Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(4):894-905. doi:10.7150/thno.21168 This issue Cite
Research Paper
Combined delivery of sorafenib and a MEK inhibitor using CXCR4-targeted nanoparticles reduces hepatic fibrosis and prevents tumor development
Yun-Chieh Sung1,2,†, Ya-Chi Liu1,†, Po-Han Chao1,†, Chih-Chun Chang1, Pei-Ru Jin1, Ts-Ting Lin1, Ja-An Lin3, Hui-Teng Cheng4,5, Jane Wang2, Charles P. Lai6, Ling-Hsuan Chen1,4, Anthony Y. Wu1,6, Ting-Lun Ho1, Tsaiyu Chiang1, Dong-Yu Gao1, Dan G. Duda7, Yunching Chen1, ![]()
1. Institute of Biomedical Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan
2. Department of Chemical Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan
3. Department of Biostatistics, University of North Caroline at Chapel Hill
4. Department of Internal Medicine, National Taiwan University Hospital Hsin-Chu Branch, Hsin Chu City 30059, Taiwan
5. Research Center for Developmental Biology and Regenerative Medicine, National Taiwan University, Taipei, Taiwan
6. Institute of Atomic and Molecular Sciences (IAMS), Academia Sinica, National Taiwan University, Taipei, Taiwan
7. Steele Laboratories for Tumor Biology, Department of Radiation Oncology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA
† These authors contributed equally to this work.
Received 2017-5-24; Accepted 2017-11-29; Published 2018-1-1
Abstract

Liver damage and fibrosis are precursors of hepatocellular carcinoma (HCC). In HCC patients, sorafenib—a multikinase inhibitor drug—has been reported to exert anti-fibrotic activity. However, incomplete inhibition of RAF activity by sorafenib may also induce paradoxical activation of the mitogen-activated protein kinase (MAPK) pathway in malignant cells. The consequence of this effect in non-malignant disease (hepatic fibrosis) remains unknown. This study aimed to examine the effects of sorafenib on activated hepatic stellate cells (HSCs), and develop effective therapeutic approaches to treat liver fibrosis and prevent cancer development.
Methods: We first examined the effects of sorafenib in combination with MEK inhibitors on fibrosis pathogenesis in vitro and in vivo. To improve the bioavailability and absorption by activated HSCs, we developed CXCR4-targeted nanoparticles (NPs) to co-deliver sorafenib and a MEK inhibitor to mice with liver damage.
Results: We found that sorafenib induced MAPK activation in HSCs, and promoted their myofibroblast differentiation. Combining sorafenib with a MEK inhibitor suppressed both paradoxical MAPK activation and HSC activation in vitro, and alleviated liver fibrosis in a CCl4-induced murine model of liver damage. Furthermore, treatment with sorafenib/MEK inhibitor-loaded CXCR4-targeted NPs significantly suppressed hepatic fibrosis progression and further prevented fibrosis-associated HCC development and liver metastasis.
Conclusions: Our results show that combined delivery of sorafenib and a MEK inhibitor via CXCR4-targeted NPs can prevent activation of ERK in activated HSCs and has anti-fibrotic effects in the CCl4-induced murine model. Targeting HSCs represents a promising strategy to prevent the development and progression of fibrosis-associated HCC.
Keywords: CXCR4-targeted nanoparticle, MAPK/ERK pathway, liver fibrosis, hepatocellular carcinoma, liver metastasis
Introduction
Chronic liver injury results in inflammation, angiogenesis and excessive extracellular matrix (ECM) deposition, leading to liver fibrosis and cirrhosis, liver failure, portal hypertension, and development of hepatocellular carcinoma (HCC)[1-3]. Around 80-90% of HCC—one of the most common cancers with a high mortality rate—arises in a cirrhotic liver [4]. In the clinic, most patients are initially diagnosed with advanced fibrosis or cirrhosis, which cannot be reversed using existing therapies. Thus, anti-fibrosis therapies that aim to inhibit disease progression and suppress advanced fibrosis and cirrhosis are urgently needed [5-9]. Emerging treatments are aimed at inhibiting the activation of hepatic stellate cells (HSCs), preventing ECM deposition or accelerating the degradation of ECM to achieve anti-fibrotic effects [10].
The discovery and characterization of fibrosis-promoting molecules or pathways in HSCs have provided new insights into treating liver cirrhosis [1, 11, 12]. Small molecule drugs are being developed to target activated HSCs to inhibit the production of key structural ECM components (e.g., collagen I), block fibrogenic signaling pathways, such as PDGF/PDGF receptor beta (PDGFRβ), or suppress proangiogenic pathways such as VEGF/VEGF receptor (VEGFR) axes [5]. Indeed, sorafenib—a drug approved and widely used for liver cancer that has dual VEGFR/PDGFR inhibitory activity—was shown to exert anti-fibrotic activity in preclinical and clinical studies [13-18]. Mechanistic studies in experimental models of hepatic fibrosis showed that sorafenib treatment can decrease ECM deposition, reduce the activation of HSCs, and inhibit angiogenesis [19, 20].
However, two major challenges remain for the preventative, chronic use of sorafenib as anti-fibrosis treatment in patients. Firstly, as demonstrated in cancer studies, RAF kinase inhibitors such as sorafenib can induce paradoxical activation of the MAPK pathway in both malignant and normal stromal cells [21-24]. Activation of MAPK pathway in HSCs leads to their activation, but whether this effect is induced in activated HSCs during liver damage is unknown. Secondly, sorafenib often causes unwanted off-target effects, leading to hand-foot syndrome, diarrhea, and hypertension, as a result of non-specific uptake of the drugs by normal tissues [25]. These challenges emphasize the urgent need to determine the mechanism of resistance to sorafenib in liver fibrosis and to develop safer, more selective therapeutic strategies.
The purpose of this study was to examine the effects of sorafenib on activated HSCs, and identify molecular targets for developing effective combination therapeutic approaches to treat liver fibrosis. Furthermore, we sought to develop and test HSC-targeted nanoparticles (NPs) for the co-delivery of multiple therapeutic cargoes with the aim of enhancing anti-fibrotic effects and preventing hepatocarcinogenesis.
Materials and methods
Cells and materials
HSCs were purchased from ScienCell Research Laboratories (San Diego, CA). Cells were activated after they were cultured for 7 days on plastic dishes. Sorafenib and Selumetinib (AZD6244) was obtained from LC Laboratories (Woburn, MA). For in vivo liver metastasis studies, the Pancreatic ductal adenocarcinoma (PDAC) cells AK4.4 were stably transduced with Gluc/GFP gene by using a retroviral vector. Carbon tetrachloride (CCl4), olive oil, coumarin 6, dimethyl sulfoxide (DMSO), D-α-Tocopherol polyethylene glycol 1000 succinate (TPGS) and paraformaldehyde were purchased from Sigma-Aldrich (St, Louis, MO). 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG-Maleimide) were obtained from Avanti Polar Lipids (Alabaster, AL). poly(lactic-co-glycolic acid) (PLGA) (50/50, inherent viscosity: 0.17 dL/g) was ordered from Green Square Materials Incorporation (Taoyuan, Taiwan). Tris(2-carboxyethyl)phosphine (TCEP) disulfide reducing gels were purchased from Thermo Fisher. CTCE9908 peptide (sequence: Lys-Gly-Val-Ser-Leu-Ser-Tyr-Arg-Cys-Arg-Tyr-Ser-Leu-Ser-Val-Gly-Lys) and scramble peptide (Leu-Tyr-Ser-Val-Lys-Arg-Ser-Gly-Cys-Gly-Ser-Arg-Lys-Val-Ser-Tyr-Leu) were synthesized by Kelowna International Scientific Incorporation.
In vivo liver damage models
C3H/HeNCrNarl male mice (5 weeks) were purchased from the National Laboratory Animal Center (Taipei, Taiwan). All animals received humane care in compliance with the “Guide for the Care and Use of Laboratory Animals” published by the National Academy of Sciences, and all study procedures and protocols (animal ethic number 10457) were approved by the Animal Research Committee of National Tsing-Hua University. To induce liver fibrosis, the mice were treated with 16% (V/V) CCl4 in olive oil (2 mL/kg, p.o.) 3 times per week for 8 continuous weeks.
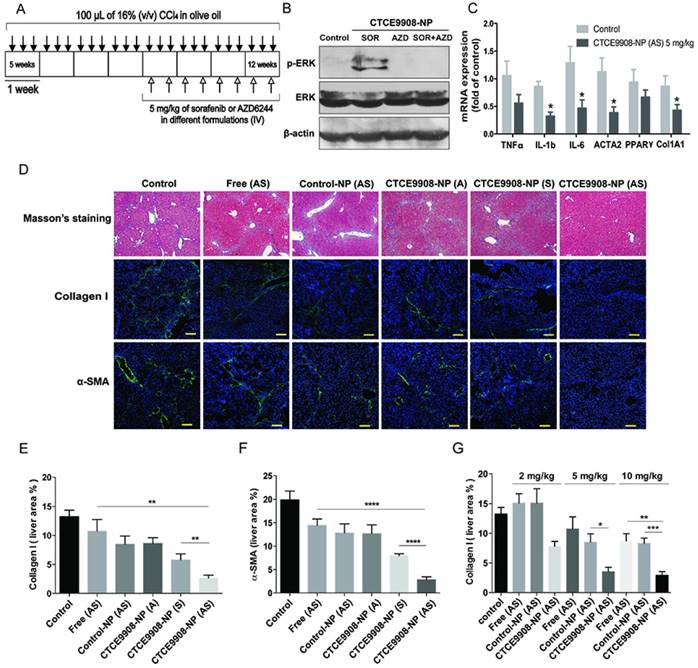
To evaluate the anti-fibrotic effect of the drug cocktail loaded in the NP formulations, sorafenib or AZD6244 (total daily dose: 5 mg/kg, two doses per week), loaded in the different formulations, was intravenously administered to mice with CCl4-induced liver fibrosis beginning 4 weeks after the start of CCl4 administration. The changes in hepatic fibrosis and vascularization were evaluated after 4 weeks of treatment (Fig. 4A). The microvascular density (MVD) was quantified by highlighting the blood vessels with Von Willebrand factor (vWF)—an endothelial cell marker—using the immunohistochemical protocols.
Sorafenib and the MEK inhibitor AZD6244 co-formulated in CTCE9908-NPs decrease liver fibrosis and suppress angiogenesis in the CCl4-induced mouse model of liver fibrosis. A, Treatment schedule of sorafenib and AZD6244 loaded in different formulations in the CCl4-induced murine model of liver damage. B, Western blot analysis showed co-delivery of sorafenib and AZD6244 suppressed ERK activation induced by sorafenib in the fibrotic livers of CCl4-treated mice. C, Sorafenib and AZD6244 loaded in CXCR4-targeted NPs inhibited mRNA expression of inflammatory and fibrotic markers such as TNF-α, IL-1b, IL-6, ACTA2, PPARγ, Col1A1 (normalized by β-actin). D, Sorafenib and AZD6244 delivered by CTCE9908-NPs (5 mg/kg, I.V., two doses per week) significantly ameliorated liver fibrosis in CCl4-treated mice, as indicated by Masson's trichrome and IF staining for collagen I, α-SMA and von Willebrand factor (vWF) (scale bar=100 μm) (n=5-15). E-F, Quantification of collagen I (E) and α-SMA (F) expression in randomly selected fields within the fibrotic livers (n=9-16). G, Sorafenib and AZD6244 loaded in different formulations reduced liver fibrosis in a dose-dependent manner, as indicated by a decrease in collagen I deposition in the fibrotic livers (n=5-16). The data are presented as mean values ± S.E.M., **p<0.01 and ***p<0.001.
NPs formulation
Nanoparticles were synthesized through a single-step nanoprecipitation method. PLGA (0.75 mg), DSPE-PEG-Maleimide (0.0846 mg), sorafenib (0.075 mg), AZD6244 (0.075 mg), TPGS (0.375 mg), and DOPC (0.0375 mg) were dissolved in 40 μL of DMSO as an organic phase. Cholesterol (0.0375 mg) was dissolved in alcohol. The organic phase mixture was added into 280 μL of deionized water drop wise. The formulations were formed with continuous stirring for 30 minutes. The ratio of the volumes of the oil and water phases was fixed at 1/7 (v/v) throughout all experiments. CTCE9908 peptides were reduced in a TCEP gel and then incubated with the nanoparticle formulations. DSPE-PEG-Maleimide was reacted with sulfhydryl groups in CTCE9908 peptides to form stable thioether bonds. Four hours after incubation, excess maleimides were quenched at the end of the reaction by adding free cysteine. To collect the peptide-modified NPs, the solution was centrifuged at 25,000 rcf for 30 min at 25 °C and re-suspended in PBS at the desired concentration for further study. Coumarin 6-loaded NPs were synthesized following the same procedure with a final weight ratio of coumarin 6 to PLGA of 1/150.
Characterization of NPs
The particle size and surface charge of the NPs were examined at room temperature by a zetasizer (300HS, Malvern Instruments Ltd., Worcestershire, UK). Transmission electron microscopy (TEM; H-7500, Hitachi High-Tech, Tokyo, Japan) was used to examine the size and morphology of the NPs. NPs were dropped on dried formvar-coated 100-mesh copper grids at room temperature and dried for two days prior to imaging.
To measure the encapsulation efficacy, the concentrations of sorafenib and AZD6244 were analyzed with a UV spectrophotometer (Multiskan, Thermo Scientific, Rockford, IL) at 270 nm and 260 nm, respectively.
Results
ERK activation after sorafenib treatment mediates HSC activation and survival
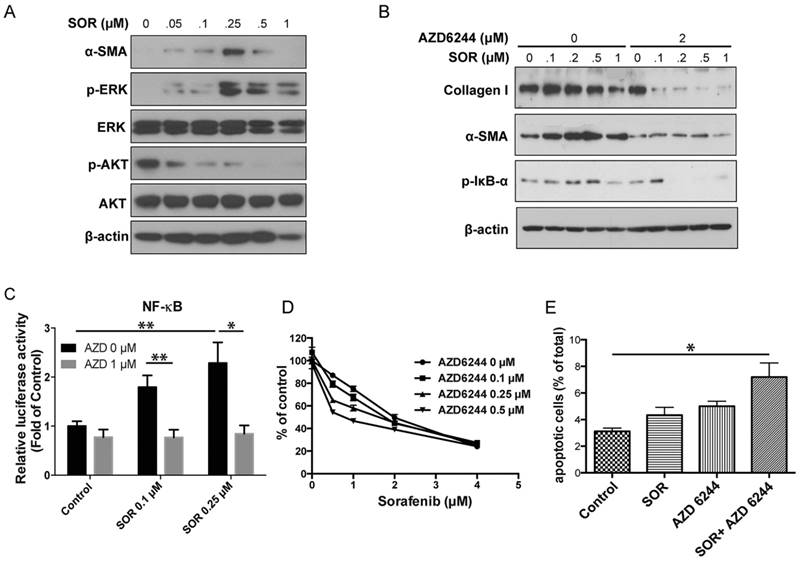
Treatment of HSCs with sorafenib at sub-micromolar concentrations increased phospo- extracellular signal-regulated kinases (ERK) levels in these cells, whereas pAKT remained strongly suppressed due to PDFGR inhibition by sorafenib (Fig. 1A and Fig. S1). Moreover, low-dose sorafenib promoted the myofibroblast differentiation of HSCs, as indicated by the increased expression of α-SMA (Fig. 1A). Thus, we next determined whether MAPK activation mediated HSC differentiation. As shown in Fig. 1B, treatment with MEK inhibitor AZD6244 alone only showed moderate suppression of HSC activation. However, MEK inhibitor AZD6244 in combination with sorafenib synergistically inhibited HSC differentiation (decreased expression of α-SMA) and activation (reduced expression of collagen I). Preventing ERK activation also resulted in reduced activation of nuclear factor kappa B (NF-κB) pathway, known to promote inflammation in the fibrotic liver microenvironment, regulate HSC activation and ensure the survival of activated HSCs (Fig. 1B-C). Indeed, AZD6244 sensitized HSCs to sorafenib treatment, and significantly enhanced the pro-apoptotic effects of sorafenib in HSCs (Fig. 1D-E).
Low doses of sorafenib triggered paradoxical activation of ERK and induced HSC activation. A, Low doses of sorafenib increased ERK activation and the expression of α-SMA in HSCs. Downregulation of AKT activity was observed after sorafenib treatment. B, The effect of MEK inhibition on HSC activation. MEK inhibition with AZD6244 (2 µM) decreased p-IkB-α and α-SMA expression and collagen content in HSCs treated with sorafenib. C, NF-κB luciferase activity from HSCs transfected with NF-κB luciferase construct. Sorafenib caused a significant increase in NF-κB activity. MEK inhibition with AZD6244 (1 µM) reversed the effect of sorafenib on NF-κB activation in HSCs (n=8-12). D, Effect of MEK inhibition on cell sensitivity to sorafenib. Inhibition of MEK activity with AZD6244 renders HSCs sensitive to sorafenib. E, Combination of sorafenib with MEK inhibitors increases apoptosis in HSCs (n=3-5). The data are given as the mean values ± S.E.M., *p<0.05 and **p<0.01.
Delivery of CXCR4-targeted NPs containing sorafenib and MEK inhibitor to activated HSCs suppresses their activation and viability in vitro
CXCR4 is a chemokine receptor induced in activated HSCs by various cellular stresses during the progression of liver fibrosis [16, 26, 27]. Thus, we developed CXCR4-targeted NPs to more selectively target activated HSCs in fibrotic livers [27]. In this study, to improve safety and efficacy of the drug cocktail, we co-formulated sorafenib and AZD6244 in CXCR4-targeted lipid-coated PLGA NPs modified with CTCE9908, a CXCR4 antagonist peptide. Electron microscopy imaging of drug-loaded NPs showed spherical morphology with an average diameter of 140 nm and a polydispersity index (PDI) of 0.4, as measured by zetasizer (Fig. 2A). The encapsulation efficacy of the drug cocktail in the CXCR4-targeted PLGA NPs was ~70%. We first examined whether the peptide modification (CTCE9908) enhanced the intracellular uptake of NPs in activated HSCs. Using coumarin 6 as a tracer molecule loaded in NPs, we found that the uptake of NPs modified with CTCE9908 (CTCE9908-NPs) was significantly enhanced in activated HSCs compared with non-targeted NPs conjugated with scramble peptides (control-NPs) (Fig. 2B-D). The addition of free CTCE9908 peptides reduced the uptake of CXCR4-targeted NPs into activated HSCs in a dose-dependent manner (Fig. 2E and Fig. S2), indicating that the nanoparticle uptake was mediated by the CXCR4 antagonist peptide (CTCE9908).
The NPs modified with CTCE9908 peptides showed enhanced cellular uptake in activated HSCs and suppressed HSC viability and activation when loaded with sorafenib and the MEK inhibitor AZD6244. A, Structures proposed for the CTCE9908-NPs with a representative TEM image (scale bar=100 nm). B, HSCs were treated with coumarin 6-loaded NPs modified with CTCE9908 peptides (CTCE9908-NPs) or scramble peptides (control-NPs) at various ratios of DSPE-PEG/peptides for 4 h (scale bar=50 μm). green, coumarin 6-loaded NPs; blue, DAPI. C-D, The cellular uptake of NPs was imaged and quantified using a Zeiss LSM 780 confocal microscope (n=7). E, The uptake of CTCE9908-NPs in HSCs was competitively inhibited by the addition of free CTCE9908 peptides in a dose-dependent manner. Cells were treated with free peptides prior to CTCE9908-NPs and analyzed for fluorescence signals by confocal microscopy (n=5-6). F, The cytotoxicity of sorafenib or AZD6244 in different formulations on HSCs was measured using the MTT assay (n=5-6). G, CTCE9908-NPs co-delivering sorafenib and AZD6244 prevented the effect of sorafenib on paradoxical activation of ERK and downregulated the expression of collagen I and α-SMA in HSCs. Free drug (AS), free-form AZD6244 and sorafenib; control-NP (-), empty NPs modified with scramble peptides; CTCE9908-NPs (-), empty NPs modified with CTCE9908 peptides; CTCE9908-NPs (A), CTCE9908-NPs loaded with AZD6244; CTCE9908-NPs (S), CTCE9908-NPs loaded with sorafenib; control-NPs (AS), AZD6244 and sorafenib loaded in NPs modified with scramble peptides. The data are given as the mean values ± S.E.M., **p<0.01 and ***p<0.001.
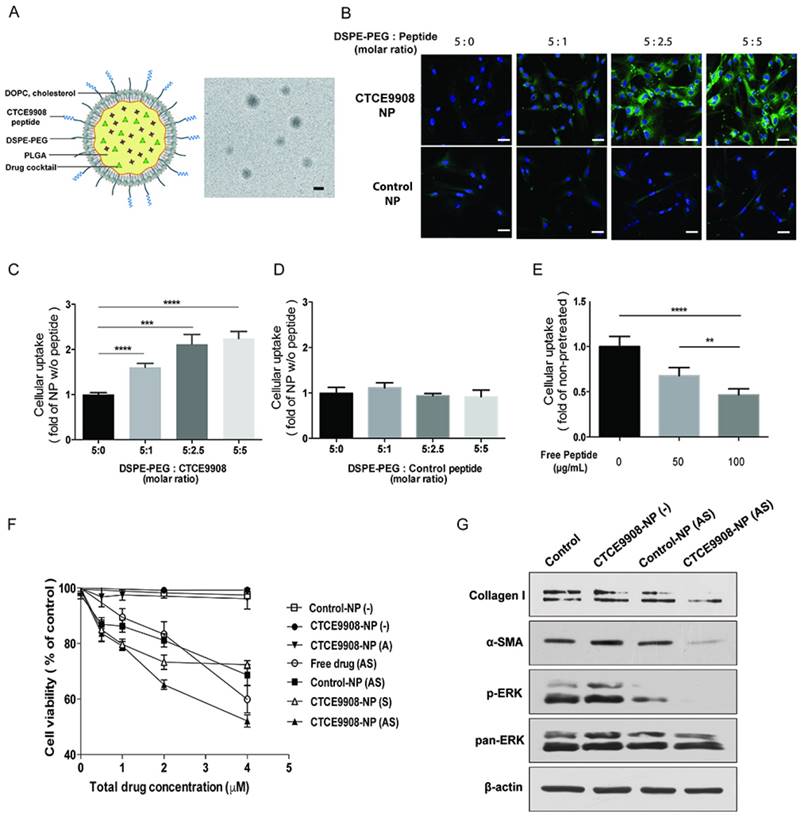
Furthermore, sorafenib and the MEK inhibitor AZD6244 co-formulated in CTCE9908-NPs exerted greater cytotoxicity on activated HSCs compared with the drug cocktail in the non-targeted NPs (control-NPs) or the single agent (sorafenib or AZD6244) delivered by CTCE9908-NPs (Fig. 2F). Moreover, sorafenib and AZD6244 co-delivered by CTCE9908-NPs prevented the paradoxical activation of ERK in HSCs and suppressed HSC differentiation and activation, as indicated by the reduced α-SMA and collagen I expression (Fig. 2G).
Systemic treatment with CXCR4-targeted NPs containing sorafenib and AZD6244 reduces liver fibrosis in mice with CCl4-induced liver damage
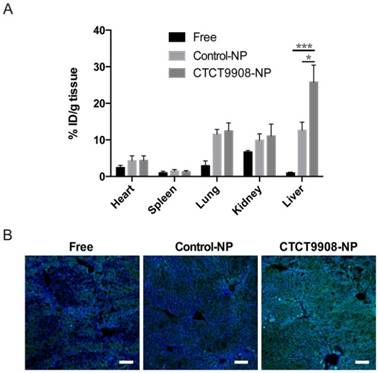
We further evaluated in vivo whether CTCE9908-NPs exhibited enhanced liver uptake in CCl4-treated mice. Coumarin 6 was used as a tracer molecule to study the biodistribution of drug-loaded NPs in mice with CCl4-induced liver fibrosis 4 h after intravenous (I.V.). injection. As shown in Fig. 3A, there was an increased uptake of CTCE9908-NPs compared to free drug or non-targeted control-NPs in the fibrotic livers of CCl4-treated mice. Similar to the in vitro results, there was increased intracellular uptake of the NPs in the fibrotic livers, indicating that the CXCR4 antagonist peptide CTCE9908 can act as a targeting moiety for specific delivery in this context (Fig. 3B). Next, we examined the anti-fibrotic effects of sorafenib and MEK inhibitors co-formulated into CXCR4-targeted NPs in CCl4-treated mice. Sorafenib or the MEK inhibitor AZD6244, loaded in different formulations, were intravenously given to mice with CCl4-induced liver fibrosis (5 mg/kg, 2 doses per week) starting at the 4th week after the first administration of CCl4, and alterations in hepatic fibrosis were evaluated after 4 weeks of treatment (Fig. 4A). When delivered using CTCE9908-NPs, sorafenib and AZD6244 significantly suppressed the activation of ERK triggered by sorafenib alone and alleviated liver inflammation and fibrosis, as measured by quantitative PCR, Masson's trichrome and immunohistochemistry (IHC) for collagen I and III (Fig. 4B-E and Fig. S3- Fig. S5). The infiltration of α-SMA-positive myofibroblasts in the fibrotic liver tissue was also significantly reduced in mice treated with the combination therapy delivered by CTCE9908-NPs compared to single drug (sorafenib or AZD6244 alone) loaded in CTCE9908-NPs (Fig. 4D-F and S3-S6). In addition, AST and ALT level significantly increased in CCl4-treated mice compared with normal control mice, indicating liver damage in CCl4-induced liver fibrosis. Sorafenib and AZD6244 co-delivered by CTCE9908-NPs reduced elevated ALT and AST, indicating that the combination treatment may facilitate liver repair (Fig. S7). Compared with the free-form drug cocktail, the CXCR4-targeted NP formulations were more effective at a lower dose (5 mg/kg) and dose-dependent inhibition of liver fibrosis was observed in mice treated with the drug cocktail loaded in the CXCR4-targeted NPs (Fig. 4D-G and Fig. S5-S6).
The NPs modified with CTCE9908 peptides enhanced coumarin 6 tissue uptake in the fibrotic livers of CCl4-treated mice. A, The tissue distribution of coumarin 6 in different formulations (n=7-8). Coumarin 6-loaded CTCE9908-NPs showed enhanced tissue uptake in the fibrotic livers 4 h after I.V. administration. B, Representative confocal images of the uptake of coumarin 6 in different formulations by the fibrotic livers. Green, coumarin 6-loaded NPs; blue, DAPI. Scale bar=100 μm. The data are given as the mean values ± S.E.M., *p<0.05 and ***p<0.001.
Co-delivery of sorafenib and AZD6244 by CXCR4-targeted NPs inhibited angiogenesis and normalized the vessel morphology in the fibrotic livers of CCl4-treated mice
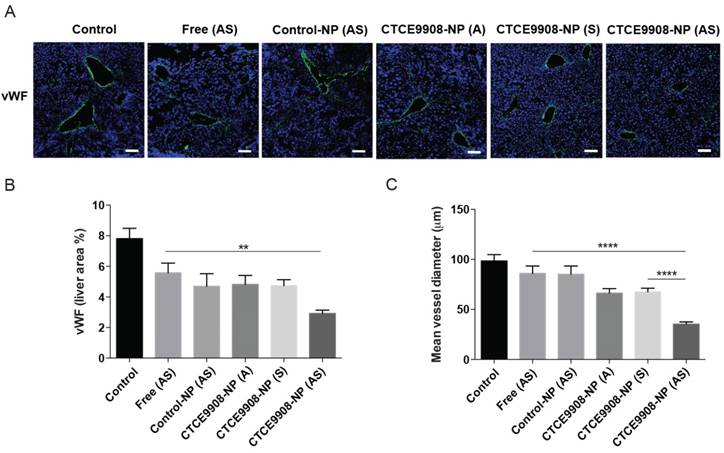
Given that sorafenib treatment could attenuate liver fibrosis and was associated with the inhibition of angiogenesis, we further examined the anti-angiogenic activity of sorafenib and AZD6244 co-formulated in CTCE9908-NPs in mice with CCl4-induced liver fibrosis. The MVD in fibrotic livers was significantly reduced in mice treated with sorafenib and MEK inhibitors co-delivered by CTCE9980-NPs compared with mice treated with the single drug alone in CTCE9908-NPs or the drug cocktail in the non-targeted control-NPs (Fig. 5A-B). Furthermore, as shown by the decreased mean vessel diameters, sorafenib and AZD6244 loaded in CTCE9908-NPs normalized the distorted morphology and size of vasculature in the fibrotic livers of mice with CCl4-induced liver fibrosis (Fig. 5A, C). Sorafenib and MEK inhibitors, which were co-formulated in the targeted NPs, suppressed angiogenesis in the fibrotic livers of CCl4-treated mice.
Sorafenib and the MEK inhibitor AZD6244 co-formulated in CTCE9908-NPs suppress angiogenesis in the fibrotic livers of CCl4-treated mice. A, Representative confocal images of vWF staining in the fibrotic livers from mice treated with different formulations (scale bar=100 μm). B-C, Sorafenib and AZD6244, delivered by CTCE9908-NPs, significantly reduced the microvascular density (B) and decreased the mean vessel diameter (C) in fibrotic livers from CCl4-treated mice, as observed by staining for vWF (n=10-26). The data are presented as mean values ± S.E.M., **p<0.01 and ***p<0.001.
Co-delivery of sorafenib and AZD6244 by CXCR4-targeted NPs prevented spontaneous HCC development in the fibrotic livers and inhibited liver metastasis
Progression of liver fibrosis increases the risk of HCC development, fuels HCC growth and influences treatment prognosis [28-31]. We next evaluated the effect of combined sorafenib and AZD6244 in a cancer prevention setting when delivered by CXCR4-targeted NPs systemically to mice with DEN and CCl4-induced liver fibrosis and inflammation-associated HCC (Fig. 6A). Sorafenib and AZD6244 loaded in CXCR4-targeted NPs significantly reduced tumor formation in the fibrotic liver as determined by significantly lower liver nodules and tumor size associated with reduced desmoplasia compared with untreated controls (Fig. 6B-D). For the combined treatment group, tumor incidence was 0.2 (95% confidence interval (CI): 0.05 and 0.80); for the control group, tumor incidence was 1.5 (95% CI: 0.95, 2.29). Tumor incidence in the combined treatment group was on average 7.3-fold (SD ± 2.1) lower than it in the control group. The tumor incidence rate was 20% in the sorafenib plus AZD6244 group (95% CI: 5.7%, 57.0%), and 80% in the control group (95% CI: 49.0%, 94.0%), with a difference in incidence rate of 60% (95% CI: 22.4%, 97.6%, p-value = 0.0035 by normal approximation z-test) (Fig. 6E). Histopathology confirmed a significant reduction of tumor lesions in livers treated with sorafenib and AZD6244 loaded in CXCR4-targeted NPs compared to the untreated controls (Fig. 6F-G and S8).
Sorafenib and the MEK inhibitor AZD6244 co-formulated in CTCE9908-NPs prevent spontaneous HCC development and PDAC liver metastasis. A, Experimental setup. B, Representative livers in a CCl4-induced spontaneous HCC model with or without treatment of sorafenib and AZD6244 loaded in CXCR4-targeted NPs. C-E, Liver tumor nodule counts (>1 mm) per mouse (C), maximal tumor sizes (mean ± S.E.M.) (D) and HCC incidence (E) in NP-treated or control mice (n=10). To investigate the number of tumor counts emerged in two groups, the generalized linear model of Poisson regression was applied. F, Histopathological analysis of livers in a CCl4-induced spontaneous HCC model with or without treatment of Sorafenib and AZD6244 loaded in CXCR4-targeted NPs (green, collagen I; blue, DAPI). Scale bar = 50 μm. G, Quantification of collagen I expression in randomly selected fields within the fibrotic livers. H, Mice were inoculated with 2x105 AK4.4 cells into the pancreas. The treatment schedule of established tumors is shown. I, Primary PDAC tumor size after treatment. J, Number of metastatic lesions was enumerated in the livers (n=6-10). K, Histopathological analysis of livers in a syngeneic orthotopic PDAC murine model with or without treatment of sorafenib and AZD6244 loaded in CXCR4-targeted NPs (red, anti-GFP; green, α-SMA; blue, DAPI). Scale bar = 50 μm. L, Quantification of α-SMA+ myofibroblast infiltration in randomly selected fields within livers of syngeneic orthotopic PDAC murine models (n=5). *p<0.05, **p<0.01 and ***p<0.001.
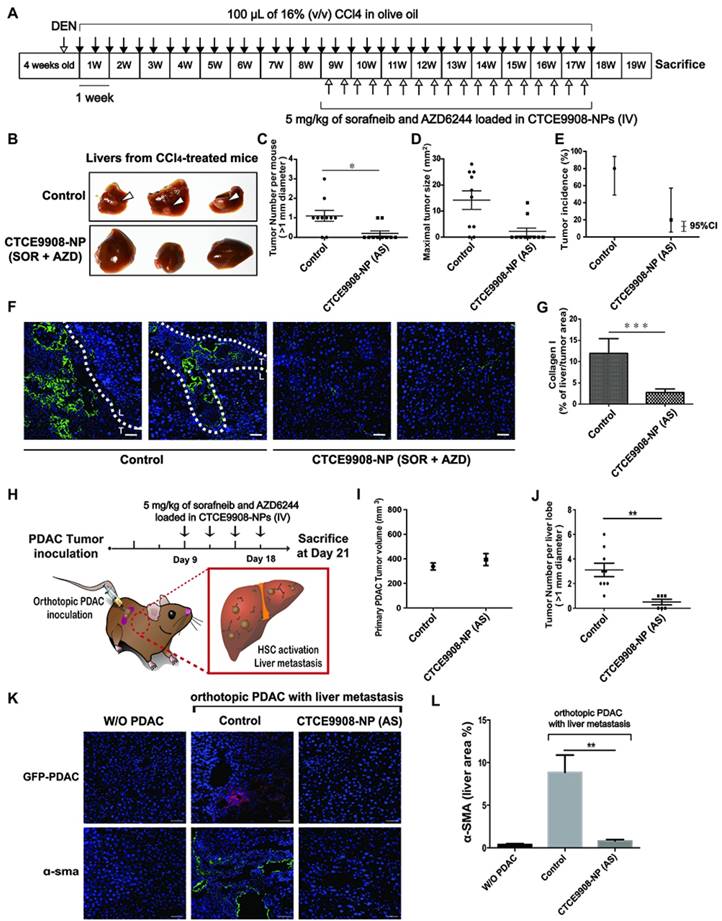
In addition to primary cancer formation, the activation of HSCs by tumor-derived soluble factors or extracellular vesicles in the liver microenvironment plays a critical role in the development of liver metastasis [32, 33]. We examined the effect of sorafenib and AZD6244 loaded in HSC-targeted NPs on the incidence of liver metastasis in mice. A spontaneous liver metastasis model was established by the inoculation of PDAC cells into pancreas of 6-8-week-old FVB mice (Fig. 6H). The treatment began on day 9 after inoculation with 2x105 AK4.4 (GFP/GLuc) cells orthotopically in mice (Fig. 6H). Mice inoculated with orthotopic PDAC developed metastatic tumor lesions in livers with increased infiltration of α-SMA-positive myofibroblasts and F4/80+ macrophages (Fig. 6I-L). In contrast, systemic administration of sorafenib and AZD6244 (5 mg/kg) loaded in CXCR4-targeted NPs on days 9, 12, 15 and 18 reduced the infiltration of α-SMA-positive myofibroblasts and F4/80+ macrophages, deceased collagen deposition in livers and suppressed the incidence of liver metastasis (Fig. 6I-L and Fig. S9-S10), without affecting the primary PDAC tumor burden (Fig. 6I). Moreover, the safety study showed that the serum levels of hepatotoxicity makers and kidney injury markers remained unchanged after treatment with sorafenib and MEK inhibitors in CTCE9908-NPs (Table 1).
Toxicity profile of sorafenib and AZD6244-loaded CTCE9908-NPs. Sera of C3H mice were collected 24 h after I.V. injections of sorafenib and AZD6244-loaded CTCE9908-NPs (5 mg/kg) for evaluation of markers for acute kidney injury and liver damage (n=4). Abbreviations: ALT: alanine aminotransferase; AST: aspartate aminotransferase; ALP: alkaline phosphatase; γ-GT: γ-Glutamyltransferase; BUN: Blood urea nitrogen; CREA: creatinine. Data are shown as mean values ± S.E.M..
| Group | AST (U/L) | ALT (U/L) | ALP (U/L) | γ-GT (U/L) | BUM (mg/dL) | CREA (mg/dL) |
|---|---|---|---|---|---|---|
| Control | 73.2±5.4 | 31.7±4.5 | 492.4±50.8 | 0.1±0.1 | 33.6±1.0 | 0.4±0.0 |
| CTCE9908-NP (AS) | 72.1±8.7 | 27.7±1.7 | 429.1±35.1 | 0.0±0.1 | 33.7±1.4 | 0.4±0.0 |
n=4-6
Discussion
Liver fibrosis is driven by abnormal activation of various signaling pathways, including MAPK, in HSCs, leading to their myofibroblast differentiation and activation, and abnormal angiogenesis. Sorafenib, a multikinase inhibitor that targets RAF/MEK/ERK pathway and PDGFR/VEGFR suppresses proliferation and activation of HSCs and angiogenesis and thus opens the possibility of using this drug to treat liver fibrosis and prevent HCC. However, previous studies from our group and others have demonstrated that RAF inhibitors such as sorafenib paradoxically induce MAPK activation in cancer cells, leading to treatment resistance [24]. These studies highlighted the potential therapeutic benefit of combining sorafenib and MEK inhibitors for treating hepatic malignancies. In this study, we further show here that low doses of sorafenib can induce paradoxical activation of ERK in HSCs. In turn, this leads to HSC activation reflected by enhanced collagen I production. NF-κB activation in HSCs also plays a crucial role in the progression of hepatic fibrosis by promoting HSC activation and proliferation, inhibiting apoptosis and increasing the secretion of chemokines and recruitment of inflammatory cells (e.g., macrophages) [30]. Although sorafenib blocking of RAF and PDGFR has potential anti-fibrotic/anti-inflammatory properties, treatment-induced ERK and NF-κB activation in HSCs can limit its therapeutic activity. Indeed, sorafenib alone only moderately attenuated hepatic fibrosis in vivo. Combining sorafenib with a MEK inhibitor (AZD6244) can prevent MAPK and NF-κB activation in HSCs and achieve synergistic anti-fibrotic efficacy. A similar effect on inhibition of HSC activation was observed after treatment of MEK inhibitor in combination with a selective PDGFR blockade (Fig. S1). Profound anti-fibrotic effects may only be achieved through targeting multiple pro-fibrotic pathways. This combination approach may have impact beyond alleviation of liver fibrosis. HSC activation also serves a key element in the creation of a pro-tumor microenvironment. Thus, the combination of sorafenib with MEK inhibitors may potentially have beneficial effects on both controlling fibrosis and preventing progression of malignant disease.
HCC is a complex disease associated with multiple risk factors and underlying conditions such as alcoholic liver disease, viral hepatitis and nonalcoholic fatty-liver disease. HCC mostly arises in the setting of cirrhosis resulting from repeated liver damage and chronic inflammation. To model the clinical feature of HCC with underlying liver cirrhosis, DEN was first administrated to trigger DNA damage and develop a mutagenic environment in liver tissues. Following administration of DEN, severe fibrotic liver damage and HCC formation were induced by repeated exposure to CCl4 [34]. Sorafenib and AZD6244 co-formulated in CXCR4-targeted NPs reduced desmoplasia and attenuated HCC formation in this clinically relevant model in the setting of carcinogenesis within cirrhotic liver tissues. However, about 15%-20% of HCC develop in the non-fibrotic or minimal fibrotic livers. For example, NASH/NAFLD-associated HCC may also derive from noncirrhotic livers in the setting of steatosis with inflammation [35]. In this study, we demonstrated the reduced expression of pro-inflammatory cytokines in livers after treatment with combined sorafenib and AZD6244. Given the anti-inflammatory properties of the drug cocktail, combined sorafenib and AZD6244 may also serve as an effective therapeutic strategy for liver diseases with an underlying pro-inflammatory microenvironment. Our previous finding showed sorafenib in combination of AZD6244 synergistically suppressed the orthotopic HCC progression without underlying cirrhosis [24]. Taken together, combination therapy with sorafenib and MEK inhibitors has potential as a preventive and therapeutic agent for multiple liver diseases, including liver cancer.
The fibrotic microenvironment not only promotes primary liver formation, but also sustains metastatic tumor growth in livers. Interestingly, we observed enhanced PDAC metastatic development in CCl4-induced fibrotic livers compared with normal livers. On the other hand, during the progression of PDAC, soluble factors or extracellular vesicles released from metastasis-associated macrophages (MAMs) or tumor cells activate HSCs and develop the fibrotic microenvironment that promotes the metastatic seeding of tumor cells. Treatment of sorafenib and AZD6244 loaded in CXCR4-targeted NPs reduced HSC activation and macrophage infiltration, leading to the suppression of liver metastatic incidence. These findings indicate that targeting the pro-fibrotic niche may serve as a potential therapeutic approach to treating PDAC liver metastasis.
In preclinical studies of HCC, MEK inhibition enhanced the anti-cancer efficacy of sorafenib [24, 36]. These agents have also been tested in phase II clinical trials in advanced HCC patients, with promising activity but frequent dose reductions due to adverse effects [37]. Side effects, including rash, fatigue, edema, gastrointestinal symptoms, neurological symptoms, and liver function impairment, are often observed in patients treated with kinase inhibitors [38]. Thus, translating this combination for treatment of liver disease in cancer prevention settings raises significant toxicity concerns. To address this issue, we developed a nano-carrier that was modified with a peptide targeting CXCR4 to co-deliver sorafenib and MEK inhibitors, and improve their bioavailability and absorption by activated HSCs in fibrotic livers. The PLGA NPs that had slow-release properties significantly extended the blood circulation of the cargo and enhanced drug accumulation in the fibrotic livers [39]. We further modified the PLGA NPs with peptides targeting CXCR4 to achieve ligand-mediated targeting to HSCs wherein CXCR4 expression was upregulated in response to activation [40]. Sorafenib and MEK inhibitors co-delivered by the CXCR4-targeted NPs prevented MAPK activation, suppressed hepatic fibrosis progression and attenuated angiogenesis in the fibrotic livers of CCl4-treated mice with no apparent toxicity. Our results demonstrate that a cocktail of sorafenib and MEK inhibitors loaded in the targeted drug carriers is a safe and promising therapeutic strategy for liver fibrosis.
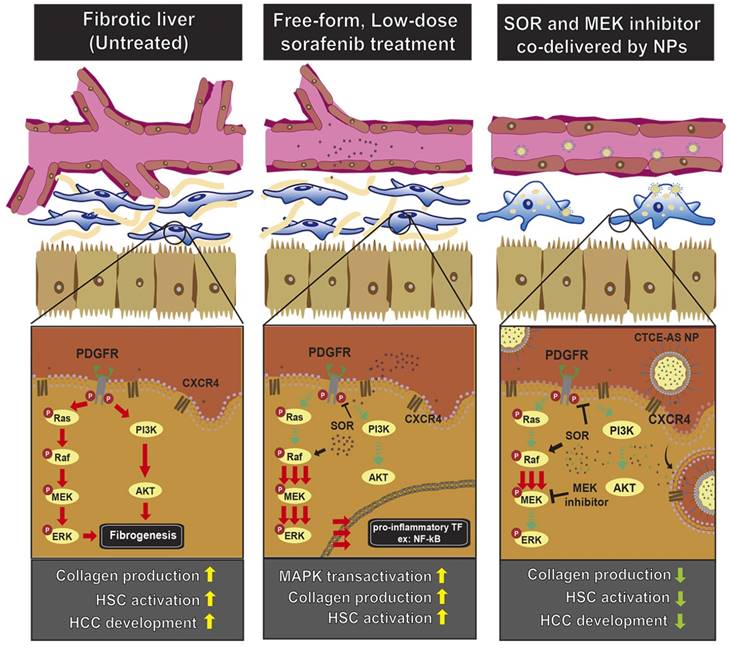
In summary, we showed that combining sorafenib with MEK inhibition is efficacious in reducing CCl4-induced liver fibrosis in mice. Moreover, combining sorafenib and MEK inhibitor-loaded NPs for more selective targeting of activated HSCs was safe and achieved significant anti-fibrotic effects and inhibited primary HCC formation and metastatic dissemination of PDAC to the fibrotic liver (Fig. 7). This novel therapeutic strategy should be further tested in models of liver fibrosis and for preventing malignant transformation and progression in the liver.
Schematic illustration of the mechanisms by which sorafenib and AZD6244-loaded CTCE9908-NPs ameliorated liver fibrosis. Sorafenib paradoxically activates ERK and increases the activation of NF-κB expression, which increases α-SMA expression and the production of collagen in HSCs. Sorafenib and the MEK inhibitor AZD6244, when co-formulated in CTCE9908-NPs, suppressed ERK activation and inhibited HSC activation, enhancing the anti-fibrotic and cancer prevention effect in a murine model of liver damage.
Supplementary Material
Supplementary materials and methods, Fig.S1 - S10.
Abbreviations
CCl4: tetrachloromethane; DMSO: dimethyl sulfoxide; DOPC: 1,2-dioleoyl-sn-glycero-3-phosphocholine; DSPE-PEG-Maleimide: 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000]; ECM: extracellular matrix; ERK: extracellular signal-regulated kinase; IHC: immunohistochemistry; HCC: hepatocellular carcinoma; HSCs: hepatic stellate cells; MAMs: metastasis-associated macrophages; MAPK: mitogen-activated protein kinase; MEK: Mitogen-activated protein kinase kinase; MVD: microvascular density; NP: nanoparticle; PDGFR: platelet-derived growth factor receptor; PBS: phosphate buffered solution; PLGA: poly(lactic-co-glycolic acid); PDAC: Pancreatic ductal adenocarcinoma; PDI: polydispersity index; PDGFR: platelet-derived growth factor receptors; TCEP: Tris(2-carboxyethyl)phosphine; TPGS: D-α-Tocopherol polyethylene glycol 1000 succinate; VEGFR: vascular endothelial growth factor receptor; vWF: von Willebrand factor.
Acknowledgements
This study was supported by Ministry of Science and Technology (MOST 104-2628-B-007-001-MY3, 105-2628-E-007-007-MY3), by Chang Gung Memorial Hospital-National Tsing Hua University Joint Research Grant (104N2744E1) and by the National Institute for Health Research (NHRI-EX106-10609BC). Dr. Duda's research is supported by the National Institutes of Health grant P01-CA080124.
Author contribution
Yun-Chieh Sung, Ya-Chi Liu, Chih-Chun Chang, Ts-Ting Lin - performed experiments, prepared Figures 3-6; Po-Han Chao - performed experiments, prepared Figures 6; Ja-An Lin - analyzed data and performed statistical analysis; Hui-Teng Cheng - supervised study; Jane Wang - supervised study; Charles P. Lai - supervised study; Ling-Hsuan Chen - performed experiments, prepared Figures 1; Peiru Jin- performed experiments, prepared Figures 1; Anthony Y. Wu - performed experiments, prepared Figures 6; Tsaiyu Chiang - performed experiments, prepared Figures 2; Dong-Yu Gao - performed experiments, prepared Figures 3; Shang-Ying Hsieh - performed experiments, prepared Figures 6; Dan G. Duda - supervised study and edited the manuscript text; Yunching Chen- performed experiments, supervised study and edited the manuscript text.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655-69
2. Fernandez M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol. 2009;50:604-20
3. Sahin H, Borkham-Kamphorst E, Kuppe C, Zaldivar MM, Grouls C, Al-samman M. et al. Chemokine Cxcl9 attenuates liver fibrosis-associated angiogenesis in mice. Hepatology. 2012;55:1610-9
4. Schlageter M, Terracciano LM, D'Angelo S, Sorrentino P. Histopathology of hepatocellular carcinoma. World J Gastroenterol. 2014;20:15955-64
5. Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123:1887-901
6. Popov Y, Schuppan D. Targeting liver fibrosis: strategies for development and validation of antifibrotic therapies. Hepatology. 2009;50:1294-306
7. Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425-36
8. Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5:167sr1
9. Schuppan D, Pinzani M. Anti-fibrotic therapy: lost in translation? J Hepatol. 2012;56(Suppl 1):S66-74
10. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209-18
11. Ray K. Liver: hepatic stellate cells hold the key to liver fibrosis. Nat Rev Gastroenterol Hepatol. 2014;11:74
12. Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61:1066-79
13. Hennenberg M, Trebicka J, Kohistani Z, Stark C, Nischalke HD, Kramer B. et al. Hepatic and HSC-specific sorafenib effects in rats with established secondary biliary cirrhosis. Lab Invest. 2011;91:241-51
14. Wang Y, Gao J, Zhang D, Zhang J, Ma J, Jiang H. New insights into the antifibrotic effects of sorafenib on hepatic stellate cells and liver fibrosis. J Hepatol. 2010;53:132-44
15. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7:3129-40
16. Chen Y, Huang Y, Reiberger T, Duyverman AM, Huang P, Samuel R. et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology. 2014;59:1435-47
17. Coriat R, Gouya H, Mir O, Ropert S, Vignaux O, Chaussade S. et al. Reversible decrease of portal venous flow in cirrhotic patients: a positive side effect of sorafenib. PLoS One. 2011;6:e16978
18. Pinter M, Sieghart W, Reiberger T, Rohr-Udilova N, Ferlitsch A, Peck-Radosavljevic M. The effects of sorafenib on the portal hypertensive syndrome in patients with liver cirrhosis and hepatocellular carcinoma-a pilot study. Aliment Pharmacol Ther. 2012;35:83-91
19. Hong F, Chou H, Fiel MI, Friedman SL. Antifibrotic activity of sorafenib in experimental hepatic fibrosis: refinement of inhibitory targets, dosing, and window of efficacy in vivo. Dig Dis Sci. 2013;58:257-64
20. Deng YR, Ma HD, Tsuneyama K, Yang W, Wang YH, Lu FT. et al. STAT3-mediated attenuation of CCl4-induced mouse liver fibrosis by the protein kinase inhibitor sorafenib. J Autoimmun. 2013;46:25-34
21. Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS. et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307-21
22. Alcala AM, Flaherty KT. BRAF inhibitors for the treatment of metastatic melanoma: clinical trials and mechanisms of resistance. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:33-9
23. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO. et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039-43
24. Chen Y, Liu YC, Sung YC, Ramjiawan RR, Lin TT, Chang CC. et al. Overcoming sorafenib evasion in hepatocellular carcinoma using CXCR4-targeted nanoparticles to co-deliver MEK-inhibitors. Sci Rep. 2017;7:44123
25. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS. et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25-34
26. Hong F, Tuyama A, Lee TF, Loke J, Agarwal R, Cheng X. et al. Hepatic stellate cells express functional CXCR4: role in stromal cell-derived factor-1alpha-mediated stellate cell activation. Hepatology. 2009;49:2055-67
27. Liu CH, Chan KM, Chiang T, Liu JY, Chern GG, Hsu FF. et al. Dual-Functional Nanoparticles Targeting CXCR4 and Delivering Antiangiogenic siRNA Ameliorate Liver Fibrosis. Mol Pharm. 2016;13:2253-62
28. Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. 2013;144:512-27
29. Hui CK, Leung N, Shek TW, Yao H, Lee WK, Lai JY. et al. Sustained disease remission after spontaneous HBeAg seroconversion is associated with reduction in fibrosis progression in chronic hepatitis B Chinese patients. Hepatology. 2007;46:690-8
30. Luedde T, Schwabe RF. NF-kappaB in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:108-18
31. Reiberger T, Chen Y, Ramjiawan RR, Hato T, Fan C, Samuel R. et al. An orthotopic mouse model of hepatocellular carcinoma with underlying liver cirrhosis. Nat Protoc. 2015;10:1264-74
32. Kang N, Gores GJ, Shah VH. Hepatic stellate cells: partners in crime for liver metastases? Hepatology. 2011;54:707-13
33. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK. et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816-26
34. Uehara T, Pogribny IP, Rusyn I. The DEN and CCl4 -Induced Mouse Model of Fibrosis and Inflammation-Associated Hepatocellular Carcinoma. Curr Protoc Pharmacol. 2014;66:14 30 1-10
35. Kawada N, Imanaka K, Kawaguchi T, Tamai C, Ishihara R, Matsunaga T. et al. Hepatocellular carcinoma arising from non-cirrhotic nonalcoholic steatohepatitis. J Gastroenterol. 2009;44:1190-4
36. Huynh H, Ngo VC, Koong HN, Poon D, Choo SP, Toh HC. et al. AZD6244 enhances the anti-tumor activity of sorafenib in ectopic and orthotopic models of human hepatocellular carcinoma (HCC). J Hepatol. 2010;52:79-87
37. Lim HY, Heo J, Choi HJ, Lin CY, Yoon JH, Hsu C. et al. A phase II study of the efficacy and safety of the combination therapy of the MEK inhibitor refametinib (BAY 86-9766) plus sorafenib for Asian patients with unresectable hepatocellular carcinoma. Clin Cancer Res. 2014;20:5976-85
38. Keefe DM, Bateman EH. Tumor control versus adverse events with targeted anticancer therapies. Nat Rev Clin Oncol. 2011;9:98-109
39. Lin Ts T, Gao DY, Liu YC, Sung YC, Wan D, Liu JY. et al. Development and characterization of sorafenib-loaded PLGA nanoparticles for the systemic treatment of liver fibrosis. J Control Release. 2016;221:62-70
40. Chow LN, Schreiner P, Ng BY, Lo B, Hughes MR, Scott RW. et al. Impact of a CXCL12/CXCR4 Antagonist in Bleomycin (BLM) Induced Pulmonary Fibrosis and Carbon Tetrachloride (CCl4) Induced Hepatic Fibrosis in Mice. PLoS One. 2016;11:e0151765
Author contact
![]() Corresponding author: Yunching Chen, PhD, Institute of Biomedical Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan, ROC; phone: 886-3-571-5131; ext: 35513; email: yunchingnthu.edu.tw
Corresponding author: Yunching Chen, PhD, Institute of Biomedical Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan, ROC; phone: 886-3-571-5131; ext: 35513; email: yunchingnthu.edu.tw