Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials & Methods
Results & Discussion
Conclusion
Acknowledgements
Supplementary Material
Abbreviations
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(4):1131-1145. doi:10.7150/thno.22078 This issue Cite
Research Paper
Biomimetic nanoparticles with enhanced affinity towards activated endothelium as versatile tools for theranostic drug delivery
Jonathan O. Martinez1* ![]() , Roberto Molinaro1,2*
, Roberto Molinaro1,2* ![]() , Kelly A. Hartman1, Christian Boada1,3, Roman Sukhovershin4, Enrica De Rosa5, Dickson Kirui5, Shanrong Zhang6, Michael Evangelopoulos1, Angela M. Carter7, James A. Bibb7, John P. Cooke4, Ennio Tasciotti1,8
, Kelly A. Hartman1, Christian Boada1,3, Roman Sukhovershin4, Enrica De Rosa5, Dickson Kirui5, Shanrong Zhang6, Michael Evangelopoulos1, Angela M. Carter7, James A. Bibb7, John P. Cooke4, Ennio Tasciotti1,8 ![]()
1. Center for Biomimetic Medicine, Houston Methodist Research Institute, 6670 Bertner Ave. Houston, TX 77030 USA.
2. Department of Cardiovascular Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, MA, USA
3. Escuela de Ingeniería y Ciencias, Tecnológico de Monterrey, Av. Eugenio Garza Sada 2501 Sur Col. Tecnológico, Monterrey, Nuevo León, México
4. Department of Cardiovascular Sciences, Houston Methodist Research Institute, Houston, TX 77030 USA
5. Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030 USA
6. Advanced Imaging Research Center, University of Texas Southwestern Medical Center, Dallas, TX 75390 USA
7. Department of Surgery, University of Alabama at Birmingham, Birmingham, AL 35294 USA
8. Houston Methodist Orthopedic and Sports Medicine, Houston Methodist Hospital, 6565 Fannin Street Houston, TX 77030 USA.
*Shared authorship
Received 2017-7-25; Accepted 2017-11-9; Published 2018-1-5
Abstract

Activation of the vascular endothelium is characterized by increased expression of vascular adhesion molecules and chemokines. This activation occurs early in the progression of several diseases and triggers the recruitment of leukocytes. Inspired by the tropism of leukocytes, we investigated leukocyte-based biomimetic nanoparticles (i.e., leukosomes) as a novel theranostic platform for inflammatory diseases.
Methods: Leukosomes were assembled by combining phospholipids and membrane proteins from leukocytes. For imaging applications, phospholipids modified with rhodamine and gadolinium were used. Leukosomes incubated with antibodies blocking lymphocyte function-associated antigen 1 (LFA-1) and CD45 were administered to explore their roles in targeting inflammation. In addition, relaxometric assessment of NPs was evaluated.
Results: Liposomes and leukosomes were both spherical in shape with sizes ranging from 140-170 nm. Both NPs successfully integrated 8 and 13 µg of rhodamine and gadolinium, respectively, and demonstrated less than 4% variation in physicochemical features. Leukosomes demonstrated a 16-fold increase in breast tumor accumulation relative to liposomes. Furthermore, quantification of leukosomes in tumor vessels demonstrated a 4.5-fold increase in vessel lumens and a 14-fold increase in vessel walls. Investigating the targeting mechanism of action revealed that blockage of LFA-1 on leukosomes resulted in a 95% decrease in tumor accumulation. Whereas blockage of CD45 yielded a 60% decrease in targeting and significant increases in liver and spleen accumulation. In addition, when administered in mice with atherosclerotic plaques, leukosomes exhibited a 4-fold increase in the targeting of inflammatory vascular lesions. Lastly, relaxometric assessment of NPs demonstrated that the incorporation of membrane proteins into leukosomes did not impact the r1 and r2 relaxivities of the NPs, demonstrating 6 and 30 mM-1s-1, respectively.
Conclusion: Our study demonstrates the ability of leukosomes to target activated vasculature and exhibit superior accumulation in tumors and vascular lesions. The versatility of the phospholipid backbone within leukosomes permits the incorporation of various contrast agents. Furthermore, leukosomes can potentially be loaded with therapeutics possessing diverse physical properties and thus warrant further investigation toward the development of powerful theranostic agents.
Keywords: Biomimetic, Leukocyte, Endothelium, Nanoparticles, Magnetic Resonance Imaging, Inflammation
Introduction
Inflammation is the body's natural response to harmful stimuli. However, chronic inflammation can lead to multiple pathologies including atherosclerosis, cancer, inflammatory bowel syndrome, and pulmonary diseases[1]. Vascular inflammation, which occurs in the early stages of these diseases, begins with activation of the endothelium in response to hemodynamic, humoral, metabolic, or pathological challenges[2]. Under these conditions, the endothelium begins to express molecules such as vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and selectins[3, 4]. The expression of these molecules promotes the recruitment, adhesion, and trans-endothelial extravasation of circulating leukocytes into the vessel wall[5]. This upregulated expression of adhesion molecules makes the vascular endothelium a promising target for early-stage diagnosis of inflammatory diseases and a useful feature for targeted drug delivery. Endothelial-targeted diagnostics and therapies could be greatly beneficial for the management of inflammatory disorders. Thus, development of specific targeting platforms capable of exploiting diverse states of the vascular endothelium for imaging or delivery of therapeutics is greatly warranted[6, 7].
NPs are promising tools to deliver poorly soluble therapeutics to specific areas, thereby limiting systemic toxicity. In addition, NPs can also function as both therapeutic and diagnostic agents (i.e., theranostics)[8]. Following systemic administration, NPs encounter several physical and biological barriers that impede their successful delivery to tumors[9]. The endothelium is a significant barrier for all systemically administered agents and functions to restrict access to NPs unless specific conditions are present (e.g., activated endothelium)[2]. In addition, the mononuclear phagocyte system (MPS) recognizes NPs as foreign and removes them from circulation, greatly reducing the amount of NP that can reach its target[10]. Furthermore, tumors exhibit additional unique barriers in the form of aberrant tissue (tumor-related vessels and matrix deposition), abnormal transport features (adverse oncotic and interstitial pressures), and cells (multidrug pumps)[11, 12].
Currently, most NPs rely on the presence of vessel fenestrations and vascular leaking, collectively referred to as the enhanced permeability and retention (EPR) effect, to overcome the endothelial barrier [13, 14]. EPR is a characteristic of unhealthy vasculature that facilitates passive accumulation of large macromolecules and NPs in solid tumors and inflamed tissues[15]. However, the heterogeneous presentation of EPR in different tumors, lack of cellular specificity, low vascular density, and limited patient data have dampened enthusiasm for EPR as an effective justification for NP targeting[16, 17]. Active targeting, or modification of the NP surface with peptides or antibodies, works in tandem with EPR to improve cell-specific targeting and tumor retention after extravasation from vasculature [18]. However, studies utilizing these strategies revealed only a modest increase in targeting efficiency in vivo [19]. This modest increase in targeting efficiency is possibly due to the formation of a protein corona on the surface of NPs that conceal the targeting moieties[20, 21]. The predominant strategy for NPs to avoid MPS uptake is to exploit the anti-opsonizing properties of polyethylene glycol (PEG)[22]. However, this strategy has been recently reconsidered after several studies showed the production of anti-PEG antibodies following repeated injections of PEGylated NPs[23, 24]. This phenomenon increases clearance of PEGylated liposomes and poses serious concerns about the long-term efficacy of PEGylated formulations[25-27]. Therefore, novel approaches are necessary to create a paradigm shift in the design of NPs to address these issues.
Recently biomimetic and bioinspired NPs have been explored as powerful alternatives for drug delivery to overcome the aforementioned barriers [28, 29]. These approaches provided unique functionalities by incorporating cellular coatings, synthesizing cell-derived nanovesicles, or mimicking the physical properties of cells to create novel NPs. These strategies successfully enabled selective binding to activated tumor endothelium[30-32], adhesion and aggregation features typical of red blood cells and platelets[33-35], and superior circulation time [36-38].
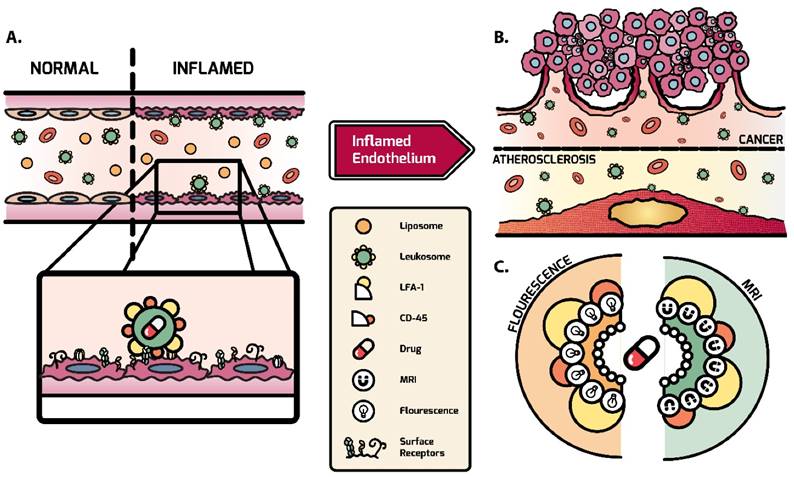
However, these approaches fell short due to their inability to reconstitute the entire profile of cell surface markers without adding excessive complexity to the fabrication process[39]. They also lacked versatility to encapsulate payloads with varying physical properties and exhibited inferior control of physicochemical parameters[39-41]. To address these issues, we developed a biomimetic NP that combines both bottom-up and top-down approaches by integrating leukocyte-derived membrane proteins into a synthetic phospholipid bilayer[42]. These specialized NPs, leukosomes, take advantage of the multifaceted and clinically tested properties of basic liposomes to enhance their stability and efficacy. Advantages of leukosomes include compatibility with diverse payloads, control of physicochemical features, avoidance of MPS, and selective targeting of various tissue types[43]. Leukosomes have previously been used to successfully target and treat local inflammation and inflammatory bowel diseases [42, 44]. Here, we demonstrate that leukosomes can be used to successfully target and image activated vasculature in breast tumors and atherosclerotic plaques (Fig. 1). Using optical imaging and intravital microscopy (IVM), we explore the ability of leukosomes to specifically target sites of inflammation. In addition, we interrogate the mechanism of targeting to elucidate the molecules critical for adherence to activated vasculature and MPS avoidance. Furthermore, we take advantage of the versatility of the leukosome formulation to demonstrate its ability to be modified for magnetic resonance imaging (MRI).
Schematic of leukosome targeting to activated endothelium. A) After systemic administration, leukosomes selectively bind to activated endothelium mediated by CD45 and LFA-1. B) Leukosomes experienced significant targeting to inflammation associated with cancer and atherosclerosis. C) The versatility of leukosomes permits the incorporation of various therapeutics and imaging modalities (e.g., fluorescence dyes and Gd).
Materials & Methods
Fabrication of NPs
NPs (liposomes and leukosomes) were assembled as previously reported[42]. Briefly, phosphocholine-based phospholipids and cholesterol (Avanti Polar Lipids) were dissolved in chloroform and the solvent was then evaporated through a rotary evaporator (BÜCHI Labortechnik AG) to form a film according to the thin layer evaporation method. Films were hydrated with mouse macrophage (J774, American Type Culture Collection) membrane proteins dispersed in water or water alone to assemble leukosomes or conventional liposomes, respectively. The lipid suspension was then extruded ten times through 200 nm pore-size cellulose acetate membranes at 45°C. For IVM studies, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl (Avanti Polar Lipids) was dissolved with the other lipids (0.1 v/v molar ratio) in chloroform before the hydration phase. For MRI studies, NPs were labeled with Gadolinium (Gd) using 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-diethylene-triamine-pentaacetic acid (Gd salt) (Avanti Polar Lipids). The Gd-labeled lipid was added to the organic phase during film formation at a 0.5 molar ratio.
Characterization of NPs
Physical characterization was performed with a Nanosizer ZS (Malvern Instruments) using dynamic light scattering to evaluate the mean diameter and poly-dispersity index (PDI). In addition, using the Nanosizer ZS, the zeta potential was measured to determine the surface charge of leukosomes and liposomes before and after labeling processes for both fluorescence (rhodamine) and MRI (Gd). Briefly, samples were diluted 1:20 in 1 ml of bi-distilled water and analyzed for average diameter, polydispersity index, and surface charge. At least five measurements per sample were performed. For cryo-transmission electron microscopy (cyro-TEM) analysis, leukosomes and liposomes were plunge-frozen on holey film grids (R2x2 Quantifoil®; Micro Tools GmbH, Jena, Germany) as previously reported[42]. Images were acquired on a JEOL 2100 electron microscope under low electron-dose conditions (5-20 electrons/Å2) using a 4,096 × 4,096 pxl CCD camera (UltraScan 895, GATAN, Inc., nominal magnifications 20,000×). For rhodamine quantification, NPs were suspended in 100% methanol to break open the bilayer structure and release the rhodamine-conjugated phospholipids into solution. This solution was then diluted 1:100 in 100% methanol and a serial dilution was made according to particle concentration (20 mM, 10 mM, 5 mM, 2.5 mM…). In order to assess fluorescence, samples were read using a plate reader (BioTek, Synergy H4 hybrid reader) and analyzed based on a standard curve for rhodamine. After purification, incorporation of Gd into NPs was evaluated using inductively coupled plasma-atomic emission spectroscopy.
Animal Care
Animal studies were performed in accordance with the guidelines of the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals based on approved protocols by Houston Methodist Research Institute's (HMRI) Institutional Animal Care and Use Committee.
4T1 Tumor Implantation
Female BALB/c (BALB/cAnNCrl; 6-8 weeks old) mice were purchased from Charles River Laboratories and maintained as previously described[45]. Murine 4T1 (American Type Culture Collection) cells were cultured in Roswell Park Memorial Institute media supplemented with 10% fetal bovine serum (Invitrogen). Upon reaching 50-70% confluency, 4T1 cells were collected and 5x105 cells per mouse were injected into the mammary fat pad of female mice to establish an orthotopic model of breast cancer. Tumors grew for 10-20 days to prepare for experiments and upon reaching 4-6 mm in diameter (200-250 mm3) were deemed appropriate for IVM imaging.
Murine Model of Atherosclerosis
8-week-old male apolipoprotein E (ApoE) -/- mice (Jackson Laboratories) were fed with a high-fat diet (Harlan Laboratories) for 12 weeks. ApoE-/- mice are devoid of ApoE, which is a subclass of high-density lipoproteins involved in cholesterol transport. These mice have elevated serum plasma cholesterol and spontaneously develop intimal lesions[46, 47]. The high fat diet accelerates the development of vascular lesions in this animal model.
Bioluminescent Imaging with Luminol
Whole animal imaging of inflammation in 4T1 tumors was assessed using luminol, as previously described[48]. A 50 mg/mL solution of luminol sodium salt (Sigma Aldrich) was prepared in phosphate buffered saline for in vivo administration. Briefly, mice (n=4) were established with tumors as previously described, with the exception that two tumors per mouse were established (i.e., 8 tumors total). Tumors were allowed to grow for 9 days and were then intraperitoneally administered with a dose of 250 mg/kg of luminol five minutes before imaging. Imaging was performed using an IVIS Spectrum with a 5 min acquisition, medium binning and an f/stop of 1. Images and analysis were exported using the Living Image software.
Intravital Microscopy (IVM)
Prior to administration of particles, mice were positioned on a heated stage for a suitable position to image tumor vessels as previously described[49, 50]. Tumor imaging was performed by exposing tumors using a skin-flap procedure. Before imaging, a bolus injection of 70 kDa fluorescein isothiocyanate dextran (50 µL in phosphate buffered saline, Invitrogen) was used to delineate the vasculature. Rhodamine-labeled liposomes and leukosomes were administered via retro-orbital injection into mice and images were captured continuously for 60 min post-injection. Images were obtained with a three-channel setup in which fluorescence was collected at 488/525 nm for fluorescein isothiocyanate-labeled dextran and at 561/579 nm for rhodamine-labeled particles. Image acquisition was performed over selected field of views with resolution of 512 × 256 pixels and an optical slice thickness of 7.1 µm. After 1 h and 6 h, animals were sacrificed and tumors were excised for ex vivo static imaging with the microscope (i.e., confocal microscopy). For blocking experiments, leukosomes were incubated with anti-lymphocyte function-associated antigen 1 (LFA-1) or anti-CD45 antibodies as previously reported[42]. Briefly, leukosomes were diluted in phosphate buffered saline and incubated separately with the above-mentioned antibodies (2.5 μg/mL) for a minimum of 30 min at room temperature. After purification through dialysis using 1000 kDa membrane filters for 1.5 h in water with mild stirring, blocked leukosomes were systemically injected for targeting studies. IVM is equipped with an upright Nikon A1R laser scanning confocal microscope with a resonance scanner, motorized and heated stage, and Nikon long-working distance 4× and 20× dry plan-apochromat objectives. It is housed within the Intravital Microscopy Core at HMRI. Images were analyzed and exported using Nikon Elements. The vessel area and the area covered by NPs were quantified with Nikon Elements. Images were analyzed from at least 3 mice to determine averages per NP type and accumulation in the lumen or vessel wall. To normalize the number of NPs to the fraction of vessels in each field of view, the fluorescence intensity of NPs per frame was divided by the vessel area of the same frame and displayed in Fig. 3F as NPs in the vessel lumen. To quantify the number of NPs that had adhered to the vessel wall, the minimum projection of several images was analyzed and the number of NPs divided by the vessel area per frame, as displayed in Fig. 3G.
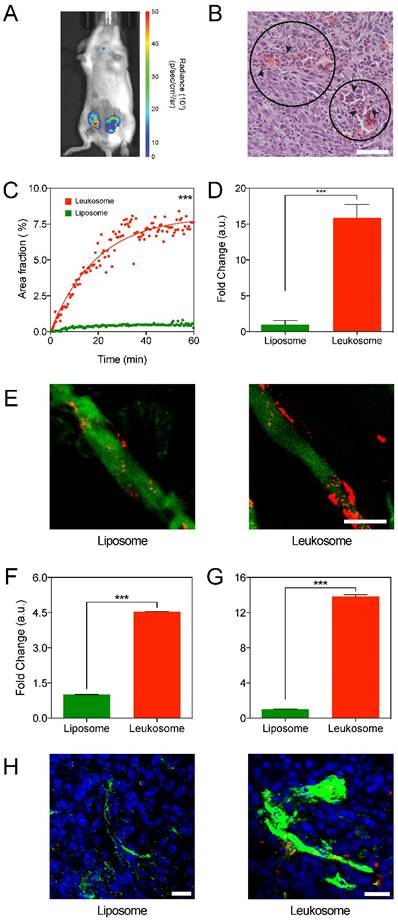
Targeting of activated tumor vasculature. A) Bioluminescence imaging of 4T1 tumors after administration with luminol to image presence of inflammation. B) H&E image (40× magnification) of a 4T1 tumor showing local necrosis with the presence of neutrophil infiltration (arrows) within the haphazard structure and distribution of blood vessels (circles); scale bar: 50 µm. C) Plot of the first 60 min of NPs accumulation in 4T1 tumors. D) Quantification of NPs in ex vivo images of tumors at 1 h after administration. E) Representative IVM images of 4T1 tumor vessels at 1 h after NP administration. NPs in red, vessels in green, scale bar: 25 µm. F,G) Quantification of NPs found within the lumen of vessels (F) and adhered to the vessel wall (G), with values normalized to vessel area. H) Representative immunofluorescence images of 4T1 tumor sections displaying the distribution of NPs (red) to CD31 (green) at 24 h after NP administration; scale bar: 50 µm. For all graphs: *** p < 0.001 and values represent the mean with error bars as s.e.m.
Histological Assessment
En-face preparation of murine aorta and microscopic analysis of plaque surfaces
After harvesting, the entire aorta from the aortic root to the iliac bifurcation was cleaned of surrounding adipose tissue, cut open longitudinally, and mounted on the dissection dish. The vascular lesions could be easily visualized and differentiated from healthy endothelium by gross examination of unstained preparations. Areas of the vessels containing plaques were imaged by using an upright Nikon A1R laser scanning confocal microscope equipped with a resonance scanner, motorized and heated stage, and Nikon long-working distance 4× and 20× dry plan-apochromat objectives (housed within the IVM Core at HMRI). Images were analyzed and exported using Nikon Elements.
H&E Staining
All samples were embedded in paraffin and sectioned sequentially from the root of the aorta following through the aortic arch. To assess the morphology of the tissue, samples were embedded in paraffin and stained using a standard Hematoxylin and Eosin (H&E) stain process. The samples were processed using the following protocol. Slides where placed in 100% xylene for three minutes three times and then transferred in a stepwise fashion from 50:50 xylene/100% ethanol to 100%, to 95%, to distilled water for three minutes each. Afterwards, the samples were placed in Gill´s hematoxylin for 5 min and then rinsed under tap water. Samples were then placed in acid alcohol briefly, washed in tap water, then placed in ammonia water and rinsed again in water. Next, samples were stained in Eosin Y for one minute and washed with tap water. After this step, the slides were washed in increasing concentrations of ethanol and then placed in xylene. Lastly, the samples were covered and mounted using cytoseal XYL and imaged using EVOS FL auto (Fischer Scientific).
Red O Staining
All samples were embedded in O.C.T. compound (Tissue-Tek) and sectioned sequentially from the root of the aorta following through the aortic arch. To assess the accumulation of lipids within the intima of the aorta, Oil Red O staining was done on cryo-sectioned tissues. 8 μm sections were cut from frozen tissues and placed on the appropriate slides to be air-dried. The slides were then fixed by briefly dipping the slide into formalin and then washing in tap water for 5 minutes. Afterwards, the slides were rinsed with 60% isopropanol. Next, the sample was stained with freshly prepared Oil Red O working solution for 15 min. The slides were then rinsed with 60% isopropanol. To visualize the nuclei, slides were stained with alum hematoxylin, dipping the slides 5 times. Following this step, the slides were rinsed with distilled water and mounted using aqueous mounting medium or glycerin jelly.
Immuno-fluorescence
Frozen 4T1 tumors were fixed in 4% paraformaldehyde followed by blocking and staining with an antibody to CD31 (BD Biosciences) overnight at 4ºC, as previously described[51]. After overnight incubation, sections were washed and an Alexa Fluor 488 secondary antibody was added for 1 h at room temperature. For CD45 and F4/80 staining, tissue sections were prepared as described above and stained with FITC-conjugated CD45 (BD Pharmingen) and FITC-conjugated F4/80 (Biolegend) overnight at 4ºC. Sections were then counterstained with 4',6-diamidino-2-phenylindole (ThermoFisher Scientific), mounted using SlowFade Diamond (ThermoFisher Scientific), and cover-slipped. Tumor sections were imaged using a Nikon A1 Confocal Imaging System housed within the Advanced Cellular and Tissue Microscopy Core at HMRI.
MRI of Phantoms
MRI scans were conducted using a 7T small animal MRI scanner (Agilent (Varian), Inc.) equipped with a 40 mm Millipede RF coil (ExtendMR LLC, Milpitas, CA). The sample was placed in the iso-center of the RF coil and the magnet. The bore temperature was kept at 25 ± 1°C by blowing hot/cold air controlled by a small animal monitoring/gating system (Small Animal Instruments, Inc., NY). Two-dimensional (2D) scout images on three orthogonal planes (axial, coronal and sagittal) were acquired to ensure the positioning. The “true” axial orientation was identified from the above three scans. Then, several images were acquired sequentially, including T1-weighted (T1WI) image, T1 map, T2-weighted (T2WI) image, and T2 map, respectively. Some common parameters were: matrix size = 128×128, field of view (FOV) = 40×40 mm2 and slice thickness = 2 mm. For T1WI imaging, a spin-echo imaging sequence (sems) was used with repetition time (TR) = 500 ms, echo time (TE) = 15 ms, and number of average = 8. For T1 mapping, a fast spin echo imaging sequence (flair) was used, with TR = 5 s, TE = 24.8 ms, number of average = 2, and TI (inversion time, ms) = 6.0, 362.3, 718.6, 1074.9, 1431.1, 1787.4, 2143.7 and 2500. For T2WI imaging, a fast spin echo imaging sequence (fsems) was used, with TR = 2.5 s, TE = 60 ms, kzero = 4, number of average = 8. Finally, for T2 mapping, a multiple echo multiple slice imaging sequence (mems) was used, with TR = 2.5 s, number of average = 2, TE = 10, 20, …, 160 ms, namely, to collect 16 echoes per TR period. Images were processed using ImageJ, a free software developed by National Institutes of Health.
Statistical Analysis
Statistics were calculated with GraphPad Prism 6 software. Statistics to determine significance for the impact of rhodamine incorporation on NP size, PDI, and charge used a two-way ANOVA with a Šidák post-hoc test for multiple comparisons. A two-tailed, unpaired t-test assuming populations with the same scatter was used to compare the amount of rhodamine and Gd incorporated, targeting of NPs, and quantification in lumen and adhered to vessels. For analysis of the real-time accumulation dynamics of NPs with IVM, nonlinear regression analysis was done using a one phase exponential association fit. The rate constants were then compared to each other using an extra sum of squares F test. A one-way ANOVA with a Dunnet post-hoc test to correct for multiple comparisons was used to analyze the antibody blocking experiments. In addition, outliers for atherosclerotic targeting of plaque and collagen were identified using a Robust regression and Outlier removal (ROUT) analysis with a Q coefficient set to 0.1%. In all cases: * p < 0.05; ** p < 0.01; *** p < 0.001. Unless otherwise noted, all values are represented as mean ± s.e.m.
Results & Discussion
NP characterization for incorporation of rhodamine
The ability to monitor the accumulation and targeting of NPs is imperative to evaluating their future potential. Fluorescence-based imaging represents a powerful tool to image cellular- and molecular-specific events at higher resolutions with faster acquisition times than current clinical imaging systems (e.g., MRI and positron emission tomography) [52]. As our goal was to evaluate the ability of the NPs to target and adhere to activated endothelium, we chose to use IVM and rhodamine-labeled lipids, permitting the inspection of real-time accumulation kinetics and adherence to vessels.
The physicochemical and biological features of liposomes and leukosomes were carefully characterized in our previous works [42, 43]. Briefly, a combination of cyro-TEM and dynamic light scattering revealed that both NP types exhibited diameters between 140-170 nm with the spherical shape commonly observed for conventional liposomes (Fig. S1). Furthermore, we validated the successful incorporation of leukocyte membrane proteins into leukosomes using differential scanning calorimetry and cyro-TEM analysis [42]. This analysis demonstrated that NPs exhibited a reduced transition temperature and an increased thickness in the bilayer after the incorporation of membrane proteins. In addition, we performed an extensive proteomic analysis of the integrated proteins, resulting in the identification of more than 300 distinct proteins. Using a combination of flow cytometry and a wheat germ agglutinin assay, we confirmed the integration, presentation, orientation, and active post-translational status of select proteins [42].
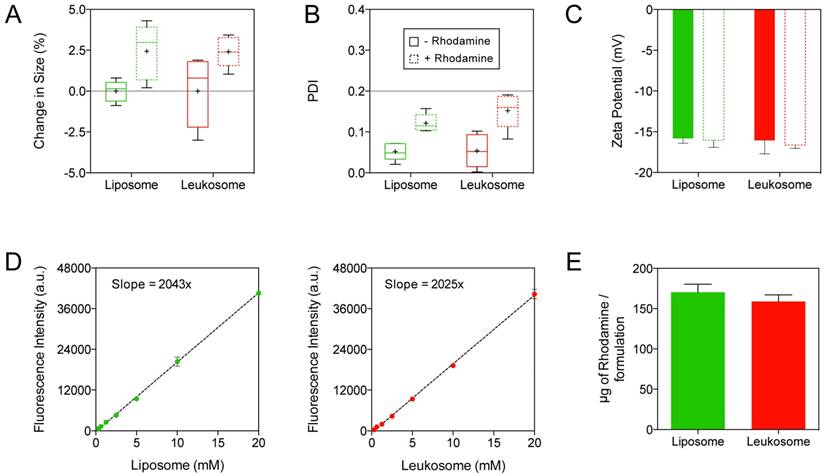
Incorporation of rhodamine-labeled lipids resulted in less than a 2.5% increase in size for both NPs (Fig. 2A). In addition, after rhodamine incorporation, the PDI of both NPs remained below 0.2 (Fig. 2B), indicating the homogeneity of the particle solution. Similarly, the surface charge of the NPs exhibited marginal alterations in comparison to unlabeled NPs (Fig. 2C). Specifically, the addition of rhodamine yielded slightly more negatively charged NP formulations (decreases of 1.5% and 3.5% for liposomes and leukosomes, respectively). Both liposomes and leukosomes exhibited a direct relationship between NP concentration and fluorescence intensity (Fig. 2D). This linear dependence is critical for accurately evaluating in vivo performance, as higher fluorescent signal will equate to greater accumulation of NPs. Using known amounts of rhodamine, we determined the correlation between mass of rhodamine and fluorescence intensity (Fig. S2) and used this information to extrapolate the amount of rhodamine in the NPs (Fig. 2E). Leukosomes successfully integrated similar amounts of rhodamine when compared to liposomes (160 vs. 170 µg/formulation for leukosomes and liposomes, respectively). In summation, this data confirmed that rhodamine labeling of NPs had minimal impact on their physicochemical features.
Integration of fluorescent rhodamine into leukosomes. A-C) Change in size (A), PDI (B), and surface charge (C) of NPs before and after incorporation of rhodamine-labeled lipids. Graphs A and B are shown as box and whisker plots with whiskers showing the min/max and the plus sign (+) corresponding to the mean of the data. D) Plot of the fluorescence intensity at various concentrations of liposome and leukosomes. A linear regression line was applied to each data set and the slope of this line is displayed. E) Quantification of the amount of rhodamine integrated into NPs. For all graphs: values represent the mean with error bars as s.e.m.
Leukosomes target the activated tumor vasculature of breast tumors
Our previous results indicated that leukosomes exhibited superior recognition and targeting of activated endothelial cells and local inflammation [42]. Expanding on these results, we explored the ability of leukosomes to target tumors and their associated vessels. For our studies, we selected the 4T1 mouse breast adenocarcinoma model. This model has shown prominent leukocyte permeation and displays mixed myeloid cell infiltrates, establishing an inflammatory microenvironment and promoting tumor progression [53, 54].
Bioluminescent imaging of 4T1 tumor-bearing mice was performed using luminol, a chemical that exhibits luminescence in the presence of phagocytes [55]. The high level of luminol expression in the tumors confirmed a significant amount phagocytic activity within the 4T1 tumor (Fig. 3A and Fig. S3), aligning with previously published research for this model [56]. Histological analysis of 4T1 tumors corroborated that an inflammatory process is present throughout the tumor tissue (Fig. 3B). Cells within this environment are characterized as necrotic (Fig. 3B, circle) due to edema, retraction of the cytoplasm, and fragmentation of nuclei (Fig. S4). Moreover, leukocyte infiltration (Fig. 3B, arrows) in areas adjacent to sites of necrosis was present, further contributing to the inflammatory tumor environment. In addition, staining of tumor sections for macrophages (F4/80) and leukocytes (CD45) confirmed their presence in this model, further corroborating the luminol and histological analysis of the inflammatory tumor environment present in 4T1 tumors (Fig. S5).
Mice with 4T1 tumors treated with liposomes and leukosomes were followed for 1 h using IVM. Analysis inspecting the kinetics of accumulation in tumors revealed that leukosomes rapidly accumulated into tumors at significantly higher concentrations than control liposomes (Fig. 3C). Fitting a one-phase exponential association curve, which describes the pseudo-first order interaction between a ligand and its receptor, revealed that leukosomes exhibited a 16-fold increase in their maximum plateau relative to liposomes (8.050 vs. 0.5085 for leukosome and liposome). These values were further validated by examining images taken from explanted tumors showing a 15-fold increase in tumor targeting at 1 h and a 9-fold increase at 6 h (Fig. 3D and Fig. S6, S7). Further examination of IVM images demonstrated that leukosomes predominately adhered to the vessel walls (Fig. 3E), whereas tumors treated with liposomes displayed minimal adherence. Quantification of NPs in tumor vessels after 1 h showed that leukosomes exhibited significant increases of 4.5- and 14-fold in the lumen of vessels (Fig. 3F) and adherence to vessel walls (Fig. 3G), respectively. Furthermore, tumor sections stained for CD31 were used to examine the interaction of NPs with endothelial cells. Immunofluorescence of tumor sections at 24 h confirmed leukosomes were closely associated with tumor endothelial cells, exhibiting extravasation and diffusion into the surrounding tumor microenvironment (Fig. 3H). Mice treated with liposomes did not demonstrate this behavior, with only minimal amounts observed in sections at 24 h. In addition, extravasation of NPs into tumors was further confirmed using IVM, where leukosomes were found deeper within tumors (Movies S1 and S2). Hence, the leukocyte membrane proteins provided leukosomes with enhanced accumulation and targeting in tumor vasculature, permitting their adherence to the wall of activated tumor vessels.
Leukocyte membrane proteins critical for leukosome targeting of tumor-associated activated endothelium
Leukocytes utilize specific adhesion proteins to recognize and adhere to activated endothelia [5]. Previously, we coupled flow cytometry and differential scanning calorimetry analysis to identify several membrane proteins that were successfully incorporated in the correct orientation on the surface of leukosomes [42]. We identified several involved in leukocyte recruitment toward activated endothelium, including both chains of LFA-1 (CD11a and CD18), P-selectin glycoprotein ligand-1, and Macrophage-1 as well as proteins involved in self-tolerance (CD45 and CD47). Among them, LFA-1 and CD45 represent two factors on the surface of leukocytes that play important roles in the targeting of inflammation. LFA-1 permits leukocytes to transiently bind to ICAM-1, which is overexpressed in activated endothelia [5, 57]. CD45 is expressed on all hematopoietic cells and plays a role in their differentiation and autoimmunity [58]. To gain a better understanding of the role these markers play in the ability of leukosomes to target activated tumor vessels and delay rapid MPS sequestration, in vivo antibody blocking experiments against LFA-1 (α-LFA-1) or CD45 (α-CD45) were performed.
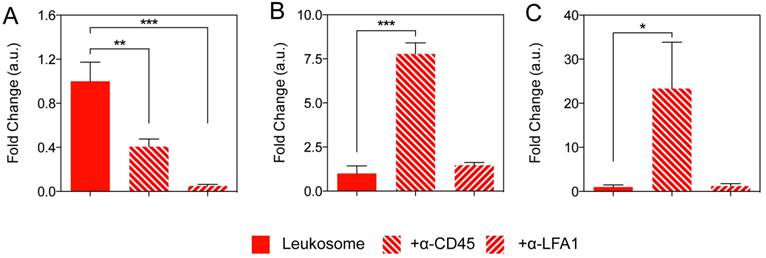
Quantification from representative IVM images showed leukosomes with blocking antibodies significantly reduced tumor targeting (Fig. 4A). Accumulation of leukosomes decreased by 60% with α-CD45 and exhibited a 95% decrease when LFA-1 was blocked, indicating both have an essential role in the leukosomes' targeting abilities. In addition, blocking of LFA-1 on leukosomes resulted in similar tumor accumulation as liposomes (Fig. S8), validating its importance in achieving site-specific targeting of tumors. Furthermore, we previously demonstrated that the transfer of LFA-1 onto biomimetic particles mediated binding to activated endothelial cells, triggered clustering and activation of ICAM-1 [57, 59]. This, in turn, prompted increased vascular permeability and significantly encouraged firm adhesion to tumor vasculature and increased tumor perfusion in vivo. Hence, LFA-1 represents a critical mediator for leukosomes to target activated endothelium within tumor vessels via LFA-1/ICAM interactions, as previously described [60, 61]. CD45 also participates in a supporting role, possibly working cooperatively with LFA-1 to mediate adhesion.
Elucidating the role of LFA-1 and CD45 for targeting and MPS avoidance. A-C) Quantification of NP accumulation in tumor (A), liver (B), and spleen (C) of mice injected with leukosome or leukosomes incubated with antibodies directed against CD45 and LFA-1. For all graphs: * p < 0.05, ** p < 0.01, *** p < 0.001 and values represent the mean with error bars as s.e.m.
A significant barrier that has impeded NPs from achieving efficient tumor accumulation is their inability to avoid uptake by the MPS [9]. Once NPs are identified, they are destined for sequestration, degradation, or elimination [19]. In tumor-bearing mice, we observed a significant decrease in liver and spleen accumulation for leukosomes (Fig. S9). We demonstrated this decreased accumulation could be, in part, due to alterations in the protein corona that is formed around the leukosomes upon systemic administration [43] or the influence of the syngeneic nature of the membrane protein source [62].
Blocking experiments in the liver and spleen revealed that only α-CD45 leukosomes yielded significant increases in liver and spleen accumulation, resulting in 7.8-fold and 23-fold increases, respectively (Fig. 4B,C). When compared to liposomes, α-CD45 leukosomes accumulated in the liver to a similar degree, but to a much lesser extent in the spleen (70% reduction, Fig. S8). This evidence suggests that the presence of other 'markers of self' on the leukosome surface, such as CD47 [63, 64] and major histocompatibility complex class I [65], are fundamental for MPS avoidance [42]. On the other hand, α-LFA-1 did not display any significant differences in liver and spleen accumulation relative to leukosomes (Fig. 4B, C) and continued to conserve the ability of leukosomes to avoid rapid sequestration by the MPS (Fig. S8). In summary, LFA-1 was primarily responsible for mediating adhesion and targeting to activated endothelium while CD45 played a vital role in bestowing leukosomes the ability to avoid rapid recognition by the immune system. Both of these properties endowed by this set of proteins are critical for leukosomes' ability to efficiently target inflammation.
Targeting inflammatory atherosclerotic plaques
To expand on the inflammatory recognition of leukosomes, we explored their ability to recognize and target an additional site of inflammation: intimal lesions in a hypercholesterolemic animal. Atherosclerosis is an inflammatory disease, the cellular interactions of which share common characteristics with other chronic inflammatory diseases [2, 66]. Atherosclerosis is characterized by early changes in the endothelium. These endothelium changes result in increased permeability, activation, up-regulation of endothelial and leukocyte adhesion molecules (e.g., VCAM-1 and ICAM-1), and infiltration of leukocytes into arterial walls [2, 66]. Combined, these changes ultimately fuel future plaque formation and progression.
ApoE-deficient mice on a high-fat diet were used to model atherogenesis. The animals are genetically devoid of ApoE, a protein that binds lipids and participates in lipid and cholesterol transport. As a result, the mice have elevated plasma cholesterol and spontaneously develop intimal lesions, which is further exacerbated by their high-fat diet. Histopathological analysis confirmed that animals used in the experiments had lesions with select features of atherosclerosis. Cross-sections of heart tissues spanning the aortic sinus and ascending aorta displayed significantly thickened tunica intima with a developed thin fibrous cap composed of inflammatory cells, activated smooth muscle cells, and extracellular matrix. They also showed a large diffuse necrotic core underneath containing cellular debris, extracellular lipid with cholesterol crystals, and foam cells. Both are features of an atherosclerotic plaque (Fig. 5A). Diffuse lipid deposition throughout the plaque was observed with more dense accumulation of foam cells in proximity to the vessel lumen (Fig. 5B), contributing to the inflammatory environment present in the lesion. This inflammatory environment, present in ApoE-deficient mice, has been previously demonstrated by several groups[46, 67-70] and corroborates the histology results presented in Fig. 5A,B.
Evaluation of lesion targeting. A) H&E image of the aortic sinus displaying advanced vascular lesions: fibrous cap with inflammatory infiltrate (fc) covering necrotic core (nc). Also seen is the tunica media (m), tunica adventitia (a), cusps of aortic valve (av); scale bar: 200 µm. B) Section of the aortic sinus stained with Oil Red O showing diffuse deposition of lipid throughout the vascular lesion (in red) and foam cells (black arrows); scale bar: 200 µm. C,D) Representative bright-field (C) and fluorescence (D) images of vascular lesions (outlined in white) treated with liposomes or leukosomes. NP are in red and auto-fluorescence of collagen in blue, scale bar: 100 µm. E,F) Quantification of NP accumulation in lesion (E) or collagen (F). For all graphs: ** p < 0.01 and values represent the mean with error bars as s.e.m.
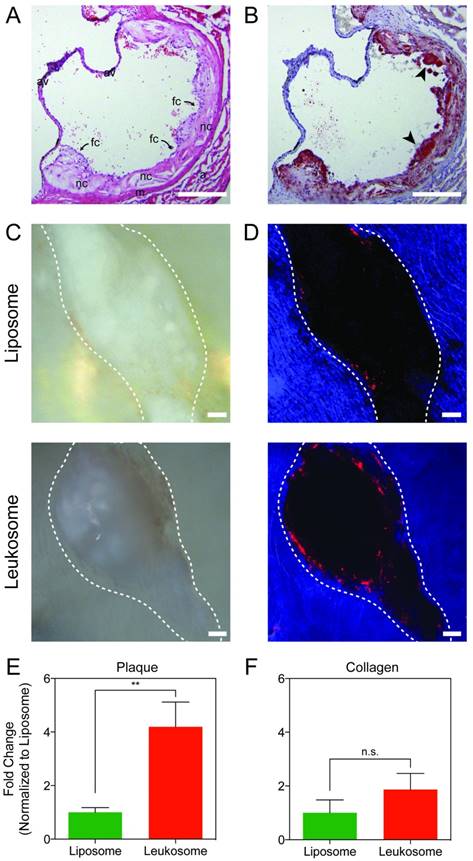
Two hours after systemic administration of NPs, the aortas of ApoE-deficient mice were collected and en face preparations of the whole vessels were made and imaged. Bright field (Fig. 5C) and fluorescence images (Fig. 5D) were used to identify plaques (Fig. 5C,D; dashed white line) from the collagen matrix (Fig. 5D, blue area) within the aorta. The collagen matrix was identified from the auto-fluorescence associated with collagen in the ultraviolet range [71, 72]. The absence of signal provided the location of vascular lesions (black), which are surrounded by an aortic collagen matrix.
Liposomes displayed minimal accumulation in the area surrounding the plaque (Fig. 5D, top). However, mice treated with leukosomes displayed prominent accumulation on the lesion surface and were found at minimal concentrations outside of this area (Fig. 5D, bottom). Quantification of these images revealed that leukosomes exhibited a 4-fold increase in targeting to the area surrounding vascular lesions in aortas of ApoE-deficient mice (Fig. 5E). In addition, quantification of areas distant from the plaque within the surrounding collagen exhibited no significant difference in accumulation between liposomes and leukosomes (Fig. 5F). Thus, the leukocyte membrane proteins on leukosome surface enabled them to significantly increase their targeting propensity for vascular lesions, accumulating in adjacent areas where other immune cells accrue during the natural progression of the disease. This data serves to further validate the targeting affinity and selectivity of leukosomes towards inflammation.
The targeting approach exhibited by leukosomes accounts for the evolving nature of inflammation. Previous strategies for targeting atherosclerosis using NPs for drug delivery or imaging relied on passive accumulation in surrounding macrophages [73]. However, as this strategy targets all macrophages equally, it results in nonspecific accumulation and is unreliable for efficient diagnostic imaging. To increase specificity, other strategies relied on targeting isolated or single molecular motifs (e.g., Type IV collagen [74], P-Selectin [75], and ICAM-1 [76]) using a post-synthesis conjugation technique to ensure biological activity of targeting molecules. However, post-synthesis conjugation introduces multiple and complex steps within the synthesis process and fails to account for the entire complexity of phenomena occurring at the vascular lesion. On the other hand, leukosomes incorporate a variety of leukocyte-membrane proteins on their surface, enabling self-tolerance (CD45 and CD47) and dynamic targeting (LFA-1, P-selectin glycoprotein ligand-1, Macrophage-1) that permit their recognition and adherence to the inflammation-based events developing at vascular lesions.
Enabling leukosomes for multi-modality imaging enhancements
Fluorescence and near infrared imaging is an emerging technology that has shown tremendous potential for minimally invasive non-ionizing imaging [52, 77]. Rhodamine-labeled leukosomes could be adapted with ease for near infrared imaging due to the versatility provided by the liposome backbone [78, 79]. However, to evaluate the true potential of leukosomes for clinical applications, we decided to test their utility for MRI.
To enable their use for MRI applications, NPs were successfully incorporated with Gd chelates by replacing the rhodamine-tagged phospholipid with one that contains Gd. Inductively coupled plasma-atomic emission spectroscopy confirmed successful incorporation of Gd and demonstrated that both types of NPs contained similar amounts of Gd (Fig. 6A). Similar to the previous results with rhodamine, we determined that 254 and 267 µg of Gd per formulation were successfully incorporated into liposomes and leukosomes, respectively (Fig. S10). Furthermore, Gd incorporation did not impact the size or stability of NPs, demonstrating equivalent changes in size and PDI compared to our previous results [42] (Fig. 6B).
Gd incorporation into NPs for MRI applications. A) Quantification of Gd entrapment within liposomes and leukosomes. B) Comparing the size (left axis and bars) and PDI (right axis and dots) stability of Gd-labeled liposomes and leukosomes after 2 and 4 weeks. C) T1WI images of phantoms containing different amounts of Gd-liposomes or Gd-leukosomes, ranging from 2-100 μM of Gd. D) Relaxometric titrations (r1 and r2) of phantoms containing different amounts of Gd-liposomes or Gd-leukosomes. E) The relaxivities, r1 and r2 values, were obtained from the linear fittings of the corresponding experimental data (as of the slopes, in unit of mM-1 s-1). For all graphs: values represent the mean with error bars as s.e.m.
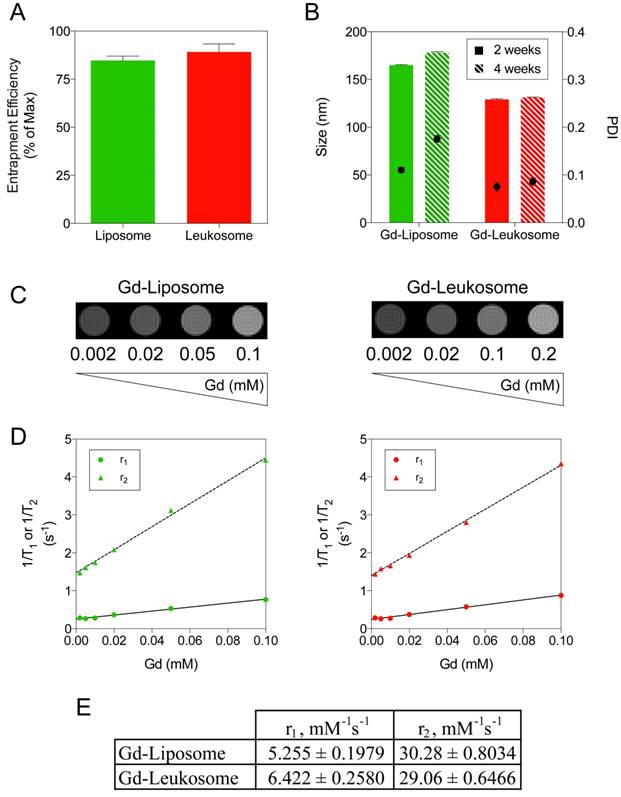
As a proof-of-concept to evaluate the use of leukosomes for MRI, we performed phantom studies inspecting various concentrations of NPs, ranging from 2-100 μM. T1WI images of both liposomes and leukosomes (Fig. 6C) displayed an increasing amount of contrast as the concentration increased, as expected with T1-based contrast agents like Gd. Computation of the r1 and r2 relaxivities for Gd-NPs (Fig. 6D) exhibited similar relaxivity values (Fig. 6E), indicating that the incorporation of proteins did not impact the contrast ability of Gd. The observed r1 relaxivities for both NPs (5.255 and 6.422 mM-1s-1 for liposomes and leukosomes, respectively) displayed a 2-fold increase in comparison to other clinically approved Gd-based contrast agents at 7T [80]. Based on the T1WI images and r1 relaxivity, the detection limit of these NPs for MRI would be 0.01 mM Gd. In addition, the observed r2 relaxivities exhibited by both NPs (30.28 and 29.06 mM-1s-1 for liposomes and leukosomes, respectively) were 5-fold larger compared to Gd-based agents used in the clinic. As Gd can be used as a T2-based agent to generate negative contrast [81], one can take advantage of this increased r2 relaxivity to run T2-weighted MRI to complement the traditional T1-weighted MRI utilized for Gd-based contrast agents.
Molecular imaging of inflammation represents a promising tool to evaluate cellular and molecular events in humans. Gd-based constructs and MRI have shown success for cardiovascular and oncology applications. Several NP-based approaches have created formulations capable of increasing the circulation time or contrast enhancement of Gd for MRI [82-84]. Furthermore, NPs themselves have emerged as powerful imaging agents due to their unique nanoscale features [85]. One example is superparamagnetic iron oxide NPs that achieved United States Food and Drug Administration approval in 1996 for the diagnosis of metastasis and liver lesions with MRI [86]. However, their success in the clinic was limited due to issues with safety, stability, and sequestration by the MPS, resulting in nonspecific accumulation [87]. Current approaches for NPs to target inflammation for MRI rely on passive accumulation in macrophages or surface decoration with recognition moieties [88]. Few consider how to circumvent the biological barriers they encounter upon systemic administration.
Over the past years, bioinspired, bioengineered, and biomimetic solutions to drug delivery have shown much promise in addressing the obstacles that have afflicted the scientific and medical communities [28, 29]. With respect to inflammatory targeting, attempts have been made to target neutrophils or use inflammatory monocytes to carry NPs past the endothelia, induce detachment from endothelia, prevent vascular inflammation, and modulate monocyte trafficking to the inflammatory site [89-91]. Leukosomes represent a promising iteration that takes advantage of both top-down and bottom-up approaches to create biomimetic particles with leukocyte features using a single synthetic process [42]. Furthermore, when compared to mice treated with PBS, leukosomes induced no significant difference in serum levels of cytokines (IL-6, TNFα, and IL-1β), histological assessment, and organ function [42]. Thus, their unique mechanism of targeting and their inherent versatility to enable clinical imaging and therapy coupled with minimal toxicity and biocompatibility issues make leukosomes a promising candidate for future clinical translation.
Conclusion
The studies described here demonstrate the ability of leukosomes to target activated endothelium and exhibit superior accumulation in tumors and vascular lesions. The targeting of leukosomes to inflamed areas was mediated by the expression of CD45 and LFA-1 on the particle surface, as illustrated by blocking agents that negated both enhanced circulation and targeting. In addition, proof-of-concept studies demonstrate the flexibility of the leukosome platform to incorporate contrast agents to enable MRI. Furthermore, as leukosomes take advantage of liposome characteristics, they allow the loading of a wide array of therapeutics with various solubility profiles and compositions (e.g., hydrophilic, hydrophobic, amphiphilic, biologics, etc.), thus enabling their future use as a novel theranostic agent.
Acknowledgements
The authors thank Dr. Masaya Takahashi for coordinating and facilitating access to the Mouse MRI Core of the Advanced Imaging Research Center located at The University of Texas Southwestern Medical Center (Dallas, TX) to perform MRI phantom experiments. In addition, we thank HMRI for access to Translational Imaging's Pre-clinical imaging (small animal) core, Advanced Cellular and Tissue Microscopy core, and IVM core for access to IVIS Spectrum, for traditional confocal scanning services, and IVM, respectively; and Manuela Sushnitha and Mary Margavio for reviewing the manuscript. This work was financially supported by: NIH (5U54CA143837, 1R21CA190024), NCI and the Office of Research on Women's Health (Grant # 1R56CA213859), William Randolph Hearst Foundation, The Cullen Trust for Health Care, Cancer Prevention & Research Institute of Texas (Award ID RP170466), and CARIPARO Foundation (Ricerca Pediatrica 2016-2018 Grant).
Supplementary Material
Supplementary figures, movie captions, and materials and methods.
Supplementary Movie S1.
Supplementary Movie S2.
Abbreviations
ApoE: apolipoprotein E; cyro-TEM: cryo-transmission electron microscopy; EPR: enhanced permeability and retention; Gd: Gadolinium; H&E: hematoxylin and eosin; HMRI: Houston Methodist Research Institute; ICAM-1: intercellular adhesion molecule-1; IVM: intravital microscopy; LFA-1: lymphocyte function-associated antigen 1; PEG: polyethylene glycol; PDI: poly-dispersity index; MPS: mononuclear phagocyte system; MRI: magnetic resonance imaging; NP: nanoparticle; VCAM-1: vascular cell adhesion molecule-1.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. The resolution of inflammation. Nature reviews Immunology. 2013;13:59-66
2. Molinaro R, Boada C, Del Rosal GM, Hartman KA, Corbo C, Andrews ED. et al. Vascular Inflammation: A Novel Access Route for Nanomedicine. Methodist Debakey Cardiovasc J. 2016;12:169-74
3. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803-15
4. Galvani S, Sanson M, Blaho VA, Swendeman SL, Obinata H, Conger H. et al. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci Signal. 2015;8:ra79
5. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678-89
6. Zhang S, Ermann J, Succi MD, Zhou A, Hamilton MJ, Cao B. et al. An inflammation-targeting hydrogel for local drug delivery in inflammatory bowel disease. Science translational medicine. 2015;7:300ra128
7. Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nature reviews Drug discovery. 2004;3:401-16
8. Xie J, Lee S, Chen X. Nanoparticle-based theranostic agents. Adv Drug Deliv Rev. 2010;62:1064-79
9. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33:941-51
10. Yang B, Han X, Ji B, Lu R. Competition Between Tumor and Mononuclear Phagocyte System Causing the Low Tumor Distribution of Nanoparticles and Strategies to Improve Tumor Accumulation. Curr Drug Deliv. 2016;13:1261-74
11. Undevia SD, Gomez-Abuin G, Ratain MJ. Pharmacokinetic variability of anticancer agents. Nat Rev Cancer. 2005;5:447-58
12. Chauhan VP, Stylianopoulos T, Boucher Y, Jain RK. Delivery of molecular and nanoscale medicine to tumors: transport barriers and strategies. Annu Rev Chem Biomol Eng. 2011;2:281-98
13. Taurin S, Nehoff H, Greish K. Anticancer nanomedicine and tumor vascular permeability; Where is the missing link? J Control Release. 2012;164:265-75
14. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387-92
15. Azzopardi EA, Ferguson EL, Thomas DW. The enhanced permeability retention effect: a new paradigm for drug targeting in infection. J Antimicrob Chemother. 2013;68:257-74
16. Nehoff H, Parayath NN, Domanovitch L, Taurin S, Greish K. Nanomedicine for drug targeting: strategies beyond the enhanced permeability and retention effect. Int J Nanomedicine. 2014;9:2539-55
17. Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC. et al. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73:2412-7
18. Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759-68
19. Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF. et al. Analysis of nanoparticle delivery to tumours. Nature Reviews Materials. 2016;1:16014
20. Salvati A, Pitek AS, Monopoli MP, Prapainop K, Bombelli FB, Hristov DR. et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat Nanotechnol. 2013;8:137-43
21. Monopoli MP, Aberg C, Salvati A, Dawson KA. Biomolecular coronas provide the biological identity of nanosized materials. Nat Nanotechnol. 2012;7:779-86
22. Milla P, Dosio F, Cattel L. PEGylation of proteins and liposomes: a powerful and flexible strategy to improve the drug delivery. Curr Drug Metab. 2012;13:105-19
23. Ishida T, Wang X, Shimizu T, Nawata K, Kiwada H. PEGylated liposomes elicit an anti-PEG IgM response in a T cell-independent manner. J Control Release. 2007;122:349-55
24. Wang X, Ishida T, Kiwada H. Anti-PEG IgM elicited by injection of liposomes is involved in the enhanced blood clearance of a subsequent dose of PEGylated liposomes. J Control Release. 2007;119:236-44
25. Gabizon AA. Stealth liposomes and tumor targeting: one step further in the quest for the magic bullet. Clin Cancer Res. 2001;7:223-5
26. Abu Lila AS, Kiwada H, Ishida T. The accelerated blood clearance (ABC) phenomenon: clinical challenge and approaches to manage. J Control Release. 2013;172:38-47
27. Ishida T, Harada M, Wang XY, Ichihara M, Irimura K, Kiwada H. Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: effects of lipid dose and PEG surface-density and chain length of the first-dose liposomes. J Control Release. 2005;105:305-17
28. Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10:521-35
29. Parodi A, Molinaro R, Sushnitha M, Evangelopoulos M, Martinez JO, Arrighetti N. et al. Bio-inspired engineering of cell- and virus-like nanoparticles for drug delivery. Biomaterials. 2017;147:155-68
30. Wang Q, Ren Y, Mu J, Egilmez NK, Zhuang X, Deng Z. et al. Grapefruit-Derived Nanovectors Use an Activated Leukocyte Trafficking Pathway to Deliver Therapeutic Agents to Inflammatory Tumor Sites. Cancer Res. 2015;75:2520-9
31. Robbins GP, Saunders RL, Haun JB, Rawson J, Therien MJ, Hammer DA. Tunable leuko-polymersomes that adhere specifically to inflammatory markers. Langmuir. 2010;26:14089-96
32. Kang T, Zhu Q, Wei D, Feng J, Yao J, Jiang T. et al. Nanoparticles Coated with Neutrophil Membranes Can Effectively Treat Cancer Metastasis. ACS Nano. 2017;11:1397-411
33. Anselmo AC, Modery-Pawlowski CL, Menegatti S, Kumar S, Vogus DR, Tian LL. et al. Platelet-like nanoparticles: mimicking shape, flexibility, and surface biology of platelets to target vascular injuries. ACS Nano. 2014;8:11243-53
34. Doshi N, Zahr AS, Bhaskar S, Lahann J, Mitragotri S. Red blood cell-mimicking synthetic biomaterial particles. Proc Natl Acad Sci U S A. 2009;106:21495-9
35. Hu CM, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D. et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526:118-21
36. Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci U S A. 2011;108:10980-5
37. Merkel TJ, Jones SW, Herlihy KP, Kersey FR, Shields AR, Napier M. et al. Using mechanobiological mimicry of red blood cells to extend circulation times of hydrogel microparticles. Proc Natl Acad Sci U S A. 2011;108:586-91
38. Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T. et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007;2:249-55
39. Luk BT, Zhang L. Cell membrane-camouflaged nanoparticles for drug delivery. J Control Release. 2015;220:600-7
40. Millan CG, Marinero ML, Castaneda AZ, Lanao JM. Drug, enzyme and peptide delivery using erythrocytes as carriers. J Control Release. 2004;95:27-49
41. Gutierrez Millan C, Colino Gandarillas CI, Sayalero Marinero ML, Lanao JM. Cell-based drug-delivery platforms. Ther Deliv. 2012;3:25-41
42. Molinaro R, Corbo C, Martinez JO, Taraballi F, Evangelopoulos M, Minardi S. et al. Biomimetic proteolipid vesicles for targeting inflamed tissues. Nat Mater. 2016;15:1037-46
43. Corbo C, Molinaro R, Taraballi F, Toledano Furman NE, Hartman KA, Sherman MB. et al. Unveiling the in Vivo Protein Corona of Circulating Leukocyte-like Carriers. ACS Nano. 2017
44. Corbo C, Cromer WE, Molinaro R, Toledano Furman NE, Hartman KA, De Rosa E. et al. Engineered biomimetic nanovesicles show intrinsic anti-inflammatory properties for the treatment of inflammatory bowel diseases. Nanoscale. 2017
45. Martinez JO, Boada C, Yazdi IK, Evangelopoulos M, Brown BS, Liu X. et al. Short and long term, in vitro and in vivo correlations of cellular and tissue responses to mesoporous silicon nanovectors. Small. 2013;9:1722-33
46. Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133-40
47. Williams H, Johnson JL, Carson KG, Jackson CL. Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2002;22:788-92
48. Pandolfi L, Furman NT, Wang X, Lupo C, Martinez JO, Mohamed M. et al. A nanofibrous electrospun patch to maintain human mesenchymal cell stemness. J Mater Sci Mater Med. 2017;28:44
49. van de Ven AL, Kim P, Ferrari M, Yun SH. Real-time intravital microscopy of individual nanoparticle dynamics in liver and tumors of live mice. Protoc exch. 2013. 2013
50. D'Apolito R, Tomaiuolo G, Taraballi F, Minardi S, Kirui D, Liu X. et al. Red blood cells affect the margination of microparticles in synthetic microcapillaries and intravital microcirculation as a function of their size and shape. J Control Release. 2015;217:263-72
51. Yokoi K, Tanei T, Godin B, van de Ven AL, Hanibuchi M, Matsunoki A. et al. Serum biomarkers for personalization of nanotherapeutics-based therapy in different tumor and organ microenvironments. Cancer Lett. 2014;345:48-55
52. Weissleder R, Pittet MJ. Imaging in the era of molecular oncology. Nature. 2008;452:580-9
53. DuPre SA, Redelman D, Hunter KW Jr. The mouse mammary carcinoma 4T1: characterization of the cellular landscape of primary tumours and metastatic tumour foci. Int J Exp Pathol. 2007;88:351-60
54. Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007;67:10019-26
55. Gross S, Gammon ST, Moss BL, Rauch D, Harding J, Heinecke JW. et al. Bioluminescence imaging of myeloperoxidase activity in vivo. Nat Med. 2009;15:455-61
56. Alshetaiwi HS, Balivada S, Shrestha TB, Pyle M, Basel MT, Bossmann SH. et al. Luminol-based bioluminescence imaging of mouse mammary tumors. J Photochem Photobiol B. 2013;127:223-8
57. Palomba R, Parodi A, Evangelopoulos M, Acciardo S, Corbo C, de Rosa E. et al. Biomimetic carriers mimicking leukocyte plasma membrane to increase tumor vasculature permeability. Sci Rep. 2016;6:34422
58. Penninger JM, Irie-Sasaki J, Sasaki T, Oliveira-dos-Santos AJ. CD45: new jobs for an old acquaintance. Nat Immunol. 2001;2:389-96
59. Parodi A, Quattrocchi N, van de Ven AL, Chiappini C, Evangelopoulos M, Martinez JO. et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8:61-8
60. Lorenz HM, Harrer T, Lagoo AS, Baur A, Eger G, Kalden JR. CD45 mAb induces cell adhesion in peripheral blood mononuclear cells via lymphocyte function-associated antigen-1 (LFA-1) and intercellular cell adhesion molecule 1 (ICAM-1). Cell Immunol. 1993;147:110-28
61. Arroyo AG, Campanero MR, Sanchez-Mateos P, Zapata JM, Ursa MA, del Pozo MA. et al. Induction of tyrosine phosphorylation during ICAM-3 and LFA-1-mediated intercellular adhesion, and its regulation by the CD45 tyrosine phosphatase. J Cell Biol. 1994;126:1277-86
62. Evangelopoulos M, Parodi A, Martinez JO, Yazdi IK, Cevenini A, van de Ven AL. et al. Cell source determines the immunological impact of biomimetic nanoparticles. Biomaterials. 2016;82:168-77
63. Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Minimal "Self" peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science. 2013;339:971-5
64. Sosale NG, Spinler KR, Alvey C, Discher DE. Macrophage engulfment of a cell or nanoparticle is regulated by unavoidable opsonization, a species-specific 'Marker of Self' CD47, and target physical properties. Curr Opin Immunol. 2015;35:107-12
65. Medzhitov R, Janeway CA Jr. Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298-300
66. Ross R. Atherosclerosis-an inflammatory disease. N Engl J Med. 1999;340:115-26
67. Getz GS, Reardon CA. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:1104-15
68. Meir KS, Leitersdorf E. Atherosclerosis in the apolipoprotein-E-deficient mouse: a decade of progress. Arterioscler Thromb Vasc Biol. 2004;24:1006-14
69. Rosenfeld ME, Polinsky P, Virmani R, Kauser K, Rubanyi G, Schwartz SM. Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler Thromb Vasc Biol. 2000;20:2587-92
70. Whitman SC. A practical approach to using mice in atherosclerosis research. Clin Biochem Rev. 2004;25:81-93
71. Minardi S, Sandri M, Martinez JO, Yazdi IK, Liu X, Ferrari M. et al. Multiscale patterning of a biomimetic scaffold integrated with composite microspheres. Small. 2014;10:3943-53
72. Corradetti B, Taraballi F, Minardi S, Van Eps J, Cabrera F, Francis LW. et al. Chondroitin Sulfate Immobilized on a Biomimetic Scaffold Modulates Inflammation While Driving Chondrogenesis. Stem Cells Transl Med. 2016;5:670-82
73. von Zur Muhlen C, von Elverfeldt D, Bassler N, Neudorfer I, Steitz B, Petri-Fink A. et al. Superparamagnetic iron oxide binding and uptake as imaged by magnetic resonance is mediated by the integrin receptor Mac-1 (CD11b/CD18): implications on imaging of atherosclerotic plaques. Atherosclerosis. 2007;193:102-11
74. Fredman G, Kamaly N, Spolitu S, Milton J, Ghorpade D, Chiasson R. et al. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci Transl Med. 2015;7:275ra20
75. Klibanov AL, Rychak JJ, Yang WC, Alikhani S, Li B, Acton S. et al. Targeted ultrasound contrast agent for molecular imaging of inflammation in high-shear flow. Contrast Media Mol Imaging. 2006;1:259-66
76. McAteer MA, Choudhury RP. Targeted molecular imaging of vascular inflammation in cardiovascular disease using nano- and micro-sized agents. Vascul Pharmacol. 2013;58:31-8
77. Weissleder R. A clearer vision for in vivo imaging. Nat Biotechnol. 2001;19:316-7
78. Li S, Goins B, Zhang L, Bao A. Novel multifunctional theranostic liposome drug delivery system: construction, characterization, and multimodality MR, near-infrared fluorescent, and nuclear imaging. Bioconjug Chem. 2012;23:1322-32
79. Jaffer FA, Libby P, Weissleder R. Optical and multimodality molecular imaging: insights into atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1017-24
80. Shen Y, Goerner FL, Snyder C, Morelli JN, Hao D, Hu D. et al. T1 relaxivities of gadolinium-based magnetic resonance contrast agents in human whole blood at 1.5, 3, and 7 T. Invest Radiol. 2015;50:330-8
81. Palekar RU, Jallouk AP, Lanza GM, Pan H, Wickline SA. Molecular imaging of atherosclerosis with nanoparticle-based fluorinated MRI contrast agents. Nanomedicine (Lond). 2015;10:1817-32
82. Sitharaman B, Kissell KR, Hartman KB, Tran LA, Baikalov A, Rusakova I. et al. Superparamagnetic gadonanotubes are high-performance MRI contrast agents. Chemical communications. 2005:3915-7
83. Toth E, Bolskar RD, Borel A, Gonzalez G, Helm L, Merbach AE. et al. Water-soluble gadofullerenes: toward high-relaxivity, pH-responsive MRI contrast agents. Journal of the American Chemical Society. 2005;127:799-805
84. Ayyagari AL, Zhang X, Ghaghada KB, Annapragada A, Hu X, Bellamkonda RV. Long-circulating liposomal contrast agents for magnetic resonance imaging. Magnetic resonance in medicine. 2006;55:1023-9
85. Martinez JO, Brown BS, Quattrocchi N, Evangelopoulos M, Ferrari M, Tasciotti E. Multifunctional to multistage delivery systems: The evolution of nanoparticles for biomedical applications. Chin Sci Bull. 2012;57:3961-71
86. Choi HS, Frangioni JV. Nanoparticles for biomedical imaging: fundamentals of clinical translation. Molecular imaging. 2010;9:291-310
87. Tasciotti E, Cabrera FJ, Evangelopoulos M, Martinez JO, Thekkedath UR, Kloc M. et al. The Emerging Role of Nanotechnology in Cell and Organ Transplantation. Transplantation. 2016;100:1629-38
88. Weissleder R, Nahrendorf M, Pittet MJ. Imaging macrophages with nanoparticles. Nat Mater. 2014;13:125-38
89. Getts DR, Terry RL, Getts MT, Deffrasnes C, Muller M, van Vreden C. et al. Therapeutic inflammatory monocyte modulation using immune-modifying microparticles. Sci Transl Med. 2014;6:219ra7
90. Wang Z, Li J, Cho J, Malik AB. Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat Nanotechnol. 2014;9:204-10
91. Chu D, Gao J, Wang Z. Neutrophil-Mediated Delivery of Therapeutic Nanoparticles across Blood Vessel Barrier for Treatment of Inflammation and Infection. ACS Nano. 2015;9:11800-11
Author contact
![]() Corresponding authors: Dr. Ennio Tasciotti, Center for Biomimetic Medicine, Houston Methodist Orthopedics & Sports Medicine, 6670 Bertner Ave, Houston, TX, 77030. Tel: +1 713-441-7319; etasciottiorg and Dr. Roberto Molinaro, Department of Medicine - Cardiovascular Medicine, Brigham and Women's Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, NRB, Boston, MA, 02115 USA. rmolinaroharvard.edu and Dr. Jonathan O. Martinez, Center for Biomimetic Medicine, Houston Methodist Research Institute, 6670 Bertner Ave, Houston, TX, 77030. jomartinezorg
Corresponding authors: Dr. Ennio Tasciotti, Center for Biomimetic Medicine, Houston Methodist Orthopedics & Sports Medicine, 6670 Bertner Ave, Houston, TX, 77030. Tel: +1 713-441-7319; etasciottiorg and Dr. Roberto Molinaro, Department of Medicine - Cardiovascular Medicine, Brigham and Women's Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, NRB, Boston, MA, 02115 USA. rmolinaroharvard.edu and Dr. Jonathan O. Martinez, Center for Biomimetic Medicine, Houston Methodist Research Institute, 6670 Bertner Ave, Houston, TX, 77030. jomartinezorg