Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(3):610-626. doi:10.7150/thno.22237 This issue Cite
Research Paper
Ligand Activation of PPARγ by Ligustrazine Suppresses Pericyte Functions of Hepatic Stellate Cells via SMRT-Mediated Transrepression of HIF-1α
Feng Zhang1,3,4, Shuai Lu5, Jianlin He2, Huanhuan Jin1, Feixia Wang1, Li Wu1,3,4, Jiangjuan Shao3,4, Anping Chen6, Shizhong Zheng1,3,4 ![]()
1. Jiangsu Key Laboratory for Pharmacology and Safety Evaluation of Chinese Materia Medica, Nanjing University of Chinese Medicine, Nanjing 210023, China.
2. Third Institute of Oceanography, State Oceanic Administration, Xiamen 361005, China.
3. Jiangsu Key Laboratory of Therapeutic Material of Chinese Medicine, Nanjing University of Chinese Medicine, Nanjing 210023, China.
4. State Key Laboratory Cultivation Base for TCM Quality and Efficacy, Nanjing University of Chinese Medicine, Nanjing 210023, China.
5. School of Science, China Pharmaceutical University, Nanjing 211198, China.
6. Department of Pathology, School of Medicine, Saint Louis University, MO 63104, USA.
Received 2017-8-5; Accepted 2017-10-22; Published 2018-1-1
Abstract

Rationale: Hepatic stellate cells (HSCs) are liver-specific pericytes regulating vascular remodeling during hepatic fibrosis. Here, we investigated how ligustrazine affects HSC pericyte functions.
Methods: Rat HSC-T6 and human HSC-LX2 cells were cultured, and multiple molecular experiments including real-time PCR, Western blot, flow cytometry, immunofluorescence, electrophoretic mobility shift assay and co-immunoprecipitation were used to elucidate the underlying mechanisms. Molecular simulation and site-directed mutagenesis were performed to uncover the target molecule of ligustrazine. Rats were intoxicated with CCl4 for evaluating ligustrazine's effects in vivo.
Results: Ligustrazine inhibited angiogenic cytokine production, migration, adhesion and contraction in HSCs, and activated PPARγ. Selective PPARγ inhibitor GW9662 potently abrogated ligustrazine suppression of HSC pericyte functions. Additionally, HIF-1α inhibitor PX-478 repressed HSC pericyte functions, and ligustrazine inhibited the transcription of HIF-1α, which was diminished by GW9662. Moreover, ligustrazine downregulation of HIF-1α was rescued by knockdown of SMRT, and ligustrazine increased PPARγ physical interaction with SMRT, which was abolished by GW9662. These findings collectively indicated that activation of PPARγ by ligustrazine led to transrepression of HIF-1α via a SMRT-dependent mechanism. Furthermore, molecular docking evidence revealed that ligustrazine bound to PPARγ in a unique double-molecule manner via hydrogen bonding with the residues Ser289 and Ser342. Site-directed mutation of Ser289 and/or Ser342 resulted in the loss of ligustrazine transrepression of HIF-1α in HSCs, indicating that interactions with both the residues were indispensable for ligustrazine effects. Finally, ligustrazine improved hepatic injury, angiogenesis and vascular remodeling in CCl4-induced liver fibrosis in rats.
Conclusions: We discovered a novel ligand activation pattern for PPARγ transrepression of the target gene with therapeutic implications in HSC pericyte biology and liver fibrosis.
Keywords: Hepatic stellate cell, Liver fibrosis, Ligustrazine, PPARγ, HIF-1α, Transrepression, Molecular simulation
Introduction
Hepatic fibrosis and cirrhosis represent global health problems due to their life-threatening complications including portal hypertension and liver failure. The pathogenesis of liver fibrosis is characterized by massive synthesis and deposition of extracellular matrix (ECM) components in the liver [1]. A pathological hallmark of this disorder is sinusoidal capillarization and angiogenesis, which is accompanied by loss of endothelium fenestrae and formation of subendothelial basement membrane. These intrahepatic vascular alterations facilitate the formation of fibrotic septa and result in reduced delivery of oxygen and nutrients, exacerbating parenchyma injuries in fibrotic liver [2]. Additionally, intrahepatic angiogenesis and endothelial dysfunction lead to increases in intrahepatic vascular resistance and thereby portal hypertension [3].
Activated hepatic stellate cells (HSCs), the most pro-fibrogenic cells in the liver, are the primary source of ECM components during fibrogenesis. In parallel to this role, HSCs are also characterized as liver-specific pericytes, making a dominant contribution to the sinusoidal structural changes during fibrosis [4]. HSCs can secrete many pro-angiogenic molecules, such as vascular endothelial growth factor-A (VEGF-A) and basic fibroblast growth factor (bFGF), which stimulate liver sinusoidal endothelial cells (LSECs) and promote a pro-angiogenic sinusoidal niche [5]. Activated HSCs also acquire enhanced capabilities of migration and adhesion to sinusoidal endothelium, strengthening the pro-angiogenic paracrine communication with LSECs and stabilizing the new vessels [6]. These processes also make HSCs have increased cellular projections around sinusoids, and consequently, the contractile nature of HSCs transforms the low-resistance vascular bed into a high-resistance and constricted sinusoidal vessel bed in the liver [6]. Collectively, HSCs share many anatomic and phenotypic similarities with pericytes in other organs, accounting for the fundamental processes of sinusoidal vascular remodeling in chronic liver diseases.
A great deal of evidence supports the notion that interventions targeting sinusoidal vascular remodeling have beneficial effects on liver fibrosis and portal hypertension [7-9]. The natural alkaloid ligustrazine has recently been reported to have therapeutic promise for hepatic fibrosis based on the observation that ligustrazine halted HSC activation by interfering with several pro-fibrogenic pathways [10, 11], and protected hepatocytes by interrupting NLRP3 inflammasome signaling [12]. Intriguingly, ligustrazine was found to inhibit angiogenesis under different pathological conditions [13, 14]. Here, we investigated the molecular mechanism and potential target for ligustrazine intervention of HSC-mediated vascular remodeling in liver fibrosis using experimental and molecular simulation approaches. Our discoveries provided novel insights into HSC pericyte biology with therapeutic implications for liver fibrosis.
Materials and Methods
Chemicals and antibodies
Ligustrazine (C8H12N2, analytical standard) and actinomycin D (C62H86N12O16, purity >98%) were obtained from Sigma-Aldrich (Saint Louis, MO, USA). GW9662 (C13H9ClN2O3, purity >99.88%) and PX-478 (C13H18Cl2N2O3, purity >97%) were obtained from Selleck Chemicals (Houston, TX, USA). These compounds were dissolved in dimethyl sulfoxide for in vitro experiments, and treatment with dimethyl sulfoxide alone was used as vehicle control. Rabbit polyclonal antibodies against VEGF-A (19003-1-AP, for WB, 1:1000 dilution), ICAM-1 (10831-1-AP, for WB, 1:1000 dilution), VCAM-1 (11444-1-AP, for WB, 1:1000 dilution), HIF-1α (20960-1-AP, for WB, 1:1000 dilution; for IF, 1:100 dilution), CD31 (11265-1-AP, for WB, 1:1000 dilution; for IHC, 1:400 dilution), CD34 (14486-1-AP, for WB, 1:1000 dilution; for IHC, 1:400 dilution), vWF (11778-1-AP, for WB, 1:1000 dilution; for IHC, 1:400 dilution), MMP-2 (10373-2-AP, for WB, 1:1000 dilution), and MMP-9 (10375-2-AP, for WB, 1:1000 dilution) were purchased from Proteintech Group (Chicago, IL, USA). Rabbit polyclonal antibodies against phospho-MLC2 (Thr18/Ser19) (#3674, for WB, 1:1000 dilution; for IF, 1:100 dilution), MLC2 (#3672, for WB, 1:1000 dilution), PPARγ (#2443, for WB, 1:1000 dilution; for IF, 1:100 dilution; for Co-IP, 1:50 dilution), Lamin B1 (#13435, for WB, 1:1000 dilution), and β-Actin (#4970, for WB, 1:1000 dilution) were purchased from Cell Signaling Technology (Danvers, MA, USA). A mouse monoclonal antibody against SMRT (sc-32298, for Co-IP, 1:50 dilution) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit monoclonal antibody against bFGF (ab16828, for WB, 1:1000 dilution) and PPARγ (ab209350, for WB, 1:1000 dilution; for IF, 1:100 dilution) were purchased from Abcam (Cambridge, UK). Horseradish peroxidase-conjugated secondary antibodies including Anti-Rabbit IgG H&L (HRP) (ab6721, for WB, 1:10000 dilution) and Anti-Mouse IgG H&L (HRP) (ab6789, for WB, 1:10000 dilution) were obtained from Abcam (Cambridge, UK).
Cell culture and transfection with siRNA or plasmids
Rat HSC-T6 and human HSC-LX2 cell lines were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). Rat primary HSCs were isolated as we previously described [15]. Rat primary LSECs were purchased from CHI Scientific, Inc. (Wuxi, China). Cells were cultured in Dulbecco's modified eagle medium with 10% fetal bovine serum, 1% antibiotics, and grown in a 5% CO2 humidified atmosphere at 37°C. SMRT siRNA (sc-36514) and NCoR siRNA (sc-36001) for human cells were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Transfection with siRNA against SMRT or NCoR, or control siRNA (sc-37007, Santa Cruz Biotechnology) was performed according to the protocols provided by the manufacturer. Briefly, cells at 60% confluence were transfected with siRNA at a final concentration of 100 nM using the Lipofectamine 2000 Transfection Reagent (Life Technologies, Grand Island, NY, USA) in medium without serum and antibiotics for 24 h. The knockdown efficiency was confirmed by real-time PCR. Recombinant expression plasmids GV230-PPARγ-wild-type and GV230-PPARγ-mutantion were constructed by Shanghai Genechem Co.,Ltd.(Shanghai, China). Their transfection was made by using the Lipofectamine 2000 Transfection Reagent in medium without serum and antibiotics for 24 h.
Enzyme-linked immunosorbent assays (ELISA)
HSC-T6 or HSC-LX2 cells were treated with vehicle or ligustrazine at indicated concentrations for 24 h. The levels of VEGF-A and bFGF in the supernatants were determined using ELISA kits (Shanghai Meilian Biology Technology Co. Ltd., Shanghai, China) according to the protocol provided by the manufacture. Briefly, 100 μL of supernatants were added to each well of the 96-well plates coated with antibodies against VEGF-A or bFGF, followed by incubation for 2 h at room temperature. 100 μL of working detector solution was loaded into each well, and the plates were incubated for an additional 1 h at room temperature before the addition of 100 μL substrate solution. The reaction was stopped by adding 50 μL of stop solution. The absorbance was read at 450 nm wavelength. Six duplicate wells were set up for each group. Values were normalized to control.
Tubulogenesis assays
HSC-T6 cells were treated with vehicle or ligustrazine at indicated concentrations for 12 h, and then were grown in fresh serum-free medium for an additional 12 h. HSC conditioned medium were collected. Next, rat primary LSECs were seeded on Matrigel and incubated with control HSC conditioned medium, or ligustrazine-treated HSC conditioned medium, or control HSC conditioned medium plus neutralizing antibodies against VEGF-A or bFGF for 6 h. Tubulogenesis was visualized at random fields under a microscope (ZEISS Axio vert. A1, Germany). Tubulogenesis was assessed by counting the number of closed intercellular compartments (closed rings or pro-angiogenic structures) according to reported methods [16]. Representative views are shown.
Boyden chamber migration assays
HSC-T6 or HSC-LX2 cells were seeded to the upper wells of polycarbonate membrane transwell inserts (8 μm pore size; Corning, USA) with serum-free medium, and treated with vehicle or different reagents at indicated concentrations. The lower chambers were filled with complete medium. In certain experiments, rat primary HSCs were seeded to the upper wells without treatment. After 24 h incubation, the polycarbonated filter was removed and the migrated cells on the lower surface were stained with crystal violet. Three images at random fields were blindly taken from each well. The number of migrated cells per field was carefully counted by naked-eye observation. The assays were performed in triplicate and representative views are shown.
Cell adhesion assays
24-well plates were coated with 200 μL fibronectin (10 μg/mL) (Aladdin Bio-Chem Technology, Shanghai, China) for each well and dried at room temperature for 2 h. HSC-T6 or HSC-LX2 cells were seeded and treated with vehicle or different reagents at indicated concentrations for 24 h. In certain experiments, rat primary HSCs were seeded into 24-well plates without treatment. Then, plates were washed three times with phosphate buffered saline (PBS) to remove the unattached cells. Adherent cells were stained with 5 μg/mL fluorescein diacetate (FDA)/PBS solution for 10 min at room temperature, and then washed with PBS two times. Three images at random fields were blindly taken from each well under a fluorescence microscope (ZEISS Axio vert. A1, Germany). The number of adherent cells per field was carefully counted by naked-eye observation. The assays were performed in triplicate and representative views are shown.
In parallel, 96-well plates were coated with 30 μL fibronectin (10 μg/mL) (Aladdin Bio-Chem Technology, Shanghai, China) for each well and dried at room temperature for 2 h. HSC-T6 or HSC-LX2 cells were seeded and treated with vehicle or ligustrazine at indicated concentrations for 24 h. Then, plates were washed three times with PBS to remove the unattached cells. Then, the medium of each well was replaced with 20 μL PBS containing 0.5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), and cells were incubated with MTT solution at 37°C for 4 h. Next, the crystals were dissolved with 200 μL dimethyl sulfoxide. The spectrophotometric absorbance at 490 nm was measured by a SPECTRAmax™ microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). Cell survival rates were calculated representing the number of adherent cells. Six duplicate wells were set up for each group.
Flow cytometry analyses of membrane distribution of adhesion molecules
Flow cytometry was used to determine the abundance of intercellular cell adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) on cell membrane. Briefly, HSC-T6 and HSC-LX2 cells were treated with vehicle or ligustrazine at indicated concentrations for 24 h. Cells were trypsinized and suspended in medium, and then incubated with Anti-ICAM-1 PerCP-eFluor® 710 or Anti-VCAM-1 Biotin (1:100 dilution; Affymetrix, Santa Clara, CA, USA) at 4°C for 24 h followed by incubation with FITC-labeled Goat Anti-Rabbit IgG (1:50 dilution; Wuhan Boster Biological Technology, Wuhan, China) for 2 h at room temperature. The fluorescence intensity was determined by flow cytometry (FACSCalibur; BD, Franklin Lakes, NJ, USA) for analysis. The experiments were performed in triplicate and representative histogram graphs are shown.
Flow cytometry analyses of intracellular Ca2+ levels
HSC-T6 and HSC-LX2 cells were treated with vehicle or ligustrazine at indicated concentrations for 24 h. Cell suspensions were incubated with the fluorescent radiometric calcium indicator fluo3-acetoxymethyl at 1 µg/mL (Sigma-Aldrich, Saint Louis, MO, USA) for 30 min at 37°C. After incubation, cells were washed twice in PBS containing bovine serum albumin (BSA, 5 mg/mL) and centrifuged at 4°C for 10 min, followed by analysis using flow cytometry (FACSCalibur; BD, Franklin Lakes, NJ, USA). The levels of intracellular Ca2+ were manifested by the fluorescence intensity of Fluo-3 for analysis. The experiments were performed in triplicate and representative histogram graphs are shown.
Cytoskeleton Staining
Cytoskeleton was visualized by F-actin staining using FITC-conjugated phalloidin (Beyotime Biotechnology, Haimen, China) or by α-Tubulin staining using Alexa Fluor 555-labled Tracker (Beyotime Biotechnology, Haimen, China). Briefly, HSC-T6 and HSC-LX2 cells were seeded on cover slips in 6-well plates, and treated with vehicle or different reagents at indicated concentrations for 24 h. Cells were fixed in PBS containing 3.7% paraformaldehyde for 10 min at room temperature, and permeabilized with 0.1% Triton X-100 in PBS for another 5 min. FITC-conjugated phalloidin or Alexa Fluor 555-labled Tracker was diluted in PBS with 0.1% Triton X-100 and 5% BSA and then added to each well. After incubation at room temperature for 60 min, the cells were washed three times with PBS containing 0.1% Triton X-100. Diamidino-phenyl-indole (DAPI) was used to stain the nuclei of cells. After incubation with DAPI for 5 min, the cells were washed three times with PBS and then visualized under a fluorescence microscope (ZEISS Axio vert. A1, Germany) at random fields. Representative views are shown.
Collagen gel contraction assays
A mixture solution containing 2 mg/mL of type I collagen (Corning, USA) was poured into 24-well plates and incubated for 30 min at 37°C to allow gelation. HSC-T6 and HSC-LX2 cells were layered on the collagen gel, and grown to 90% confluence. Then, cells were treated with vehicle or different reagents at indicated concentrations followed by detaching the gels from the walls. In certain experiments, rat primary HSCs were seeded without treatment. Photographs were taken after 24 h treatment to monitor the collagen gel change. The assays were performed in triplicate and representative views are shown. Percentages of original gel area in each well were quantified using Image J software.
Electrophoretic mobility shift assays (EMSA)
HSC-T6 and HSC-LX2 cells were seeded in 6-well plates and then treated with vehicle or ligustrazine at indicated concentrations for 24 h. Then the nuclear extracts were prepared using the NE-PER Nuclear Extraction Kit (Thermo Scientific, Waltham, MA, USA) according to the protocol. The extracted nuclear proteins were incubated with biotin-labeled PPARγ probe using the Lightshift Chemiluminescent EMSA Kit (Thermo Scientific) and subject to polyacrylamide gel electrophoresis as we previously described [17]. The probe was a double-stranded nucleotide oligomer of 5'-CAAAACTAGGTCAAAGGTCA-3' and 5'-TGACCTTTGACCTAGTTTTG-3'.
Co-immunoprecipitation
LX2 cells were treated with vehicle, ligustrazine, and/or GW9662 at indicated concentrations for 24 h. Cells were lysed at 4°C in RIPA buffer containing protease inhibitor. Cell lysates adjusted to 1 mg/mL protein were precleared by immunoprecipitation-grade antibodies against peroxisome proliferator-activated receptor γ (PPARγ) or silencing mediator of retinoid acid and thyroid hormone (SMRT). After gentle rocking at 4°C overnight, Protein A/G PLUS-Agarose (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was added to the lysate/antibody mixture, and incubated with gentle agitation at 4°C for 4 h. Then the immunoprecipitates were collected by centrifugation, washed three times with cell lysis buffer, then boiled for 5 min with the same volume of 2× loading buffer (62.5 mM Tris-HCl, pH 6.8, 2% w/v SDS, 10% glycerol, 50 mM DTT, and 0.01% w/v bromophenol blue). Proteins were resolved by 10% SDS-PAGE, and subject to Western blot analyses.
Molecular simulation studies
Molecular docking was performed using the GLIDE software (Schrödinger, LLC, New York, NY, USA). The X-ray crystal structure of PPARγ (PDB code: 5JI0) complexed with rosiglitazone was retrieved from the PDB database. The Protein Preparation Wizard in the GLIDE software was used to prepare the structure of PPARγ, which included assigning bond orders and water orientations, removing water molecules, adding hydrogens, and creating zero-order bonds to metals and disulfide bonds. The protein was then minimized using the OPLS3 force field with a default constraint of 0.30 Å root-mean-square deviation. A receptor grid was created before GLIDE docking. The grid box was limited to a size of 20 Å at the active site. Three-dimensional structure of ligustrazine was generated using LigPrep module from the Schrödinger with the default settings. For GLIDE docking, the Standard Precision mode was carried out and the parameters of scaling factor and partial charge cutoff were set at the default values 0.80 and 0.15, respectively. Top ten ranking conformations for ligustrazine were chosen in the output tab to set the output numbers. Images of the docking conformations were subsequently prepared using Pymol. Additionally, the method of FTsite was used to predict other possible binding sites of ligustrazine on the surface of the PPARγ protein using the default parameters. FTsite is freely available as a web-based server with high accuracy detection of ligand binding sites on unbound protein structures [18].
Site-directed mutagenesis
PCR-based site-directed mutagenesis was carried out to individually or simultaneously mutate the residues Ser289 and Ser342 to alanine in human PPARγ gene (NCBI accession: NM_005037). The PCR product was subcloned between the Xho I and Kpn I sites of the mammalian expression vector GV230 (element sequence: CMV-MCS-EGFP-SV40-Neomycin) provided by Shanghai Genechem Co., Ltd. (Shanghai, China). Mutagenic primers were listed in Table S1. PPARγ-mutant plasmids were generated using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) according to the manufacturer's instructions with suitable oligonucleotides. All mutations were confirmed on positive colonies by DNA sequencing.
Animal procedures and treatments
All experimental procedures were approved by the Institutional and Local Committee on the Care and Use of Animals of Nanjing University of Chinese Medicine, and all animals received humane care according to the National Institutes of Health (USA) guidelines. Male Sprague-Dawley rats (200-250 g body weight) were obtained from Shanghai Slac Laboratory Animal Co., Ltd. (Shanghai, China). A mixture of CCl4 (0.1 mL/100 g body weight) and olive oil [1:1 (v/v)] was used to induce liver fibrosis in rats. Forty-eight rats were randomly divided into six groups (n=8). Rats in group 1, as a negative control, were not administrated CCl4 but were intraperitoneally injected with olive oil every other day during weeks 1-8. Rats in groups 2-6 were intraperitoneally injected with CCl4 every other day during weeks 1-8, of which the group 2 was set as the model control. From the beginning of week 5, rats in groups 3-5 were orally administrated with ligustrazine suspended in sterile PBS at 50, 100, and 200 mg/kg/d, respectively, during weeks 5-8, and rats in group 6, as a positive control, were orally given colchicine tablets (Yifeng Pharmacy, Nanjing, China) at 0.5 mg/kg during weeks 5-8. Meanwhile, the rats in groups 1 and 2 were orally administrated with an equal volume of PBS for vehicle control during weeks 5-8. Notably, the doses of ligustrazine were determined by preliminary experiments, and the dose of colchicine was converted from the clinical dose. At the end of the experiments, primary HSCs were isolated from two rats in each group for examining pericyte functions in vitro, and the remaining six rats in each group were sacrificed after being anesthetized by intraperitoneal injection of pentobarbital (50 mg/kg). Rat livers were isolated for histopathological and molecular exanimations.
Liver histopathology and collagen examinations
Harvested liver tissues were fixed in 10% neutral buffered formalin and embedded in paraffin. Hematoxylin-eosin (HE) staining was used for pathological assessment according to standard methods. Masson staining and Sirius Red staining were used for collagen exanimation according to standard methods. Images were blindly taken at random fields under a microscope (ZEISS Axio vert. A1, Germany). Representative views are shown.
Immunohistochemistry
Liver tissue sections were incubated with primary antibodies against CD31, CD34 and vWF for immunohistochemical evaluation using standard methods. Images were blindly taken at random fields under a microscope (ZEISS Axio vert. A1, Germany). Representative views are shown.
Immunofluorescence analyses
For staining liver tissues, thin sections (5 μm) were blocked with 1% bovine serum albumin, and then incubated with primary antibodies overnight at 4°C. After three washes with PBS, sections on slides were incubated with fluorescence-conjugated secondary antibodies at room temperature for 2 h. Sections incubated with secondary antibodies alone were used as negative control. For staining with cells, HSC-T6 and HSC-LX2 cells were seeded in 6-well plates, and treated with vehicle or different reagents at indicated concentrations for 24 h. Cells were incubated with primary and secondary antibodies in succession, similar to the above described protocol. Finally, DAPI was used to stain the nuclei of cells. Images were blindly taken at random fields under a microscope (ZEISS Axio vert. A1, Germany). Representative views are shown.
Real-time PCR
Total RNA was prepared from HSCs or liver tissues using Trizol reagent (Sigma, Saint Louis, MO, USA). The integrity of total RNA was assessed using 1.5% agarose gel electrophoresis for detecting the 28S and 18S rRNA bands (2:1 ratio), and the A260/A280 value (1.9-2.0) was used to evaluate the purity of total RNA. Total RNA was subject to reverse transcription to cDNA using the TransScript All-in One First-Strand cDNA Synthesis SuperMix for qPCR (One-Step gDNA Removal) Kits provided by TransGen Biotech Co., Ltd. (Beijing, China) according to the protocols. The contaminating genomic DNA in RNA templates was eliminated using the gDNA Remover contained in the reverse transcription kits. Real-time PCR was performed using the SYBR Green Master Mix (Vazyme Biotech Co., Ltd., Nanjing China) according to the protocol. Fold changes in the mRNA levels of target genes related to the invariant control glyceraldehyde phosphate dehydrogenase (GAPDH) were calculated as suggested [19]. The primers designed to span gene introns (GenScript Co., Ltd., Nanjing, China) are listed in Table S2.
Western blot analyses
Whole cell protein extracts were prepared from HSCs or liver tissues with RIPA buffer. In certain experiments, nuclear proteins were separated using a Bioepitope Nuclear and Cytoplasmic Extraction Kit (Bioworld Technology Co., Ltd., Saint Louis Park, MN, USA) according to the protocol. Protein detection and band visualization were performed as we previously described [11]. β-Actin was used as an invariant control for equal loading of total proteins, and Lamin B1 was for nuclear proteins. Representative blots are shown.
Statistical analyses
Data are presented as mean ± SD and were analyzed using GraphPad Prism 5 (San Diego, CA, USA). The significance of difference was determined by one-way ANOVA with the post-hoc Dunnett's test for in vitro experiments and Holm-Sidak test for in vivo experiments. Values of P<0.05 were considered to be statistically significant.
Results
Ligustrazine inhibits production of pro-angiogenic cytokines in HSCs
We initially examined two key pro-angiogenic cytokines in HSC-T6 and HSC-LX2 cells treated with ligustrazine. Ligustrazine concentration-dependently decreased the mRNA and protein levels of VEGF-A and bFGF in HSCs (Figure 1A-B), and reduced the supernatant levels of VEGF-A and bFGF (Figure 1C). To determine how these pro-angiogenic cytokines affected LSEC-mediated angiogenesis, we evaluated the effects of HSC-T6 conditioned medium on the tube formation capacity of rat LSECs. The results showed that LSEC tubulogenesis on Matrigel was remarkably suppressed by incubation with the conditioned medium from ligustrazine-treated HSCs, and was attenuated by incubation with the HSC medium containing neutralizing antibodies against VEGF-A or bFGF (Figure 1D). Together, these data indicated that ligustrazine reduced the production of pro-angiogenic cytokines in HSCs, leading to suppression of LSEC-driven angiogenesis.
Ligustrazine inhibits production of pro-angiogenic cytokines in HSCs. (A) Real-time PCR analyses of mRNA expression of pro-angiogenic cytokines in HSCs treated with ligustrazine for 24 h. Significance: *P<0.05 versus control, **P<0.01 versus control. (B) Western blot analyses of protein expression of pro-angiogenic cytokines in HSCs treated with ligustrazine for 24 h. (C) ELISA for supernatant levels of pro-angiogenic cytokines in HSCs treated with ligustrazine for 24 h. Significance: *P<0.05 versus control, **P<0.01 versus control. (D) Primary rat LSECs were seeded on Matrigel and incubated with control HSC-T6 conditioned medium, or ligustrazine (20 μM)-treated HSC-T6 conditioned medium, or control HSC-T6 conditioned medium plus neutralizing antibodies (Ab) against VEGF-A or bFGF for 6 h. LSEC-mediated tube formation was evaluated by counting the number of closed intercellular compartments per field (100× magnification, scale bar: 100 μm). Significance: *P<0.05 versus control, #P<0.05 versus ligustrazine.
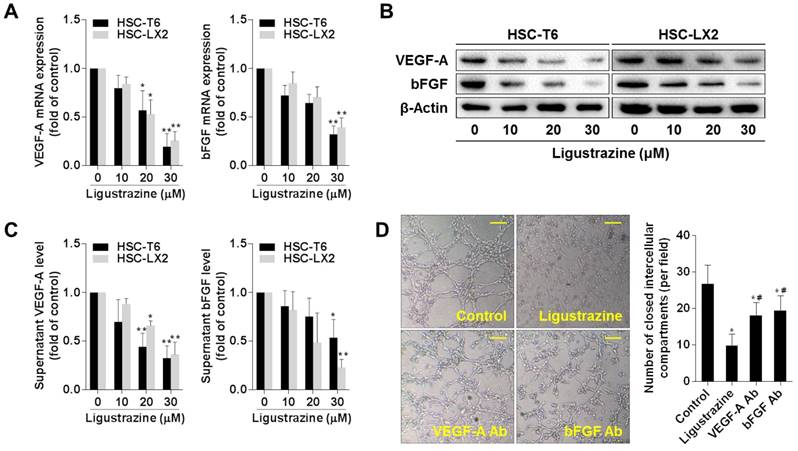
Ligustrazine suppresses HSC migration and adhesion
Enhanced migration and adhesion of HSCs potentiate these cells to more effectively embrace the endothelial cell layer of the sinusoids with thorn-like microprojections, which is essential for vascular tube maturation and integrity [20]. Boyden chamber assays showed that ligustrazine concentration-dependently inhibited the migration of HSCs (Figure 2A). The adhesion capacity of HSCs was also restricted by ligustrazine, as evidenced by the FDA morphology staining (Figure 2B) and MTT quantitative assay (Figure 2C). Furthermore, ligustrazine downregulated the expression of ICAM-1 and VCAM-1 in HSCs, two important molecules regulating cell adhesion (Figure 2D). Consistently, flow cytometry analyses demonstrated that the membrane abundance of ICAM-1 and VCAM-1 was also decreased by ligustrazine in HSCs (Figure 2E, Figure S1). Collectively, these data suggested that ligustrazine suppressed HSC migration and adhesion.
Ligustrazine suppresses HSC migration and adhesion. (A) Boyden chamber assay for evaluating migration of HSCs treated with ligustrazine for 24 h (100× magnification, scale bar: 20 μm). The number of migrated cells per field was counted. Significance: **P<0.01 versus control. (B) FDA staining for evaluating adhesion of HSCs treated with ligustrazine for 24 h (100× magnification, scale bar: 20 μm). The number of adherent cells per field was counted. Significance: *P<0.05 versus control, **P< 0.01 versus control. (C) MTT assay for evaluating adhesion of HSCs treated with ligustrazine for 24 h. Significance: *P<0.05 versus control, **P<0.01 versus control. (D) Western blot analyses of protein expression of adhesion molecules in HSCs treated with ligustrazine for 24 h. (E) Flow cytometry analyses of membrane distribution of adhesion molecules determined by fluorescence intensity in HSCs treated with ligustrazine for 24 h. Significance: *P<0.05 versus control, **P<0.01 versus control.
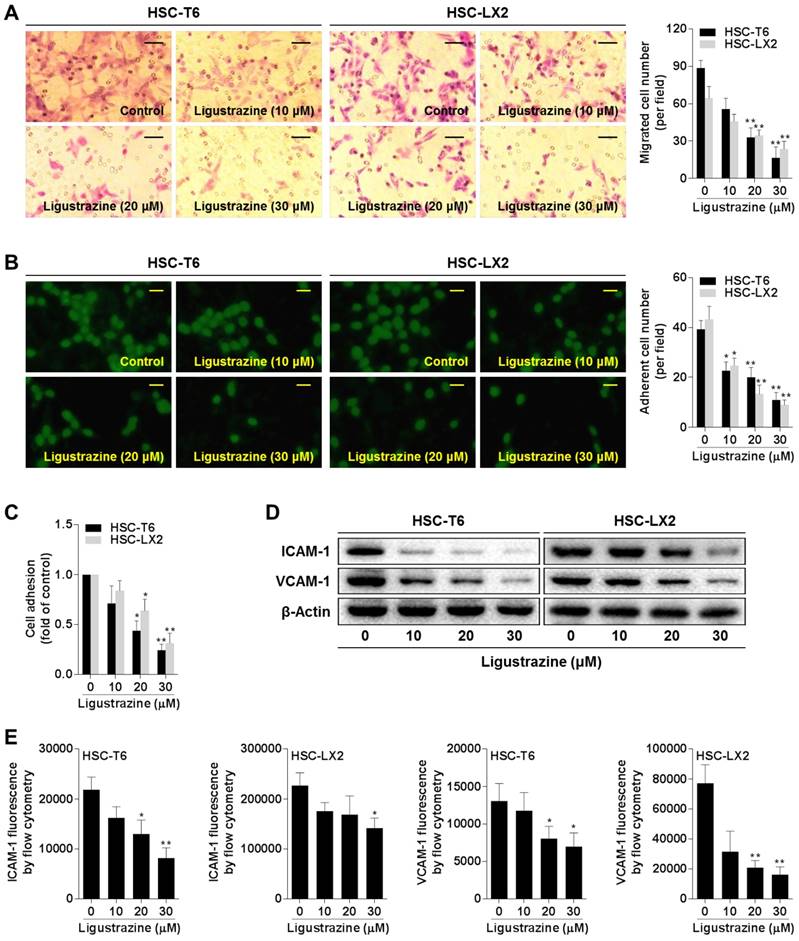
Ligustrazine restricts HSC contraction
The anatomical location of HSCs and their capacity to contract or relax in response to various vasoactive mediators indicate HSCs as principal cells regulating sinusoidal blood flow and portal hypertension [6]. Cytoskeletons provide valuable information on the organization of cell contractile machinery [21]. We used FITC-phalloidin to stain the microfilaments represented by F-actin, and Alexa Fluor 555-labled tubulin tracker to stain the microtubules represented by α-tubulin, respectively, in HSCs. Ligustrazine at 20 μM remarkably reduced the formation of actin stress fibers and inhibited the assembly of tubulin, disturbing the dense bundles of stress fibers aligning along the major axis of the cell (Figure 3A-B). Collagen gel contraction assays provided clearer evidence that ligustrazine repressed the contractibility of HSCs (Figure 3C). Given that myosin light chain 2 (MLC2) is a central mediator of cell contraction [22], we examined this molecule and found that ligustrazine decreased the phosphorylation of MLC2 in HSCs (Figure 3D). Consistent results were recaptured by immunofluorescence analyses (Figure 3E). Moreover, ligustrazine decreased the intracellular Ca2+ levels in HSCs, as demonstrated by flow cytometry analyses (Figure 3F, Figure S2). Taken together, these discoveries revealed that ligustrazine inhibited HSC contraction.
Ligustrazine restricts HSC contraction. (A) F-actin staining using FITC-conjugated phalloidin for evaluating contraction of HSCs treated with ligustrazine for 24 h (400× magnification, scale bar: 10 μm). (B) α-Tubulin staining using Alexa Fluor 555-labled Tracker for evaluating contraction of HSCs treated with ligustrazine for 24 h (400× magnification, scale bar: 10 μm). (C) Collagen gel assays for evaluating contraction of HSCs treated with ligustrazine for 24 h. Percentages of original gel area were quantified. Significance: *P<0.05 versus control. (D) Western blot analyses of phospho-MLC2 in HSCs treated with ligustrazine for 24 h. (E) Immunofluorescence analyses of phospho-MLC2 in HSCs treated with ligustrazine for 24 h (400× magnification, scale bar: 10 μm). (F) Flow cytometry analyses of intracellular Ca2+ levels determined by fluorescence intensity in HSCs treated with ligustrazine for 24 h. Significance: *P<0.05 versus control, **P<0.01 versus control.
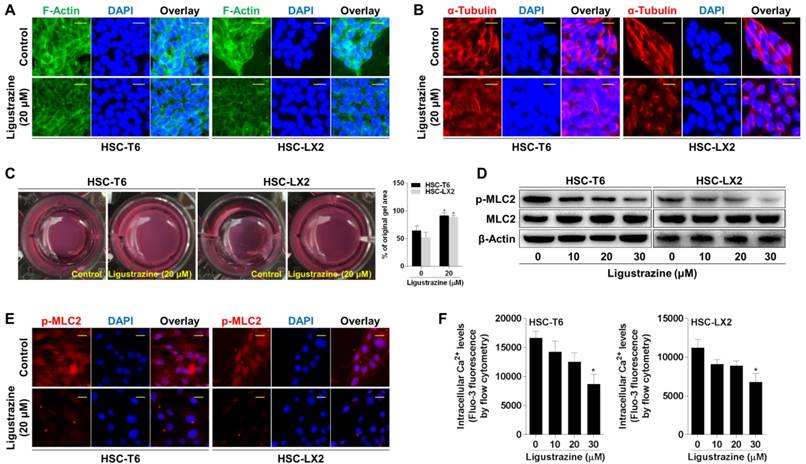
Activation of PPARγ is a prerequisite for ligustrazine suppression of HSC pericyte functions
Given that PPARγ is highlighted as a target molecule controlling the pro-fibrogenic biology of HSCs [23, 24], we explored the role of this nuclear receptor in ligustrazine suppression of HSC pericyte functions. Western blot data demonstrated that ligustrazine increased the nuclear abundance of PPARγ in HSCs (Figure 4A). Immunofluorescence analyses gave consistent results showing ligustrazine-driven nuclear distribution of PPARγ within HSCs (Figure 4B). EMSA results revealed that ligustrazine facilitated the binding of PPARγ to DNA sequences (Figure 4C). Additionally, ligustrazine upregulated the mRNA levels of fatty acid transporter (FATP), a well-established target gene of PPARγ [25] (Figure 4D). These discoveries collectively suggested ligustrazine activation of PPARγ in HSCs. Subsequently, we used PPARγ selective inhibitor GW9662 [26] to test the involvement of PPARγ in the current experimental context. GW9662 rescued ligustrazine inhibition of pro-angiogenic cytokine expression in HSCs (Figure 4E). GW9662 also abolished ligustrazine suppression of HSC migration and adhesion (Figure 4F-G). Furthermore, ligustrazine inhibition of HSC contraction was also abrogated by GW9662 (Figure 4H). Altogether, these findings indicated a PPARγ activation-dependent mechanism underlying ligustrazine suppression of HSC pericyte functions.
Activation of PPARγ is a prerequisite for ligustrazine suppression of HSC pericyte functions. (A) Western blot analyses of PPARγ abundance in nuclear lysates in HSCs treated with ligustrazine for 24 h. (B) Immunofluorescence analyses of PPARγ nuclear translocation in HSCs treated with ligustrazine for 24 h (400× magnification, scale bar: 10 μm). (C) EMSA for examining PPARγ binding to DNA sequences in HSCs treated with ligustrazine for 24 h. (D) Real-time PCR analyses of mRNA expression of PPARγ target gene FATP in HSCs treated with ligustrazine for 24 h. Significance: *P<0.05 versus control. (E) Western blot analyses of protein expression of pro-angiogenic cytokines in HSCs treated with ligustrazine at 20 μM and/or GW9662 at 1 μM for 24 h. (F) Boyden chamber assay for evaluating migration of HSCs treated with ligustrazine at 20 μM and/or GW9662 at 1 μM for 24 h (100× magnification, scale bar: 20 μm). The number of migrated cells per field was counted. Significance: *P<0.05 versus control, #P<0.05 versus ligustrazine. (G) FDA staining for evaluating adhesion of HSCs treated with ligustrazine at 20 μM and/or GW9662 at 1 μM for 24 h (100× magnification, scale bar: 20 μm). The number of adherent cells per field was counted. Significance: *P<0.05 versus control, #P<0.05 versus ligustrazine. (H) Collagen gel assays for evaluating contraction of HSCs treated with ligustrazine at 20 μM and/or GW9662 at 1 μM for 24 h. Percentages of original gel area were quantified. Significance: *P<0.05 versus control, #P<0.05 versus ligustrazine. In some panels of this figure, ligustrazine is abbreviated as ligu.
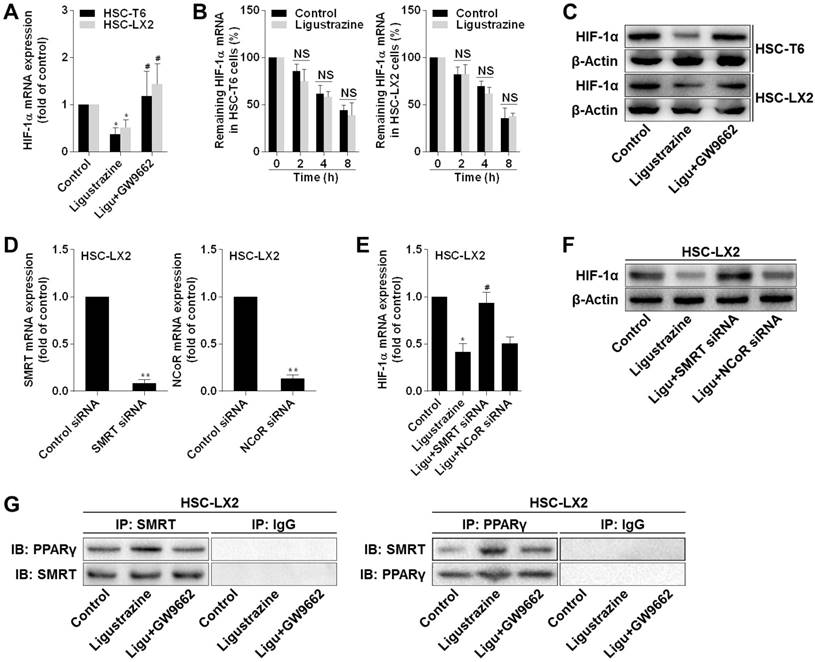
Activation of PPARγ by ligustrazine represses transcription of hypoxia inducible factor-1α (HIF-1α) in a SMRT-dependent manner in HSCs
We investigated the underlying molecular mechanisms focusing on HIF-1α because this molecule plays a pivotal role in both physiological and pathological angiogenesis [27]. Pharmacological inhibition of HIF-1α by PX-478 decreased pro-angiogenic cytokine expression, inhibited the migration and adhesion capacities, and limited the contractile machinery in HSCs (Figure S3A-D), demonstrating a critical role of HIF-1α in regulation of HSC pericyte functions. Subsequently, we observed that ligustrazine decreased the mRNA expression of HIF-1α in HSCs, but this effect was abolished by GW9662 (Figure 5A). We then measured the stability of HIF-1α mRNA in HSCs in the presence of transcription inhibitor actinomycin D. Real-time PCR analyses showed that no significant difference in HIF-1α mRNA levels was found between control HSCs and ligustrazine-treated HSCs at each time point (Figure 5B). Additionally, HIF-1α protein abundance was downregulated by ligustrazine, but was rescued by GW9662 (Figure 5C). These data collectively indicated that ligustrazine transcriptionally inhibited HIF-1α expression dependent on activation of PPARγ, leading to suppression of HSC pericyte functions. Next, we explored the linking molecule involved in PPARγ transrepression of HIF-1α. It has been delineated that PPARγ could recruit the co-repressors NCoR (nuclear receptor co-repressor protein) or SMRT to repress gene transcription [28, 29]. Interestingly, we observed that transfection with SMRT siRNA, but not NCoR siRNA, significantly rescued ligustrazine downregulation of HIF-1α at both mRNA and protein levels in HSC-LX2 cells (Figure 5D-F), suggesting the involvement of SMRT in the current context. Indeed, co-immunoprecipitation results showed that ligustrazine increased the physical interaction between PPARγ and SMRT in HSC-LX2 cells, but GW9662 abrogated ligustrazine enhancement of PPARγ binding to SMRT (Figure 5G). Altogether, these discoveries indicated that activation of PPARγ by ligustrazine resulted in transrepression of HIF-1α in a SMRT-dependent mechanism in HSCs.
Activation of PPARγ by ligustrazine represses transcription of HIF-1α in a SMRT-dependent manner in HSCs. (A) Real-time PCR analyses of mRNA expression of HIF-1α in HSCs treated with ligustrazine at 20 μM and/or GW9662 at 1 μM for 24 h. Significance: *P<0.05 versus control, #P<0.05 versus ligustrazine. (B) Real-time PCR analyses of decay of HIF-1α mRNA at different time points in HSCs treated with ligustrazine at 20 μM in the presence of transcription inhibitor actinomycin D (5 μg/mL). NS represents non-significant differences. (C) Western blot analyses of protein expression of HIF-1α in HSCs treated with ligustrazine at 20 μM and/or GW9662 at 1 μM for 24 h. (D) Real-time PCR analyses of mRNA expression of SMRT and NCoR in HSC-LX2 cells transfected with their corresponding siRNA for 24 h. Significance: **P<0.01 versus control siRNA. (E) Real-time PCR analyses of mRNA expression of HIF-1α in HSC-LX2 cells treated with ligustrazine at 20 μM and/or transfected with SMRT siRNA or NCoR siRNA for 24 h. Significance: *P<0.05 versus control, #P<0.05 versus ligustrazine. (F) Western blot analyses of protein expression of HIF-1α in HSC-LX2 cells treated with ligustrazine at 20 μM and/or transfected with SMRT siRNA or NCoR siRNA for 24 h. (G) Co-Immunoprecipitation for examining physical interactions between PPARγ and SMRT in HSC-LX2 cells treated with ligustrazine at 20 μM and/or transfected with SMRT siRNA for 24 h. In some panels of this figure, ligustrazine is abbreviated as ligu.
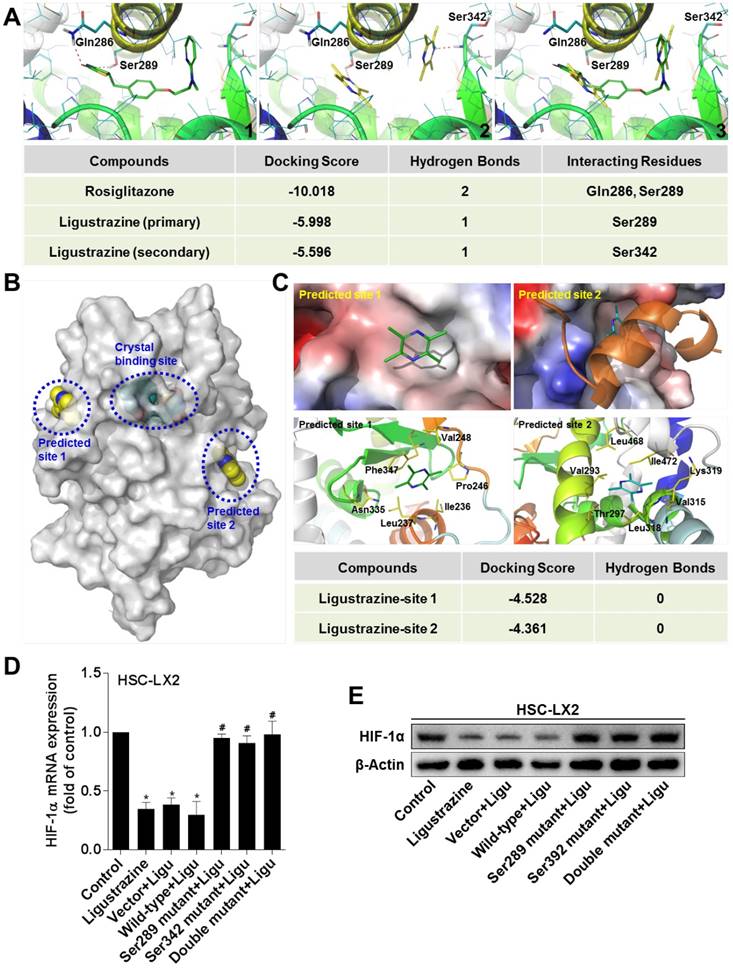
Ligustrazine binds to the ligand-binding domain of PPARγ in a unique double-molecule manner via direct interactions with the residues Ser289 and Ser342, which is necessary for transrepression of HIF-1α in HSCs
The experimental data above raised a possibility that PPARγ could be a direct target molecule of ligustrazine in HSCs. We used molecular simulation approaches to test this hypothesis. The docking results showed that ligustrazine could be embedded, in a double-molecule manner, into the canonical ligand-binding cavity of PPARγ, which is exactly the binding pocket of thiazolidinedione agonists represented by rosiglitazone. Interestingly, ligustrazine, unlike rosiglitazone, used two molecules to form hydrogen bonds with the residues Ser289 and Ser342, respectively, in the crystal binding cavity of PPARγ, whereas rosiglitazone physically interacted with the residues Ser289 and Gln286 (Figure 6A), suggesting that ligustrazine and rosiglitazone might have different binding modes to activate the receptor even though they shared the same binding cavity. Additionally, we explored other possible binding sites for ligustrazine in PPARγ, and two predicted sites on the surface of the protein were obtained (Figure 6B). However, the docking results revealed that ligustrazine could not form any hydrogen bond with the residues in the two predicted sites (Figure 6C). This reinforced the notion that the canonical thiazolidinedione binding cavity was responsible for ligustrazine activation of PPARγ. Subsequently, we used site-directed mutagenesis strategies to confirm whether interaction with the resides Ser289 and/or Ser342 was necessary for ligustrazine effects. We observed that ligustrazine suppression of HIF-1α mRNA and protein expression was significantly abrogated in HSC-LX2 cells transfected with PPARγ plasmids bearing Ser289 and/or Ser342 mutation to alanine (Figure 6D-E). Taken together, these discoveries suggested a unique ligand-binding pattern for ligustrazine activation of PPARγ leading to the currently observed biological consequences.
Ligustrazine binds to PPARγ in a unique double-molecule manner via direct interactions with the residues Ser289 and Ser342, which is necessary for transrepression of HIF-1α in HSCs. (A) Interaction modes of rosiglitazone (1) and ligustrazine (2) with the binding pocket of PPARγ. The superposition of binding conformations of rosiglitazone and ligustrazine is shown in (3). The red dashed lines indicate hydrogen bonds with the residues in the binding cavity of PPARγ. (B) Use of FTsite predicted two possible binding sites on the surface of PPARγ, in addition to the canonical crystal binding site. (C) Docking ligustrazine into the predicted site 1 and site 2. No hydrogen bond was formed with the residues in the two predicted sites. (D) HSC-LX2 cells were transfected with recombinant plasmids GV230-PPARγ-wild-type or GV230-PPARγ-mutantion at Ser289 and/or Ser342 for 24 h followed by treatment with ligustrazine at 20 μM for 24 h. Real-time PCR analyses of mRNA expression of HIF-1α. Significance: *P<0.05 versus control, #P<0.05 versus vector+ligustrazine. (E) HSC-LX2 cells were transfected with recombinant plasmids GV230-PPARγ-wild-type or GV230-PPARγ-mutantion at Ser289 and/or Ser342 for 24 h followed by treatment with ligustrazine at 20 μM for 24 h. Western blot analyses of protein expression of HIF-1α. In some panels of this figure, ligustrazine is abbreviated as ligu.
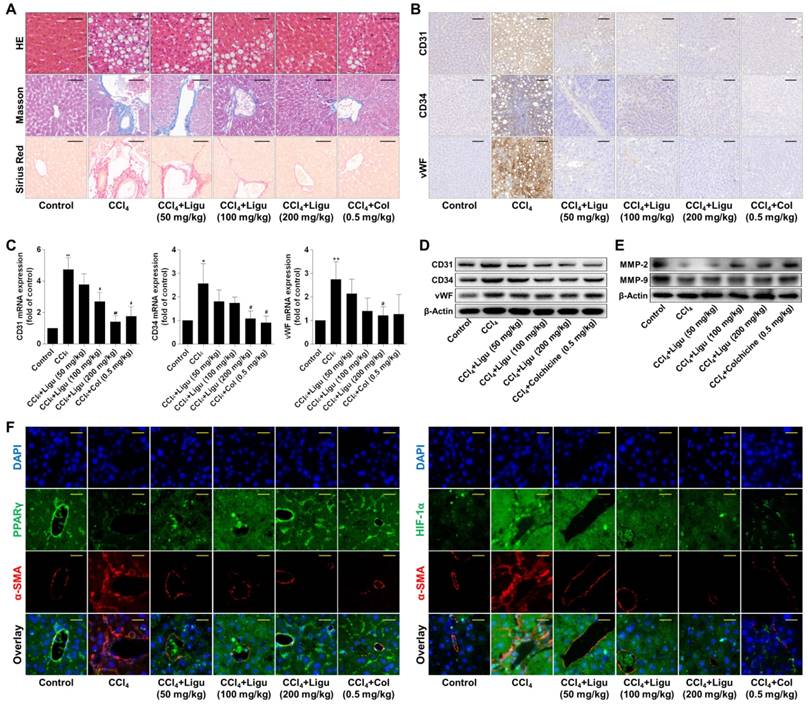
Ligustrazine ameliorates liver fibrotic injury and vascular remodeling associated with suppression of HSC pericyte functions in rats intoxicated with CCl4
Evidence in vivo showed that hepatic lobules maintained normal structure with less collagen deposition in the liver of vehicle control rats; however, significant hepatocyte necrosis and fibrous septa accompanied by massive collagen accumulation were observed in the liver of CCl4-intoxicated rats (Figure 7A). After treatment with ligustrazine at various doses or colchicine, hepatic pathological alterations were improved and collagen contents were reduced to different extents (Figure 7A). In addition, endothelial markers CD31, CD34 and vWF were remarkably upregulated in fibrotic liver compared to the vehicle control, but their expression was decreased by ligustrazine or colchicine (Figure 7B). Consistently, the elevated mRNA and protein levels of CD31, CD34 and vWF in rat fibrotic liver were decreased by ligustrazine or colchicine (Figure 7C-D). Moreover, a hallmark of hepatic pathological vascular remodeling is the formation of vessel basement membrane, which can be degraded by matrix metalloproteinases (MMPs) [30]. Here, the expression of MMP-2 and MMP-9 was downregulated in the liver of model rats compared to the vehicle control, but their expression was restored by ligustrazine and colchicine (Figure 7E). Furthermore, immunofluorescence analyses of PPARγ and HIF-1α with α-smooth muscle actin (α-SMA, a well-known HSC marker) demonstrated that ligustrazine upregulated PPARγ expression and decreased HIF-1α expression in rat fibrotic liver (Figure 7F). On the other hand, primary HSCs were isolated for examining pericyte functions. Compared to the vehicle control, HSCs from fibrotic model rats showed significantly increased production of pro-angiogenic cytokines, and enhanced capabilities of migration, adhesion and contraction; however, these pericyte functions were remarkably repressed in primary HSCs isolated from ligustrazine-treated rats (Figure S4A-D), which was consistent with the results in the culture system. Altogether, these data suggested that ligustrazine improved liver fibrosis and vascular remodeling in vivo and confirmed that suppression of HSC pericyte functions contributed to ligustrazine effects.
Ligustrazine ameliorates liver fibrotic injury and vascular remodeling in rats intoxicated with CCl4. (A) Liver sections were stained with HE, Masson reagents, and Sirius Red reagents for histological and collagen examinations (200× magnification, scale bar: 100 μm). (B) Immunohistochemical analyses of endothelial markers in liver tissues (200× magnification, scale bar: 100 μm). (C) Real-time PCR analyses of mRNA expression of endothelial markers in liver tissues. Significance (post-hoc Holm-Sidak test): *P<0.05 versus control, **P<0.01 versus control, #P<0.05 versus CCl4, ##P<0.01 versus CCl4. (D) Western blot analyses of protein expression of endothelial markers in liver tissues. (E) Western blot analyses of protein expression of MMP-2 and MMP-9 in liver tissues. (F) Immunofluorescence analyses of PPARγ and HIF-1α in liver tissues (400× magnification, scale bar: 50 μm). Staining with α-SMA was used to indicate HSCs. In this figure, ligustrazine is abbreviated as ligu, and colchicine is abbreviated as col.
Discussion
Ligustrazine inhibited multiple facets of HSC pericyte functions. These observations provoked us to investigate the molecular mechanisms and target for ligustrazine effects. Nuclear receptors are ligand-activated transcription factors that act as sensors for a broad range of natural and synthetic ligands. Targeting nuclear receptors offers exciting new perspectives for treatment of liver diseases [31]. In the current investigation, ligustrazine activated PPARγ by stimulating its nuclear translocation in HSCs. Ligustrazine activation of PPARγ was similarly observed in RAW264.7 cells for reducing lipid accumulation [32]. We then used the compound GW9662 to test the role of PPARγ in the current setting. GW9662, at relatively low concentrations, functions as a potent PPARγ antagonist with high selectivity over PPARα and PPARβ/δ by covalently modifying a cysteine residue in the ligand binding site of the receptor, and thus it is a useful tool for elucidating the role of PPARγ in biological processes [26]. For example, GW9662 at concentrations lower than 10 μM has been utilized to characterize PPARγ as a potential target of curcumin in several types of cells [33-36]. In the current study, we treated HSCs with GW9662 at 1 μM, at which it could selectively target PPARγ. We observed that GW9662 potently abolished ligustrazine suppression of HSC pericyte functions, suggesting that activation of PPARγ was required for ligustrazine actions.
Our further investigations highlighted HIF-1α as a linking molecule in ligustrazine effects. Pharmacological blockade of HIF-1α significantly repressed pro-angiogenic cytokine expression, migration, adhesion and contraction in HSCs, demonstrating a critical role for HIF-1α in controlling HSC pericyte functions. These results were consistent with our previous data that HIF-1α was involved in hedgehog regulation of HSC-mediated angiogenesis in hepatic fibrosis [37]. Although HIF-1α may undergo ubiquitin/acetylation-mediated proteasomal degradation under normoxia, its transcription and translation can be regulated by signaling pathways in an oxygen-independent manner [38]. This notion impelled us to ask whether and how HIF-1α was controlled by ligustrazine-activated PPARγ in HSCs. We found that ligustrazine suppressed the transcription of HIF-1α, which was considerably diminished by GW9662, suggesting transrepression of HIF-1α by ligustrazine-activated PPARγ. It is known that certain co-repressors such as NCoR and SMRT mediate PPARγ transrepression of target genes, which is commonly ligand-independent [24, 39]. Our loss-of-function analyses reveled that SMRT was involved in ligustrazine inhibition of HIF-1α in HSCs. We further observed that ligustrazine promoted PPARγ to physically interact with SMRT, which was abrogated by GW9662, suggesting a ligand-dependent manner for PPARγ physical interaction with SMRT in HSCs. These results indicated a novel role for SMRT in transrepression of HIF-1α by ligustrazine-activated PPARγ, and extended current molecular understanding of nuclear receptor-mediated transrepression of target genes.
Based on the experimental observations, we speculated that activation of PPARγ and the pharmacological consequences could be due to physical interaction with the receptor by ligustrazine. This speculation was supported by molecular docking evidence showing that ligustrazine could bind to the canonical ligand-binding pocket of PPARγ. Interestingly, two novel characteristics distinct from that of PPARγ full agonist rosiglitazone were observed in the current setting: i) a double-molecule binding mode; ii) formation of hydrogen bonds with the residue Ser342, in addition to Ser289. The first difference could be explained by the relatively small size of the ligustrazine molecule: the two molecules could simultaneously embed into the active cavity and constitute a confirmation for receptor activation in concert. This represented a new binding pattern for ligand activation of PPARγ, challenging the prevalent view of one-on-one ligand-receptor interactions with respect to their bound conformations. On the other hand, the recognition that PPARγ may adopt multiple conformations to accommodate the changes of ligand conformations [40] could account for the different binding residues involved in ligustrazine interaction with PPARγ. Importantly, mutagenesis assays provided clear evidence that interaction with both the residues were required for ligustrazine effects. Given that rosiglitazone also interacted with Ser289, we speculated that interaction with Ser289 was conserved for ligand activation of PPARγ, but interaction with Ser342 could be uniquely essential for transrepression of HIF-1α by ligustrazine-activated PPARγ in HSCs. These results also confirmed the notion that binding patterns of agonists of PPARγ affect the recruitment of co-activators and co-repressors, playing an important role in modulating target gene expression [41]. Furthermore, molecular simulation evidence could also explain why GW9662 antagonized ligustrazine effects. It has been delineated that GW9662 covalently reacted with the Cys285 residue in the PPARγ ligand-binding domain, which is closely adjacent to the Ser289 residue [42]. Accordingly, we speculated that GW9662 could competitively interrupt the formation of hydrogen bonds between the pyrazine nitrogen of ligustrazine and the Ser289 residue in the active cavity of PPARγ, resulting in the loss of ligustrazine suppression of HSC pericyte functions. Altogether, these results indicated ligustrazine as a full agonist of PPARγ and a valuable chemical tool, implicating novel molecular and structural mechanisms for ligand-dependent transrepression by PPARγ.
Our current data also indicated a negative regulation of HIF-1α by PPARγ. Although this phenomenon has been described in some other pathological circumstances such as pulmonary hypertension [43], inconsistent results should be noted. For example, joint upregulation of PPARγ and HIF-1α was observed in primary neonatal mouse cardiomyocytes [44] and hepatoma HepG2 cells [45], and PPARγ was characterized as a downstream target gene of HIF-1α in the two studies. These observations, combined with our current discoveries, raised a possibility that there could be a negative feedback loop controlling the two molecules, that is, HIF-1α may induce PPARγ expression as a response of cells to hypoxia or pathological stress, then the overexpressed or activated PPARγ, in turn, represses HIF-1α transcription to limit amplification of the response. Which arm of the regulatory loop dominates the cellular responses could be dependent on pathological conditions or external stimuli. In the current study, we conceived that the negative feedback loop could be implemented by ligustrazine activation of PPARγ, resulting in suppression of HIF-1α expression in HSCs.
We subsequently used a classical CCl4-induced liver fibrosis model in rats to establish the in vivo correlation. In this model, olive oil was used as a solvent for CCl4 injection solution because it is a commonly available oily solvent with better quality than other types of edible oils. Although dietary intake of CCl4 could make the model closer to the clinical situation, we here adopted intraperitoneal injection of CCl4 given that it is a quantitative and well-controlled manner guaranteeing homogeneity of disease progression, and that hepatic responses to chronic CCl4 insult in rats have been shown to be superficially similar to human cirrhosis [46]. Moreover, our experimental procedures represented a therapeutic strategy by establishing liver fibrosis prior to treatment with ligustrazine. Noteworthy, injection with CCl4 was not halted during the ligustrazine treatment period because much evidence has indicated that spontaneous reversal of liver fibrotic injury could occur once the toxicant is removed [47]. In that case, it would be difficult to accurately evaluate the anti-fibrotic effects of ligustrazine. Furthermore, colchicine was used as a positive control drug because both basic and clinical studies have demonstrated that colchicine has therapeutic effects on liver fibrosis through reducing collagen synthesis and deposition [48-50]. Colchicine does not target PPARγ, but interestingly, it also improved hepatic vascular architecture in current investigations. We inferred that inhibition of collagen production could contribute to the correction of the vascular microenvironment in fibrotic liver. These findings also suggested a potential link between collagen dynamics and HSC pericyte functions and related sinusoidal remodeling, which has therapeutic implications for liver fibrosis.
Although the expression of PPARγ was decreased in activated HSCs in vitro and in vivo, this nuclear receptor has been recognized as a key negative regulator of HSC transdifferentiation [23, 24]. Currently, little is known about the role of PPARγ in HSC pericyte biology and its therapeutic implications. We here demonstrated clearly that ligustrazine activation of PPARγ led to suppression of HSC pericyte functions and attenuation of sinusoidal vascular remodeling. For suppression of HSC pericyte functions and inhibition of HSC transdifferentiation, it would be difficult to judge which effect was dominant for the overall antifibrotic effects of ligustrazine, despite the notion that HSC activation is a pivotal event during liver fibrosis. We speculated that both the effects could work in concert to produce the potent antifibrotic efficacy in vivo. Furthermore, although the nuclear transcriptional function of PPARγ facilitated by ligustrazine resulted in transrepression of HIF-1α and suppression of HSC pericyte functions, the potential role of ligustrazine regulation of PPARγ expression in this context could not be excluded, especially given that ligustrazine upregulation of PPARγ expression was delineated in HSCs [51] and some other pathological conditions [52, 53]. Increased protein abundance of PPARγ may facilitate its nuclear transcriptional activity. We thus speculated that ligustrazine suppression of HSC pericyte functions could also be partially due to upregulation of PPARγ expression. For this reason, siRNA-mediated knockdown of PPARγ expression could validate this speculation and come to similar results as current discoveries. These investigations would help to more exactly elucidate the role of PPARγ in HSC pericyte biology.
In conclusion, ligustrazine activation of PPARγ repressed HIF-1α transcription via a SMRT-dependent mechanism, leading to suppression of HSC pericyte functions and reduction of vascular remodeling in liver fibrosis. These pharmacological consequences could be due to a unique binding mode for ligustrazine interaction with PPARγ. We discovered novel mechanisms for PPARγ regulation of HSC pathophysiology and added valuable insights into structure-based design of PPARγ-based anti-fibrotic agents.
Abbreviations
α-SMA: α-smooth muscle actin; bFGF: basic fibroblast growth factor; BSA: bovine serum albumin; DAPI: diamidino-phenyl-indole; ECM: extracellular matrix; ELISA: enzyme-linked immunosorbent assays; EMSA: electrophoretic mobility shift assays; FATP: fatty acid transporter; FDA: fluorescein diacetate; HE: hematoxylin-eosin; HIF-1α: hypoxia inducible factor-1α; HSCs: hepatic stellate cells; ICAM-1: intercellular cell adhesion molecule-1; LSECs: liver sinusoidal endothelial cells; MMPs: matrix metalloproteinases; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; NCoR: nuclear receptor co-repressor protein; PBS: phosphate buffered saline; PPARγ: peroxisome proliferator-activated receptor γ; SMRT: silencing mediator of retinoid acid and thyroid hormone; VCAM-1: vascular cell adhesion molecule-1; VEGF-A: vascular endothelial growth factor-A.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31401210, 31571455, 31600653, and 81600483), the Natural Science Foundation of Jiangsu Province (BK20140955), the Open Project Program of Jiangsu Key Laboratory for Pharmacology and Safety Evaluation of Chinese Materia Medica (JKLPSE201601 and JKLPSE201502), the Project of the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Qing Lan Project of Jiangsu Province.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Greuter T, Shah VH. Hepatic sinusoids in liver injury, inflammation, and fibrosis: new pathophysiological insights. J Gastroenterol. 2016;51:511-9
2. Elpek GO. Angiogenesis and liver fibrosis. World J Hepatol. 2015;7:377-91
3. Garbuzenko DV, Arefyev NO, Belov DV. Mechanisms of adaptation of the hepatic vasculature to the deteriorating conditions of blood circulation in liver cirrhosis. World J Hepatol. 2016;8:665-72
4. Lee JS, Semela D, Iredale J. et al. Sinusoidal remodeling and angiogenesis: a new function for the liver-specific pericyte? Hepatology. 2007;45:817-25
5. Marrone G, Shah VH, Gracia-Sancho J. Sinusoidal communication in liver fibrosis and regeneration. J Hepatol. 2016;65:608-17
6. Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: new targets for the treatment of portal hypertension? J Hepatol. 2010;53:976-80
7. Tugues S, Fernandez-Varo G, Munoz-Luque J. et al. Antiangiogenic treatment with sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology. 2007;46:1919-26
8. Hennenberg M, Trebicka J, Stark C. et al. Sorafenib targets dysregulated Rho kinase expression and portal hypertension in rats with secondary biliary cirrhosis. Br J Pharmacol. 2009;157:258-70
9. Mejias M, Garcia-Pras E, Tiani C. et al. Beneficial effects of sorafenib on splanchnic, intrahepatic, and portocollateral circulations in portal hypertensive and cirrhotic rats. Hepatology. 2009;49:1245-56
10. Zhang X, Zhang F, Kong D. et al. Tetramethylpyrazine inhibits angiotensin II-induced activation of hepatic stellate cells associated with interference of platelet-derived growth factor beta receptor pathways. FEBS J. 2014;281:2754-68
11. Zhang F, Zhang Z, Kong D. et al. Tetramethylpyrazine reduces glucose and insulin-induced activation of hepatic stellate cells by inhibiting insulin receptor-mediated PI3K/AKT and ERK pathways. Mol Cell Endocrinol. 2014;382:197-204
12. Zhang F, Jin H, Wu L. et al. Ligustrazine disrupts lipopolysaccharide-activated NLRP3 inflammasome pathway associated with inhibition of Toll-like receptor 4 in hepatocytes. Biomed Pharmacother. 2016;78:204-9
13. Jia Y, Wang Z, Zang A. et al. Tetramethylpyrazine inhibits tumor growth of lung cancer through disrupting angiogenesis via BMP/Smad/Id-1 signaling. Int J Oncol. 2016;48:2079-86
14. Cai X, Chen Z, Pan X. et al. Inhibition of angiogenesis, fibrosis and thrombosis by tetramethylpyrazine: mechanisms contributing to the SDF-1/CXCR4 axis. PLoS One. 2014;9:e88176
15. Zhang Z, Guo Y, Zhang S. et al. Curcumin modulates cannabinoid receptors in liver fibrosis in vivo and inhibits extracellular matrix expression in hepatic stellate cells by suppressing cannabinoid receptor type-1 in vitro. Eur J Pharmacol. 2013;721:133-40
16. Caliceti C, Aquila G, Pannella M. et al. 17beta-estradiol enhances signalling mediated by VEGF-A-delta-like ligand 4-notch1 axis in human endothelial cells. PLoS One. 2013;8:e71440
17. Lian N, Jiang Y, Zhang F. et al. Curcumin regulates cell fate and metabolism by inhibiting hedgehog signaling in hepatic stellate cells. Lab Invest. 2015;95:790-803
18. Ngan CH, Hall DR, Zerbe B. et al. FTSite: high accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics. 2012;28:286-7
19. Schmittgen TD, Zakrajsek BA, Mills AG. et al. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal Biochem. 2000;285:194-204
20. Hellerbrand C. Hepatic stellate cells-the pericytes in the liver. Pflugers Arch. 2013;465:775-8
21. Abreu-Blanco MT, Watts JJ, Verboon JM. et al. Cytoskeleton responses in wound repair. Cell Mol Life Sci. 2012;69:2469-83
22. Hong F, Haldeman BD, Jackson D. et al. Biochemistry of smooth muscle myosin light chain kinase. Arch Biochem Biophys. 2011;510:135-46
23. Zhang F, Lu Y, Zheng S. Peroxisome proliferator-activated receptor-gamma cross-regulation of signaling events implicated in liver fibrogenesis. Cell Signal. 2012;24:596-605
24. Zhang F, Kong D, Lu Y. et al. Peroxisome proliferator-activated receptor-gamma as a therapeutic target for hepatic fibrosis: from bench to bedside. Cell Mol Life Sci. 2013;70:259-76
25. Zhou J, Febbraio M, Wada T. et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology. 2008;134:556-67
26. Leesnitzer LM, Parks DJ, Bledsoe RK. et al. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry. 2002;41:6640-50
27. Zimna A, Kurpisz M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. Biomed Res Int. 2015;2015:549412
28. Li P, Fan W, Xu J. et al. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell. 2011;147:815-26
29. Bloch M, Prock A, Paonessa F. et al. High-mobility group A1 protein: a new coregulator of peroxisome proliferator-activated receptor-gamma-mediated transrepression in the vasculature. Circ Res. 2012;110:394-405
30. Yao Q, Lin Y, Li X. et al. Curcumin ameliorates intrahepatic angiogenesis and capillarization of the sinusoids in carbon tetrachloride-induced rat liver fibrosis. Toxicol Lett. 2013;222:72-82
31. Trauner M, Halilbasic E. Nuclear receptors as new perspective for the management of liver diseases. Gastroenterology. 2011;140:1120-5 e1-12
32. Zhu J, Teng YH, Wang PE. et al. Effect of ligustrazine on reverse cholesterol transport in foam cells. China J Chinese Materia Medica. 2014;39:1255-9
33. Dong SZ, Zhao SP, Wu ZH. et al. Curcumin promotes cholesterol efflux from adipocytes related to PPARgamma-LXRalpha-ABCA1 passway. Mol Cell Biochem. 2011;358:281-5
34. Liu ZJ, Liu W, Liu L. et al. Curcumin Protects Neuron against Cerebral Ischemia-Induced Inflammation through Improving PPAR-Gamma Function. Evid Based Complement Alternat Med. 2013;2013:470975
35. Liu ZJ, Liu HQ, Xiao C. et al. Curcumin protects neurons against oxygen-glucose deprivation/reoxygenation-induced injury through activation of peroxisome proliferator-activated receptor-gamma function. J Neurosci Res. 2014;92:1549-59
36. Li HY, Yang M, Li Z. et al. Curcumin inhibits angiotensin II-induced inflammation and proliferation of rat vascular smooth muscle cells by elevating PPAR-gamma activity and reducing oxidative stress. Int J Mol Med. 2017;39:1307-16
37. Zhang F, Hao M, Jin H. et al. Canonical hedgehog signalling regulates hepatic stellate cell-mediated angiogenesis in liver fibrosis. Br J Pharmacol. 2017;174:409-23
38. Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol. 2002;64:993-8
39. Nofsinger RR, Li P, Hong SH. et al. SMRT repression of nuclear receptors controls the adipogenic set point and metabolic homeostasis. Proc Natl Acad Sci U S A. 2008;105:20021-6
40. Yu S, Xu HE. Couple dynamics: PPARgamma and its ligand partners. Structure. 2012;20:2-4
41. Jang JY, Koh M, Bae H. et al. Structural basis for differential activities of enantiomeric PPARgamma agonists: Binding of S35 to the alternate site. Biochim Biophys Acta. 2017;1865:674-81
42. Ohtera A, Miyamae Y, Yoshida K. et al. Identification of a New Type of Covalent PPARgamma Agonist using a Ligand-Linking Strategy. ACS Chem Biol. 2015;10:2794-804
43. Wang Y, Lu W, Yang K. et al. Peroxisome proliferator-activated receptor gamma inhibits pulmonary hypertension targeting store-operated calcium entry. J Mol Med (Berl). 2015;93:327-42
44. Krishnan J, Suter M, Windak R. et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009;9:512-24
45. Zhao YZ, Liu XL, Shen GM. et al. Hypoxia induces peroxisome proliferator-activated receptor gamma expression via HIF-1-dependent mechanisms in HepG2 cell line. Arch Biochem Biophys. 2014;543:40-7
46. Yanguas SC, Cogliati B, Willebrords J. et al. Experimental models of liver fibrosis. Arch Toxicol. 2016;90:1025-48
47. Snowdon VK, Fallowfield JA. Models and mechanisms of fibrosis resolution. Alcohol Clin Exp Res. 2011;35:794-9
48. Poo JL, Feldmann G, Moreau A. et al. Early colchicine administration reduces hepatic fibrosis and portal hypertension in rats with bile duct ligation. J Hepatol. 1993;19:90-4
49. Kershenobich D, Vargas F, Garcia-Tsao G. et al. Colchicine in the treatment of cirrhosis of the liver. N Engl J Med. 1988;318:1709-13
50. Nikolaidis N, Kountouras J, Giouleme O. et al. Colchicine treatment of liver fibrosis. Hepatogastroenterology. 2006;53:281-5
51. Zhang F, Kong DS, Zhang ZL. et al. Tetramethylpyrazine induces G0/G1 cell cycle arrest and stimulates mitochondrial-mediated and caspase-dependent apoptosis through modulating ERK/p53 signaling in hepatic stellate cells in vitro. Apoptosis. 2013;18:135-49
52. He X, Zheng Z, Yang X. et al. Tetramethylpyrazine attenuates PPAR-gamma antagonist-deteriorated oxazolone-induced colitis in mice. Mol Med Rep. 2012;5:645-50
53. Chen J, Tian J, Ge H. et al. Effects of tetramethylpyrazine from Chinese black vinegar on antioxidant and hypolipidemia activities in HepG2 cells. Food Chem Toxicol. 2016
Author contact
![]() Corresponding author: Dr Shizhong Zheng, Jiangsu Key Laboratory for Pharmacology and Safety Evaluation of Chinese Materia Medica, Nanjing University of Chinese Medicine, 138 Xianlin Avenue, Nanjing 210023, Jiangsu, China. Tel: 86-025-85811246; Email: 300631edu.cn, nytwscom
Corresponding author: Dr Shizhong Zheng, Jiangsu Key Laboratory for Pharmacology and Safety Evaluation of Chinese Materia Medica, Nanjing University of Chinese Medicine, 138 Xianlin Avenue, Nanjing 210023, Jiangsu, China. Tel: 86-025-85811246; Email: 300631edu.cn, nytwscom