Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(2):486-504. doi:10.7150/thno.22059 This issue Cite
Research Paper
MiR-31-5p-ACOX1 Axis Enhances Tumorigenic Fitness in Oral Squamous Cell Carcinoma Via the Promigratory Prostaglandin E2
Yi-Hsuan Lai1, Hsuan Liu1,2,3,4, Wei-Fan Chiang5,6, Ting-Wen Chen3, Lichieh Julie Chu3,7, Jau-Song Yu1,2,3,7, Shu-Jen Chen8, Hua-Chien Chen8, Bertrand Chin-Ming Tan1,3,9,10 ![]()
1. Graduate Institute of Biomedical Sciences, College of Medicine, Chang Gung University, Guishan, Taoyuan, Taiwan, ROC
2. Department of Cell and Molecular Biology, College of Medicine, Chang Gung University, Guishan, Taoyuan, Taiwan, ROC
3. Molecular Medicine Research Center, Chang Gung University, Guishan, Taoyuan, Taiwan, ROC
4. Division of Colon and Rectal Surgery, Linkou Medical Center, Chang Gung Memorial Hospital, Taoyuan, Taiwan, ROC
5. Department of Oral & Maxillofacial Surgery, Chi-Mei Medical Center, Liouying, Tainan, Taiwan, ROC
6. School of Dentistry, National Yang Ming University, Beitou, Taipei, Taiwan, ROC
7. Liver Research Center, Linkou Medical Center, Chang Gung Memorial Hospital, Taoyuan, Taiwan, ROC
8. ACT Genomics, Co., Ltd., Neihu, Taipei, Taiwan, ROC
9. Department of Biomedical Sciences, College of Medicine, Chang Gung University, Guishan, Taoyuan, Taiwan, ROC
10. Department of Neurosurgery, Linkou Medical Center, Chang Gung Memorial Hospital, Taoyuan, Taiwan, ROC
Received 2017-7-24; Accepted 2017-10-24; Published 2018-1-1
Abstract

During neoplastic development, a multitude of changes in genome-encoded information are progressively selected to confer growth and survival advantages to tumor cells. microRNAs-mRNAs regulatory networks, given their role as a critical layer of robust gene expression control, are frequently altered in neoplasm. However, whether and how these gene perturbations impact metabolic homeostasis remains largely unresolved.
Methods: Through targeted miRNA expression screening, we uncovered an oral squamous cell carcinoma (OSCC)-associated miRNAome, among which miR-31-5p was identified based on extent of up-regulation, functional impact on OSCC cell migration and invasion, and direct regulation of the rate-limiting enzyme in peroxisomal β-oxidation, ACOX1.
Results: We further found that both miR-31-5p and ACOX1 underpin, in an antagonistic manner, the overall cellular lipidome profiles as well as the migratory and invasive abilities of OSCC cells. Interestingly, the extracellular levels of prostaglandin E2 (PGE2), a key substrate of ACOX1, were controlled by the miR-31-5p-ACOX1 axis, and were shown to positively influence the extent of cell motility in correlation with metastatic status. The promigratory effect of this metabolite was mediated by an elevation in EP1-ERK-MMP9 signaling. Of note, functional significance of this regulatory pathway was further corroborated by its clinicopathologically-correlated expression in OSCC patient specimens.
Conclusions: Collectively, our findings outlined a model whereby misregulated miR-31-5p-ACOX1 axis in tumor alters lipid metabolomes, consequently eliciting an intracellular signaling change to enhance cell motility. Our clinical analysis also unveiled PGE2 as a viable salivary biomarker for prognosticating oral cancer progression, further underscoring the importance of lipid metabolism in tumorigenesis.
Keywords: miRNA-mRNA network, ACOX1, lipid metabolism, oral squamous cell carcinoma, prostaglandins
Introduction
MicroRNAs (miRNAs) are non-coding, single-stranded RNAs of 18-24 nucleotides that serve as pivotal gene regulators in mammals and other multicellular organisms [1, 2]. miRNAs may silence gene expression at the post-transcriptional level through either perfect or imperfect binding to the 3' untranslated region (3' UTR) of messenger RNAs [2]. To date, 2,588 different mature human miRNAs have been identified and published in miRBase database and reportedly target more than 30% of the protein-coding genes. Given their extensive involvement in many biological processes, including proliferation, apoptosis, differentiation, and metabolic homeostasis, mutations or mis-expression of miRNAs have been associated with the progression of various types of human cancers [1, 3].
In an effort to map, from a systems perspective, the miRNA-mRNA networks underlying tumor pathogenesis, we designed and performed a targeted expression profiling of miRNAomes on the basis of in-house clinical specimens as well as a public cancer database. We focused this systems approach on oral cancer for the following reasons: 1) With approximately 300,373 new cases in year 2012, this globally prevalent cancer stands as a growing health concern [4, 5]. Despite progress in diagnosis and therapy strategies in the last three decades, the five-year survival rate in patients has not been significantly improved due to aggressive local invasion, distant metastasis, and even recurrence [6]. 2) A unique etiology - tobacco usage, betel quid chewing, alcohol consumption, infection with high-risk genotypes of human papillomavirus (HPV), and dietary intake - has been linked to oral squamous cell carcinoma (OSCC), the major subtype (>90%) of oral cancer [7, 8]. In line with this disease complexity, multiple dysregulated molecular networks underlie OSCC neoplastic development, including the epidermal growth factor receptor (EGFR), Ras, NF-κB, STAT, Wnt/β-catanin, TGF-β, and PI3K-AKT-mTOR signaling pathways [9]. However, the contribution of the transcriptome and/or metabolome alteration to OSCC incidence or progression has not yet been fully explored. 3) Given the anatomical sites of the disease and also because of the reportedly high stability of miRNAs in bodily fluids, cumulative studies have emphasized the potential of these small RNAs as non-invasive biomarkers for predictive monitoring of OSCC [10-12]. For example, plasma and salivary levels of miR-31-5p are elevated in OSCC and could be abrogated after tumor resection [13, 14].
Our approach uncovered several aberrantly expressed miRNAs, some of which have been identified previously by numerous independent studies [10, 12], thus strongly supporting the feasibility of our experimental paradigm. Among them, miR-21-5p is a frequently over-expressed oncogenic miRNA in OSCC as well as head and neck squamous cell carcinoma (HNSCC) samples. miR-21-5p plays crucial roles in cancer cell survival and resistance to anticancer agents, and its increased expression is related to poor prognosis and reduced survival rate in patients with HNSCC [11, 15]. miR-31-5p is another significantly up-regulated miRNA in oral cancer. In HNSCC, aberrantly elevated expression of miR-31-5p has been demonstrated to directly target the 3' UTR of factor-inhibiting hypoxia-inducible factor (FIH), an inhibitor of hypoxia-inducible factor (HIF), leading to activation of HIF downstream pathway under normoxic conditions [11, 16]. We then examined how this altered miRNAome could systematically impact the transcriptome and tumor biology. Interestingly, aside from oncogenic signaling and cell proliferation, an enrichment of predicted targets in fatty acid and peroxisome-associated metabolic pathways was evident, suggesting that distinct and hitherto unknown cellular changes may arise as a consequence of these OSCC-associated miRNA alterations.
Peroxisomes are single membrane-enclosed organelles that can be found in virtually all eukaryotic cells. Previous studies have demonstrated an increased number of peroxisomes in tissues with active lipid metabolism, such as liver and adipose tissues [17]. The primarily metabolic functions of mammalian peroxisomes include β-oxidation of a broad range of fatty acids, particularly the very long chain fatty acids (VLCFAs), α-oxidation of branched chain fatty acids, biosynthesis of etherphospholipids and bile acids, and regulation of redox homeostasis [17]. Both the peroxisomal and mitochondrial β-oxidation undergo four identical steps for degradation of acyl-CoA: (1) desaturation of acyl-CoA; (2) hydration of trans-2-enoyl-CoA; (3) dehydrogenation of 3-hydroxyacyl-CoA; (4) thiolysis of β-ketoacyl-CoA to form acetyl-CoA and a new fatty acyl-CoA with two fewer carbons [17, 18]. However, the peroxisomal β-oxidation is distinct from its mitochondrial counterpart in several aspects of enzymology and catabolic substrates. For instance, mitochondrial β-oxidation contributes to the catabolism of a bulk of lipid substrates, such as short, medium and long chain fatty acids, whereas the VLCFAs are exclusively oxidized in peroxisomes. Additionally, peroxisomal β-oxidation system in mammals also engages in the metabolic degradation of 2-methyl branched chain fatty acids, bile acid intermediates, polyunsaturated fatty acids (PUFA), dicarboxylic fatty acids, xenobiotics, and eicosanoids (i.e. prostaglandins and leukotrienes) [18, 19]. Furthermore, the first β-oxidation step in mitochondria is catalyzed by acyl-CoA dehydrogenase (ACAD) and concomitantly reduces FAD+ to FADH2, which are used to produce ATP through the electron transfer chain, whereas the initial step of peroxisomal lipid metabolism is carried out by acyl-CoA oxidase (ACOX), also culminating in formation of FADH2. However, instead of acting as an ATP generating source, FADH2 in peroxisomes is further re-oxidized, with its electrons transferred to oxygen and producing H2O2 as a by-product [17, 18, 20], suggesting a role of peroxisomal lipid metabolism in redox regulation.
There are at least three acyl-CoA oxidases encoded by the mammalian genomes - ACOX1 (with two alternative isoforms, a/b or I/II), ACOX2, ACOX3 and ACOX4 [19] - which are different in tissue distribution and substrate specificity. Unlike other peroxisomal acyl-CoA oxidases, ACOX1 is commonly expressed in most tissues and is specific for the desaturation of straight chain CoA esters of VLCFAs, dicarboxylic acids, PUFAs, and prostaglandins. In contrast, the 2-methyl branched compounds, such as pristanoyl-CoA, cholestanoyl-CoA and CoA esters of other bile acids in S configuration, are preferentially oxidized by ACOX2 [19]. ACOX1 deficiency is connected with a lethal human disease, called pseudoneonatal adrenoleukodystrophy (P-NALD). The biochemical symptoms of this autosomal recessive disorder are accumulated VLCFAs and enlarged peroxisomes [21]. Patients with P-NALD often exhibit neurodevelopmental abnormalities, including hypotonia and seizures, and die early. Recently, it has become evident that activation of ER stress contributes to the progression of hepatocarcinogenesis in homozygous ACOX1 knockout mice [22]. While this implies a tumor-suppressive role for this oxidase, precise functional implication of ACOX1-mediated metabolism in human cancers remains largely elusive.
In this study, we identified ACOX1 as a new target of miR-31-5p and further connected this microRNA to the maintenance of proper lipid metabolic profiles. Our results further revealed that this functional antagonism underpins the OSCC cell migratory and invasive capacity. Mechanistically, PGE2, one of the substrates of ACOX1, acts as a downstream mediator of these oncogenic changes by conferring an elevated promigratory ERK-MMP9 signaling. Clinicopathological significance of this regulatory pathway was further reinforced by distinct expression alterations of the molecular components in OSCC patient specimens, which are in correspondence with their functional consequences. In summary, our findings demonstrated that miR-31-5p acts as a tumor-promoting miRNA in OSCC and outlined a downstream regulatory network whereby tumor cell motility is favored by altered lipid metabolomes. Our studies also uncovered a potentially viable biomarker in salivary PGE2 for OSCC disease monitoring.
Materials and Methods
miRNA expression profiling analysis
The expression levels of 270 miRNAs were determined using the stem-loop RT-PCR method. First, total RNA was extracted from normal and cancerous specimens of oral origin. 1 μg of total RNA from tissues was subsequently subjected to a reverse transcription reaction with pooled miRNA-specific stem-looped RT primers. The reversely transcribed cDNA was diluted and analyzed by SYBR-based real-time PCR on an Applied Biosystems 7900 Fast PCR System (Foster City, CA, USA). After global median normalization, the expression levels of individual miRNAs were expressed as 40 minus the value of the threshold cycle (Ct), defined as the cycle number where the SYBR green intensity reaches a threshold of 0.2. For screening the differentially expressed miRNAs, three-way analysis of variance (ANOVA) was performed and the false discovery rate (FDR)-based multiple testing correction (q-value) was used to adjust the statistical confidence measures as previously described [23]. All the data processing and statistical analyses described above were performed with Partek Genomics Suite 6.5 (Partek Inc.; St. Louis, MO, USA). The data have been submitted to the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE103931.
Cell culture
Three human OSCC cell lines were used in this study: OECM-1, a gingival epidermoid carcinoma line from a Taiwanese patient, was maintained in Dulbecco's Modified Eagle Medium (DMEM), while tongue squamous cell lines SCC25 and SCC4 were cultured in DMEM/F12 medium. These OSCC cell lines were supplemented with 10% heat-inactivated fetal bovine serum (FBS; Biological industries; Cromwell, CT, USA), 1X non-essential amino acids and 1 mM sodium pyruvate. Except for OECM-1, the complete growth media for SCC25 and SCC4 require additional supplement of 400 ng/mL hydrocortisone (Sigma-Aldrich; St. Louis, MO, USA). Human gingival epithelial S-G cells were cultured in DMEM with 10% heat-inactivated FBS. All cells were incubated in fully humidified air containing 5% carbon dioxide at 37°C. Unless otherwise specified, all culture media and reagents were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Reagents
Locked nucleic acids (LNA) against miR-31-5p and scramble control were purchased from Exiqon A/S (Vedbaek, Denmark), and the small interfering RNAs (siRNAs) against ACOX1 were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Lipofectamine 2000, Lipofectamine RNAiMAX, and all RT reagents were purchased from Invitrogen (Carlsbad, CA, USA). PGE2 and PGE2 monoclonal ELISA kit were purchased from Cayman Chemical (Ann Arbor, MI, USA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
RNA extraction and quantitative reverse transcription PCR (RT-qPCR)
Total RNA was isolated from cells by the TRIzol reagent (Thermo Fisher Scientific) according to manufacturer's instructions. The concentration of extracted RNA was determined using NanoDrop spectrophotometer. For miRNA RT reaction, 1 μg of total RNA was first converted into cDNA by Superscript III reverse transcriptase (Invitrogen), in which miRNA-specific stem-looped RT primers were used. RT reaction was done under the following condition: 16°C for 30 min, followed by 50 cycles of 20°C for 30 s, 42°C for 30 s and 50°C for 1 s. The enzyme was then inactivated at 70°C for 10 min. For mRNA, 1 μg of total RNA was used as templates to synthesize first-strand cDNA using oligo-dT primers. RT reaction was carried out at 42°C for 90 min and then 70°C for 15 min. For the quantitative determination of the reversely transcribed products, real-time PCR was performed on an Applied Biosystems 7500 Fast PCR System (Foster City, CA, USA) using the standard SYBR green method under the following condition: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. For expression analysis, the Ct values of mRNA targets and miRNA were first normalized respectively to those of the eukaryotic translation elongation factor 1 alpha 1 (EEF1A1) and U6 genes in the same samples. The resulting ∆Ct was further utilized to assess the relative gene expression between different experimental groups, with the data presented as fold change to control. Sequences of primers used in real-time PCR assay are listed in Table S1.
Transient transfection and lentiviral-based infection
For transient transfection of LNA, 10 nM LNAs directed against miR-31-5p or control LNA (scramble) were transfected into cultured OSCC cells using Lipofectamine 2000 according to the manufacturer's instructions. To deplete endogenous ACOX1, two siRNAs against ACOX1 or negative control (si-Ctrl) were transfected into cells through Lipofectamine RNAiMAX. For co-transfection experiments with miR-31-5p antisense oligomers and ACOX1 siRNA, the combination of 10 nM LNAs and 10 nM siRNAs was used, together with Lipofectamine 2000 according to the manufacturer's instructions. All functional analyses were performed after 24 h of transfection.
Double-stranded oligonucleotides encoding miR-31-5p or control (LacZ) sequence were synthesized and inserted into pcDNA-GW/EmGFP-miR vectors. The cloning primers utilized here are listed in Table S1. Those vectors were further transferred into lentiviral expression plasmids using Gateway recombination technology according to the manufacturer's instructions. The resulting constructs were co-transfected with packaging vectors into HEK293-FT cells to generate lentiviral particles, which were further used to infect cells for establishing stable expression. All materials were purchased form Invitrogen.
Dual luciferase reporter assay
The luciferase reporter vectors for the wild-type or mutant ACOX1 3' UTR containing the miR-31-5p binding site were constructed into pMIR-REPORT luciferase system (Thermo Fisher Scientific). The primers used here are shown in Table S1. Mutated ACOX1 3' UTR was generated by replacement of the target sequence UCUUGC to AAGCUU using two-step mutagenesis PCR. The vectors expressing firefly luciferase reporter fused with wild-type or mutant ACOX1 3' UTR were co-transfected with miR-31-5p antisense oligomers or scramble control into OSCC cells. After 24 h, cells were harvested and the two distinct luciferase activities were measured using Dual-luciferase reporter assay according to the manufacturer's instructions (Promega; Madison, WI, USA). Renilla luciferase activity was used to normalize the extent of firefly luciferase activity.
Prediction and pathway analysis of miR-31-5p putative targets
For miR-31-5p target prediction, the 3' UTR sequences of 17,330 human protein-coding genes were retrieved from the UCSC genome database (https://genome-asia.ucsc.edu/index.html), and the sequence of mature hsa-miR-31-5p was downloaded from miRBase database (http://www.mirbase.org/). Target prediction and context score analyses were performed using the targetscan_50.pl and targetscan_50_context_scores.pl Perl scripts were downloaded from the TargetScan website (http://www.targetscan.org/vert_50/). A sum context score of <0.2 was used to filter for the highly probable targets. Consequently, a total of 1,213 putative targets (irrespective of species conservation) were recovered. These targets were further subjected to pathway enrichment analysis using online DAVID functional annotation tools (https://david.ncifcrf.gov) and GeneGo MetaCore software (Thomson Reuters; New York, NY, USA).
Cell proliferation and colony formation assays
For the growth curve assay, OSCC cells were seeded in 96-well plates. The cells were then fixed with 3.7% formaldehyde and stained with 4',6-diamidino-2-phenylindole (DAPI) on indicated days. Cell numbers were determined using IN Cell Analyzer 1000 imaging system (GE Healthcare Life Sciences; Buckinghamshire, UK). For colony formation assay, OSCC cells were seeded at a low cell density of 100 cells (SCC25, OECM-1 and SCC4) into 6-well plates and maintained in complete medium for the indicated days (SCC25: 9 to 13 days; OECM-1: 9 days; SCC4: 13 days). The cells were then washed, fixed, and stained with crystal violet. Colony forming activity was determined by quantifying the visible colonies using UVP BioSpectrum Imaging System.
In vitro migratory and invasive assays
For the wound healing-based migration assay, OSCC cells were seeded and grown in 6-well plates with complete medium to allow monolayer cell formation. The cells were scratched with a sterile pipette tip to create artificial wounds [24]. At indicated time points, images of the healed wound were photographed by ZEISS Axio Observer A1 microscope and the wound area was determined using ZEISS AxioVision 4.6 microscope software.
The transwell migration and invasion assays were carried out using Boyden chambers with transwell inserts in 24-well plates (Corning; Corning, NY, USA). For the invasion assay, we pre-coated the transwell inserts with Matrigel Basement Membrane Matrix (BD Biosciences; San Jose, CA, USA). The indicated OSCC transfectants were resuspended in serum-free medium and seeded into the upper transwell inserts, with the supplement of 10% fetal bovine serum-containing culture media in the bottom chamber. After 14 h for OECM-1 and 24 h for SCC25, the numbers of cells migrated or invaded towards the bottom chamber were determined by crystal violet staining and quantified using IN Cell Developer Toolbox.
Western blotting
Western blotting was conducted after separation of polypeptides using SDS-PAGE. Proteins on gel were transferred to PVDF membrane (Merck Millipore; Billerica, MA, USA). The membrane was further incubated with indicated primary and appropriate secondary antibodies. Antibodies against ACOX1 were obtained from Proteintech Group (Chicago, IL, USA). Phosphorylated form of ERK1/2 and ERK1 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and BD Biosciences, respectively. Antibodies targeting MMP3, MMP10 and phospho-Akt (Thr308) were also purchased from Santa Cruz Biotechnology. Antibodies against phospho-Akt (Ser473) were obtained from Cell Signaling Technology (Danvers, MA, USA). MMP2 and MMP9 antibodies were purchased from Abcam (Cambridge, Cambridgeshire, UK). An anti-GAPDH antibody (BioWorld; St. Louis Park, MN, USA) was used as control. HRP-conjugated secondary antibodies against rabbit IgG or mouse IgG (GeneTex) were incubated with membrane for 1 h at room temperature. Immunobands were detected using chemiluminescent HRP substrates (ECL; Merck Millipore) and captured by UVP BioSpectrum 600 Imaging System. The intensity of bands was quantified by Image J software.
Lipidomic analyses: UPLC-MS metabolomics, lipid bodies staining and PGE2 quantification
For Ultra Performance Liquid Chromatography (UPLC)-MS metabolomics analysis, global metabolites were extracted from OSCC cell lysates using 80% methanol. Folch method (ddH2O : CHCl3 : Methanol = 3 : 8 : 4) was implemented for further lipid metabolites extraction. Subsequently, UPLC was performed and mass spectrometry was operated in negative-ion (ESI-) mode. Data analysis was accomplished using MetaboAnalyst tool and Partek Genomics Suite software.
For lipid droplet staining, cultured OSCC cells were fixed on coverslips and incubated with a mixture of Alexa Fluor 488-conjudated BODIPY (Lipid droplet), Alexa Fluor 555-conjugated wheat germ agglutinin and DAPI for 1 h at room temperature prior to washing and mounting. The concentrations of total free fatty acids in plasma were measured by using the Non-Esterified Fatty Acids (NEFA) assay kit (Randox Laboratories; Crumlin, Antrim, UK), based on the manufacturer's instructions. PGE2 levels in saliva or conditioned media were detected using PGE2 monoclonal ELISA kit according to the manufacturer's instructions. Each subject signed an informed consent form approved by the Institutional Review Board of Chi-Mei Medical Center, permitting the use of plasma and saliva samples for this study.
Gelatin zymography
To measure the gelatinase activity of the experimentally manipulated cell culture, 2 × 106 cells were first switched to serum-free DMEM/F12. After 24 h, the conditioned media were collected and concentrated by Amicon Ultra-15 centrifugal filter (Merck Millipore). Subsequently, all samples were run on 8% SDS-PAGE with 0.1% gelatin. Gels were washed twice with 2.5% Tritox X-100 for 30 min at room temperature, and then incubated with development buffer at 37°C overnight. Lastly, gels were stained with Coomassie blue to determine the MMP2 and MMP9 activity.
Statistical analysis
All data are presented as mean ± SEM (standard error of the mean). Statistical analyses were carried out by using GraphPad Prism 6 (San Diego, CA, USA). Pearson correlation analysis was utilized to evaluate the correlation between two groups. Two-tailed Student's t-test was used to determine the statistical significance between independent samples. p-values less than 0.05 were considered statistically significant. Degrees of significance (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant) are depicted in the respective figure legends.
Results
Systematic expression profiling uncovers miR-31-5p as a highly expressed miRNA in both OSCC and HNSCC
To provide further insights into the physiological relevance of miRNAs in tumor malignancy, we first sought to compile, from a systems perspective, the miRNA-mRNA networks underlying OSCC pathogenesis. Our targeted, qRT-PCR-based expression profiling of 270 miRNAs in 49 human oral tissue samples (including 19 normal and 30 OSCC specimens) previously identified 49 differentially expressed miRNAs (p-value < 0.01 and fold change > 2) in OSCC tissues - 11 of these were significantly up-regulated in OSCC tissues (Figure S1A-S1C and Table S2). To confirm our findings, we also analyzed HNSCC and OSCC microRNA deep sequencing datasets from The Cancer Genome Atlas (TCGA) database, and subsequently identified 40 markedly up-regulated miRNAs (p-value < 0.05 and fold change > 2) in 43 HNSCC tissues (Figure 1A) and 47 over-expressed miRNAs (p-value < 0.05 and fold change > 4) in 28 OSCC subjects (Figure S2), as compared with their paired normal specimens. Six of these were commonly over-expressed in both in-house OSCC samples and public TCGA HNSCC specimens (Figure 1B). To elucidate how this altered miRNAome could systemically impact tumor biology, we performed computational miRNA target prediction for these six miRNAs and subsequently utilized pathway analysis (Figure S3 and Table S3). Interestingly, an enrichment of predicted targets in several KEGG pathways was found, with different types of lipid metabolic pathways shown as the top ranked terms, suggesting distinct cellular changes may arise as a consequence of these OSCC-associated miRNA alterations.
Systems profiling identifies miRNAs mis-regulated in HNSCC. (A) Hierarchical clustering of 43 normal/tumor paired HNSCC samples based on the expression levels of 40 significantly upregulated miRNAs. (B) Venn diagram shows the extent of overlap in differentially over-expressed miRNAs between in-house OSCC samples and public TCGA HNSCC data. The six commonly over-expressed miRNAs are also indicated. (C) Expression levels (in log2 reads per million, or RPM, value) of miR-31-5p in 42 HNSCC tumor tissues (red) and their matched normal tissues (blue) (upper panel), as well as in 28 OSCC samples (red) and their paired normal controls (blue) (lower panel). Paired samples are connected by lines. Corresponding box plots are also shown alongside. (D) Expression levels of miR-31-5p were analyzed in normal (n = 43) vs. HNSCC samples (Stage I, n = 25; Stage II, n = 73; Stage III, n = 70; Stage IV, n = 228) (upper panel), and in normal (n = 43) vs. OSCC samples (Stage I, n = 19; Stage II, n = 53; Stage III, n = 50; Stage IV, n = 142) (lower panel) according to AJCC stages. ***, p < 0.001, Student's t-test.
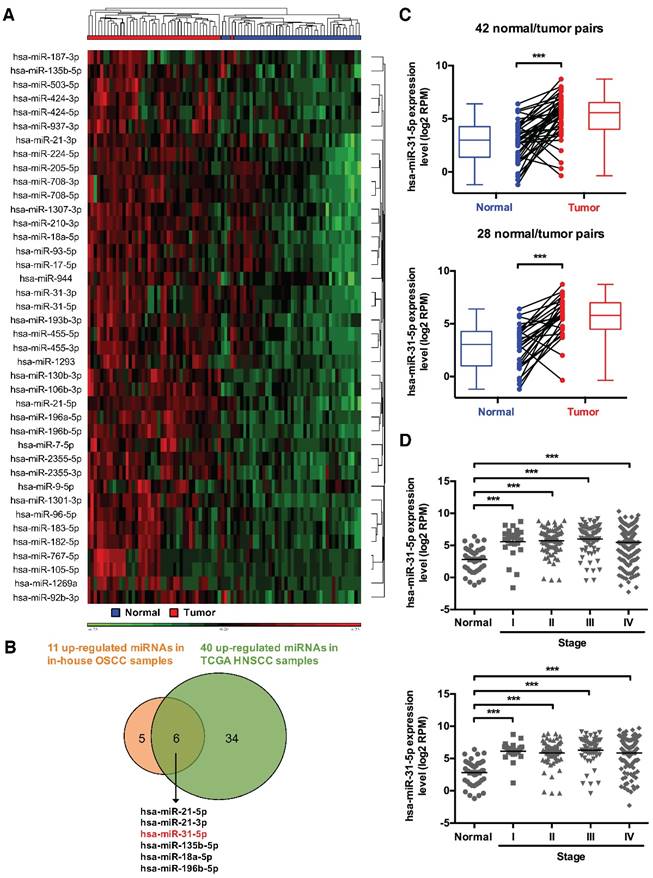
We selected miR-31-5p as the target miRNA for further functional studies in this study on the basis of the following criteria: 1) Among the six miRNAs, miR-31-5p exhibited the greatest extent of expression up-regulation as well as the highest abundance in the OSCC tissues (Figure S1D and Table S2). 2) The tumor-associated elevation of miR-31-5p was coincidently evident in both the TCGA OSCC and HNSCC miRNA expression datasets (Figure 1C). In addition, analysis of clinical relevance of miR-31-5p expression with regard to disease stages revealed that, while miR-31-5p was significantly increased in the tumor tissues, its levels remained invariably high across all four stages of HNSCC and OSCC patient samples (Figure 1D), indicating that this altered elevation may be an early event in neoplastic progression of OSCC and HNSCC. 3) Pathway analyses of the presumptive targets, based on either KEGG (Figure 2A and Table S4) or GeneGo/MetaCore (Figure S4), showed enrichment in the regulation of lipid or fatty acid metabolism. To test whether these hypothetical molecular changes in metabolism are verifiable in a clinicopathological context, we monitored the levels of free fatty acids in the plasma of OSCC patients. Interestingly, we found that OSCC plasma exhibited higher levels of free fatty acids (Figure 2B), thus supporting our in silico functional predictions of this miRNA.
ACOX1, the rate-limiting enzyme in peroxisomal β-oxidation, is directly targeted by miR-31-5p. (A) Schematic depiction of the distribution of putative miR-31-5p targets in two fatty acid metabolism pathways. The candidate targets are highlighted in red. (B) The concentrations of total free fatty acids in 16 normal and 70 OSCC patient plasma specimens were detected by the NEFA kit as mentioned in Methods. (C) mRNA levels of ACOX1 in SCC25 and OECM-1 cells were measured by qRT-PCR from RNAs extracted from miR-31-5p-depleted (upper panel) or over-expressing (lower panel) cells. The data are expressed as fold change relative to corresponding controls. (D) Protein levels of ACOX1 were detected by western blotting in SCC25 and OECM-1 cells treated with anti-miR-31-5p oligomers or scramble control (left panel), and in SCC25 cells infected with lentivirus containing miR-31-5p and control (LacZ) vector (right panel). GAPDH was used as a loading control. (E) Predicted duplex formation between hsa-miR-31-5p and human ACOX1 3' UTR (Wt). The nucleotides highlighted in bold (positions 2945 to 2951) indicate the seed region of miR-31-5p. The underline points out the mutated miR-31-5p target site in the ACOX1 3' UTR (Mut) construct. SCS25 and OECM-1 cells were transfected with a luciferase reporter construct that contains wild type (Wt) or mutated (Mut) ACOX1 3' UTR, together with anti-miR-31-5p or anti-scramble oligomers. Luciferase activity was determined 24 h after transfection. (F) Expression levels (in log2 normalized raw count) of ACOX1 in 41 HNSCC tumor tissues (red) and their matched normal tissues (blue). Paired samples are connected by lines, with corresponding box plots also shown. (G) Correlation between the expression levels of miR-31-5p and ACOX1 in 39 normal (blue) and tumor (red) paired HNSCC samples. Pearson's r analysis was performed. The bar graphs in (C) and (E) show mean ± SEM from three independent experiments (ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001; Student's t-test).
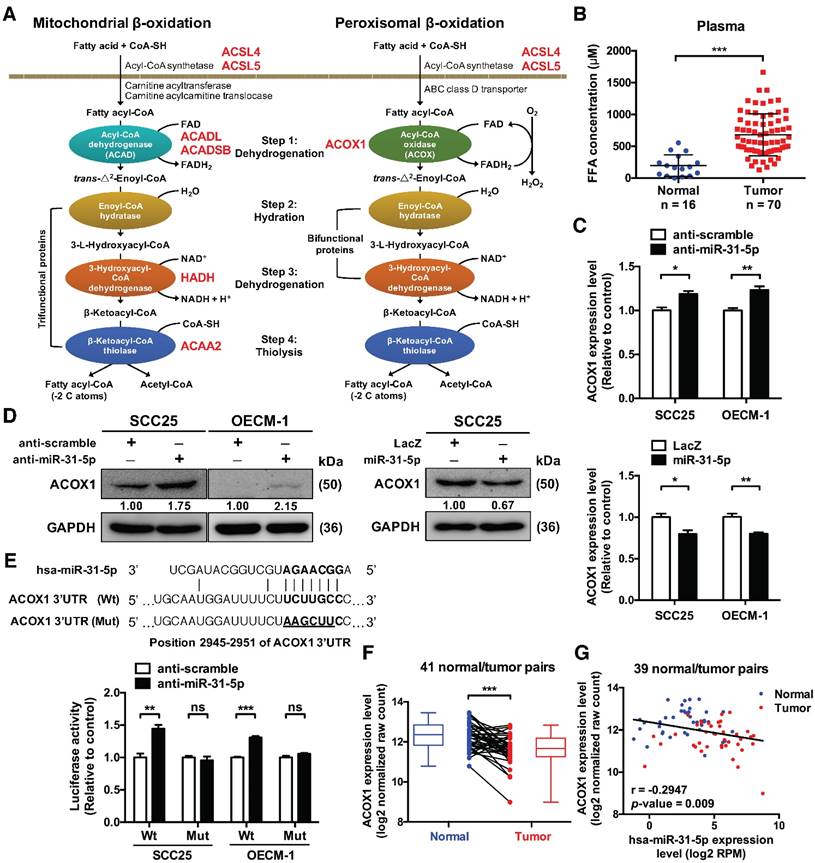
miR-31-5p directly targets ACOX1, the first and rate-limiting enzyme of β-oxidation in peroxisomes
In line with our observations, several reports in the literature have revealed that expression of miR-31-5p is frequently up-regulated in different cancer types, including HNSCC [16]. In order to further dissect the biological roles of miR-31-5p in OSCC, we first performed miR-31-5p expression profiling in four OSCC cell lines by stem-loop RT-PCR, which revealed that it was considerably elevated in all four OSCC cell lines compared with normal gingival epithelioid S-G cell lines (Figure S5A). Next, to examine whether the aforementioned lipid metabolism-associated candidates were indeed regulated by miR-31-5p, we analyzed their mRNA and protein levels in miR-31-5p mis-expressed OSCC cells (Figure S5B-S5D) using real-time PCR and western blotting, respectively. This expression profiling uncovered ACOX1, the first enzyme in peroxisomal lipid metabolism, as a possible target: depletion of miR-31-5p using antisense oligonucleotides increased both mRNA and protein levels of ACOX1 in various cultured OSCC cells (Figure 2C-2D and S5E) and also in a dose-dependent manner (Figure S5F). By contrast, lentiviral-based ectopic expression of miR-31-5p led to expression suppression of ACOX1 (Figure 2C-2D). Knockdown or over-expression of miR-31-5p did not seem to influence the expression levels of other predicted targets associated with lipid metabolism (Figure S5E-S5F), suggesting miR-31-5p does not target these 3' UTR despite the presence of targeting sequences.
The presence of a binding site for miR-31-5p in the 3' UTR of ACOX1 suggests a direct regulation (Figure 2E). We tested this possibility by performing luciferase reporter assay on constructs harboring the wild-type or a binding site-mutated 3' UTR. Our results revealed that depleting endogenous miR-31-5p in OSCC cells elevated the luciferase activity of the wild type, but not the mutant ACOX1 3' UTR construct (Figure 2E), strengthening the direct targeting by miR-31-5p. Additionally, deep sequencing data from TCGA database further corroborated this regulatory relationship: the ACOX1 expression was down-regulated in HNSCC samples relative to their paired normal samples (Figure 2F), and further was inversely correlated in expression with miR-31-5p in the HNSCC normal/tumor paired specimens (Figure 2G). Collectively, these results connected the tumor-associated up-regulation of miR-31-5p with lower level of ACOX1 in HNSCC clinical specimen.
miR-31-5p regulates the reprogramming of lipid metabolism in OSCC via targeting ACOX1
ACOX1 is known to desaturate straight chain lipid substrates, such as the CoA esters of long chain fatty acids, very long chain fatty acids, dicarboxylic acids, polyunsaturated fatty acids and prostaglandins [18, 19]. The negative regulation of ACOX1 thus signifies that miR-31-5p may be linked to tumor-associated lipid metabolic alterations. To test this hypothesis, we set out to assess various lipidomic features of cells in the context of miR-31-5p or ACOX1 mis-expression. First, a mass spectrometry-based, untargeted lipid metabolite analysis was conducted on SCC25 cells with miR-31-5p or ACOX1 knockdown (Figure 3A). We found that several long chain fatty acids, including myristic acid (C14:0), palmitic acid (C16:0) and stearic acid (C18:0), were notably reduced upon inactivation of miR-31-5p in the cells (Figure 3B, left), whereas the levels of two C18 unsaturated fatty acids exhibited no significant change (Figure S6A, top). While the UPLC-MS data showed that reduced expression of ACOX1 did not affect the abundance of C14, C16 and C18 free fatty acids (Figure 3B, left), certain phospholipids and sphingolipids, especially those derived from free fatty acids with 16- or 18-carbon chain, underwent significant increases in the same knockdown culture (Figure 3B, right). These alterations could hypothetically imply that the free fatty acids accumulated in the absence of ACOX1 might be rapidly utilized to synthesize new components of plasma membrane for cell proliferation and migration. Notably, we discovered that several phosphatidylinositols (PI), phosphatidylglycerol (PG) and sphingomyelin were dramatically repressed upon ACOX1 depletion (Figure 3B and S6A), whereas the miR-31-5p inactivation exhibited a discernibly reverse outcome. Together, these cellular lipidomic profile alterations highlighted that the miR-31-5p-ACOX1 axis may be linked to reprogramming of lipid metabolism pathways in OSCC (Figure S6B-S6C).
The miR-31-5p-ACOX1 axis reprograms the fatty acid metabolism of OSCC cells. (A-B) SCC25 cells were transfected with scramble or miR-31-5p antisense oligomers, or treated with control or ACOX1 siRNAs (single or mixture of two). Protein levels of ACOX1 were determined using western blotting, with GAPDH as a loading control (A). The intensity of lipid metabolites was measured via UPLC-MS, and the levels of selected lipid metabolites are shown as mean ± SEM from at least three independent repeats (B). (C-D) Alexa Fluor 488-conjudated BODIPY (Lipid droplet) and Fluor 555-conjugated wheat germ agglutinin (WGA) staining in SCC25 after inactivation of miR-31-5p or suppression of ACOX1. Scale bar, 20 μm. Representative images (C) and the quantitative data (D) are also shown. (E) SCC25 cells were transfected with two ACOX1 siRNAs (si-ACOX1 #1 or si-ACOX1 #2) or control (si-Ctrl) for 24 h. The concentrations of PGE2 in conditioned media were determined via ELISA assay. (F) PGE2 detection assay was performed to assess PGE2 production in SCC25 cells with the indicated combination of co-transfection. The data shown in (D) to (F) were derived from biological triplicates and are expressed as mean ± SEM. (*, p < 0.05; **, p < 0.01; ***, p < 0.001)
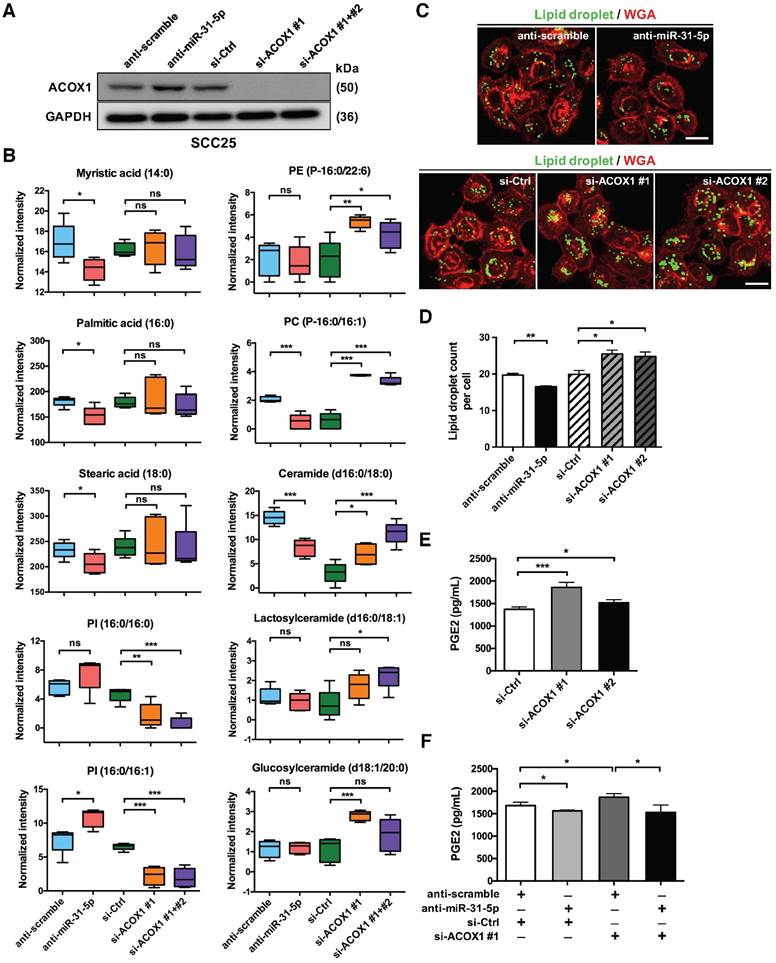
Second, given that certain peroxisomal metabolic enzymes are enriched in and functionally linked to lipid droplets [17, 25], and also owing to the association of lipid droplet biogenesis with cancer progression [26], we turned our attention to the distribution of these organelles. Lipid droplets are phospholipid monolayer-enclosed subcellular organelles composed of neutral lipids, and can be detected based on this lipid-rich characteristic. Lipid-specific BODIPY staining illustrated that depletion of endogenous miR-31-5p diminished the levels of cellular lipid droplets. By contrast, there was a prominent accumulation of lipid droplets in the ACOX1 knockdown cells (Figure 3C-3D). These data therefore confirmed the link of peroxisomal β-oxidation with lipid droplets and further implied that miR-31-5p, by targeting ACOX1, could regulate the formation of these lipid bodies.
Finally, it has been reported that PGE2-CoA is one of the substrates of ACOX1 [27]. Via transport into the extracellular space, intracellular PGE2 is also known to activate various pro-tumorigenesis signaling pathways. Therefore, we next sought to assess the PGE2 levels in the conditioned media (CM) of ACOX1-depleted OSCC cells. We found that siRNA abrogation of ACOX1 expression significantly enhanced the PGE2 levels in the CM (Figure 3E). Conversely, inhibition of miR-31-5p reduced the concentration of PGE2. In addition, simultaneous knockdown of miR-31-5p in the ACOX1-depleted cells reversed the induction of PGE2 production in response to ACOX1 down-regulation (Figure 3F), implying that the amount of PGE2 in the extracellular space is regulated by the miR-31-5p-ACOX1 axis in OSCC. Viewed together, these observations implicated miR-31-5p in the ACOX1-depednent reprogramming of lipid metabolism in OSCC.
Suppression of ACOX1 accelerates OSCC cell migration and invasion
To further decipher the biological consequence of miR-31-5p-mediated functions in OSCC, we carried out several functional studies based on gain- and loss-of-function manipulation. Toward this end, mis-expression of miR-31-5p did not influence the cell proliferation and colony formation capacity in three independent OSCC cell lines (Figure S7A-S7D). However, in vitro migratory assay showed that loss of miR-31-5p notably suppressed the migratory ability of OSCC cells, and that ectopic expression of miR-31-5p exhibited an adverse effect on cell migration (Figure 4A-4B and S7E). These results were further supported by the Boyden chamber migration assay - miR-31-5p expression was correlated with the migratory and invasive extent of OSCC cells (Figure 4C-4D and S7F-S7G). Collectively, these findings indicated that miR-31-5p acts positively on the motility of OSCC cells, a function that is independent of cell growth.
miR-31-5p and ACOX1 reciprocally modulate the OSCC migratory and invasive capability. (A-B) Wound healing migration assay was performed to assess the migratory ability of SCC25 upon knockdown or ectopic expression of miR-31-5p. Photographs were taken at indicated time points after cells were scratched, with the representative images shown in (A). Scale bar, 100 μm. Wound area was measured and normalized to 0 h (B). Data are shown as mean ± SEM from at least three independent experiments (**, p < 0.01; Student's t-test). (C-D) Transwell migration and Matrigel invasion assays were carried out after knockdown or stable over-expression of miR-31-5p in SCC25 cells. The representative photomicrographs are shown (C). Scale bar, 200 μm. Average migratory area per field was quantified and expressed as fold change relative to control (D). (E-F) SCC25 cells were transfected with ACOX1 siRNAs or control siRNA, and subjected to wound healing migration (E) and transwell migratory (F) assays. The bar graphs in (D) to (F) show mean ± SEM from three independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
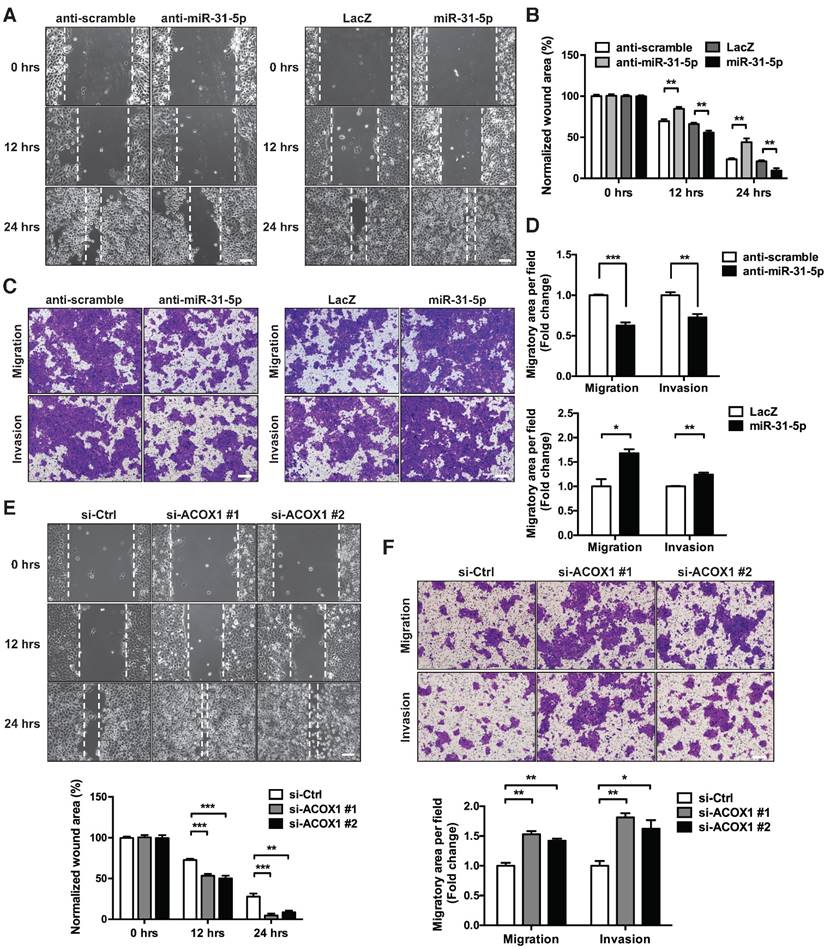
We also examined whether the down-regulation of ACOX1 in tumor recapitulates similar oncogenic attributes. Repressing endogenous ACOX1 mRNA and protein levels in two OSCC cell lines, SCC25 and OECM-1 (Figure S8A-S8B), facilitated the in vitro migratory capacity of these cells on the basis of a wound healing assay (Figure 4E and S8C); however, this ACOX1 abrogation had seemingly no effect on cell proliferation and growth (Figure S8D-S8E). To further ascertain this functional role, we employed Matrigel invasion assay to monitor OSCC invasive capability, and subsequently observed that ACOX1 attenuation considerably stimulated the migratory and invasive abilities of two different OSCC lines (Figure 4F and S8F). These data were reminiscent of the results found in OSCC cells over-expressing miR-31-5p, and further supported the suggested connection of the miR-31-5p-ACOX1 axis to OSCC malignancy.
PGE2 promotes OSCC cell migration and invasion through activating the ERK-MMP9 cascade
Having established that ACOX1 down-regulation underlies cell motility, we next speculated the contribution of lipid metabolites to these features. While few studies directly link the lipid metabolites to cell mobility, PGE2 is reportedly linked to tumor cell proliferation, survival and migration [28]. To further elucidate its role, we directly stimulated OSCC cells with PGE2 and examined their migratory capacity and invasiveness. Since the concentration of PGE2 in SCC25 CM was measured to be 4 nM, we consequently treated the OSCC cells with a higher dose (10 nM) to assess its effect. Our results revealed that this PGE2 treatment triggered the in vitro migratory and invasive activities of OSCC cells (Figure 5A).
The miR-31-5p-ACOX1 axis-mediated up-regulation of prostaglandin E2 enhances the migration and invasiveness of OSCC cells. (A) SCC25 cells were treated with or without PGE2 (0.01 µM), or co-treated with PGE2 and EP1 receptor antagonist (SC-51322; 0.02 µM), and analyzed by transwell migration assay. Representative photomicrographs are shown (left panel; scale bar, 200 μm). Average migratory area per field was quantified and expressed as fold change relative to control (right panel). (B) SCC25 cells were serum-starved and stimulated with PGE2 (upper panel) or with the combination of PGE2 and SC-51322 (lower panel) for the indicated times. Protein levels of p-ERK, ERK1, p-AKT (Ser473), p-AKT (Thr308) and AKT were determined via western blotting, with GAPDH as a loading control. (C) SCC25 cells were stimulated with or without 0.01 µM PGE2 for 24 h. The expression levels of various MMP molecules were assessed by qRT-PCR (upper panel). Relative expression of MMP9 in SCC25 cells was determined upon stimulation of PGE2 alone or together with SC-51322 (lower panel). (D) Incubation of SCC25 cells with conditioned media from ACOX1-depleted or control cells for 24 h, followed by assessment of their migratory and invasive activities. Quantitative results in (A), (C) and (D) correspond to mean ± SEM from three independent experiments. (E) The concentrations of PGE2 in 50 normal and 50 OSCC salivary specimens were determined by ELISA (left panel). The receiver operating characteristic (ROC) curve was utilized to distinguish OSCC from non-tumor groups (middle panel). For tumor state-associated alterations, the abundance of PGE2 in saliva from normal (n = 50) and OSCC samples sub-grouped according to AJCC stages (Stage I, n = 12; Stage II, n = 8; Stage III, n = 10; Stage IV, n = 20) was further plotted (right panel). Statistical analysis in this figure was done by Student's t-test (ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001).
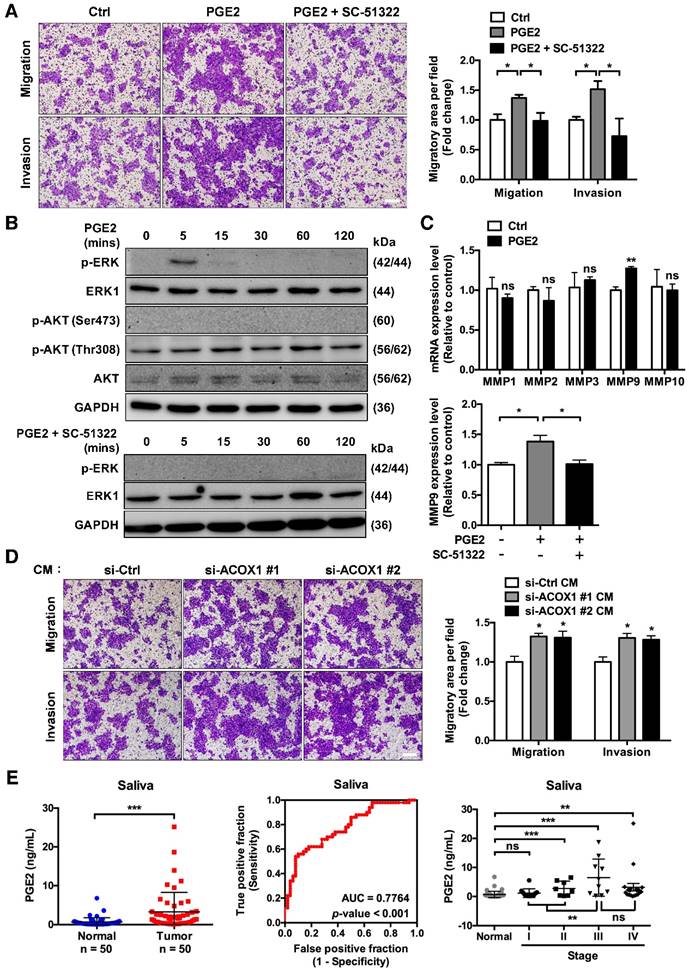
Cumulative evidence has suggested a role of PGE2 in tumor cell motility: PGE2 is known to promote colorectal cancer (CRC) cell migration and invasion through the β-arrestin-SRC-EGFR-PI3K-Akt cascade, while its role in pancreatic cancer cells is via ERK-ETS1-matrix metalloproteinase 2 (MMP2) signaling [29, 30]. We then set out to delineate the PGE2-dependent signaling associated with OSCC cell migration. Subsequent immunoblotting analyses showed a rapid but transient activation of ERK: phospho-ERK signal was detectable at 5 min following PGE2 stimulation but restored to basal level at 30 min (Figure 5B, top). In contrast, levels of phospho-Akt (Thr308 or Ser473) were either invariable or undetectable, respectively (Figure 5B, top), indicating that AKT-associated signaling was not activated in PGE2-treated OSCC cells. Next, given that PGE2 up-regulates the expression of certain matrix metalloproteinases in immunocytes and cancer cells [30-32], we subsequently examined the mRNA expression levels of various MMP molecules, such as MMP1, MMP2, MMP3, MMP9 and MMP10, in PGE2-stimulated OSCC cells. We discovered that PGE2 treatment markedly triggered an increase in MMP9 relative to control but not the other MMPs, at both the mRNA (Figure 5C, top) and protein (Figure S9) levels. Next, given that stimulation of SCC4 cells with PGE2 induces the expression of one of its cognate receptors (EP1) but not the other three receptor subtypes [33], we assessed the role of EP1 in PGE2 downstream signaling by subjecting cells to an EP1 receptor-specific antagonist (SC-51322). Our results showed that treatment with SC-51322 significantly antagonized the PGE2-induced cell migration and invasion (Figure 5A), ERK activation (Figure 5B, bottom) and MMP9 expression (Figure 5C, bottom and S9). Together, these observations were thus in line with the scenario that PGE2 promotes OSCC cell migration through an EP1-ERK-MMP9 signaling cascade.
To further substantiate this notion, we next performed migratory assay on OSCC cells cultured with the CM derived from ACOX1-silenced cells. Interestingly, addition of this CM evidently promoted migration and invasiveness of OSCC cells (Figure 5D), implying extracellular medium conditioned by ACOX1 down-regulation may accelerate OSCC cell migration. Owing to this possible oncogenic implication of the extra-tumor environment, we next addressed the possible clinical relevance of PGE2 secretion by quantifying the amounts of PGE2 in saliva specimens from OSCC patients and comparing them to healthy controls. Based on ELISA measurements, the salivary PGE2 levels were notably increased in OSCC vs. normal samples (Figure 5E, left). Furthermore, the ROC analysis validated that this salivary secretion may be a promising biomarker - AUC = 0.776, sensitivity = 0.70, and specificity = 0.68 using 0.566 ng/mL as the cut-off concentration for differentiating OSCC from non-tumorous group (p-value < 0.001) (Figure 5E, middle). When accounting for disease stages, we further found a slight but significant elevation of PGE2 levels during OSCC progression, and its production was maximal in stage III OSCC patients, characterized by local metastasis (Figure 5E, right). Collectively, these results illustrated the potential value of secreted PGE2 as a powerful biomarker for diagnosing OSCC progression.
miR-31-5p-ACOX1 axis enhances OSCC cell migration via up-regulating ERK-MMP9 signaling
Upon establishing the connection between PGE2 and ERK-MMP9 signaling, we next aimed to corroborate the implication of miR-31-5p-ACOX1 axis in this functional regard. To this end, inhibiting the expression of miR-31-5p in OSCC cells significantly attenuated the level of ERK1/2 phosphorylation (Figure S10A-S10B). By contrast, siRNA-mediated ACOX1 suppression in two OSCC cell lines activated the ERK pathway (Figure S10A-S10B). In terms of MMP expression, our data showed that ectopic expression of miR-31-5p significantly elevated the expression of MMP9 in OSCC cells (Figure S10C, left), while depletion of endogenous miR-31-5p conversely lowered MMP9 levels (Figure S10C, middle). Moreover, in SCC25 cells harboring ACOX1-specific siRNAs, we detected a discernible rise in MMP9 expression (Figure S10C, right).
To further substantiate the functional antagonism between miR-31-5p and ACOX1, we next carried out co-knockdown of miR-31-5p and ACOX1 in SCC25 cells. Consistent with earlier observations, we first found that miR-31-5p inhibition led to reduction in the levels and activities of MMP9 (Figure 6A-6B) and activated ERK (Figure 6C), as well as a lessened migration capability (Figure 6D), in comparison with control cells. In contrast, loss of ACOX1 had an opposite effect on these functional attributes. Simultaneous knockdown of ACOX1 in the miR-31-5p-depleted cells antagonized the alterations exerted by single knockdown of miR-31-5p: the down-regulation of phospho-ERK and MMP9 was reversed upon co-treatment of ACOX1 siRNAs (Figure 6A-6C), and the reduced migration and invasiveness potential as a consequence of miR-31-5p knockdown was rescued by ACOX1 inactivation (Figure 6D). Considered together, these results pinpointed miR-31-5p-ACOX1 axis as the key upstream step in the promigratory ERK-MMP9 signaling pathway.
ACOX1 interferes with the promigratory ERK-MMP9 signaling in OSCC cells. (A) Expression levels of miR-31-5p, ACOX1 and MMP9 in SCC25 transfected with the indicated combination of antisense oligomers (miR-31-5p) and siRNAs (ACOX1). (B) Gelatinase zymograms of lysates from SCC25 cells with the indicated co-transfection conditions. Silver staining was used as a loading control. (C) Immunoblotting of the indicated proteins for cells shown in (A). (D) SCC25 cells with the indicated transfection combinations were examined for transwell migration and Matrigel invasion activities. Average migratory area per field was determined and expressed as fold change relative to control. Scale bar, 200 μm. The bar graphs in (A) and (D) show mean ± SEM from three independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant). (E) Correlation between the expression levels of miR-31-5p and MMP9 (top) and between those of ACOX1 and MMP9 (bottom) in 39 normal (blue) and tumor (red) paired HNSCC samples, as determined by Pearson's r analysis. (F) Schematic illustration of the mechanistic model for the role of miR-31-5p-ACOX1 axis in oral squamous cell carcinoma.
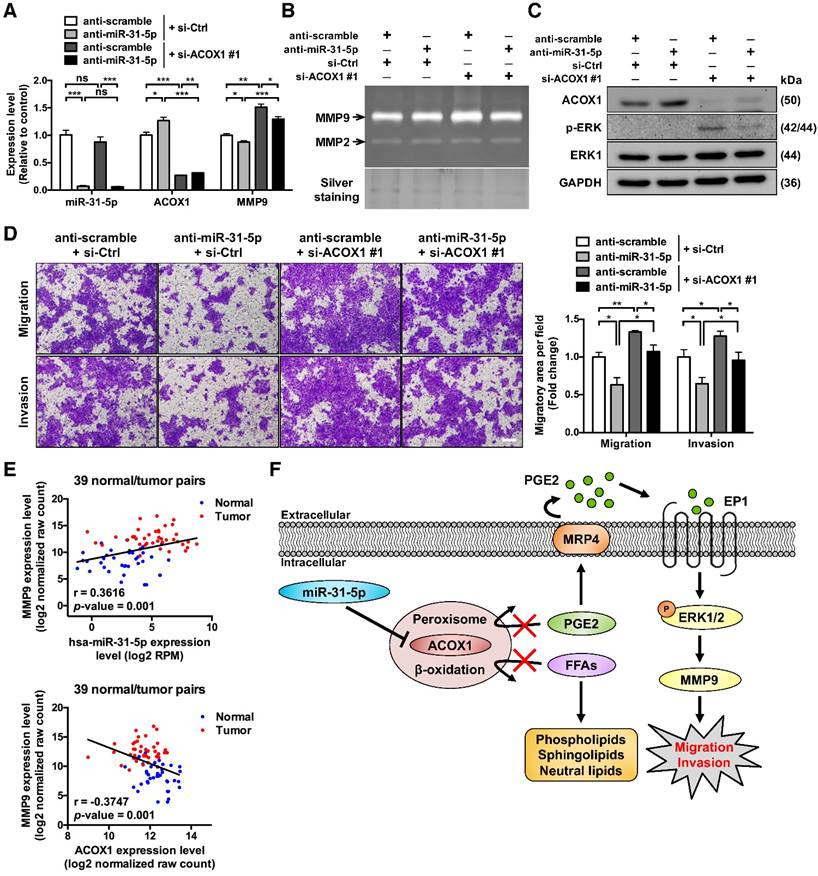
To further strengthen the clinical relevance of this regulatory pathway, we analyzed the HNSCC microRNA and mRNA deep sequencing data archived in TCGA. The expression levels of miR-31-5p showed a positive correlation with those of MMP9 (Figure 6E, top), whereas ACOX1 levels were negatively associated with MMP9 in the HNSCC specimens (Figure 6E, bottom). Moreover, we sought to elucidate the clinical significance of the levels of ACOX1 and MMP9 in patients with metastatic HNSCC. To this end, we analyzed two independent datasets, including the online Oncomine microarray and TCGA deep sequencing datasets, and discovered that the ACOX1 expression was distinctly down-regulated in HNSCC samples as compared with normal samples in both datasets, irrespective of the extent of local metastasis (Figure S10D-S10E, top). Interestingly, we further discovered that the expression levels of ACOX1 were progressively decreased in HNSCC patients with higher-grade lymph node metastasis, in line with our observations that ACOX1 silencing raised the migratory and invasive potential of OSCC cells. Concordantly, the MMP9 levels were dramatically elevated in HNSCC relative to normal specimens, and also exhibited a rise in correspondence to lymph node invasion (Figure S10D-S10E, bottom). In light of our data, we propose a model whereby the elevated expression of miR-31-5p in OSCC directly contributes to the down-regulation of ACOX1, consequently leading to accumulation of un-degradable lipid metabolites. In this reprogrammed lipidome, PGE2 represents a key determinant in OSCC cell migration and invasion and exerts its pro-tumorigenic role through activating EP1-ERK-MMP9 signaling (Figure 6F).
Discussion
While the critical roles of miRNAs in carcinogenesis have been demonstrated in the literature, a systems characterization of miRNA-mRNA networks in oral cancer pathogenesis has not been sufficiently established. A detailed investigation of the OSCC-associated miRNAs and their regulated networks may provide important mechanistic insights into the progression and treatment of this disease. In this regard, our present work advanced the understanding of OSCC biology by presenting the following lines of findings. 1) We catalogued an OSCC-associated altered miRNAome and further uncovered distinct cellular pathways potentially targeted by this miRNAs-mediated regulatory network. 2) The putative link of these regulatory molecules to lipid metabolism was substantiated by the delineation of a miR-31-5p-ACOX1 axis, which was further demonstrated to closely modulate cellular lipidomes as well as the migratory and invasive capacity of cancer cells. 3) PGE2, one of the key substrates of the ACOX1-mediated peroxisomal β-oxidation, acted as the key mediator of miR-31-5p function by augmenting the ERK-MMP9 signaling. Importantly, clinicopathological relevance of PGE2 was further corroborated by the observed elevation in saliva, which is in association with local invasion of OSCC and also indicative of its value as a promising disease biomarker. Viewed together, our findings have outlined a miR-31-5p-driven, tumor-promoting regulatory network that has considerable implications in the lipid metabolomic alterations associated with OSCC tumor biology.
Elevated miR-31-5p is a contributor to progression of advanced OSCC
In line with our observations, previous studies have reported that miR-31-5p is frequently up-regulated in multiple cancer types, including HNSCC [16], colorectal cancer [34] and lung cancer [35]. In contrast, its down-regulation in tumor has also been documented based on expression reduction in gastric cancer [36] and prostate cancer [37], and homozygous deletion of miR-31-5p host gene (MIR31HG) in mesothelioma [38]. Intriguingly, in esophageal cancer, miR-31-5p is distinctly altered between the two major subtypes of this malignancy, with its expression up-regulated in esophageal squamous cell carcinoma (ESCC) [39] but down-regulated in esophageal adenocarcinoma (EAC) [40]. In line with its frequent up-regulation, miR-31-5p is oncogenic in several squamous cell-derived malignancies, including OSCC, ESCC and skin squamous cell carcinoma. However, the findings that miR-31-5p is significantly over-expressed in lung adenocarcinoma and colorectal cancer (more than 90% of cases are adenocarcinoma) is seemingly contradictory to the expression profiles observed in EAC [35, 41, 42]. Viewed together, these findings imply a complex, tumor-dependent regulation and possibly dual role of this miRNA in tumor malignancy. To this end, several studies have mechanistically dissected this tumor-associated dysregulation of miR-31-5p. For instance, the expression levels of both miR-31-5p and MIR31HG are diminished in triple-negative breast cancer (TNBC) cell lines through promoter hypermethylation [43]. In addition, the promoter region of MIR31HG contains multiple potential binding sites for C/EBP-β, which exerts positive regulation towards cigarette smoke-induced MIR-31HG and miR-31-5p transcription [44]. In OSCC, it has further been shown that miR-31-5p elevation is mediated through the EGFR-AKT-C/EBP-β regulatory axis [45].
Despite its up-regulation in OSCC, the clinical relevance of miR-31-5p has not been characterized in depth. For patients exhibiting oral potentially malignant disorder (OPMD) features, an elevated miR-31-5p expression is more likely to result in malignant progression [46]. Furthermore, recent work by Hung et al. has implicated miR-31-5p in the early stage of oral carcinogenesis by showing that miR-31-5p cooperates with hTERT to immortalize normal oral keratinocytes [47]. These results are then consistent with our findings that enhanced miR-31-5p expression occurs in early stage of OSCC progression, which are often characterized by fast proliferation. However, our functional analyses revealed that miR-31-5p stimulates OSCC migratory and invasive capabilities without altering the rate of cell growth (Figure 4A-4D and S7). It is noteworthy that a progressive up-regulation of miR-31-5p is associated with advanced clinicopathological stages in lung adenocarcinoma, and that higher miR-31-5p levels in patients with lung adenocarcinoma also correlate with lymph node metastasis [48] and poor five-year overall survival [35]. Recently, work by Lu et al. has found that miR-31-5p promotes the oncogenicity and stemness of HNSCC via directly targeting the 3' UTR of AT-rich interacting domain 1A (ARID1A), leading to transactivation of pluripotency genes such as Nanog, OCT4 and Sox2 [49]. In line with this pro-tumorigenic regulatory network, HNSCC tumors carrying an elevated expression of miR-31-5p, together with a higher expression of pluripotency genes and lower level of ARID1A, display poorer survival and prognosis. Collectively, these observations provide support for multiple roles of miR-31-5p at distinct stages of carcinogenesis; it may function in early steps of OSCC development as a driver to promote cell immortalization and transformation, whereas its invariably increased level in late aggressive stages may contribute to other oncogenic attributes, such as migration and invasion.
The connection between miRNA and tumor-associated lipid metabolism
The importance of miRNAs in lipid homeostasis and tumor-related lipidomic alteration has recently emerged. In this capacity, liver-specific miR-122-5p is reportedly associated with hepatic cholesterol and lipid metabolism, as miR-122-5p inactivation triggers down-regulation of plasma cholesterol and triglycerides in high-fat diet mice [50]. Mechanistically, miR-122-5p is known to mediate translational inhibition of the cholesterol 7α-hydroxylase (CYP7A1), the first and rate-limiting enzyme in cholesterol-derived bile acid synthesis. Concordantly, treatment of miR-122-5p inhibitors in human HepG2 cells leads to bile acids elevation, further supporting the role of miR-122-5p in CYP7A1-dependent plasma cholesterol concentrations [50, 51]. miR-122-5p homozygous knockout mice are also shown to manifest steatohepatitis and fibrosis features, as well as spontaneous hepatocellular carcinoma (HCC) [52]. Our present study has strengthened this emerging lipidomic role of miRNAs by uncovering a novel regulator with critical implication in tumor-associated lipid metabolomes. We further showed that miR-31-5p targets a key enzyme in peroxisome and that this miR-31-5p-ACOX1 axis may constitute an oncogenic switch in cancer progression.
Tumor-associated lipid alteration
Given the critical roles of lipids in many cellular processes, dysregulation of lipid metabolism has been conceivably linked to cancer [53]. An important finding of our current study is the identification of PGE2 as key mediator of the promigratory miR-31-5p-ACOX1 pathway and also as a promising salivary biomarker for monitoring OSCC progression. Interestingly, a recent salivary metabolomics study on OSCC patients has unveiled a panel of five metabolites - lactic acid, γ-aminobutyric acid, valine, phenylalanine and n-eicosanoic acid - as altered metabolites in the OSCC vs. non-cancerous samples. Among them, the concentrations of lactic acid and n-eicosanoic acid are elevated but those of the other three showed significant reduction [54]. While these findings underscore the significance of metabolic reprogramming in malignancy, the functional implications of these metabolites and their tumorigenic contributions remain presently unclear. Aside from PGE2, our own UPLC-MS-based lipidomic analyses also uncovered several altered metabolites in the ACOX1-depleted cells - elevated levels of phosphatidylethanolamine (PE), phosphatidylcholine (PC), ceramide, glucosylceramide and lactosylceramide, in contrast to the decline of certain phosphatidylinositols (Figure S6C) - raising the possibility that additional lipid metabolites may be in effect. Notably, several recent studies have also revealed the link of lipidomic changes in cellular transformation. Work by Lv et al. highlighted a rise in serum levels of palmitic acid (C16:0), stearic acid (C18:0) and linoleic acid (C18:2) in patients with breast cancer, and further identified three types of free fatty acid (C16:0, C18:2 and total free fatty acids) as novel biomarkers for breast cancer detection [55]. Similarly, Cheng et al. reported higher levels of phosphatidylethanolamine and phosphatidylcholine in breast cancer patients vs. normal groups [56]. Together with our findings, these reports therefore attest to the scenario that excess fatty acids are utilized to synthesize phospholipids during carcinogenesis. How this reprogrammed lipidomes might affect oncogenic signaling and ultimately impact tumor biology remains an unresolved issue and warrants further investigation.
Balanced PGE2 synthesis and degradation is pivotal in OSCC oncogenesis
Prostaglandins (PGs) are biologically active lipids and have been linked to tissue homoeostasis, inflammation and cancer development. Given these widespread and important functions, levels of PGs are dynamically controlled by a myriad of enzymes and pathways. The biosynthesis of PGs is successively carried out by three types of enzymes: cytoplasmic phospholipase A2 (cPLA2), cyclooxygenases (COXs) and a group of PG synthases [28, 57]. First, arachidonic acids (AA) are liberated from membrane phospholipids via cPLA2. Two COX proteins, COX-1 and COX-2, catalyze the subsequent conversion of free AAs to PGG2 and then PGH2. Finally, specific PG synthases are responsible for metabolizing the PGH2 to their corresponding prostaglandins. For instance, PGE synthase (PGES) mediates the production of PGE2, the predominant product derived from AA. Once synthesized, the PGs are exported to the extracellular space by specific multidrug resistance-associated proteins (MRPs) and subsequently exert their physiological functions via interacting with their cognate receptors. Conversely, the metabolic breakdown of PGs is initiated through a two-step process: their uptake is catalyzed by PG transporter (PGT) [57, 58], which is followed by, in the case of PGE2, rapid inactivation to 15-keto-PGE2 by 15-hydroxyprostaglandin dehydrogenase (15-PGDH) and continuously to 13, 14-dihydro-15-keto-PGE2 by prostaglandin reductase (PTGR2) [59]. Furthermore, these inactivated PGE2 are further degraded through two cycles of peroxisomal β-oxidation to tetranor-PGE metabolites (tetranor-PGEMs), the major urinary metabolites found in mammals [60, 61]. Multiple studies have ascribed ACOX1 as the primary oxidase for this process, by showing that synthetic PGE2-CoA is oxidized exclusively by rat liver ACOX1 [27, 62].
In line with the pivotal role of PGE2 in tumor progression as revealed by our study, recent reports have ascribed tumorigenic relevance to various components of the PGE2-asscoiated pathway. To this end, the upstream synthetic enzymes, COX-2 and PGES, are reportedly up-regulated in many human malignancies, including HNSCC [63]. Moreover, treatment with COX antagonists is known to significantly inhibit cell invasion and lessen the expression and activity of MMP2 and MMP9 in HNSCC cells [64]. In terms of the downstream signaling of PGE2, for which there are four cognate receptors (EP1-EP4), only EP1 is notably up-regulated in HNSCC compared with paired non-tumoral mucosa [63], which possibly could be attributed to PGE2 induction [33]. EP4 receptor might also contribute to metastatic behavior, as treatment with EP4 antagonists significantly inhibits breast cancer metastasis [65]. While the functional selectivity of distinct EP receptors in various cancer types remains a currently unresolved issue, blockade of the EP receptors and/or the PGE2 biosynthetic pathway by corresponding antagonists may serve as a new treatment strategy for PGE2-positive tumors.
Abbreviations
OSCC: oral squamous cell carcinoma; ACOX: acyl-CoA oxidase; Ct: threshold cycle; ESCC: esophageal squamous cell carcinoma; HNSCC: head and neck squamous cell carcinoma; HPV: human papillomavirus; LNA: locked nucleic acids; miRNAs: microRNAs; PGE2: prostaglandin E2; SEM: standard error of the mean; MMPs: matrix metalloproteinases; VLCFAs: very long chain fatty acids; PUFAs: polyunsaturated fatty acids; 3' UTR: 3' untranslated region; TCGA: The Cancer Genome Atlas; CM: conditioned media.
Acknowledgements
We are grateful to Dr. Mei-Ling Cheng and the members of the Metabolomics Core Laboratory at Healthy Aging Research Center of Chang Gung University for assistance with untargeted lipid metabolomics analysis. We are also thankful for the high-throughput cell image acquisition system (IN Cell Analyzer 1000) and image analysis software (IN Cell Investigator) provided by the Core Instrument Center of Chang Gung University. This work was supported by grants from the Ministry of Science and Technology of Taiwan (MOST103-2632-B-182-001, MOST104-2320-B-182-029-MY3, MOST104-2632-B-182-001, MOST105-2314-B-182-061-MY4, MOST105-2632-B-182-001, and MOST106-2320-B-182-035-MY3), Chang Gung Memorial Hospital (CMRPD1F0571 and CMRPG3D1514 to HL; CMRPD3E0153, CMRPD1F0442, and BMRP960 to BC-MT), and the Ministry of Education, Taiwan. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Funding for open access charge: Ministry of Science and Technology of Taiwan.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nature reviews Cancer. 2006;6:259-69
2. Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nature reviews Cancer. 2015;15:321-33
3. Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Developmental biology. 2007;302:1-12
4. Chi AC, Day TA, Neville BW. Oral cavity and oropharyngeal squamous cell carcinoma-an update. CA: a cancer journal for clinicians. 2015;65:401-21
5. Global Burden of Disease Cancer C, Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M. et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015;1:505-27
6. Wang B, Zhang S, Yue K, Wang XD. The recurrence and survival of oral squamous cell carcinoma: a report of 275 cases. Chinese journal of cancer. 2013;32:614-8
7. Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nature reviews Cancer. 2011;11:9-22
8. Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral oncology. 2009;45:301-8
9. Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral oncology. 2009;45:324-34
10. Wu BH, Xiong XP, Jia J, Zhang WF. MicroRNAs: new actors in the oral cancer scene. Oral oncology. 2011;47:314-9
11. Tu HF, Lin SC, Chang KW. MicroRNA aberrances in head and neck cancer: pathogenetic and clinical significance. Current opinion in otolaryngology & head and neck surgery. 2013;21:104-11
12. Shiiba M, Uzawa K, Tanzawa H. MicroRNAs in Head and Neck Squamous Cell Carcinoma (HNSCC) and Oral Squamous Cell Carcinoma (OSCC). Cancers. 2010;2:653-69
13. Liu CJ, Kao SY, Tu HF, Tsai MM, Chang KW, Lin SC. Increase of microRNA miR-31 level in plasma could be a potential marker of oral cancer. Oral diseases. 2010;16:360-4
14. Liu CJ, Lin SC, Yang CC, Cheng HW, Chang KW. Exploiting salivary miR-31 as a clinical biomarker of oral squamous cell carcinoma. Head & neck. 2012;34:219-24
15. Li J, Huang H, Sun L, Yang M, Pan C, Chen W. et al. MiR-21 indicates poor prognosis in tongue squamous cell carcinomas as an apoptosis inhibitor. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:3998-4008
16. Liu CJ, Tsai MM, Hung PS, Kao SY, Liu TY, Wu KJ. et al. miR-31 ablates expression of the HIF regulatory factor FIH to activate the HIF pathway in head and neck carcinoma. Cancer research. 2010;70:1635-44
17. Lodhi IJ, Semenkovich CF. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell metabolism. 2014;19:380-92
18. Van Veldhoven PP. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. Journal of lipid research. 2010;51:2863-95
19. Baes M, Van Veldhoven PP. Mouse models for peroxisome biogenesis defects and beta-oxidation enzyme deficiencies. Biochimica et biophysica acta. 2012;1822:1489-500
20. Wanders RJ. Peroxisomes, lipid metabolism, and peroxisomal disorders. Molecular genetics and metabolism. 2004;83:16-27
21. Ferdinandusse S, Denis S, Hogenhout EM, Koster J, van Roermund CW, L IJ. et al. Clinical, biochemical, and mutational spectrum of peroxisomal acyl-coenzyme A oxidase deficiency. Human mutation. 2007;28:904-12
22. Huang J, Viswakarma N, Yu S, Jia Y, Bai L, Vluggens A. et al. Progressive endoplasmic reticulum stress contributes to hepatocarcinogenesis in fatty acyl-CoA oxidase 1-deficient mice. The American journal of pathology. 2011;179:703-13
23. Noble WS. How does multiple testing correction work? Nature biotechnology. 2009;27:1135-7
24. Chen YJ, Chang JT, Lee L, Wang HM, Liao CT, Chiu CC. et al. DSG3 is overexpressed in head neck cancer and is a potential molecular target for inhibition of oncogenesis. Oncogene. 2007;26:467-76
25. Binns D, Januszewski T, Chen Y, Hill J, Markin VS, Zhao Y. et al. An intimate collaboration between peroxisomes and lipid bodies. The Journal of cell biology. 2006;173:719-31
26. Bozza PT, Viola JP. Lipid droplets in inflammation and cancer. Prostaglandins, leukotrienes, and essential fatty acids. 2010;82:243-50
27. Van Veldhoven PP, Vanhove G, Assselberghs S, Eyssen HJ, Mannaerts GP. Substrate specificities of rat liver peroxisomal acyl-CoA oxidases: palmitoyl-CoA oxidase (inducible acyl-CoA oxidase), pristanoyl-CoA oxidase (non-inducible acyl-CoA oxidase), and trihydroxycoprostanoyl-CoA oxidase. The Journal of biological chemistry. 1992;267:20065-74
28. Wang D, Dubois RN. Eicosanoids and cancer. Nature reviews Cancer. 2010;10:181-93
29. Buchanan FG, Wang D, Bargiacchi F, DuBois RN. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. The Journal of biological chemistry. 2003;278:35451-7
30. Ito H, Duxbury M, Benoit E, Clancy TE, Zinner MJ, Ashley SW. et al. Prostaglandin E2 enhances pancreatic cancer invasiveness through an Ets-1-dependent induction of matrix metalloproteinase-2. Cancer research. 2004;64:7439-46
31. Bu X, Zhao C, Dai X. Involvement of COX-2/PGE(2) Pathway in the Upregulation of MMP-9 Expression in Pancreatic Cancer. Gastroenterology research and practice. 2011;2011:214269
32. Yen JH, Kocieda VP, Jing H, Ganea D. Prostaglandin E2 induces matrix metalloproteinase 9 expression in dendritic cells through two independent signaling pathways leading to activator protein 1 (AP-1) activation. The Journal of biological chemistry. 2011;286:38913-23
33. Yang SF, Chen MK, Hsieh YS, Chung TT, Hsieh YH, Lin CW. et al. Prostaglandin E2/EP1 signaling pathway enhances intercellular adhesion molecule 1 (ICAM-1) expression and cell motility in oral cancer cells. The Journal of biological chemistry. 2010;285:29808-16
34. Sun D, Yu F, Ma Y, Zhao R, Chen X, Zhu J. et al. MicroRNA-31 activates the RAS pathway and functions as an oncogenic MicroRNA in human colorectal cancer by repressing RAS p21 GTPase activating protein 1 (RASA1). The Journal of biological chemistry. 2013;288:9508-18
35. Edmonds MD, Boyd KL, Moyo T, Mitra R, Duszynski R, Arrate MP. et al. MicroRNA-31 initiates lung tumorigenesis and promotes mutant KRAS-driven lung cancer. The Journal of clinical investigation. 2016;126:349-64
36. Wang H, Zhang X, Liu Y, Ni Z, Lin Y, Duan Z. et al. Downregulated miR-31 level associates with poor prognosis of gastric cancer and its restoration suppresses tumor cell malignant phenotypes by inhibiting E2F2. Oncotarget. 2016;7:36577-89
37. Lin PC, Chiu YL, Banerjee S, Park K, Mosquera JM, Giannopoulou E. et al. Epigenetic repression of miR-31 disrupts androgen receptor homeostasis and contributes to prostate cancer progression. Cancer research. 2013;73:1232-44
38. Ivanov SV, Goparaju CM, Lopez P, Zavadil J, Toren-Haritan G, Rosenwald S. et al. Pro-tumorigenic effects of miR-31 loss in mesothelioma. The Journal of biological chemistry. 2010;285:22809-17
39. Zhang T, Wang Q, Zhao D, Cui Y, Cao B, Guo L. et al. The oncogenetic role of microRNA-31 as a potential biomarker in oesophageal squamous cell carcinoma. Clin Sci (Lond). 2011;121:437-47
40. Leidner RS, Ravi L, Leahy P, Chen Y, Bednarchik B, Streppel M. et al. The microRNAs, MiR-31 and MiR-375, as candidate markers in Barrett's esophageal carcinogenesis. Genes Chromosomes Cancer. 2012;51:473-9
41. Liu X, Sempere LF, Ouyang H, Memoli VA, Andrew AS, Luo Y. et al. MicroRNA-31 functions as an oncogenic microRNA in mouse and human lung cancer cells by repressing specific tumor suppressors. The Journal of clinical investigation. 2010;120:1298-309
42. Yang MH, Yu J, Chen N, Wang XY, Liu XY, Wang S. et al. Elevated microRNA-31 expression regulates colorectal cancer progression by repressing its target gene SATB2. PloS one. 2013;8:e85353
43. Augoff K, McCue B, Plow EF, Sossey-Alaoui K. miR-31 and its host gene lncRNA LOC554202 are regulated by promoter hypermethylation in triple-negative breast cancer. Molecular cancer. 2012;11:5
44. Xi S, Yang M, Tao Y, Xu H, Shan J, Inchauste S. et al. Cigarette smoke induces C/EBP-beta-mediated activation of miR-31 in normal human respiratory epithelia and lung cancer cells. PloS one. 2010;5:e13764
45. Lu WC, Kao SY, Yang CC, Tu HF, Wu CH, Chang KW. et al. EGF up-regulates miR-31 through the C/EBPbeta signal cascade in oral carcinoma. PloS one. 2014;9:e108049
46. Hung KF, Liu CJ, Chiu PC, Lin JS, Chang KW, Shih WY. et al. MicroRNA-31 upregulation predicts increased risk of progression of oral potentially malignant disorder. Oral oncology. 2015
47. Hung PS, Tu HF, Kao SY, Yang CC, Liu CJ, Huang TY. et al. miR-31 is upregulated in oral premalignant epithelium and contributes to the immortalization of normal oral keratinocytes. Carcinogenesis. 2014;35:1162-71
48. Meng W, Ye Z, Cui R, Perry J, Dedousi-Huebner V, Huebner A. et al. MicroRNA-31 predicts the presence of lymph node metastases and survival in patients with lung adenocarcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:5423-33
49. Lu WC, Liu CJ, Tu HF, Chung YT, Yang CC, Kao SY. et al. miR-31 targets ARID1A and enhances the oncogenicity and stemness of head and neck squamous cell carcinoma. Oncotarget. 2016
50. Flowers E, Froelicher ES, Aouizerat BE. MicroRNA regulation of lipid metabolism. Metabolism: clinical and experimental. 2013;62:12-20
51. Song KH, Li T, Owsley E, Chiang JY. A putative role of micro RNA in regulation of cholesterol 7alpha-hydroxylase expression in human hepatocytes. Journal of lipid research. 2010;51:2223-33
52. Tsai WC, Hsu SD, Hsu CS, Lai TC, Chen SJ, Shen R. et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. The Journal of clinical investigation. 2012;122:2884-97
53. Zhang F, Du G. Dysregulated lipid metabolism in cancer. World journal of biological chemistry. 2012;3:167-74
54. Wei J, Xie G, Zhou Z, Shi P, Qiu Y, Zheng X. et al. Salivary metabolite signatures of oral cancer and leukoplakia. International journal of cancer Journal international du cancer. 2011;129:2207-17
55. Lv W, Yang T. Identification of possible biomarkers for breast cancer from free fatty acid profiles determined by GC-MS and multivariate statistical analysis. Clinical biochemistry. 2012;45:127-33
56. Cheng M, Bhujwalla ZM, Glunde K. Targeting Phospholipid Metabolism in Cancer. Frontiers in oncology. 2016;6:266
57. Menter DG, Dubois RN. Prostaglandins in cancer cell adhesion, migration, and invasion. International journal of cell biology. 2012;2012:723419
58. Kochel TJ, Fulton AM. Multiple drug resistance-associated protein 4 (MRP4), prostaglandin transporter (PGT), and 15-hydroxyprostaglandin dehydrogenase (15-PGDH) as determinants of PGE2 levels in cancer. Prostaglandins & other lipid mediators. 2015;116-117:99-103
59. Wu YH, Ko TP, Guo RT, Hu SM, Chuang LM, Wang AH. Structural basis for catalytic and inhibitory mechanisms of human prostaglandin reductase PTGR2. Structure. 2008;16:1714-23
60. Diczfalusy U, Alexson SE. Identification of metabolites from peroxisomal beta-oxidation of prostaglandins. Journal of lipid research. 1990;31:307-14
61. Hamberg M, Samuelsson B. On the metabolism of prostaglandins E 1 and E 2 in man. The Journal of biological chemistry. 1971;246:6713-21
62. Schepers L, Casteels M, Vamecq J, Parmentier G, Van Veldhoven PP, Mannaerts GP. Beta-oxidation of the carboxyl side chain of prostaglandin E2 in rat liver peroxisomes and mitochondria. The Journal of biological chemistry. 1988;263:2724-31
63. Camacho M, Leon X, Fernandez-Figueras MT, Quer M, Vila L. Prostaglandin E(2) pathway in head and neck squamous cell carcinoma. Head & neck. 2008;30:1175-81
64. Koontongkaew S, Monthanapisut P, Saensuk T. Inhibition of arachidonic acid metabolism decreases tumor cell invasion and matrix metalloproteinase expression. Prostaglandins & other lipid mediators. 2010;93:100-8
65. Fulton AM, Ma X, Kundu N. Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer research. 2006;66:9794-7
Author contact
![]() Corresponding author: Bertrand Chin-Ming Tan, Graduate Institute of Biomedical Sciences and Department of Biomedical Sciences, College of Medicine, Chang Gung University, 259 Wenhua 1st Road, Guishan, Taoyuan 333, Taiwan, ROC. Tel.: +886-3-211-8800; Fax: +866-3-211-8700; E-mail: btancgu.edu.tw
Corresponding author: Bertrand Chin-Ming Tan, Graduate Institute of Biomedical Sciences and Department of Biomedical Sciences, College of Medicine, Chang Gung University, 259 Wenhua 1st Road, Guishan, Taoyuan 333, Taiwan, ROC. Tel.: +886-3-211-8800; Fax: +866-3-211-8700; E-mail: btancgu.edu.tw