Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(2):384-398. doi:10.7150/thno.22222 This issue Cite
Research Paper
Desacetylvinblastine Monohydrazide Disrupts Tumor Vessels by Promoting VE-cadherin Internalization
Xueping Lei1,2*, Minfeng Chen1,2*, Maohua Huang1,2, Xiaobo Li1,2, Changzheng Shi3, Dong Zhang3, Liangping Luo3, Youwei Zhang4, Nan Ma1,2, Heru Chen1, Huafeng Liang1,2, Wencai Ye1,2 ![]() , Dongmei Zhang1,2
, Dongmei Zhang1,2 ![]()
1. College of Pharmacy, Jinan University, Guangzhou 510632, China;
2. Guangdong Province Key Laboratory of Pharmacodynamic Constituents of Traditional Chinese Medicine and New Drugs Research, Jinan University, Guangzhou 510632, China;
3. The First Affiliated Hospital of Jinan University, Guangzhou 510632, China;
4. Department of Pharmacology, Case Comprehensive Cancer Center, School of Medicine, Case Western Reserve University, Cleveland, Ohio 44106, USA.
* These authors contributed equally to this work.
Received 2017-8-3; Accepted 2017-10-13; Published 2018-1-1
Abstract

Vinca alkaloids, the well-known tubulin-binding agents, are widely used for the clinical treatment of malignant tumors. However, little attention has been paid to their vascular disrupting effects, and the underlying mechanisms remain largely unknown. This study aims to investigate the vascular disrupting effect and the underlying mechanisms of vinca alkaloids.
Methods: The capillary disruption assay and aortic ring assay were performed to evaluate the in vitro vascular disrupting effect of desacetylvinblastine monohydrazide (DAVLBH), a derivate of vinblastine, and the in vivo vascular disrupting effect was assessed on HepG2 xenograft model using magnetic resonance imaging, hematoxylin and eosin staining and immunohistochemistry. Tubulin polymerization, endothelial cell monolayer permeability, western blotting and immunofluorescence assays were performed to explore the underlying mechanisms of DAVLBH-mediated tumor vascular disruption.
Results: DAVLBH has potent vascular disrupting activity both in vitro and in vivo. DAVLBH disrupts tumor vessels in a different manner than classical tubulin-targeting VDAs; it inhibits microtubule polymerization, promotes the internalization of vascular endothelial cadherin (VE-cadherin) and inhibits the recycling of internalized VE-cadherin to the cell membrane, thus increasing endothelial cell permeability and ultimately resulting in vascular disruption. DAVLBH-mediated promotion of VE-cadherin internalization and inhibition of internalized VE-cadherin recycling back to the cell membrane are partly dependent on inhibition of microtubule polymerization, and Src activation is involved in DAVLBH-induced VE-cadherin internalization.
Conclusions: This study sheds light on the tumor vascular disrupting effect and underlying mechanisms of vinca alkaloids and provides new insight into the molecular mechanism of tubulin-targeting VDAs.
Keywords: Vascular disrupting agents, vinca alkaloids, desacetylvinblastine monohydrazide, tubulin polymerization, VE-cadherin internalization.
Introduction
Vascular disrupting agents (VDAs) are potential anti-cancer agents that selectively destroy the pre-existing tumor vasculature and cause rapid and pronounced shutdown of blood flow in tumors, resulting in a large area of tumor ischemia and central necrosis [1, 2]. VDAs have been shown to possess potential preclinical activity in many types of cancer, including ovarian cancer, non-small cell lung carcinoma and sarcoma [3]. Because of resistant cells at the interface of tumor and normal tissue (viable rim) where rapid re-growth occurs, VDAs have been frequently used in combination with radiotherapy or chemotherapy in clinical studies [4]. Tubulin-binding VDAs are the most important group of VDAs. So far, extensive studies have analyzed the effects of the tubulin-binding VDA combretastatin and its derivatives, which bind to the colchicine-binding site on tubulin. Several derivatives of combretastatin, such as combretastatin A4 phosphate (CA4-P) and AVE8062, have been developed and applied in preclinical and clinical trials [4-6].
Vinca alkaloids, including vinblastine, vincristine, vinflunine, vinorelbine and vindesine, are tubulin-binding agents that have been widely used as antitumor therapy in the treatment of breast cancer, osteosarcoma and acute lymphocytic leukemia, either as a single agent or in combination with other drugs [7-9]. They have been considered cell cycle-specific cytotoxic drugs that bind to the vinblastine-binding site on tubulin, prevent microtubule polymerization, cause metaphase arrest and inhibit mitosis in cancer cells, leading to cell death [10, 11]. Vinca alkaloids also exert potent antiangiogenic effects by inhibiting endothelial cell motility [12], suppressing Rac1 and Cdc42 activity [13], down-regulating vascular endothelial growth factor receptor-2 expression [14] or disturbing EB1 localization [15]. However, little attention has been paid to the vascular disrupting effects of vinca alkaloids; there are a few reports that vinblastine and vinflunine cause profound and chronic reductions in blood flow in murine tumors [16-19]. Furthermore, the underlying mechanism by which vinca alkaloids disrupting vessels is still largely unknown.
Endothelial cell hyperpermeability is a major reason for VDA-induced tumor vascular disruption [20]. Vascular endothelial cadherin (VE-cadherin) is the endothelial-specific transmembrane component that localizes exclusively at endothelial cell-cell contacts; it maintains endothelial permeability and vessel integrity by homophilic interactions and association with catenins, including β-catenin and p120-catenin [21, 22]. VE-cadherin contains nine tyrosine residues that are mainly unphosphorylated in quiescent vessels and in confluent endothelial cell cultures [23]. The suppression of VE-cadherin phosphorylation is involved in the vascular disrupting effect of tubulin-binding VDAs. VDAs that bind to the colchicine-binding site on tubulin, such as CA4-P, JG-03-14 and TR-644, can inhibit VE-cadherin phosphorylation at Tyr685, which interferes with VE-cadherin/β-catenin-mediated cell-cell interactions, resulting in increased endothelial cell permeability and vascular disruption [24-26]. However, when endothelial cells are stimulated with VEGF and other factors, VE-cadherin is phosphorylated at Tyr658, and the VE-cadherin/β-catenin complex disengages to promote VE-cadherin internalization, which increases endothelial cell permeability [27-30]. Whether VE-cadherin internalization is involved in the vascular disrupting effect of tubulin-binding VDAs needs to be further investigated.
In the present study, we screened a panel of vinca alkaloids using a capillary disruption assay and found that desacetylvinblastine monohydrazide (DAVLBH) possessed the most potent vascular disrupting activity. DAVLBH disrupts vessels both in vitro and in vivo, and these effects are associated with the promotion of VE-cadherin internalization and the inhibition of internalized VE-cadherin recycling to the endothelial cell membrane.
Materials and methods
Reagents
DAVLBH (purity > 98%) was synthesized according to previously described methods [31]. DAVLBH was dissolved in DMSO to produce a 20 mM stock solution and was stored at -20°C protected from light. Vinblastine, vincristine, vinorelbine, vinflunine, paclitaxel and the Src selective inhibitor PP1 were from Selleck Chemicals (Houston, TX). Matrigel was purchased from Corning (Bedford, MA). Gd-DTPA was from Bayer Schering Pharma (Berlin, Germany). Antibodies against β-tubulin, VE-cadherin, p-VE-cadherinY658, Src, p-SrcTyr416, β-actin, Rab11a, clathrin and LAMP were obtained from Cell Signaling Technology (Danvers, MA). The antibody against CD31 was obtained from R&D Systems (Minneapolis, MN). The antibody against BV9 was from Merck Millipore (Darmstadt, Germany). Alexa Fluor 546 Donkey anti-Goat IgG and Alexa Fluor 488 Donkey anti-Rabbit IgG were purchased from Life Technologies (Invitrogen, Eugene, OR). The endothelial cell medium (ECM) supplement with 5% fetal bovine serum (FBS, Gibco BRL, Grand Island, NY), 1% penicillin/streptomycin solution, 10 µg/mL BSA, 5 µg/mL apo-transferrin, 5 mg/mL insulin, 10 ng/mL EGF, 2 ng/mL FGF-2, 2 ng/mL VEGF, 2 ng/mL IGF-I, 1 mg/mL hydrocortisone and 10-7 M retinoic acid was purchased from ScienCell Research Laboratories (San Diego, CA). The tubulin polymerization assay kit was obtained from Cytoskeleton (Denver, CO). Vindesine, rhodamine phalloidin and other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Cells and cell culture
HepG2 hepatocellular carcinoma cells were purchased from American Type Culture Collection (ATCC, Manassas, Virginia) and cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Human umbilical vein endothelial cells (HUVECs) were from ScienCell Research Laboratories and were cultured in ECM. The cells were maintained in humidified environment containing 5% CO2 at 37°C.
Animals
Adult male Sprague-Dawley rats (weighing 220-240 g) were obtained from Guangdong Medical Experimental Animal Center (Guangzhou, China). Male BALB/c nu/nu mice (4-6 weeks old) were purchased from Huafukang Bioscience Co. Ltd. (Beijing, China). All animals were raised in an SPF environment with constant temperature and humidity and a 12 h light cycle. In addition, all animal experimental procedures were conducted under supervision of the Laboratory Animal Ethics Committee of Jinan University (Guangzhou, China).
Capillary disruption
The capillary disruption assay was performed as previously described with some modifications [25]. The newly formed capillaries were treated with or without various concentrations of DAVLBH for 4 h. The capillaries were photographed before and after DAVLBH treatment using an Olympus IX70 inverted microscope. The number of capillaries was calculated using Image-Pro Plus 6.0.
Aortic ring assay
The vascular disrupting effect was also evaluated with a modified aortic ring assay [32]. Briefly, 1.0-1.5 mm long aorta rings were placed in matrigel-coated 96-well plates and covered with another 50 µL of matrigel. When the microvessels developed to the point of branching, the rings were treated with or without different concentrations of DAVLBH for 4 h, and photographs were taken before and after treatment. The number of microvessels was calculated using Image-Pro Plus 6.0.
In vitro tubulin polymerization
The in vitro tubulin polymerization assay was performed with a tubulin polymerization assay kit following the instructions of the manufacturer. Tubulin proteins were suspended in reaction buffer (containing 10% glycerol and 1 mM GTP) with or without the indicated concentration of DAVLBH in 96-well plates. The fluorescence value was measured at 360 nm excitation and 420 nm emission; the fluorescence signal indicates microtubule formation from tubulin heterodimers. Fluorescence measurements were obtained at 2 min intervals for a total of 70 min. The negative control was 0.3% DMSO, and 3 µM paclitaxel served as a positive control.
Endothelial cell monolayer permeability
The endothelial cell monolayer permeability assay was performed in transwell inserts (with a polycarbonate filter, 0.4 mm pore; Corning Costar, Cambridge, MA) as described previously [33]. HUVEC monolayers in the upper inserts were treated with or without different concentrations of DAVLBH for 4 h; then, 1 mg/mL FITC-dextran (average MW 40,000; Sigma-Aldrich, USA) was added to the upper inserts, which were incubated for 30 min. The medium in the bottom chamber was pipetted into 96-well plates, and the wells were then evaluated in a fluorescence plate reader with an excitation wavelength of 488 nm and an emission wavelength of 520 nm.
Immunofluorescence
HUVECs were cultured in small confocal laser dishes overnight and then treated with or without DAVLBH for 4 h. Then, the cells were fixed with 4% paraformaldehyde for 0.5 h and permeabilized with 0.1% Triton X-100. Next, the cells were incubated overnight with the primary antibody, labeled with the appropriate Alexa Fluor secondary antibody for 1 h at room temperature, and stained with DAPI for 5 min. And the cells were stained with rhodamine phalloidin to identify the F-actin. The images were captured by a laser scanning confocal microscope (LSM 800, ZEISS) with a 63× objective. The colocalization analysis was conducted with Imaris Colocalization Software (Bitplane AG, Zurich, Switzerland).
VE-cadherin internalization
The VE-cadherin internalization assay was performed in accordance with a previous report [34]. The antibody against the VE-cadherin extracellular domain (BV9) was dialyzed into ECM containing 20 mM Hepes and 3% BSA. HUVECs were incubated with BV9 for 30 min at 4°C, washed with ice-cold PBS to remove unbound antibody and transferred to 37°C for 30 min. The cells were then treated with or without DAVLBH for 4 h and washed with acid PBS (pH 2.7, containing 25 mM glycine and 3% BSA) for 15 min to remove cell surface-bound antibody while retaining internalized antibody. Then, the cells were fixed and processed for immunofluorescence assays.
Western blot
HUVECs were harvested and lysed with RIPA buffer. The cell extracts were subjected to western blot analysis as previously described [35]. The trypsinization assay, which was performed as previously described, was used to distinguish cell surface and intracellular pools of VE-cadherin [34]. In brief, HUVECs treated with or without DAVLBH were rinsed and then incubated in trypsin-EDTA to remove cell surface VE-cadherin. Then, the cells were collected and lysed with RIPA buffer for western blot assay. As a negative control, cells were harvested in parallel using RIPA buffer without trypsinization.
Tumor xenografts
HepG2 cells (2×106) suspended in 200 µL of a 50% matrigel mixture were inoculated subcutaneously into the armpits of nude mice. When the tumor volume reaching approximately 400 mm3, the tumor-bearing mice were intravenously (i.v.) injected with saline or 0.75 mg/kg DAVLBH every two days. The mice were then examined for tumor growth and body weight changes. Tumors were measured using a slide caliper (Mitutoyo, Tokyo, Japan), and tumor volume was calculated using the following formula: a2 × b × 0.5, where a refers to the smaller diameter, and b is the diameter perpendicular to a. In addition, the mice were subjected to magnetic resonance imaging (MRI) after DAVLBH administration at various time points (0 h, 4 h, 24 h and 52 h).
MRI analysis
MRI was performed on a 1.5 T MR system (GE Healthcare Signa HDxt, Milwaukee, WI) with an eight-channel wrist coil. The mice were anesthetized with pentobarbital sodium, a 24 G catheter was inserted in the tail vein, and the mice were imaged in the supine position. Each mouse was scanned with T1-weighted imaging (T1WI), T2-weighted imaging (T2WI), dynamic contrast-enhanced MRI (DCE-MRI) and intravoxel incoherent motion diffusion-weighted imaging (IVIM-DWI) sequences. T1WI sequences were acquired with the following parameters: repetition/echo time (TR/TE) 400/17.6 ms, matrix size 256 × 192, field of view (FOV) 70 mm × 49 mm, slice thickness 2 mm, slice gap 0.2 mm, number of slices 8, number of excitations (NEX) 2. The T2WI sequences were acquired with the following parameters: TR/TE 2040/77.6 ms, matrix size 256 × 192, FOV 70 mm × 56 mm, slice thickness 2 mm, slice gap 0.2 mm, number of slices 8, and NEX 2. DCE-MRI was performed with a 3D fast spoiled gradient-recalled echo (3D-FSPGR), and the images were acquired at a temporal resolution of three seconds before and after the injection of 0.1 mmol/kg Gd-DTPA (Magnevist, Bayer Schering Pharma, Berlin, Germany) via the tail vein catheter, followed by an injection of 0.3 mL saline for flushing. The detailed parameters were as follows: TR/TE 40/2.4 ms, matrix size 128 × 96, FOV 70 mm × 56 mm, slice thickness 2 mm, number of slices 6, slice gap 0.2 mm, flip angle 35°, and NEX 1. The baseline T1 map was assessed using a variable flip angle T1 mapping method (flip angles: 3°, 6°, 9°, 12°, 15° and 35°). The IVIM-DWI was performed following a free-breathing single-shot echo-planar imaging pulse sequence (TR 4000 ms, TE 91.8 ms, slice thickness 2.0 mm, slice gap 0.2 mm, matrix size 128 × 96, and FOV 10×7 cm2) with diffusion gradients applied in three orthogonal directions (12 b values: 25, 50, 75, 100, 150, 200, 400, 600, 800, 1000, 1200, and 1500 s/mm2). All data were analyzed at the post-processing workstation (AW 2.0, GE Healthcare). The regions of interest were drawn by outlining the entire tumor boundary or the dorsal normal muscle on the contrast enhancement images, and the Ktrans and standard apparent diffusion coefficient (ADC) values were automatically calculated.
Histology and immunohistochemistry
Five mice in each group were sacrificed at each MRI examination time point, and the tumors were removed and fixed with 4% paraformaldehyde for 24 h. Then, the tumors were embedded in paraffin and cut into 5 μm-thick sections. The sections were stained with hematoxylin-eosin (H&E) following standard procedures. For immunohistochemistry, the sections were incubated with anti-CD31 antibody overnight at 4°C and then with HRP-conjugated secondary antibodies. The proteins were detected using a DAB kit, followed by counterstaining with hematoxylin. The images were visualized and recorded with an Olympus BX 53 microscope.
Statistical analysis
The data are expressed as the mean ± SEM, and the data were analyzed with GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA). Significant differences were evaluated using a two-tailed unpaired t test or one-way ANOVA followed by Tukey's test. A difference was considered significant when P < 0.05.
Results
DAVLBH disrupts the newly-formed capillaries in vitro and ex vivo
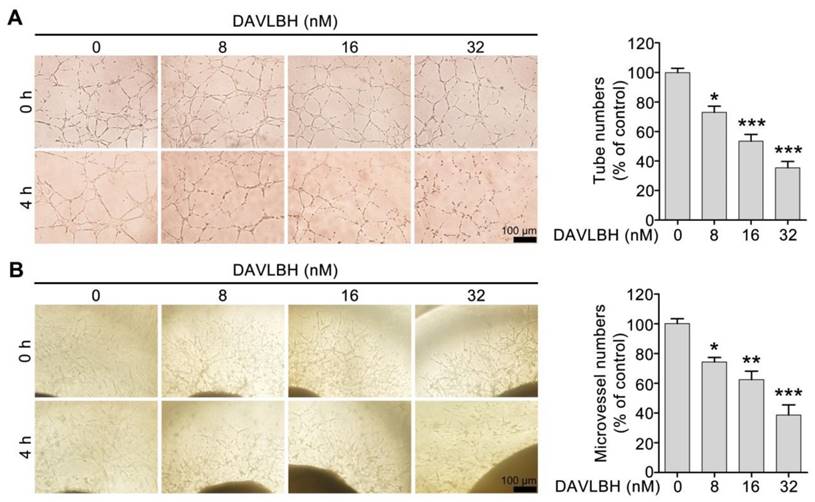
We first screened six vinca alkaloids, including vinblastine, vincristine, vinflunine, vinorelbine, vindesine and DAVLBH, using a capillary disruption assay to select a representative agent to investigate the vascular disrupting effect of vinca alkaloids. DAVLBH exhibited the most potent vascular disrupting activity on newly formed capillaries (Figure S1) and was therefore selected as the representative agent to further explore the vascular disrupting effect of vinca alkaloids. Within the non-cytotoxic concentrations (ranging from 8 to 32 nM), DAVLBH disrupted pre-established HUVEC tubes in a dose-dependent manner, as evidenced by the formation of disorganized sheaths (Figure 1A and Figure S2). Furthermore, DAVLBH caused the collapse of pre-established microvessels sprouting from rat aortic rings, as evidenced by the visible, small, broken particles; microvessels in the control group showed negligible change (Figure 1B). Taken together, these results suggest that DAVLBH significantly disrupts newly-formed capillaries both in vitro and ex vivo.
DAVLBH disrupts the newly-formed capillaries in vitro and ex vivo. (A) DAVLBH disrupted pre-established endothelial tubular networks in a dose-dependent manner. The formed endothelial cell tubes were treated with or without various concentrations of DAVLBH for 4 h. The tubular networks were photographed before and after treatment. (B) DAVLBH disrupted the sprouted microvessels in rat aortic rings. The rat aortas were cultured in matrigel-coated 96-well plates, and the sprouted microvessels were treated with or without various concentrations of DAVLBH for 4 h. The images were taken before and after DAVLBH treatment. Three independent experiments were performed, and representative images are shown. Scale bar, 100 µm. Quantitative data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with the control group (one-way ANOVA with Tukey's post hoc comparison).
DAVLBH disrupts tumor blood vessels in HepG2 xenografts
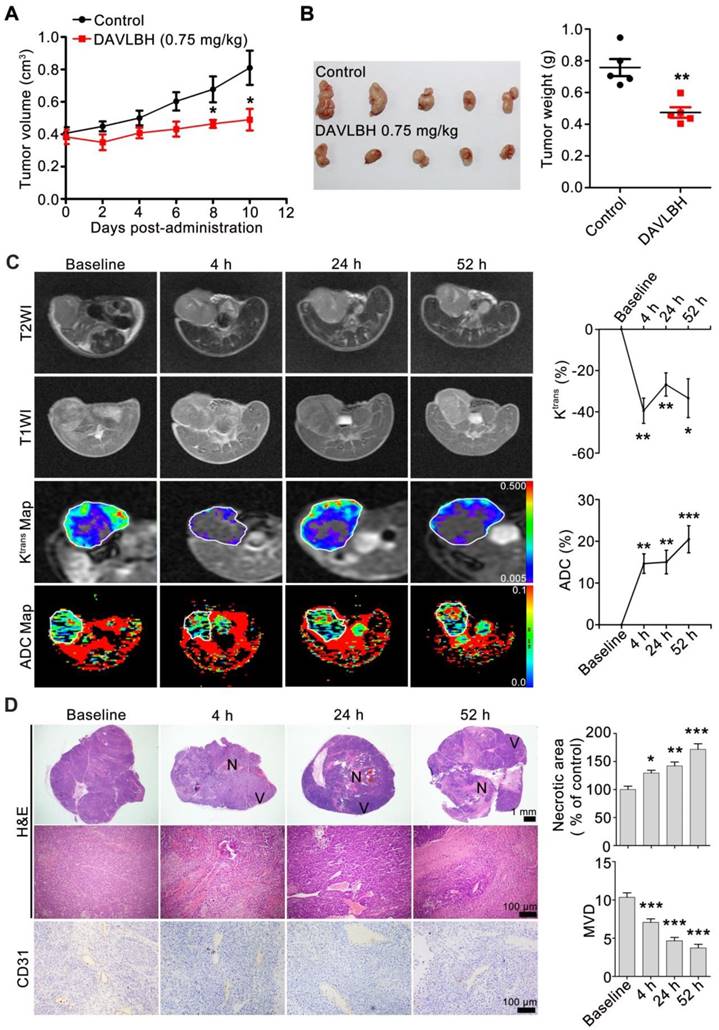
To explore the in vivo vascular disrupting activity of DAVLBH, DCE-MRI and diffusion-weighted magnetic resonance imaging (DW-MRI) experiments were performed on HepG2 xenografts. Our results showed that DAVLBH (0.75 mg/kg) treatment significantly suppressed the growth of HepG2 xenografts, with an inhibitory rate of approximately 40% (Figure 2A, B). MRI analysis showed that DAVLBH treatment for 4 h resulted in a rapid reduction in the Ktrans value, which is a constant indicating tissue perfusion or permeability [36]. The Ktrans value recovered stably but remained under the baseline, and a second administration of DAVLBH reduced Ktrans once again. In addition, DAVLBH treatment caused a rapid increase ADC value (Figure 2C), a constant that reflecting the changes in cellularity and water diffusion in proportion to tissue necrosis [37]. By contrast, DAVLBH had minimal effects on the Ktrans and ADC values in normal muscle (Figure S3). The MRI results were confirmed by pathological examination. DAVLBH treatment for 4, 24 and 52 h created a deficiency in CD31-positive endothelial cells and caused erythrocyte aggregation in the tumor core (Figure 2D), indicating that DAVLBH can rapidly disrupt tumor vessels. As a result, DAVLBH treatment led to a large area of necrosis, while an obvious viable rim were observed in the tumor periphery (Figure 2D). Altogether, DAVLBH selectively disrupts tumor vessels and inhibits the growth of HepG2 xenografts.
DAVLBH inhibits tumor growth by disrupting tumor vessels. (A) DAVLBH inhibited HepG2 xenograft growth. Once the tumors were established (approximately 400 mm3), mice bearing HepG2 xenografts received i.v. injections of saline or 0.75 mg/kg DAVLBH once every two days for a total of 5 doses. Tumor volume was plotted. The data are presented as the mean ± SEM. *P < 0.05 compared with the control group. (B) Tumors were removed and imaged at the end of the treatment period (left panel, n = 5), and tumor weight was calculated (right panel, n = 5). The data are presented as the mean ± SEM. **P < 0.01 compared with the control group. (C) MRI monitoring of vascular disruption by DAVLBH in HepG2 xenografts. Mice treated with DAVLBH were subjected to MRI at 0, 4, 24 and 52 h after the first drug administration. Representative images and quantification of Ktrans and ADC values are shown (n = 5), the white rings indicate the tumors. (D) Representative images of tumor sections stained with H&E and CD31. Tumors were removed at each MRI examination time point. Quantification of the images is shown (n = 5). N: central necrosis; V: viable rim. MVD: microvessel density. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus baseline (one-way ANOVA with Tukey's post hoc comparison).
DAVLBH increases endothelial cell permeability by promoting VE-cadherin internalization
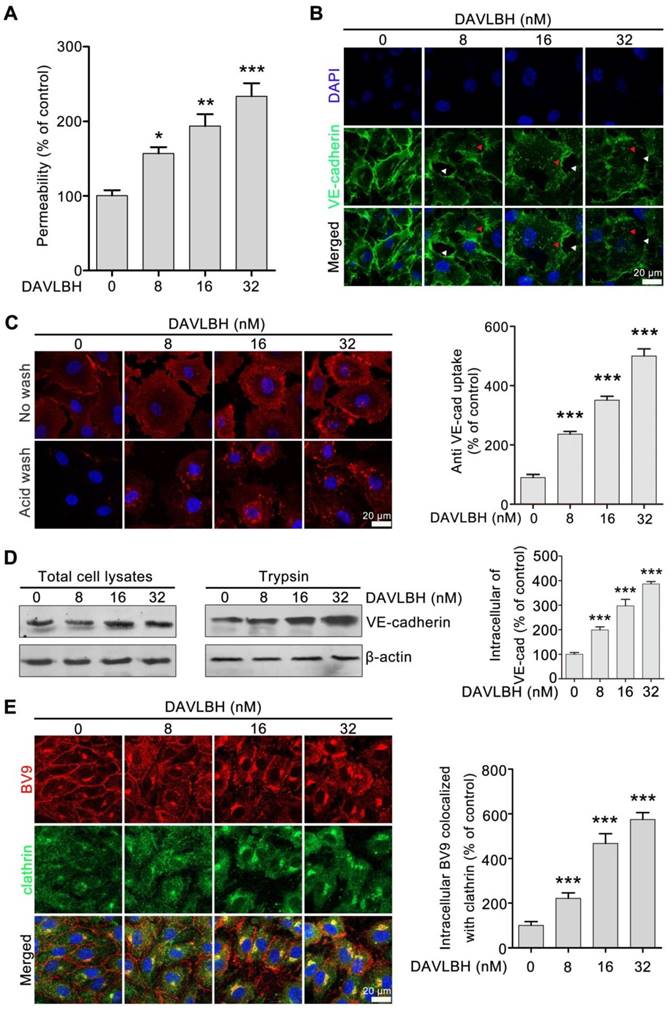
Given that endothelial hyperpermeability contributes to vascular leakage [21, 25], we next investigated whether DAVLBH-mediated disruption of tumor vessels is associated with its effect on endothelial permeability. Our results showed that DAVLBH significantly increased the permeability of a HUVEC monolayer in a dose-dependent manner (Figure 3A). VE-cadherin plays an important role in maintaining endothelial cell permeability [21]. In the control group, VE-cadherin was present at tight junctions between neighboring HUVECs. By contrast, DAVLBH treatment caused a discontinuous distribution and intracellular accumulation of VE-cadherin in HUVECs in a dose-dependent manner (Figure 3B), indicating that DAVLBH may promote the internalization of VE-cadherin. An internalization assay was conducted to directly visualize intracellular VE-cadherin. BV9 is a monoclonal antibody that recognizes the extracellular fragment of VE-cadherin, and BV9 antibody that remains after acid washing represents intracellular VE-cadherin [34]. Our results showed that the accumulation of intracellular VE-cadherin in HUVECs was increased sharply after DAVLBH treatment (Figure 3C). The trypsinization assay confirmed that DAVLBH promoted the internalization of VE-cadherin but had a negligible effect on total VE-cadherin levels (Figure 3D). Furthermore, we observed that internalized VE-cadherin colocalized with clathrin, indicating that DAVLBH induced VE-cadherin internalization in a clathrin-mediated manner (Figure 3E). Altogether, DAVLBH promotes clathrin-mediated VE-cadherin internalization, resulting in increasing endothelial cell permeability.
DAVLBH increases endothelial cell permeability by promoting the internalization of VE-cadherin. (A) DAVLBH increased the permeability of the endothelial cell monolayer. (B) DAVLBH disrupted VE-cadherin distribution. HUVECs were treated with or without various concentrations of DAVLBH for 4 h, labeled with VE-cadherin and analyzed by confocal microscopy. The white arrowheads indicate loss of tight junctions between neighboring cells, and the red arrowheads indicate the intracellular accumulation of VE-cadherin. (C) DAVLBH promoted the internalization of VE-cadherin in HUVECs. HUVECs labeled with the BV9 antibody were treated with or without various concentrations of DAVLBH, and the cells were then washed with or without acid PBS. Representative images and quantification of intracellular BV9 are shown (n = 3). (D) DAVLBH-induced VE-cadherin internalization was detected by western blot. After treatment with or without various concentrations of DAVLBH, trypsinized and non-trypsinized HUVECs were lysed, and the proteins were analyzed by western blot. Quantification of intracellular VE-cadherin is shown (n = 3). (E) DAVLBH induced VE-cadherin internalization in a clathrin-mediated manner. HUVECs treated with or without various concentrations of DAVLBH were labeled with anti-BV9 and anti-clathrin antibodies to visualize the colocalization of BV9 and clathrin. Representative images and quantification of BV9/clathrin colocalization are shown (n = 3). Scale bar, 20 µm. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with the control group (one-way ANOVA with Tukey's post hoc comparison).
DAVLBH inhibits internalized VE-cadherin recycling back to the endothelial cell membrane and promotes VE-cadherin degradation
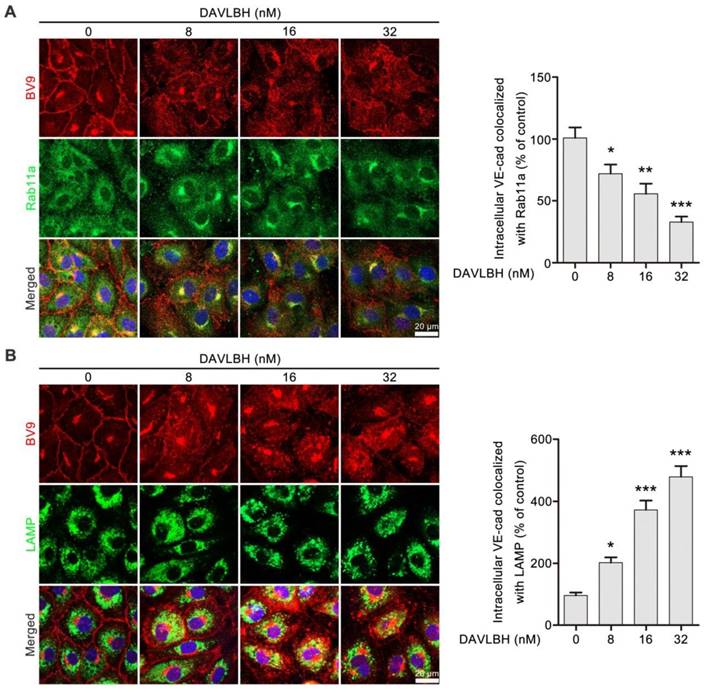
Internalized VE-cadherin can be either recycled back to the cell membrane to maintain VE-cadherin integrity or degraded in lysosomes, which leads to endothelial cell barrier dysfunction [38]. An immunofluorescence assay was performed to explore the fate of internalized VE-cadherin in HUVECs after DAVLBH treatment. Rab11a is required for VE-cadherin recycling [39, 40]. DAVLBH treatment increased intracellular VE-cadherin levels but decreased colocalization of BV9 and Rab11a, indicating that DAVLBH inhibited the recycling of internalized VE-cadherin back to the cell membrane (Figure 4A). In addition, DAVLBH increased the colocalization of BV9 and LAMP (a lysosome marker [41]) (Figure 4B), indicating DAVLBH promoted the degradation of VE-cadherin. Taken together, DAVLBH inhibits the recycling of internalized VE-cadherin back to the cell membrane and promotes the degradation of internalized VE-cadherin.
DAVLBH inhibits VE-cadherin recycling and promotes VE-cadherin degradation. (A) DAVLBH inhibited Rab11a-mediated VE-cadherin recycling to the cell membrane. After treatment with or without various concentrations of DAVLBH, HUVECs labeled with anti-Rab11a and anti-BV9 antibodies were analyzed by confocal microscopy to visualize BV9 and Rab11a colocalization. Representative images and quantification of BV9/Rab11a colocalization are shown (n = 3). (B) DAVLBH promoted VE-cadherin degradation. HUVECs treated with or without various concentrations of DAVLBH were labeled with anti-BV9 and anti-LAMP antibodies to visualize the colocalization of BV9 and LAMP. Representative images and quantification of colocalized BV9/LAMP are shown (n = 3). Scale bar, 20 µm. Data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with the control group (one-way ANOVA with Tukey's post hoc comparison).
DAVLBH inhibits microtubule polymerization and activates Src
As a derivative of vinblastine, DAVLBH has been reported to have powerful microtubule-destabilizing effects [42]. We confirmed the tubulin polymerization effect of DAVLBH using a tubulin polymerization assay. In the control group, the self-assembly of microtubules was increased in a time-dependent manner. Paclitaxel promoted microtubule formation, while DAVLBH inhibited tubulin polymerization in a dose-dependent manner (Figure 5A). Consistently, DAVLBH caused a dramatic dose-dependent disruption of microtubules in HUVECs, resulting in diffuse microtubules in a dose-dependent manner, as visualized by β-tubulin and F-actin staining (Figure 5B). We next investigated whether DAVLBH-meditated VE-cadherin internalization is due to the inhibition of microtubule polymerization, and an immunofluorescence assay was conducted to visualize the early changes in β-tubulin and VE-cadherin. Our results showed that DAVLBH suppressed the polymerization of β-tubulin within 15 min, at which point VE-cadherin was still organized, and minimal intracellular VE-cadherin was observed within 30 min, indicating that DAVLBH-mediated inhibition of microtubule polymerization occurs prior to VE-cadherin internalization (Figure 5C). Src is a key regulator of VE-cadherin internalization; thus, we explored whether DAVLBH can activate Src to promote VE-cadherin internalization [27]. DAVLBH promoted the phosphorylation of Src at Tyr416 and VE-cadherin at Y658 in a time- and dose-dependent manner (Figure 5D). Src was activated at 30 min, after microtubule damage occurred and before the phosphorylation of VE-cadherin (Figure 5C, D). Taken together, these results indicated that DAVLBH-induced VE-cadherin internalization may attribute to the inhibition of microtubule polymerization and that Src activation may play an important role in mediating VE-cadherin internalization.
DAVLBH inhibits tubulin polymerization and promotes Src activation. (A) DAVLBH inhibited tubulin polymerization in vitro. Purified porcine brain tubulin was incubated with reaction buffer with or without different concentrations of DAVLBH. DMSO was the negative control, and paclitaxel was the positive control. Tubulin polymerization was monitored by fluorescence (Ex: 360 nm, Em: 420 nm) once every two minutes for 70 min at 37°C. (B) DAVLBH inhibits microtubule polarization in HUVECs in a dose-dependent manner. HUVECs treated with or without various concentrations of DAVLBH for 4 h were labeled with a β-tubulin antibody and rhodamine-phalloidin to visualize β-tubulin and F-actin, respectively. (C) DAVLBH inhibits microtubule polymerization before it induced VE-cadherin disorganization. HUVECs treated with or without DAVLBH (16 nM) for different times were stained with β-tubulin and VE-cadherin antibodies. Representative images are shown. (D) DAVLBH induced the phosphorylation of Src at Try416 and VE-cadherin at Y658. HUVECs were exposed to DAVLBH, and proteins were collected for western blot analysis. Scale bar, 20 µm.
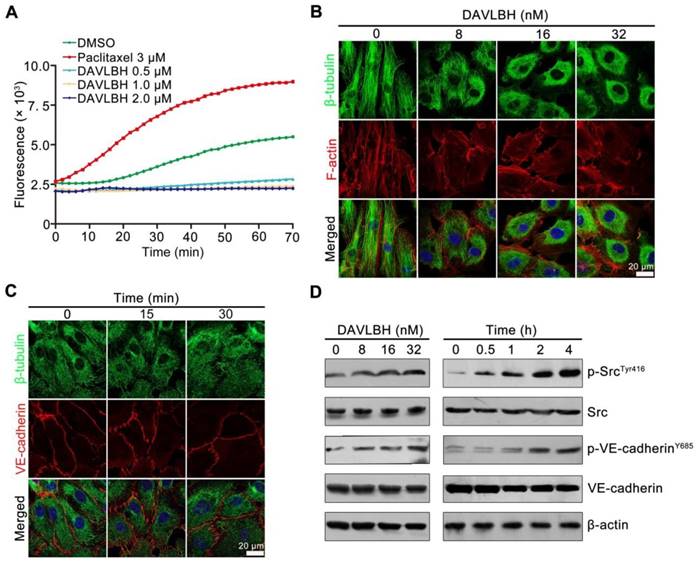
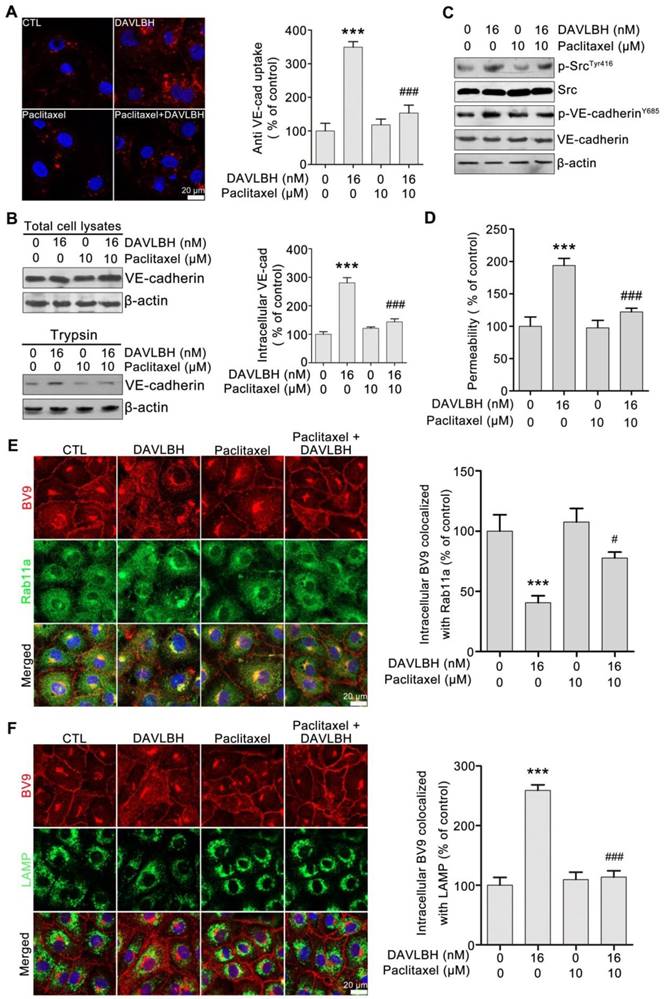
DAVLBH-mediated VE-cadherin internalization and inhibition of internalized VE-cadherin recycling to the cell membrane are partly dependent on the inhibition of microtubule polymerization
To investigate whether DAVLBH-induced inhibition of microtubule polymerization is associated with VE-cadherin internalization, HUVECs were pretreated with paclitaxel, an alternative microtubule stabilizing agent, and VE-cadherin internalization was evaluated. Paclitaxel pretreatment obviously attenuated DAVLBH-induced VE-cadherin internalization compared with DAVLBH treatment alone (Figure 6A). The trypsinization assay confirmed these results, as evidenced by the decrease of intracellular VE-cadherin in the paclitaxel pretreatment group (Figure 6B). In addition, paclitaxel pretreatment attenuated the phosphorylation of Src at Try416 and VE-cadherin at Y658 (Figure 6C) and weakened DAVLBH-induced endothelial hyperpermeability (Figure 6D). We also showed that the inhibitory effect of DAVLBH on internalized VE-cadherin recycling back to the cell membrane was diminished by paclitaxel pretreatment (Figure 6E), which also attenuated the DAVLBH-induced degradation of VE-cadherin (Figure 6F). Taken together, the effects of DAVLBH on VE-cadherin internalization and internalized VE-cadherin recycling back to the membrane are partly dependent on the failure of microtubule polymerization.
DAVLBH-induced VE-cadherin internalization partly depends on the inhibition of microtubule polymerization. (A) Paclitaxel suppressed DAVLBH-induced VE-cadherin internalization. HUVECs labeled with anti-BV9 were treated or not with DAVLBH for 4 h with or without paclitaxel pretreatment; then, the cells were washed with acid PBS and labeled with an Alexa Fluor secondary antibody. Representative images and image quantification are shown (n = 3). (B) Paclitaxel suppressed DAVLBH-induced VE-cadherin internalization. HUVECs pretreated with or without paclitaxel were treated with or without DAVLBH for 4 h, and the cells were lysed in RIPA with or without trypsin/EDTA to collect the intracellular and total VE-cadherin fractions, respectively. Quantification of intracellular VE-cadherin is shown (n = 3). (C) Paclitaxel inhibited the DAVLBH-induced phosphorylation of Src at Try416 and VE-cadherin at Y658. HUVECs pretreated with or without paclitaxel were treated with or without DAVLBH for 4 h. (D) Paclitaxel attenuated DAVLBH-mediated endothelial cell monolayer hyperpermeability. HUVEC monolayers pretreated with or without paclitaxel were treated with or without DAVLBH for 4 h. (E) Paclitaxel reversed the DAVLBH-mediated inhibition of internalized VE-cadherin recycling back to the cell membrane. HUVECs pretreated with or without paclitaxel were treated with or without DAVLBH for 4 h. Quantification of BV9/Rab11a colocalization is shown (n = 3). (F) Paclitaxel attenuated DAVLBH-induced lysosomal degradation of internalized VE-cadherin. HUVECs pretreated with or without paclitaxel were treated with or without DAVLBH for 4 h. Quantification of BV9/LAMP colocalization is shown (n = 3). Scale bar, 20 µm. The data are presented as mean ± SEM. ***P < 0.001 compared with the control group; #P < 0.05 and ###P < 0.001 compared with the DAVLBH group (one-way ANOVA with Tukey's post hoc comparison).
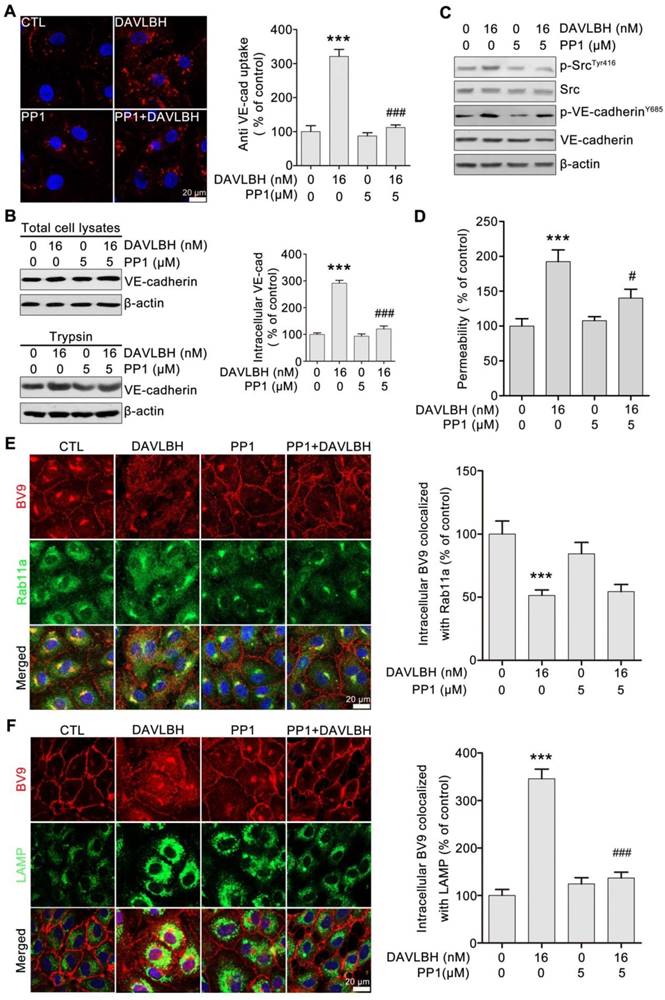
DAVLBH-induced VE-cadherin internalization partly depends on Src activation
To gain further insights into the role of Src activation in DAVLBH-induced VE-cadherin internalization, we pretreated HUVECs with PP1, a selective Src inhibitor, and then evaluated the effect of DAVLBH on VE-cadherin internalization. Our results showed that the DAVLBH-induced intracellular accumulation of VE-cadherin was diminished following pretreatment with PP1 (Figure 7A). The trypsinization assay confirmed these results; DAVLBH increased intracellular VE-cadherin levels, but PP1 pretreatment attenuated this effect (Figure 7B). In addition, PP1 pretreatment diminished the DAVLBH-induced phosphorylation of Src at Tyr416 and VE-cadherin at Y658 (Figure 7C). We also found that DAVLBH-induced HUVEC hyperpermeability was weakened by PP1 pretreatment (Figure 7D). However, PP1 had a negligible effect on the DAVLBH-induced inhibition of internalized VE-cadherin recycling back to the cell membrane (Figure 7E), but DAVLBH-induced VE-cadherin degradation was diminished by PP1 pretreatment (Figure 7F). Taken together, these data indicated that Src activation is critical for DAVLBH-induced VE-cadherin internalization but not for VE-cadherin recycling.
DAVLBH-mediated VE-cadherin internalization partly depends on Src activation. (A) The Src inhibitor PP1 suppressed DAVLBH-induced VE-cadherin internalization. HUVECs labeled with the BV9 antibody were treated or not with DAVLBH for 4 h with or without PP1 pretreatment; then, the cells were washed with acid PBS and labeled with an Alexa Fluor secondary antibody. Representative images and quantification of internalized VE-cadherin are shown (n = 3). (B) PP1 suppressed DAVLBH-induced VE-cadherin internalization. HUVECs pretreated with or without PP1 were treated with or without DAVLBH, and the cells were then lysed in RIPA with or without trypsin/EDTA to collect the intracellular and total VE-cadherin fractions, respectively. Quantification of intracellular VE-cadherin is shown (n = 3). (C) PP1 weakened the DAVLBH-induced phosphorylation of Src (Try416) and VE-cadherin (Y658). HUVECs pretreated with or without PP1 were treated with or without DAVLBH for 4 h. (D) PP1 attenuated DAVLBH-mediated endothelial cell monolayer hyperpermeability. HUVEC monolayers pretreated with or without PP1 were treated with or without DAVLBH for 4 h. (E) PP1 had a negligible effect on DAVLBH-induced recycling of internalized VE-cadherin back to the cell membrane. HUVECs pretreated with or without PP1 were treated with or without DAVLBH for 4 h. Quantification of BV9/Rab11a colocalization is shown (n = 3). (F) PP1 attenuated DAVLBH-induced lysosomal degradation of internalized VE-cadherin. HUVECs pretreated with or without PP1 were treated with or without DAVLBH for 4 h. Quantification of BV9/LAMP colocalization is shown (n = 3). The data are presented as mean ± SEM. ***P < 0.001 compared with the control group; #P < 0.05 and ###P < 0.001 compared with the DAVLBH group (one-way ANOVA with Tukey's post hoc comparison). Scale bar, 20 µm.
Discussion
Although vinca alkaloids have been widely used in preclinical and clinical cancer chemotherapy, their vascular disrupting effect and the underlying mechanism remain largely unexplored. In this study, we showed that a representative vinca alkaloid, DAVLBH, disrupted tumor vessels via a distinct mechanism from that of classical VDAs such as CA4-P (Figure S4). DAVLBH induced microtubule destabilization to activate Src, promoted VE-cadherin internalization and inhibited the recycling of internalized VE-cadherin to the endothelial cell membrane, thus resulting in endothelial hyperpermeability and vascular disruption.
The different effects of DAVLBH and CA4-P (colchicine-site binding VDAs) on VE-cadherin phosphorylation may be due to their distinct effects on Src activation. Src is the key regulator of VE-cadherin phosphorylation [43]. The inhibition of microtubule polymerization can either activate or inhibit Src phosphorylation, which may occur in a cell type-specific manner [44, 45]. In HUVECs, CA4-P inhibited Src activation, while vinca alkaloids, including vinblastine, vincristine, vinflunine, vinorelbine, vindesine and DAVLBH, significantly promoted Src activation (Figure S5). These different effects may be attributed to their different binding sites on tubulin, but the relationship between the inhibition of microtubule polymerization and Src activation remains unknown. Apart from Src, VE-cadherin phosphorylation is regulated by a complicated mechanism including physiological (haemodynamic forces, p-120 catenin, S-nitrosylation) and pathophysiological conditions such as external permeability factors (VEGF, TNF or histamine) [27, 46-48]. DAVLBH and CA4-P may have distinct effect on these factors. Further research is required to ascertain the underlying mechanism by which DAVLBH- and CA4-P-mediated inhibition of microtubule polymerization has different effects on Src activation and VE-cadherin phosphorylation.
Microtubule dynamics are vital for the local concentrations of E-cadherin and N-cadherin at cell-cell contacts [49-51]. However, little is known about the relationship between microtubules and the local concentration of VE-cadherin, which is highly homologous to E-cadherin and N-cadherin and shares intracellular partners with them, including p120-catenin and β-catenin [52]. Our results showed that DAVLBH-mediated inhibition of microtubule polymerization contributes to the local concentration of VE-cadherin through promoting VE-cadherin internalization and inhibiting internalized VE-cadherin recycling back to cell membrane, result in decrease of VE-cadherin distribution at the cell membrane. On the one hand, DAVLBH-induced microtubule damage led to the VE-cadherin internalization through promoting Src and VE-cadherin phosphorylation, declined VE-cadherin distribution at the cell membrane. On the other hand, microtubule dynamic is also vital for the function of Rab11a, the crucial factor for the VE-cadherin recycling process [39]. Rab11a is transported along microtubules to the cell periphery in the process of recycling internalized plasma membrane proteins back to the cell membrane [53-55]; microtubule damage can result in dispersal of Rab11a throughout the cytoplasm [56]. Once DAVLBH inhibited microtubule polymerization, Rab11a dispersed throughout the cytoplasm and lacked a carrier for transport to the cell periphery; thus, the internalized VE-cadherin could not recycle back to the cell membrane and VE-cadherin location at the cell membrane decreased. In this regard, our study shows that microtubule dynamics are crucial for VE-cadherin distribution and provides new clues regarding the relationship between microtubules and VE-cadherin trafficking.
The VDAs binding to different binding sites of tubulin may exert distinct effects on the cellular morphology. Although both the colchicine-site binding VDAs and vinca alkaloid-site binding VDAs can induce microtubule polymerization in HUVECs, the colchicine-site binding VDAs, such as CA4-P, promote actin stress fibers formation, and conversely, the vinca alkaloid-site binding VDAs, such as DAVLBH, leads to the disorganization of F-actin structures (Figure S6). Rho-kinases are important upstream regulators of actin reorganization and actomyosin contractility, linking outside stimulators to cytoskeletal dynamics [57, 58]. We hypothesize that the different effects of DAVLBH and CA4-P on the formation of actin stress fibers may be associated with the fact that the colchicine-site binding agents, such as CA4-P, OXi8006 and nocodazole, can activate the Rho/Rho-kinase [59-61], while the vinca alkaloid-site binding agents, such as DAVLBH and vinblastine, cause inhibition of Rho/Rho-kinase [13, 62]. However, the underlying mechanism mediating their different effects on the formation of actin stress fiber is still unclear and needs to be further explored.
Recently, studies on VDAs have mainly concentrated on CA4-P or its derivatives. However, the significant therapeutic value of these colchicine-binding VDAs is limited by their intrinsic toxicity, particularly their cardiac toxicity [63, 64]. In addition, the biological potency of these VDAs is relatively low; they have vascular disrupting effects at doses higher than 25 mg/kg [65, 66]. By contrast, 0.75 mg/kg DAVLBH can significantly disrupt tumor vessels but has no obvious effect on blood flow in normal tissues. The dose of DAVLBH used to disrupt tumor vessels is much lower than its maximum tolerated dose (MTD, 4.0 mg/kg in mice), thereby updating the idea that vinca alkaloids disrupt tumor vessels at a dose close to their MTD and indicating that vinca alkaloids could be developed into VDAs for the treatment of malignant tumors [27-30]. Furthermore, DAVLBH is the parent drug of the folate-conjugated prodrug EC145, which has been shown to possess potent antitumor activity in folate receptor-overexpressing tumors with well tolerated regimens in clinical trials [67, 68]. In the current study, we report a novel vascular disrupting effect of DAVLBH and reveal its underlying mechanism, which may lead to adjustments and improvements in the clinical application of EC145. However, as DAVLBH single treatment caused an obvious viable rim, which may be a risk for rapid recurrence after cessation of treatment, it would elicit enhanced antitumor effect when combined with radiotherapy or chemotherapy.
In conclusion, we report the vascular disrupting effect of DAVLBH and its underlying mechanism, which provides new evidence for the development of vinca alkaloids as VDAs for the treatment of malignant tumors and sheds new light on the underlying mechanism of tubulin-binding VDAs.
Abbreviations
VDAs: vascular disrupting agents; DAVLBH: desacetylvinblastine monohydrazide; Vascular endothelial cadherin: VE-cadherin; CA4-P: combretastatin A4 phosphate; HUVECs: human umbilical vein endothelial cells; ECM: endothelial cell medium; T1WI: t1-weighted imaging; T2WI: t2-weighted imaging; DCE-MR: dynamic contrast-enhanced MRI; IVIM-DWI: intravoxel incoherent motion diffusion weighted imaging; ADC: apparent diffusion coefficient.
Supplementary Material
Supplementary method (Annexin V/PI assay) and Figure S1-S6.
Acknowledgements
This work was supported by the Science and Technology Program of China (2017ZX09101003-008-008), the National Science Foundation of China (81573455), the Natural Science Foundation of Guangdong Province (S2013050014183 and 2013CXZDA006), the Program for New Century Excellent Talents in University and Guangdong Pear River Scholar Funded Scheme (D.M. Zhang).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer. 2005;5:423-35
2. Strecker TE, Odutola SO, Lopez R. et al. The vascular disrupting activity of OXi8006 in endothelial cells and its phosphate prodrug OXi8007 in breast tumor xenografts. Cancer Lett. 2015;369:229-41
3. Baguley BC. Preclinical efficacy of vascular disrupting agents in non-small-cell lung cancer. Clin Lung Cancer. 2011;12:81-6
4. Clemenson C, Chargari C, Deutsch E. Combination of vascular disrupting agents and ionizing radiation. Crit Rev Oncol Hematol. 2013;86:143-60
5. Kanthou C, Tozer GM. Microtubule depolymerizing vascular disrupting agents: novel therapeutic agents for oncology and other pathologies. Int J Exp Pathol. 2009;90:284-94
6. Sessa C, Lorusso P, Tolcher A. et al. Phase I safety, pharmacokinetic and pharmacodynamic evaluation of the vascular disrupting agent ombrabulin (AVE8062) in patients with advanced solid tumors. Clin Cancer Res. 2013;19:4832-42
7. Attard G, Greystoke A, Kaye S. et al. Update on tubulin-binding agents. Pathol Biol (Paris). 2006;54:72-84
8. Ng JS. Vinflunine: review of a new vinca alkaloid and its potential role in oncology. J Oncol Pharm Pract. 2011;17:209-24
9. Moudi M, Go R, Yien CY. et al. Vinca alkaloids. Int J Prev Med. 2013;4:1231-5
10. Lee CT, Huang YW, Yang CH. et al. Drug delivery systems and combination therapy by using vinca alkaloids. Curr Top Med Chem. 2015;15:1491-500
11. Duffin J. Poisoning the spindle: serendipity and discovery of the anti-tumor properties of the Vinca alkaloids. Pharm Hist. 2002;44:64-76
12. Vacca A, Iurlaro M, Ribatti D. et al. Antiangiogenesis is produced by nontoxic doses of vinblastine. Blood. 1999;94:4143-55
13. Bijman MN, van Nieuw Amerongen GP, Laurens N. et al. Microtubule-targeting agents inhibit angiogenesis at subtoxic concentrations, a process associated with inhibition of Rac1 and Cdc42 activity and changes in the endothelial cytoskeleton. Mol Cancer Ther. 2006;5:2348-57
14. Meissner M, Pinter A, Michailidou D. et al. Microtubule-targeted drugs inhibit VEGF receptor-2 expression by both transcriptional and post-transcriptional mechanisms. J Invest Dermatol. 2008;128:2084-91
15. Honore S, Pagano A, Gauthier G. et al. Antiangiogenic vinflunine affects EB1 localization and microtubule targeting to adhesion sites. Mol Cancer Ther. 2008;7:2080-9
16. Hill SA, Lonergan SJ, Denekamp J. et al. Vinca alkaloids: anti-vascular effects in a murine tumour. Eur J Cancer. 1993;29A:1320-4
17. Holwell SE, Hill BT, Bibby MC. Anti-vascular effects of vinflunine in the MAC 15A transplantable adenocarcinoma model. Br J Cancer. 2001;84:290-5
18. Hill SA, Sampson LE, Chaplin DJ. Anti-vascular approaches to solid tumour therapy: evaluation of vinblastine and flavone acetic acid. Int J Cancer. 1995;63:119-23
19. Baguley BC, Holdaway KM, Thomsen LL. et al. Inhibition of growth of colon 38 adenocarcinoma by vinblastine and colchicine: evidence for a vascular mechanism. Eur J Cancer. 1991;27:482-7
20. Belzacq-Casagrande AS, Bachelot F, De Oliveira C. et al. Vascular disrupting activity and the mechanism of action of EHT 6706, a novel anticancer tubulin polymerization inhibitor. Invest New Drugs. 2013;31:304-19
21. Harris ES, Nelson WJ. VE-cadherin: at the front, center, and sides of endothelial cell organization and function. Curr Opin Cell Biol. 2010;22:651-8
22. Gavard J. Endothelial permeability and VE-cadherin: a wacky comradeship. Cell Adh Migr. 2014;8:158-64
23. Lampugnani MG, Corada M, Andriopoulou P. et al. Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J Cell Sci. 1997;110(17):2065-77
24. Vincent L, Kermani P, Young LM. et al. Combretastatin A4 phosphate induces rapid regression of tumor neovessels and growth through interference with vascular endothelial-cadherin signaling. J Clin Invest. 2005;115:2992-3006
25. Dalyot-Herman N, Delgado-Lopez F, Gewirtz DA. et al. Interference with endothelial cell function by JG-03-14, an agent that binds to the colchicine site on microtubules. Biochem Pharmacol. 2009;78:1167-77
26. Porcu E, Viola G, Bortolozzi R. et al. TR-644 a novel potent tubulin binding agent induces impairment of endothelial cells function and inhibits angiogenesis. Angiogenesis. 2013;16:647-62
27. Sidibe A, Imhof BA. VE-cadherin phosphorylation decides: vascular permeability or diapedesis. Nat Immunol. 2014;15:215-7
28. Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121:2115-22
29. Wessel F, Winderlich M, Holm M. et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol. 2014;15:223-30
30. Semina EV, Rubina KA, Sysoeva VY. et al. Novel mechanism regulating endothelial permeability via T-cadherin-dependent VE-cadherin phosphorylation and clathrin-mediated endocytosis. Mol Cell Biochem. 2014;387:39-53
31. Reddy JA, Dorton R, Westrick E. et al. Preclinical evaluation of EC145, a folate-vinca alkaloid conjugate. Cancer Res. 2007;67:4434-42
32. Deng ZT, Feng T, Wang P. et al. Effects of the novel vascular targeting agent MDS-11P on tumor vascularity and its antitumor activity. Biochem Pharmacol. 2011;82:1832-42
33. Setyawati MI, Tay CY, Chia SL. et al. Titanium dioxide nanomaterials cause endothelial cell leakiness by disrupting the homophilic interaction of VE-cadherin. Nat Commun. 2013;4:1673
34. Xiao K, Allison DF, Buckley KM. et al. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol. 2003;163:535-45
35. Shi JM, Bai LL, Zhang DM. et al. Saxifragifolin D induces the interplay between apoptosis and autophagy in breast cancer cells through ROS-dependent endoplasmic reticulum stress. Biochem Pharmacol. 2013;85:913-26
36. Lickliter JD, Francesconi AB, Smith G. et al. Phase I trial of CYT997, a novel cytotoxic and vascular-disrupting agent. Br J Cancer. 2010;103:597-606
37. Thoeny HC, Ross BD. Predicting and monitoring cancer treatment response with diffusion-weighted MRI. J Magn Reson Imaging. 2010;32:2-16
38. Benn A, Bredow C, Casanova I. et al. VE-cadherin facilitates BMP-induced endothelial cell permeability and signaling. J Cell Sci. 2016;129:206-18
39. Yan Z, Wang ZG, Segev N. et al. Rab11a Mediates Vascular Endothelial-Cadherin Recycling and Controls Endothelial Barrier Function. Arterioscler Thromb Vasc Biol. 2016;36:339-49
40. Su W, Kowalczyk AP. The VE-cadherin cytoplasmic domain undergoes proteolytic processing during endocytosis. Mol Biol Cell. 2017;28:76-84
41. Kaneko Y, Sullivan R, Dailey T. et al. Kainic Acid-Induced Golgi Complex Fragmentation/Dispersal Shifts the Proteolysis of Reelin in Primary Rat Neuronal Cells: An In Vitro Model of Early Stage Epilepsy. Mol Neurobiol. 2016;53:1874-83
42. Owellen RJ, Hartke CA, Dickerson RM. et al. Inhibition of tubulin-microtubule polymerization by drugs of the Vinca alkaloid class. Cancer Res. 1976;36:1499-502
43. Wang Y, Jin G, Miao H. et al. Integrins regulate VE-cadherin and catenins: dependence of this regulation on Src, but not on Ras. Proc Natl Acad Sci U S A. 2006;103:1774-9
44. Schmid-Alliana A, Menou L, Manie S, Schmid-Antomarchi H. et al. Microtubule integrity regulates src-like and extracellular signal-regulated kinase activities in human pro-monocytic cells. Importance for interleukin-1 production. J Biol Chem. 1998;273:3394-400
45. Head BP, Patel HH, Roth DM. et al. Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J Biol Chem. 2006;281:26391-9
46. Orsenigo F, Giampietro C, Ferrari A. et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun. 2012;3:1208
47. Hatanaka K, Simons M, Murakami M. Phosphorylation of VE-cadherin controls endothelial phenotypes via p120-catenin coupling and Rac1 activation. Am J Physiol Heart Circ Physiol. 2011;300:H162-72
48. Guequen A, Carrasco R, Zamorano P. et al. S-nitrosylation regulates VE-cadherin phosphorylation and internalization in microvascular permeability. Am J Physiol Heart Circ Physiol. 2016;310:H1039-44
49. Stehbens SJ, Paterson AD, Crampton MS. et al. Dynamic microtubules regulate the local concentration of E-cadherin at cell-cell contacts. J Cell Sci. 2006;119:1801-11
50. Mary S, Charrasse S, Meriane M. et al. Biogenesis of N-cadherin-dependent cell-cell contacts in living fibroblasts is a microtubule-dependent kinesin-driven mechanism. Mol Biol Cell. 2002;13:285-301
51. Bulgakova NA, Grigoriev I, Yap AS. et al. Dynamic microtubules produce an asymmetric E-cadherin-Bazooka complex to maintain segment boundaries. J Cell Biol. 2013;201:887-901
52. Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26:441-54
53. Horgan CP, McCaffrey MW. Rab GTPases and microtubule motors. Biochem Soc Trans. 2011;39:1202-6
54. Delevoye C, Goud B. Rab GTPases and kinesin motors in endosomal trafficking. Methods Cell Biol. 2015;130:235-46
55. Takahashi S, Kubo K, Waguri S. et al. Rab11 regulates exocytosis of recycling vesicles at the plasma membrane. J Cell Sci. 2012;125:4049-57
56. Casanova JE, Wang X, Kumar R. et al. Association of Rab25 and Rab11a with the apical recycling system of polarized Madin-Darby canine kidney cells. Mol Biol Cell. 1999;10:47-61
57. Buchsbaum RJ. Rho activation at a glance. J Cell Sci. 2007;120:1149-52
58. Mulloy JC, Cancelas JA, Filippi MD. et al. Rho GTPases in hematopoiesis and hemopathies. Blood. 2010;115:936-47
59. Kanthou C, Tozer GM. The tumor vascular targeting agent combretastatin A-4-phosphate induces reorganization of the actin cytoskeleton and early membrane blebbing in human endothelial cells. Blood. 2002;99:2060-9
60. Strecker TE, Odutola SO, Lopez R. et al. Charlton-Sevcik AK. et al. The vascular disrupting activity of OXi8006 in endothelial cells and its phosphate prodrug OXi8007 in breast tumor xenografts. Cancer Lett. 2015;369:229-41
61. Birukova AA, Smurova K, Birukov KG. et al. Microtubule disassembly induces cytoskeletal remodeling and lung vascular barrier dysfunction: role of Rho-dependent mechanisms. J Cell Physiol. 2004;201:55-70
62. Lei X, Chen M, Nie Q. et al. In vitro and in vivo antiangiogenic activity of desacetylvinblastine monohydrazide through inhibition of VEGFR2 and Axl pathways. Am J Cancer Res. 2016;6:843-58
63. van Heeckeren WJ, Bhakta S, Ortiz J. et al. Promise of new vascular-disrupting agents balanced with cardiac toxicity: is it time for oncologists to get to know their cardiologists? J Clin Oncol. 2006;24:1485-8
64. Subbiah IM, Lenihan DJ, Tsimberidou AM. Cardiovascular toxicity profiles of vascular-disrupting agents. Oncologist. 2011;16:1120-30
65. Tozer GM, Prise VE, Wilson J. et al. Combretastatin A-4 phosphate as a tumor vascular-targeting agent: early effects in tumors and normal tissues. Cancer Res. 1999;59:1626-34
66. Salmon HW, Siemann DW. Effect of the second-generation vascular disrupting agent OXi4503 on tumor vascularity. Clin Cancer Res. 2006;12:4090-4
67. Dosio F, Milla P, Cattel L. EC-145, a folate-targeted Vinca alkaloid conjugate for the potential treatment of folate receptor-expressing cancers. Curr Opin Investig Drugs. 2010;11:1424-33
68. Zhao R, Diop-Bove N, Goldman ID. Enhanced receptor-mediated endocytosis and cytotoxicity of a folic acid-desacetylvinblastine monohydrazide conjugate in a pemetrexed-resistant cell line lacking folate-specific facilitative carriers but with increased folate receptor expression. Mol Pharmacol. 2014;85:310-21
Author contact
![]() Corresponding authors: Professor Dongmei Zhang, College of Pharmacy, Jinan University, 601 Huangpu Avenue. Wes, Guangzhou 510632, China; Phone: +86-20-85222653; E-mail: dmzhang701edu.cn. or Professor Wencai Ye, College of Pharmacy, Jinan University, 601 Huangpu Avenue. Wes, Guangzhou 510632, China; Phone: +86-20-85220004; E-mail: chywccom.
Corresponding authors: Professor Dongmei Zhang, College of Pharmacy, Jinan University, 601 Huangpu Avenue. Wes, Guangzhou 510632, China; Phone: +86-20-85222653; E-mail: dmzhang701edu.cn. or Professor Wencai Ye, College of Pharmacy, Jinan University, 601 Huangpu Avenue. Wes, Guangzhou 510632, China; Phone: +86-20-85220004; E-mail: chywccom.