research & therapy. 2015;6:1
56. Pessina A, Bonomi A, Coccè V, Invernici G, Navone S, Cavicchini L. et al. Mesenchymal stromal cells primed with paclitaxel provide a new approach for cancer therapy. PLoS One. 2011;6:e28321
57. Levy O, Brennen WN, Han E, Rosen DM, Musabeyezu J, Safaee H. et al. A prodrug-doped cellular Trojan Horse for the potential treatment of prostate cancer. Biomaterials. 2016;91:140-50
58. Levy O, Zhao W, Mortensen LJ, LeBlanc S, Tsang K, Fu M. et al. mRNA-engineered mesenchymal stem cells for targeted delivery of interleukin-10 to sites of inflammation. Blood. 2013;122:e23-e32
Author contact
![]() Corresponding author: Prof. Fan Yang, Ph.D., 300 Pasteur Drive, Edwards R105, Stanford, CA, 94305-5341; Phone: 650-725-7128; Fax: 650-723-9370
Corresponding author: Prof. Fan Yang, Ph.D., 300 Pasteur Drive, Edwards R105, Stanford, CA, 94305-5341; Phone: 650-725-7128; Fax: 650-723-9370