Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(1):45-60. doi:10.7150/thno.20885 This issue Cite
Research Paper
Therapeutic Effects of Targeted PPARɣ Activation on Inflamed High-Risk Plaques Assessed by Serial Optical Imaging In Vivo
Jah Yeon Choi1,*, Jiheun Ryu2,*, Hyun Jung Kim1,*, Joon Woo Song1, Joo Hee Jeon1, Dae-Hee Lee3, Dong Joo Oh1, Dae-Gab Gweon2, Wang-Yuhl Oh2, Hongki Yoo4, ![]() , Kyeongsoon Park5,
, Kyeongsoon Park5, ![]() , Jin Won Kim1,
, Jin Won Kim1, ![]()
1. Multimodal Imaging and Theranostic Lab, Cardiovascular Center, Korea University Guro Hospital, Seoul, Republic of Korea, 152-703
2. Department of Mechanical Engineering, KAIST, Daejeon, Republic of Korea, 305-701
3. Division of Medical Oncology, Department of Internal Medicine, Korea University Guro Hospital, Seoul, Republic of Korea, 152-703
4. Department of Biomedical Engineering, Hanyang University, Seoul, Republic of Korea, 133-791
5. Department of Systems Biotechnology, College of Biotechnology and Natural Resources, Chung-Ang University, Anseong-si, Gyeonggi-do, Republic of Korea, 17546
*JYC, JR, HJK contributed equally to this work.
Received 2017-5-5; Accepted 2017-9-27; Published 2018-1-1
Abstract

Rationale: Atherosclerotic plaque is a chronic inflammatory disorder involving lipid accumulation within arterial walls. In particular, macrophages mediate plaque progression and rupture. While PPARγ agonist is known to have favorable pleiotropic effects on atherogenesis, its clinical application has been very limited due to undesirable systemic effects. We hypothesized that the specific delivery of a PPARγ agonist to inflamed plaques could reduce plaque burden and inflammation without systemic adverse effects.
Methods: Herein, we newly developed a macrophage mannose receptor (MMR)-targeted biocompatible nanocarrier loaded with lobeglitazone (MMR-Lobe), which is able to specifically activate PPARγ pathways within inflamed high-risk plaques, and investigated its anti-atherogenic and anti-inflammatory effects both in in vitro and in vivo experiments.
Results: MMR-Lobe had a high affinity to macrophage foam cells, and it could efficiently promote cholesterol efflux via LXRα-, ABCA1, and ABCG1 dependent pathways, and inhibit plaque protease expression. Using in vivo serial optical imaging of carotid artery, MMR-Lobe markedly reduced both plaque burden and inflammation in atherogenic mice without undesirable systemic effects. Comprehensive analysis of en face aorta by ex vivo imaging and immunostaining well corroborated the in vivo findings.
Conclusion: MMR-Lobe was able to activate PPARγ pathways within high-risk plaques and effectively reduce both plaque burden and inflammation. This novel targetable PPARγ activation in macrophages could be a promising therapeutic strategy for high-risk plaques.
Keywords: Macrophage, Targeted, Plaque, PPARγ, Lobeglitazone, Serial imaging
Introduction
Atherosclerosis is recognized as a chronic inflammatory disorder associated with lipid accumulation within arterial walls [1]. Since macrophages play a pivotal role in plaque vulnerability, macrophage-abundant atheroma tends to become destabilized, and finally rupture, causing thrombotic complications. Although high-risk plaque can be treated with lipid-lowering agents, cardiovascular events still remain the leading cause of death worldwide [2].
Peroxisome proliferator-activated receptor gamma (PPARγ) is a nuclear receptor, functioning as a ligand-activated transcriptional regulator of genes that control lipid and glucose metabolism [3, 4]. Since PPARγ activation upregulates the regulators of cholesterol efflux in macrophages [3, 4] and reduces inflammation in atheroma by inhibiting nuclear factor-kappa B activity [4-8], this pathway could be an attractive target for treating atherosclerosis. While a few reports have demonstrated that PPARγ agonists could stabilize high-risk plaques [9-12], PPARγ agonist therapy for atherosclerosis is still challenging due to adverse systemic effects including weight gain, edema, congestive heart failure, and bone fracture [13-15].
The recent advent of smart targeted drug delivery has emerged as a promising strategy to overcome these hurdles and enhance therapeutic effects with a minimum of side effects [16-21]. Despite promising results in oncology [22-24], the targetable strategy in atherosclerosis has been explored in only a few studies, providing limited benefits such as anti-inflammatory effects [21, 25-27].
To address these unmet needs, we developed a novel PPARγ activation strategy targeting plaque macrophages to treat inflamed high-risk atheroma. As mannose receptor is highly expressed in the macrophages within thin-cap fibroatheroma (TCFAs) of human coronary arteries, it has been utilized as a target for PET and optical imaging of plaque inflamemation [28, 29]. Here, we loaded PPARγ agonist, lobeglitzone, into a macrophage mannose receptor (MMR) targeted nanocarrier based on biocompatible glycol chitosan (GC). By combining customized in vivo serial optical imaging, comprehensive ex vivo analysis, and in vitro assays, we evaluated whether our targeted therapeutic strategy enhances specific activation of the PPARγ pathways in inflamed high-risk plaques and could effectively reduce both plaque burden and inflammatory activity.
Materials and Methods
Expanded materials and methods are presented in Supplementary Materials.
Synthesis of MMR-Lobe
Macrophage mannose receptor (MMR)-targeting carriers were prepared based on thiolated glycol chitosan, as a lobeglitazone delivery system, as described in the previous study [29]. Briefly, thiolated glycol chitosan and mannose-polyethylene glycol-maleimide (MAN-PEG-MAL) were reacted in phosphate-buffered saline (PBS) for 20 h, then lyophilized to yield MAN-PEG-GC. To fabricate self-assembled structures, MAN-PEG-GC and cholesteryl chloroformate were reacted in anhydrous dimethyl sulfoxide (DMSO): dimethylforamide (DMF) co-solvent (3:1) containing triethylamine for 24 h.
For in vivo imaging and monitoring of drug efficacy in atherosclerotic plaques, the carriers were labelled with cyanine 5.5-NHS ester (MMR-Cy5.5). Therapeutic MMR-Lobe were prepared as follows: MMR-targeting carriers and lobeglitazone were completely dissolved in DMSO by stirring for 3 h, dialyzed against distilled water using a dialysis membrane (MWCO 6,000-8,000 Da) for 1 day, and lyophilized for 2 days to obtain MMR-Lobe.
Characterization of MMR-Lobe
To evaluate the shape and size of MMR-Lobe, 1 mg of MMR-Lobe was dispersed in distilled water (1 mL). Then, they were analyzed using a Zetasizer 3000 instrument (Malvern Instruments) and an Energy Filtering Transmission Electron Microscope (EF-TEM, LEO 912AB OMEGA, Carl Zeiss, Germany).
The drug loading efficiency and loading content of lobeglitazone in MMR-Lobe were calculated by first dissolving MMR-Lobe (1 mg) in 1 mL of acetonitrile/H2O/formic acid (60:40:0.25). The amount of lobeglitazone loaded in MMR-Lobe was determined by high-performance liquid chromatography (HPLC) (Agilent 1260 series) as described in Supplementary Materials.
To study the in vitro release profile of lobeglitazone from MMR-Lobe, the lyophilized MMR-Lobe (1 mg) was dispersed in PBS (1 mL, pH 7.4). The dispersed MMR-Lobe was then placed in a dialysis membrane (MWCO 6,000-8,000 Da) which was immersed in PBS (pH 7.4) followed by gentle shaking in a water bath (37ºC) oscillating 100 times/min. The medium was replaced with fresh medium at predetermined intervals. The amount of lobeglitazone released from MMR-Lobe was determined by HPLC in the same manner described above for determining drug loading efficiency.
To determine the uptake of MMR probes in macrophage derived foam cells, RAW 264.7 cells were stimulated with low-density lipoprotein (LDL, 100 µg/mL) and lipopolysaccharide (LPS, 200 ng/mL). After 24 h, cells were replaced with fresh media containing various concentrations of MMR-Cy5.5 (25, 50, and 100 μg/mL) for 1 h. The cells were washed two times with PBS to remove MMR-Cy5.5 that were not internalized, and were fixed with 4% paraformaldehyde solution for 30 min. After nuclei of the cells were stained with DAPI for 10 min, cellular uptake was visualized using intravital multi-photon laser-scanning confocal microscopy (LSM 780 Meta NLO, Carl Zeiss, Germany).
In vitro assays
RAW 264.7 cells were cultured in Dulbecco's modified Eagle medium (DMEM; GenDEPOT) supplemented with 10% heat-inactivated fetal bovine serum (GenDEPOT), 100 U/mL penicillin and 100 μg/mL streptomycin (GenDEPOT). The cells were incubated in an atmosphere of 5% CO2 at 37ºC and were subcultured every 2 days.
To measure the production of tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), and matrix metallopeptidase 9 (MMP-9) by RAW 264.7 cells, the cells were incubated with increasing concentrations of lobeglitazone, dose-equivalent MMR-Lobe, MMR vehicle, or saline for 6 h. The cells were challenged with LPS (200 ng/mL) for 3 h, and then TNF-α, IL-6, and MMP-9 levels in the supernatant were measured using enzyme-linked immunosorbent assay (ELISA).
To determine the expression of ABCA1, ABCG, and LXRα, RAW 264.7 cells were plated at a density of 5 × 106 cells per 100 mm dish. Cells were pretreated with lobeglitazone, MMR-Lobe, MMR vehicle, or saline. After incubation for 9 h, western blot analysis and real-time PCR were performed in the lysed cells.
To determine the effects of MMR-Lobe on foam cell formation, RAW 264.7 cells were incubated with increasing concentrations of lobeglitazone, dose-equivalent MMR-Lobe, MMR vehicle, or saline. After stimulation with low-density lipoprotein (LDL, 100 µg/mL) and LPS (200 ng/mL), quantitation of Oil Red O (ORO) stained areas was performed by histomorphometirc analysis with Image J software. The stained area was measured and averaged for 30 cells in 10 fields per dish.
To accurately estimate total cholesterol levels in macrophage foam cells treated with lobeglitazone or MMR-Lobe compared with control, RAW 264.7 cells were plated at a density of 1 x 106 cells in each 6 cm culture dish. Cells were incubated with increasing concentrations of lobeglitazone, dose-equivalent MMR-Lobe, MMR vehicle, or saline. After stimulation with low-density lipoprotein (LDL, 100 µg/mL) and LPS (200 ng/mL), cells were washed with cold PBS two times prior to lysis. Total cholesterol in macrophage foam cells in each group was extracted with 200 µL of a mixture of chloroform/isopropanol/NP-40 (7:11:0.1). Total cholesterol was measured by commercial assay kit (Cell Biolabs, Inc., San Diego, CA, USA), following the manufacturer's protocol.
In vivo experimental design
The therapeutic effects of MMR-Lobe were evaluated in apoE-/- mice (B6. 129P2-Apoetm1Unc/J) purchased from Jackson Laboratory. The apoE-/- mice consumed a high cholesterol diet containing 16.0% fat, 21.0% protein, 46.0% carbohydrate, and 1.25% cholesterol (Research Diets Inc., USA) for 8 weeks from 8 to 16 weeks of age. After baseline in vivo imaging of carotid plaques, the apoE-/- mice with established carotid plaques were randomized into 4 groups: MMR-Lobe treatment, lobeglitazone per se gavage, MMR vehicle, and saline group. The mice of each group were subsequently treated for 4 weeks with intravenous injection of MMR-Lobe (7 mg/kg, 2 injections per week, n = 6), oral administration of lobeglitazone (2 mg/kg per day, n = 6), MMR vehicle (2 MMR vehicle injections per week, n = 6), or placebo (2 saline injections per week, n = 6). Mice were maintained on a normal diet during the treatment. After 4 weeks of treatment, the in vivo imaging was performed in the same carotid plaque with identical imaging settings to determine the serial changes of carotid plaques features including size and inflammation. After the 2nd in vivo imaging, a comprehensive analysis including ex vivo imaging and immunohistopathology was performed. The Institutional Animal Care and Use Committee of Korea University (KUIACUC-20150820-1) approved the study protocol.
In vivo serial imaging using customized multi-channel confocal intravital imaging
A multi-channel confocal intravital fluorescence microscope (MCC-IVFM) was customized to visualize murine carotid plaques and macrophages in vivo. Briefly, optimized features of the MCC-IVFM include upright epi-fluorescence configuration, fast image acquisition rate of 4 frames/s, wide field-of-view of 2.4 mm × 2.4 mm, and flexible access to the carotid artery using a 5-axis staging platform. To visualize carotid angiogram and plaque macrophage distribution simultaneously, 488 nm and 633 nm excitation lasers were used for fluorescein isothiocyanate (FITC)-dextran and MMR-Cy5.5, respectively. Baseline and serial imaging of each mouse were performed under identical imaging settings. Also, bright-field images were acquired to co-register the imaging fields of 2 IVFM data sets.
To estimate the changes in MMR-NIRF signals between baseline and serial images of each apoE-/- mouse, MMR-Cy5.5 (10 mg/kg) was injected via tail vein 48 h prior to imaging. Immediately before imaging, FITC-dextran was administered via tail vein as a fluorescent agent for angiography. After surgical exposure of the carotid artery, apoE-/- mice underwent our customized intravital imaging. To accurately quantify MMR-Cy5.5 signals from macrophages, the average level of auto-fluorescence from the vasculature was pre-determined by imaging normal carotid arteries of four CL57BL/6 mice without injecting fluorescent agents. The auto-fluorescence signal was used as a background threshold level to determine whether or not each pixel value contained MMR-Cy5.5 signals. Plaque area was determined by filling defect area on a FITC-dextran vessel angiogram. Macrophage areas were calculated by counting the number of effective pixels, that have higher macrophage-derived signals above the background threshold within the plaque region. Macrophage signal intensity was calculated from the mean signal intensity of the macrophage in the plaques.
Ex vivo en face fluorescence reflectance imaging and Oil Red O staining
Near-infrared signals from en face preparations of whole aortas were measured using fluorescence reflectance imaging (FRI, Davinch-K Co., Ltd, Korea). Then, en face preparations of whole aortas stained with ORO were used to quantify aortic plaque burden by measuring the ORO-stained area.
Immunofluorescence staining
Immunofluorescence stainings were performed on consecutive cross-sections of aortic sinus. The following primary antibodies for PPARγ (1:50, rabbit anti-PPARγ, Novus Biological, USA), LXRα (1:50, rabbit anti-LXRα, Abcam, UK), ABCA1 (1:500, rabbit anti-ABCA1, Abcam, UK), ABCG1 (1:50, rabbit anti-ABCG1, Abcam, UK), and MMP-9 (1:500, rabbit anti-MMP9, Abcam, UK) were used. After incubating overnight at 4℃ and washing with PBS for 15 min, aortic sinus sections were incubated for 2 h at 37℃ with appropriate secondary antibodies: Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:100, Jackson ImmuneResearch Laboratories, USA), Alexa Fluor 594-conjugated goat anti-rabbit IgG (1:100, Jackson ImmuneResearch Laboratories, USA), AMCA-conjugated goat anti-rabbit IgG (1:100, Jackson ImmuneResearch Laboratories, USA). The sections were observed under a confocal laser scanning microscope (LSM700, Carl Zeiss, Germany) with identical windowing.
Histopathology and immunohistochemistry
Histomorphometry and immunostaining analyses were performed on consecutive cross-sections of imaging - targeted carotid plaques (total 72 sections; 3 sections of largest plaque area per mouse, 6 mice per group, were analyzed) to validate the in vivo findings, and also aortic sinus (total 960 cross sections; 40 sections per mice, 6 mice per group, were analyzed) to quantitatively compare the treatment effects between groups. To detect lipid accumulation and macrophage contents, ORO staining and Mac3 staining were performed, respectively. For classically activated macrophage analysis, CD86 staining was performed for each consecutive plaque.
Statistical analysis
All quantitative experiments were performed in triplicate. Statistical analyses were performed with GraphPad Prism (v5.3; GraphPad Software, San Diego, CA). Data are expressed as mean ± SD. Mean values were compared between four groups by a Kruskal-Wallis non-parametric one-way analysis of variance (ANOVA), and between two-groups by a Mann-Whitney test. The Wilcoxon matched-pairs signed-ranks test was used to determine differences between the first and second images from the same murine carotid plaque. P values less than 0.05 were considered statistically significant.
Results
Synthesis and characteristics of MMR-Lobe
A synthetic process and schematic illustration of the MMR-Lobe are presented in Figure 1A and 1B. MMR-targeting carriers were fabricated by chemically conjugating hydrophilic thiolated glycol chitosan with mannose-polyethylene glycol-maleimide to target macrophage mannose receptors. Then, it was reacted with the hydrophobic molecule, cholesteryl chloroformate to form self-assembled particles. The hydrophobic PPARγ agonist, lobeglitazone, was then incorporated into the hydrophobic inner core of the MMR-targeted carrier (MMR-Lobe). The morphological properties of MMR-Lobe were investigated by transmission electron microscopy which identified self-assembled spherical particles with a diameter ranging from 300-350 nm (Figure 1C).
Characterization of macrophage mannose receptor targeted PPARɣ agonist, lobeglitazone (MMR-Lobe). (A) The synthetic molecular structure of MMR-Lobe. (B) Schematic illustration of MMR-Lobe. (C) Representative transmission electron microscopy image of MMR-Lobe, showing a monodisperse population (scale bar, 500 nm). The size distribution of MMR-Lobe demonstrates an average diameter of 300-350 nm. (D) In vitro release of lobeglitazone from MMR-Lobe. (E) Confocal microscopy images of MMR-Cy5.5 internalization in macrophage foam cells.
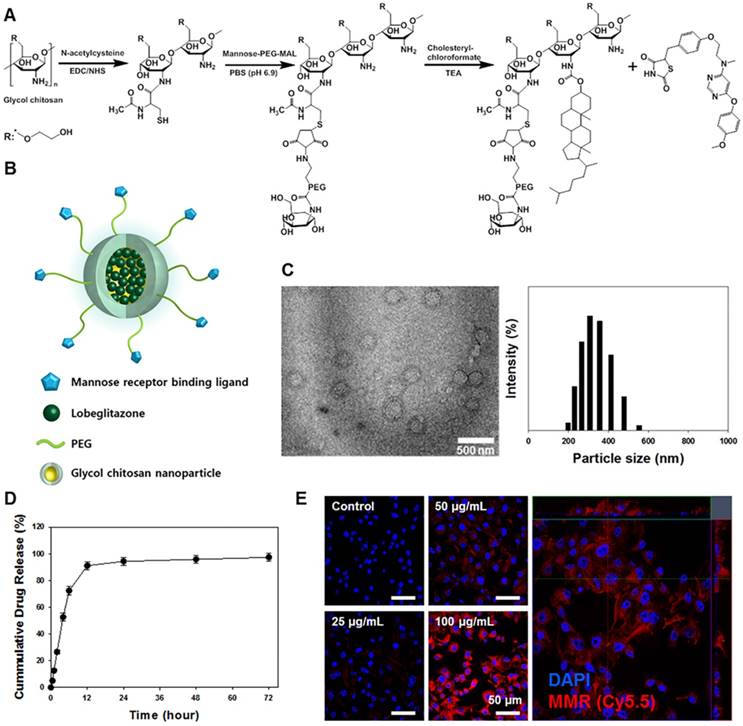
There were no significant changes in serum concentration of inflammatory cytokines such as TNF-α, IL-6, and MMP-9 after MMR-carrier injection (Figure S1). The calculated loading efficiency and content of lobeglitazone were 95.7% ± 4.21%, and 28.7% ± 1.26%, respectively. Lobeglitazone release from MMR-Lobe was studied over time, and the results showed a rapid release of 91.0% ± 3.06% within the first 12 h, followed by a sustained release (Figure 1D).
To characterize the uptake of MMR carriers into macrophage foam cells, we induced foam cell formation by adding LDL (100 μg/mL) and LPS (200 ng/mL) to RAW 264.7 cells and incubated with MMR-Cy5.5 for 1 h. After fixation, confocal fluorescence microscopy revealed that the particles were indeed internalized by macrophage foam cells in a dose-dependent manner (Figure 1E). To address the specificity of MMR-Lobe, we performed a blocking study with D-mannose and mannan, which bind mannose receptors. The fluorescence intensity of MMR-Cy5.5 decreased in macrophage-derived foam cells when pre-treated with D-mannose or mannan, indicating that MMR-probe can more specifically target mannose receptors (Figure S2).
In vitro mechanism assays: enhancement of cholesterol efflux and suppression of inflammation activity
To determine whether PPARγ activation by MMR-Lobe inhibits inflammation in macrophages, we incubated RAW 264.7 cells with increasing concentrations of lobeglitazone, dose-equivalent MMR-Lobe, MMR vehicle, or saline. We then stimulated the cells with LPS and assessed expression of the inflammatory cytokines TNF-α, IL-6, and MMP-9 using ELISA. The levels of these cytokines significantly decreased in macrophages treated with MMR-Lobe (Figure 2A). Lobeglitazone and MMR-Lobe were comparably effective in attenuating inflammatory cytokines at all concentrations.
In vitro assays of MMR-Lobe. (A) Anti-inflammatory effects of MMR-Lobe and lobeglitazone treatment in RAW 264.7 cells. ELISA showed a decrease in TNF-α, IL-6, and MMP-9 production in both MMR-Lobe and lobeglitazone-treated cells as compared with those in the saline control and MMR vehicle groups, in a dose-dependent manner. (B) Effects of MMR-Lobe and lobeglitazone on cholesterol efflux. RT-PCR and western blot analysis showed an increase in ABCA1, ABCG1, and LXRα expressions in RAW 264.7 cells treated by MMR-Lobe in a dose-dependent manner. ABCA1: ATP-binding cassette transporters A1; ABCG1: ATP-binding cassette transporters G1; LXRα: Liver X receptor alpha. (C) Inhibition of foam cell formation in RAW 264.7 cells by MMR-Lobe. (D) Cellular cholesterol level in MMR-Lobe and lobeglitazone compared to control and MMR vehicle. Results were obtained from 3 separate experiments; * P < 0.05, ** P < 0.01, by ANOVA followed by Mann-Whitney test.
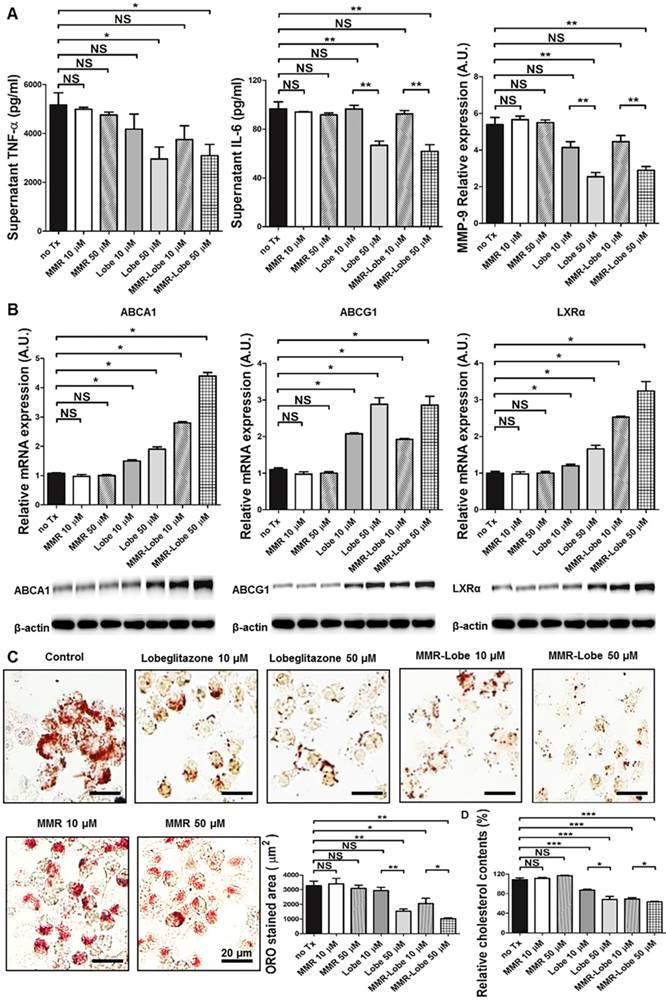
To investigate whether MMR-Lobe increases cholesterol efflux in macrophages, we likewise incubated RAW 264.7 cells with increasing concentrations of lobeglitazone, dose-equivalent MMR-Lobe, MMR vehicle, or saline. As ATP-binding cassette transporters A1 and G1 (ABCA1, ABCG1) are essential regulators of cholesterol efflux and Liver X receptor alpha (LXRα) is a nuclear receptor that induces ABCA1 and ABCG1 transcription, the expression of ABCA1, ABCG1, and LXRα were evaluated by RT-PCR and western blotting. Both RNA and protein levels of these molecules significantly increased in macrophages treated with MMR-Lobe in a dose dependent manner. Lobeglitazone and MMR-Lobe were equally effective in enhancing ABCG1 expression; but, intriguingly, MMR-Lobe was more effective than lobeglitazone in enhancing expression of ABCA1 and LXRα at high concentration (Figure 2B).
To assess the effects of MMR-Lobe on macrophage foam cell formation, we incubated RAW 264.7 derived foam cell with increasing concentrations of lobeglitazone, dose-equivalent MMR-Lobe, MMR vehicle, or saline. Treatment with MMR-Lobe significantly and dose-dependently decreased foam cell formation. At low concentration, the effect of MMR-Lobe on foam cell inhibition was comparable to that of lobeglitazone. But at high concentration, foam cell formation was more prominently attenuated by MMR-Lobe treatment compared to lobeglitazone treatment (Figure 2C). To investigate whether this inhibitory effect of MMR-Lobe on macrophage foam cells is mediated by targeting mannose receptor, we performed a blocking study with D-mannose and mannan. Foam cell formation increased in MMR Lobe-treated macrophages when mannose receptors were blocked with D-mannose or mannan (Figure S3).
To quantitatively estimate the cholesterol contents in macrophage foam cells treated with lobeglitazone and MMR-Lobe, we incubated RAW 264.7-derived foam cells with increasing concentrations of lobeglitazone, dose-equivalent MMR-Lobe, MMR vehicle, or saline. After washing with PBS two times, cellular cholesterol was extracted and measured. The level of cholesterol in macrophage foam cells was significantly lower in the MMR-Lobe group compared to controls (Figure 2D).
In vivo serial imaging assessment of carotid plaques
To assess the anti-atherogenic and anti-inflammatory effects of MMR-Lobe in vivo, we performed serial imaging of carotid plaques in apoE-/- mice using a customized multi-channel confocal laser scanning microscopy before and after treatment with MMR-Lobe, lobeglitazone, or saline (Figure 3A and 3B).
In vivo serial imaging of carotid plaques in ApoE-/- mice. (A) Schematic of the serial imaging of carotid plaques in ApoE-/- mice. (B) Schematic illustration of multi-channel confocal intravital fluorescence microscopy. Representative in vivo optical imaging of carotid plaques at baseline and after treatment in the MMR-Lobe (C), lobeglitazone per se (D), MMR vehicle (E) and control groups (F). Vessel angiograms obtained from the fluorescein isothiocyanate (FITC)-dextran channel clearly identified the plaques. Macrophage contents within the plaques were imaged using the cyanine 5.5 channel. The plaque burden and macrophage signals markedly decreased over the 4 weeks of treatment in MMR-Lobe-treated mice as compared to the other groups. Arrows indicate carotid bifurcation.
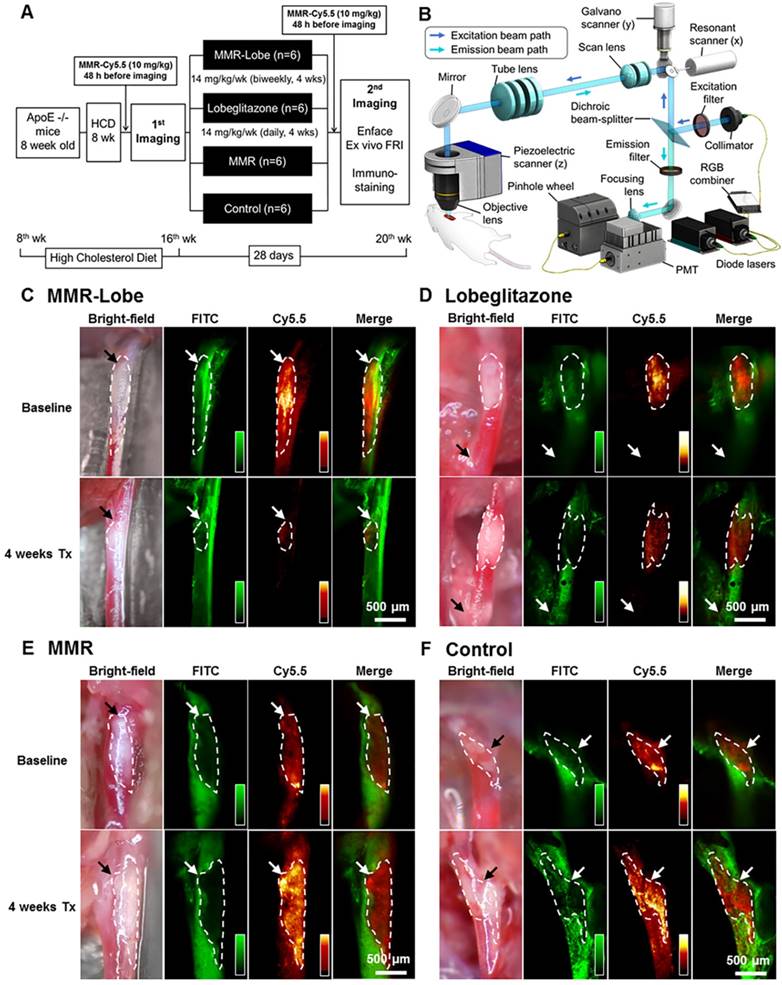
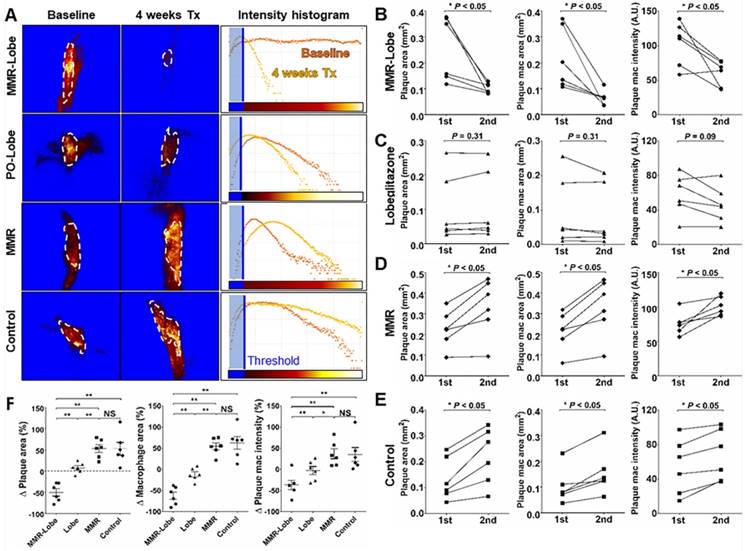
In in vivo images, plaque areas were determined as a filling defect in the fluorescein isothiocyanate (FITC) angiogram, and macrophage signals were evaluated using the Cy5.5 fluorescence channel which was described in a previous study [29]. To accurately quantify the macrophage signals, we applied threshold levels that were obtained by averaging the levels of auto-fluorescence from four CL57BL/6 mice. Macrophage areas were determined based on the threshold within the plaque, and macrophage signal intensities were calculated from the mean signal intensity in the plaque macrophages (Figure S4). In the mice treated with MMR-Lobe, all parameters, such as carotid plaque size, plaque macrophage area, and plaque macrophage signal intensity, markedly decreased over the 4 weeks of treatment (plaque size decreased by 42.5% compared to baseline, P = 0.031; plaque macrophage area decreased by 56.4%, P = 0.030; plaque macrophage signal intensity decreased by 50.4%, P = 0.031) (Figure 3C, 4A, 4B and 4F). In contrast, plaque size, plaque macrophage area, and plaque macrophage signal intensity increased over 4 weeks in the control group (plaque size increased by 52.6%, P = 0.030; plaque macrophage area increased by 62.7%, P = 0.031; plaque macrophage signal intensity increased by 45.5%, P = 0.030) (Figure 3F, 4A, 4E, and 4F) and in the MMR vehicle group (plaque size increased by 52.9%, P = 0.031; plaque macrophage area increased by 54.4%, P = 0.031; plaque macrophage signal intensity increased by 36.9%, P = 0.031) (Figure 3E, 4A, 4D, and 4F). In the mice treated with lobeglitazone per se, these parameters did not show significant changes over the treatment period (plaque size increased by 7.3%, P = 0.313; plaque macrophage area decreased by 11.7%, P = 0.313; and plaque macrophage signal intensity decreased by 17.9%, P = 0.094) (Figure 3D, 4A, 4C, and 4F).
Ex vivo imaging validation of whole aorta en face
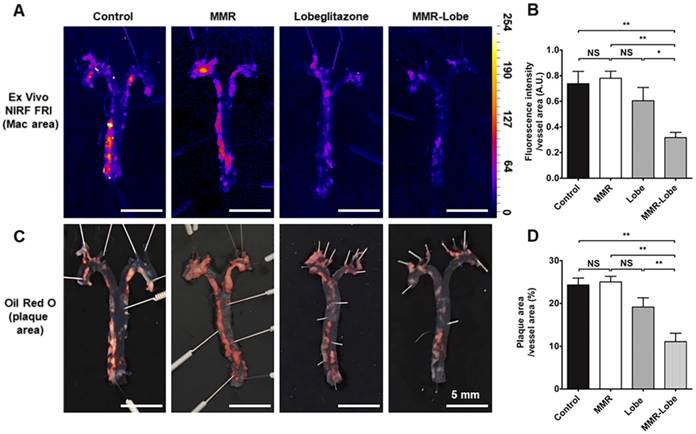
To estimate the effects of MMR-Lobe treatment on systemic atheroma burden and inflammation, we took the en face aortas of apoE-/- mice after serial intravital imaging. The MMR-Lobe treatment group, but not MMR vehicle or lobeglitazone per se group, showed a prominent reduction in inflammatory burden of the aorta en face as determined by fluorescence reflectance imaging ex vivo (FRI) (Figure 5A and B). Additionally, the MMR-Lobe group, but not MMR vehicle or lobeglitazone per se group, showed a potent decrease in plaque burden as measured by ORO staining (Figure 5C and D).
Quantitative analysis of serial therapeutic effects on carotid plaques. (A) Representative example of quantitative analysis in four treatment groups. (B) In the MMR-Lobe group, plaque size, plaque macrophage area, and macrophage signal decreased dramatically over the 4 weeks of treatment. (C) In the lobeglitazone per se group, plaque size and macrophage content did not show significant changes over time. In the MMR vehicle (D) and control (E) groups, all parameters including plaque size, plaque macrophage area, and plaque macrophage signal increased over the 4 weeks of treatment; n = 6 mice per group; * P < 0.05, ** P < 0.01, by Wilcoxon matched-pairs signed-ranks test. (F) Collectively, plaque size, plaque macrophage area, and plaque macrophage signal prominently decreased in the MMR-Lobe treatment group, as compared to the lobeglitazone per se, MMR vehicle, and control groups; n = 6 mice per group; * P < 0.05, ** P < 0.01, by ANOVA followed by Mann-Whitney test.
Ex vivo imaging and quantitative analysis of en face whole vessels. (A) Representative images of ex vivo fluorescence reflectance imaging of the aorta en face after 4 weeks of treatment. (B) Fluorescence intensity analysis. The MMR-Lobe treatment group showed a significant reduction in inflammatory activity compared with lobeglitazone per se, MMR vehicle, or control groups; n = 6 mice per group. (C) Representative ORO staining images of the aorta en face after 4 weeks of experimental treatment. (D) Quantification of plaque burden of the aorta en face. The plaque burden in MMR-Lobe treatment group prominently decreased compared with lobeglitazone per se, MMR vehicle, or control; Data are presented as mean ± SD. n = 6 mice per group. * P < 0.05, ** P < 0.01, by ANOVA followed by Mann-Whitney test.
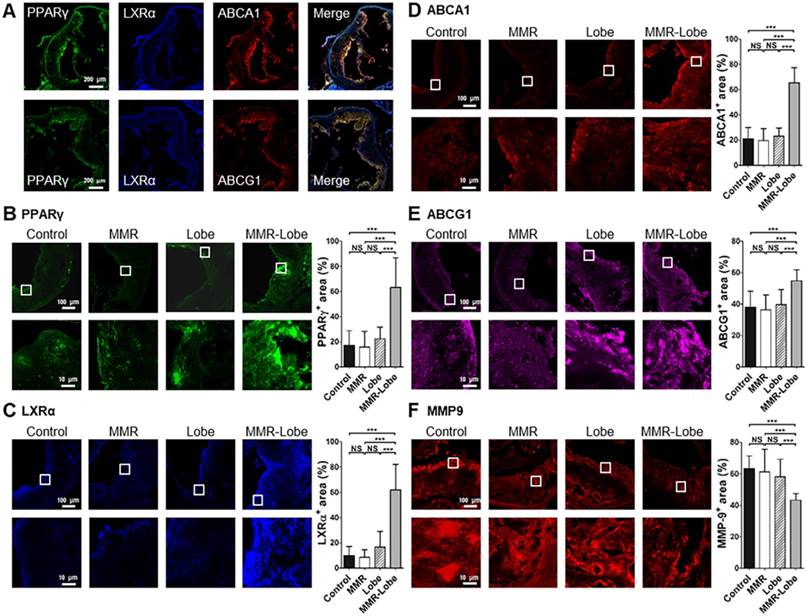
Comparative activation of PPARγ, LXRα, ABCA1, ABCG1 and expression of MMP-9 in aortic atheroma
To determine whether MMR-Lobe activates PPARγ pathways in the atheroma, immunofluorescence imaging of PPARγ, LXRα, ABCA1 and ABCG1 in the aortic plaques was analyzed. PPARγ immunoreactivities in the plaques of MMR-Lobe-treated mice co-localized well with LXRα, ABCA1 and ABCG1 activations (Figure 6A and Figure S5). The double immunofluorescence staining for macrophage and PPAR pathway markers revealed that the macrophage immunoreactivities were well co-localized with PPARγ, LXRα, ABCA1, ABCG1 but not MMP9 (Figure S6). Intriguingly, activation of PPARγ, LXRα, ABCA1 and ABCG1 significantly increased in MMR-Lobe-treated mice as compared to lobeglitazone per se, MMR vehicle, or control. (Figure 6B-6E). Additionally, the expression of MMP-9 was significantly attenuated in MMR-Lobe-treated mice compared with other groups (Figure 6F).
Comparison of PPARγ, LXRα, ABCA1, ABCG1 activity and MMP-9 expression in MMR-Lobe group vs. lobeglitazone per se, MMR vehicle, or control groups. (A) Representative images of aortic plaques co-stained with antibodies against PPARγ (green), LXRα (blue), ABCA1 (red), and ABCG1(red) in the MMR-Lobe treated group. Quantitative analysis of PPARγ (B), LXRα (C), ABCA1 (D), ABCG1 (E) activation and MMP-9 expression (F). The co-expressions of PPARγ, LXRα, ABCA1, and ABCG1 in the plaques were strongly enhanced by MMR-Lobe treatment, but the expression of MMP-9 decreased significantly in the MMR-Lobe-treated mice. Data are presented as mean ± SD. n = 6 mice per group; * P < 0.05, ** P < 0.01, *** P < 0.001 by ANOVA followed by Mann-Whitney test.
Immunohistological validation of the plaques at imaged carotid artery and the aortic sinus
Total 72 sections (3 consecutive sections of the largest imaged carotid plaque per mouse, with 6 mice per group) were analyzed. In line with the in vivo imaging results, immunostainings of the carotid plaques of apoE-/- mice in the MMR-Lobe treatment group showed a prominent reduction of lipid accumulation and macrophage infiltration as compared to those of control, MMR vehicle, and lobeglitazone treatment groups; 54.2% reduction vs. control group, 50.6% reduction vs. MMR vehicle, and 44.1% reduction vs. lobeglitazone group in the percentage of ORO-staining areas (Figure 7A and 7B); 72.6% reduction vs. control group, 69.8% reduction vs. MMR vehicle, and 41.2% reduction vs. lobeglitazone group in the percentage of Mac3-staining areas, respectively (Figure 7A and 7C).
Comprehensive immunohistologic analysis of carotid and aortic plaques. (A) Representative images of immunohistochemical staining and fluorescence microscopy of carotid plaques. (B) Lipid contents were assessed by Oil Red O (ORO) staining. (C) Macrophage contents were analyzed by Mac3 immunostaining. Data are presented as mean ± SD. n = 6 mice per group; *P < 0.05, ** P < 0.01, by ANOVA followed by Mann-Whitney test. (D) Representative images of the aortic root lesions subjected to hematoxylin and eosin (H&E) staining, ORO staining, immunostaining for pan-macrophage marker Mac3 and classically activated macrophage marker CD86 in each group. Quantification results of H&E (E), ORO (F), Mac3 (G), and CD86 (H) stained area. Data are presented as mean ± SD. n = 6 mice per group; * P < 0.05, ** P < 0.01, by ANOVA followed by Mann-Whitney test.
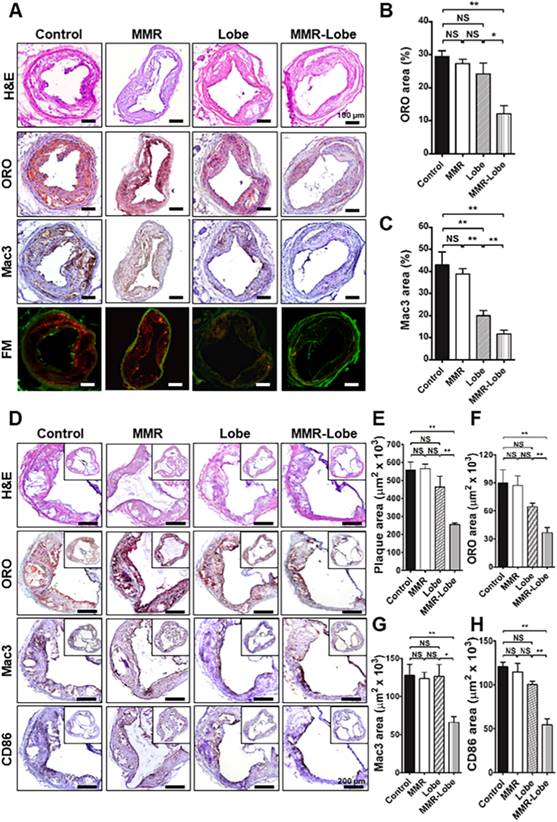
To further validate the effects of MMR-Lobe on systemic atheroma, we comprehensively compared atherosclerotic burden at aortic sinus level among the treatment groups. 960 cross-sections in total were analyzed, covering the entire aortic root with 40 cross-sections of the aortic sinus area per mouse (6 mice per group). The largest plaque area with the three valve leaflets was adopted for morphological analysis. Aortic plaque lesions were significantly smaller in the mice treated with MMR-Lobe than those of control, MMR vehicle, and lobeglitazone-treated mice (54.1% vs. control, P = 0.002, 54.8% vs. MMR vehicle, P = 0.002, and 45.0% vs. lobeglitazone, P = 0.005, respectively) as determined by H & E staining (Figure 7D and 7E). Consistently, immunostainings of the MMR-lobe group showed lower levels of lipid contents, macrophages, and even activated macrophages as compared to those of control, MMR vehicle, and lobeglitazone groups; 59.3% vs. control, P = 0.022, 58.0% vs. MMR vehicle, P = 0.008, and 43.2% vs. lobeglitazone, P = 0.005, reductions in lipids by ORO stainings (Figure 7D and 7F); 51.8% vs. control, P = 0.001, 50.1% vs. MMR vehicle, P < 0.001, and 51.3% vs. lobeglitazone, P = 0.035, reductions in macrophages by Mac3 stainings, respectively (Figure 7D and 7G); 54.9% vs. control, P = 0.005, 52.6% vs. MMR vehicle, P = 0.003, and 45.8% vs. lobeglitazone, P = 0.008, reductions in classically activated macrophages by CD86 staining (Figure 7D and 7H).
Metabolic parameters after MMR-Lobe treatment
There were no significant differences in body weight changes before and after 4 weeks of treatment among four groups. Blood glucose levels, and lipid profiles were similar among the groups. Although statistically insignificant, HDL-cholesterol in the lobeglitazone per se group tended to be higher than the other groups (Table 1).
Metabolic parameters. Data are expressed as mean ± SD for 6 mice per group. NS; not significant.
| Control (n=6) | MMR (n=6) | Lobeglitazone (n=6) | MMR-Lobe (n=6) | P value | |
|---|---|---|---|---|---|
| Body weight change (%) | 12.5 ± 9.0 | 9.1 ± 4.7 | 14.8 ± 9.1 | 10.6 ± 6.5 | NS |
| Glucose (mg/dL) | 147.0 ± 25.3 | 152.8 ± 19.0 | 143.0 ± 13.8 | 157.4 ± 27.7 | NS |
| Total cholesterol (mg/dL) | 275.3 ± 92.7 | 296.5 ± 27.9 | 328.4 ± 58.9 | 290.0 ± 26.5 | NS |
| Triglyceride (mg/dL) | 37.8 ± 14.6 | 30.8 ± 10.8 | 37.2 ± 13.7 | 34.4 ± 5.0 | NS |
| HDL cholesterol (mg/dL) | 77.7 ± 6.8 | 82.0 ±3.6 | 87.3 ± 6.4 | 77.2 ± 8.2 | NS |
| LDL cholesterol (mg/dL) | 204.7 ± 86.5 | 203.8 ±27.7 | 250.3 ± 42.0 | 217.6 ± 20.9 | NS |
Discussion
The present study demonstrates robust therapeutic efficacy of novel targeted PPARγ activation on inflamed high-risk plaques. We developed a biocompatible nanocarrier containing a new PPARγ agonist, lobeglitazone, which was designed for binding to mannose receptors of macrophages (MMR-Lobe), and thus, allowed for specific activation of the PPARγ pathways within the inflamed atheroma. Notably, serial in vivo optical imaging showed that the treatment of injectable MMR-Lobe significantly reduced plaque burden and inflammation in atherogenic mice without undesired systemic influences. The comprehensive assessment of en face whole aortas by ex vivo imaging and immunohistological analysis well corroborated the serial imaging findings in vivo. The in vitro assays and immunofluorescence findings revealed the underlying mechanism of our MMR-Lobe therapy, such as enhancement of cholesterol efflux through PPARγ-, LXRα-, ABCA1, ABCG1 dependent pathways and prominent suppression of inflammation activity in macrophage foam cells.
PPARγ agonist is known to enhance insulin sensitivity and decrease hepatic glucose production in type 2 diabetes mellitus. Besides their glucose lowering efficacy, these drugs have favorable effects on lipid profile and inflammatory parameters in a dose dependent manner [30]. Despite a prospective randomized clinical trial showing a reduction of cardiovascular events in type 2 diabetic patients [31-33] and several studies demonstrating beneficial effects on atherogenesis [3, 34-38], the translational application of high-dose PPARγ agonists for atherosclerosis treatment has still been limited due to systemic adverse effects [13-15].
As a targeted therapeutic strategy to solve this obstacle, poly (lactic-co-glycolic acid) (PLGA)-mediated rosiglitazone was reported to increase regulators of cholesterol efflux in bone marrow-derived macrophages; however, in vivo effects regarding plaque size or composition were lacking [39]. Likewise, PLGA-mediated delivery of pioglitazone significantly decreased the degree of buried fibrous cap and local protease activity [25], but failed to achieve significant reduction in plaque size and macrophage accumulation. In the current study, we loaded a new PPARγ agonist, lobeglitazone, into the MMR-targeted GC carrier (MMR-Lobe) to specifically activate PPARγ pathways in high-risk plaques. Lobeglitazone is a new PPARγ agonist with more potent affinity for PPARγ receptors than other ones including pioglitazone and rosiglitazone [40]. Hydrophobically-modified glycol chitosan (GC) was used as a carrier vehicle because it is biocompatible, biodegradable, and rarely immunogenic and its general properties and in vivo biodistribution were well established in the previous studies [29, 41-43]. In brief, as GC carrier is composed of a hydrophilic shell and hydrophobic inner core, it can efficiently encapsulate various hydrophobic agents with high loading efficiency, and enhance their solubility and stability in vivo [42, 44, 45]. Biodistribution experiments showed relatively high florescence signals in the spleen and liver, which could be accounted by the abundant distribution of macrophages expressing mannose receptors in both organs. MMR probe also was uptaken by plaque macrophages and emitted strong NIRF signals [29]. In the present study, while lobeglitazone per se treatment modestly attenuated inflammation in the atheroma, intriguingly, 4 week treatment of MMR-Lobe caused a marked reduction of both plaque burden and inflammation activity without major systemic disturbances. To the best of our knowledge, our result is the first in vivo evidence to demonstrate prominent therapeutic effects of targeted PPARγ activation on the atheroma, including regression of plaque burden and suppression of inflammatory activity.
Mechanistically, MMR-Lobe induced cholesterol efflux in RAW 264.7 cells through the upregulation of cholesterol efflux regulatory proteins such as ABCA1 and ABCG1 via nuclear receptor LXRα, and suppressed inflammatory cytokines in macrophage foam cells. In immunofluorescence analysis of the plaques, co-activation of PPARγ, LXRα, ABCA1, and ABCG1 were strongly enhanced by MMR-Lobe treatment, whereas the expression of MMP-9 decreased significantly in MMR-Lobe-treated mice as compared to those in the lobeglitazone per se group or controls. These findings are ascribed to the outstanding affinity of MMR-Lobe to plaque macrophages achieved by combining effects of both macropinocytosis and receptor mediated endocytosis, resulting in higher concentration of the PPARγ agonist inside macrophage foam cells than lobeglitazone per se.
While previous studies were limited by their analysis of ex vivo findings [21, 25, 39], our research serially evaluated the in vivo effects of MMR-Lobe on carotid plaques in the same individuals using customized real-time optical imaging. The in vivo imaging of the murine carotid plaque was optimized by a manual 5-axis staging platform with vessel fixation and a monitored axial piezoelectric scanner. The ability to quantitatively and serially estimate plaque macrophage distribution in each individual is expected to provide much more relevant comparison regarding drug efficacy, requiring far fewer experimental animals than conventional ex vivo analysis. Furthermore, to quantitate the macrophage-derived NIRF signals, we applied a thresholding algorithm based on averaged auto-fluorescence. Considering that plaque target-to-background ratio (pTBR) has a potential limitation such as intra- and inter-individual variability depending on the selection of target region-of-interest (ROI) area [10], automatic quantitative comparison of plaque macrophages in our study could be more reliable and accurate.
The present study has some limitations. First, the dose of p.o. lobeglitazone may not be completely comparable with the dose of the intravenously administered lobeglitazone contained in MMR-Lobe. However, the oral bioavailability of lobeglitazone was reported to be ~95% [46]. Second, neither tissue concentration nor serum concentration of lobeglitazone was measured. Further studies are required to clarify the in vivo pharmacokinetics of MMR-Lobe in mice and larger animals as well for translational applications. Third, although mannose receptors are present in high-risk plaque phenotypes such as TCFA [28], neovascularization, and intraplaque hemorrhages [47, 48], they could be associated with alternatively activated macrophages. Thus, there is still concern regarding whether our strategy targeting MMR would have sustainable beneficial effects on plaque stabilization. Plaque instability might be the consequence of an imbalance between pro-inflammatory effects of classically activated macrophages and beneficial effects of alternatively activated macrophages. The macrophages comprise heterogeneous cells that adapt their functional phenotype in response to their microenvironment [48]. As alternatively activated macrophage differentiation accelerates cholesterol uptake [49], and exposure to oxidized LDL renders those cells more pro-inflammatory [50], our MMR-Lobe strategy is expected to effectively stabilize the plaque by inhibition of foam cell formation via augmented cholesterol efflux, and sequential attenuation of inflammatory activity. Interestingly, in current immunohistochemistry analysis, not only Mac3 but also CD86, a classically activated macrophage marker, decreased significantly in the MMR-Lobe group. Considering the possibility of a heterogeneous spectrum of macrophage phenotypes between anti- and pro-inflammatory poles, we speculate that MMR-Lobe treatment has overall favorable effects on inflamed plaques, albeit further research regarding the long-term influence of MMR-Lobe treatment on functional polarization of plaque macrophages is needed.
The clinical implementation of MMR-Lobe would be relevant to both acute and chronic stages of coronary artery disease. By extrapolating from the results of our study, we anticipate that intravenous administration of MMR-Lobe could rapidly suppress plaque inflammation by modulating the macrophage inflammatory cascade in patients with acute coronary syndrome, along with the current standard therapy. Even in patients with chronic coronary artery disease, MMR-Lobe treatment could be considered as an alternative for plaque regression in addition to the use of standard medications including statin. Finally, when conjugated with NIRF dye, targeted PPARγ activation by MMR-Lobe could be a promising theranostic strategy, which can simultaneously detect and treat the inflamed plaques using an appropriate in vivo imaging system such as OCT-NIRF integrated catheter [29, 51-53].
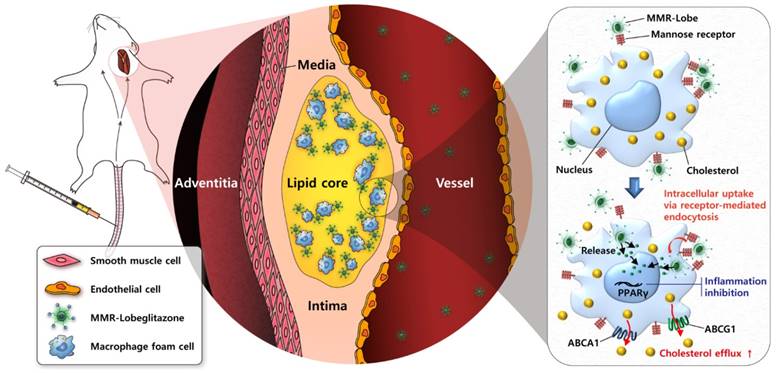
In conclusion, the present study has demonstrated robust anti-atherogenic and anti-inflammatory therapeutic effects of a novel plaque macrophage targeted, PPARɣ activation strategy on inflamed plaques (Figure 8). Given the prominent efficacy of MMR-Lobe in murine atheroma, this novel strategy could be a promising therapeutic approach for high-risk plaques.
Schematic illustration of targeted MMR-Lobe effects. After intravenous injection, circulating MMR-Lobe specifically binds to plaque macrophages and is taken up by macrophage foam cells. Once inside the plaque macrophages, MMR-Lobe releases lobeglitazone and enhances PPARγ activity, thus resulting in reduction of inflammation activity and enhancement of cholesterol efflux via ABCA1 and ABCG1.
Abbreviations
ABCA1: ATP-binding cassette transporters A1; ABCG1: ATP-binding cassette transporters G1; GC: glycol chitosan; LXRα: Liver X receptor alpha; MMR: macrophage mannose receptor; NIRF: near infrared fluorophore; PPARγ: Peroxisome proliferator-activated receptor gamma; TCFAs: thin-cap fibroatheromas.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF-2015R1A2A2A07027863 to J.W.K., K.S.P., H.Y.) and Korea University Future Research Grant. The authors thank Professor Soon Young Hwang for her expert advice on statistical assessments.
Supplementary Material
Supplementary methods and figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129-38
2. Ridker PM, Mora S, Rose L, Group JTS. Percent reduction in LDL cholesterol following high-intensity statin therapy: potential implications for guidelines and for the prescription of emerging lipid-lowering agents. Eur Heart J. 2016;37:1373-9
3. Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP. et al. PPAR-α and PPAR-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nature medicine. 2001;7:53-8
4. Li AC, Binder CJ, Gutierrez A, Brown KK, Plotkin CR, Pattison JW. et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARα, β/δ, and γ. Journal of Clinical Investigation. 2004;114:1564-76
5. Brown JD, Plutzky J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation. 2007;115:518-33
6. Fajas L, Auboeuf D, Raspe E, Schoonjans K, Lefebvre AM, Saladin R. et al. The organization, promoter analysis, and expression of the human PPARgamma gene. The Journal of biological chemistry. 1997;272:18779-89
7. Law RE, Goetze S, Xi XP, Jackson S, Kawano Y, Demer L. et al. Expression and function of PPARgamma in rat and human vascular smooth muscle cells. Circulation. 2000;101:1311-8
8. Corzo C, Griffin PR. Targeting the Peroxisome Proliferator-Activated Receptor-gamma to Counter the Inflammatory Milieu in Obesity. Diabetes Metab J. 2013;37:395-403
9. Patel CB, De Lemos JA, Wyne KL, McGuire DK. Thiazolidinediones and risk for atherosclerosis: pleiotropic effects of PPar gamma agonism. Diab Vasc Dis Res. 2006;3:65-71
10. Chang K, Francis SA, Aikawa E, Figueiredo JL, Kohler RH, McCarthy JR. et al. Pioglitazone suppresses inflammation in vivo in murine carotid atherosclerosis: novel detection by dual-target fluorescence molecular imaging. Arterioscler Thromb Vasc Biol. 2010;30:1933-9
11. Vucic E, Dickson SD, Calcagno C, Rudd JH, Moshier E, Hayashi K. et al. Pioglitazone modulates vascular inflammation in atherosclerotic rabbits noninvasive assessment with FDG-PET-CT and dynamic contrast-enhanced MR imaging. JACC Cardiovasc Imaging. 2011;4:1100-9
12. Lim S, Lee K-S, Lee JE, Park HS, Kim KM, Moon JH. et al. Effect of a new PPAR-gamma agonist, lobeglitazone, on neointimal formation after balloon injury in rats and the development of atherosclerosis. Atherosclerosis. 2015;243:107-19
13. Freid J, Everitt D, Boscia J. Rosiglitazone and hepatic failure. Ann Intern Med. 2000;132:164
14. Nesto RW, Bell D, Bonow RO, Fonseca V, Grundy SM, Horton ES. et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. October 7, 2003. Circulation. 2003;108:2941-8
15. Graham DJ, Ouellet-Hellstrom R, MaCurdy TE, Ali F, Sholley C, Worrall C. et al. Risk of acute myocardial infarction, stroke, heart failure, and death in elderly Medicare patients treated with rosiglitazone or pioglitazone. Jama. 2010;304:411-8
16. Decuzzi P, Pasqualini R, Arap W, Ferrari M. Intravascular delivery of particulate systems: does geometry really matter? Pharm Res. 2009;26:235-43
17. Kubo M, Egashira K, Inoue T, Koga J, Oda S, Chen L. et al. Therapeutic neovascularization by nanotechnology-mediated cell-selective delivery of pitavastatin into the vascular endothelium. Arterioscler Thromb Vasc Biol. 2009;29:796-801
18. Decuzzi P, Godin B, Tanaka T, Lee SY, Chiappini C, Liu X. et al. Size and shape effects in the biodistribution of intravascularly injected particles. Journal of controlled release: official journal of the Controlled Release Society. 2010;141:320-7
19. Oda S, Nagahama R, Nakano K, Matoba T, Kubo M, Sunagawa K. et al. Nanoparticle-mediated endothelial cell-selective delivery of pitavastatin induces functional collateral arteries (therapeutic arteriogenesis) in a rabbit model of chronic hind limb ischemia. J Vasc Surg. 2010;52:412-20
20. Chen L, Nakano K, Kimura S, Matoba T, Iwata E, Miyagawa M. et al. Nanoparticle-mediated delivery of pitavastatin into lungs ameliorates the development and induces regression of monocrotaline-induced pulmonary artery hypertension. Hypertension. 2011;57:343-50
21. Katsuki S, Matoba T, Nakashiro S, Sato K, Koga J, Nakano K. et al. Nanoparticle-mediated delivery of pitavastatin inhibits atherosclerotic plaque destabilization/rupture in mice by regulating the recruitment of inflammatory monocytes. Circulation. 2014;129:896-906
22. Fan B, Kang L, Chen L, Sun P, Jin M, Wang Q. et al. Systemic siRNA Delivery with a Dual pH-Responsive and Tumor-targeted Nanovector for Inhibiting Tumor Growth and Spontaneous Metastasis in Orthotopic Murine Model of Breast Carcinoma. Theranostics. 2017;7:357-76
23. Fan F, Yu Y, Zhong F, Gao M, Sun T, Liu J. et al. Design of Tumor Acidity-Responsive Sheddable Nanoparticles for Fluorescence/Magnetic Resonance Imaging-Guided Photodynamic Therapy. Theranostics. 2017;7:1290-302
24. Gao N, Bozeman EN, Qian W, Wang L, Chen H, Lipowska M. et al. Tumor Penetrating Theranostic Nanoparticles for Enhancement of Targeted and Image-guided Drug Delivery into Peritoneal Tumors following Intraperitoneal Delivery. Theranostics. 2017;7:1689-704
25. Nakashiro S, Matoba T, Umezu R, Koga JI, Tokutome M, Katsuki S. et al. Pioglitazone-Incorporated Nanoparticles Prevent Plaque Destabilization and Rupture by Regulating Monocyte/Macrophage Differentiation in ApoE-/- Mice. Arterioscler Thromb Vasc Biol. 2016
26. Duivenvoorden R, Tang J, Cormode DP, Mieszawska AJ, Izquierdo-Garcia D, Ozcan C. et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun. 2014;5:3065
27. Kim H, Kim Y, Kim IH, Kim K, Choi Y. ROS-responsive activatable photosensitizing agent for imaging and photodynamic therapy of activated macrophages. Theranostics. 2013;4:1-11
28. Tahara N, Mukherjee J, de Haas HJ, Petrov AD, Tawakol A, Haider N. et al. 2-deoxy-2-[18F]fluoro-D-mannose positron emission tomography imaging in atherosclerosis. Nat Med. 2014;20:215-9
29. Kim JB, Park K, Ryu J, Lee JJ, Lee MW, Cho HS. et al. Intravascular optical imaging of high-risk plaques in vivo by targeting macrophage mannose receptors. Sci Rep. 2016;6:22608
30. Pfutzner A, Marx N, Lubben G, Langenfeld M, Walcher D, Konrad T. et al. Improvement of cardiovascular risk markers by pioglitazone is independent from glycemic control: results from the pioneer study. J Am Coll Cardiol. 2005;45:1925-31
31. Dormandy JA, Charbonnel B, Eckland DJA, Erdmann E, Massi-Benedetti M, Moules IK. et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. The Lancet. 2005;366:1279-89
32. Erdmann E, Song E, Spanheimer R, van Troostenburg de Bruyn AR, Perez A. Observational follow-up of the PROactive study: a 6-year update. Diabetes Obes Metab. 2014;16:63-74
33. Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M. et al. Pioglitazone after Ischemic Stroke or Transient Ischemic Attack. N Engl J Med. 2016;374:1321-31
34. Duan SZ, Usher MG, Mortensen RM. Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circ Res. 2008;102:283-94
35. Marx N. Antidiabetic PPARgamma-Activator Rosiglitazone Reduces MMP-9 Serum Levels in Type 2 Diabetic Patients With Coronary Artery Disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;23:283-8
36. Charo IF. Macrophage polarization and insulin resistance: PPARgamma in control. Cell Metab. 2007;6:96-8
37. Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82-6
38. Akiyama TE, Sakai S, Lambert G, Nicol CJ, Matsusue K, Pimprale S. et al. Conditional Disruption of the Peroxisome Proliferator-Activated Receptor Gene in Mice Results in Lowered Expression of ABCA1, ABCG1, and apoE in Macrophages and Reduced Cholesterol Efflux. Molecular and Cellular Biology. 2002;22:2607-19
39. Di Mascolo D, C JL, Aryal S, Ramirez MR, Wang J, Candeloro P. et al. Rosiglitazone-loaded nanospheres for modulating macrophage-specific inflammation in obesity. Journal of controlled release: official journal of the Controlled Release Society. 2013;170:460-8
40. Lee HW, Kim BY, Ahn JB, Kang SK, Lee JH, Shin JS. et al. Molecular design, synthesis, and hypoglycemic and hypolipidemic activities of novel pyrimidine derivatives having thiazolidinedione. Eur J Med Chem. 2005;40:862-74
41. Kean T, Thanou M. Biodegradation, biodistribution and toxicity of chitosan. Adv Drug Deliv Rev. 2010;62:3-11
42. Hwang HY, Kim IS, Kwon IC, Kim YH. Tumor targetability and antitumor effect of docetaxel-loaded hydrophobically modified glycol chitosan nanoparticles. Journal of controlled release: official journal of the Controlled Release Society. 2008;128:23-31
43. Park K, Kim JH, Nam YS, Lee S, Nam HY, Kim K. et al. Effect of polymer molecular weight on the tumor targeting characteristics of self-assembled glycol chitosan nanoparticles. Journal of controlled release: official journal of the Controlled Release Society. 2007;122:305-14
44. Min KH, Park K, Kim YS, Bae SM, Lee S, Jo HG. et al. Hydrophobically modified glycol chitosan nanoparticles-encapsulated camptothecin enhance the drug stability and tumor targeting in cancer therapy. Journal of controlled release: official journal of the Controlled Release Society. 2008;127:208-18
45. Min HS, You DG, Son S, Jeon S, Park JH, Lee S. et al. Echogenic Glycol Chitosan Nanoparticles for Ultrasound-Triggered Cancer Theranostics. Theranostics. 2015;5:1402-18
46. Lee JH, Noh CK, Yim CS, Jeong YS, Ahn SH, Lee W. et al. Kinetics of the Absorption, Distribution, Metabolism, and Excretion of Lobeglitazone, a Novel Activator of Peroxisome Proliferator-Activated Receptor Gamma in Rats. J Pharm Sci. 2015;104:3049-59
47. Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X. et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol. 2012;59:166-77
48. Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12:10-7
49. Oh J, Riek AE, Weng S, Petty M, Kim D, Colonna M. et al. Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. The Journal of biological chemistry. 2012;287:11629-41
50. van Tits LJ, Stienstra R, van Lent PL, Netea MG, Joosten LA, Stalenhoef AF. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: a crucial role for Kruppel-like factor 2. Atherosclerosis. 2011;214:345-9
51. Yoo H, Kim JW, Shishkov M, Namati E, Morse T, Shubochkin R. et al. Intra-arterial catheter for simultaneous microstructural and molecular imaging in vivo. Nat Med. 2011;17:1680-4
52. Lee S, Lee MW, Cho HS, Song JW, Nam HS, Oh DJ. et al. Fully integrated high-speed intravascular optical coherence tomography/near-infrared fluorescence structural/molecular imaging in vivo using a clinically available near-infrared fluorescence-emitting indocyanine green to detect inflamed lipid-rich atheromata in coronary-sized vessels. Circ Cardiovasc Interv. 2014;7:560-9
53. Kim S, Lee MW, Kim TS, Song JW, Nam HS, Cho HS. et al. Intracoronary dual-modal optical coherence tomography-near-infrared fluorescence structural-molecular imaging with a clinical dose of indocyanine green for the assessment of high-risk plaques and stent-associated inflammation in a beating coronary artery. Eur Heart J. 2016
Author contact
![]() Corresponding authors: Jin Won Kim, M.D., Ph.D., F.A.C.C. Address: Cardiovascular Center, Korea University Guro Hospital, 80, Guro-dong, Guro-gu, Seoul 152-703, Republic of Korea. Tel: 82-2-2626-3021, Fax: 82-2-863-1109, E-mail: kjwmmac.kr and Kyeongsoon Park, Ph.D. Address: Department of Systems Biotechnology, College of Biotechnology and Natural Resources, Chung-Ang University, Anseong-si, Gyeonggi-do, 17546, Korea, Tel: +82-31-670-3357. Fax: 82-31-675-1381. E-mail: kspark1223ac.kr and Hongki Yoo, Ph.D. Address: Department of Biomedical Engineering, Hanyang University, 222 Wangsimni-ro, Seongdong-gu, Seoul 133-791, Republic of Korea. Tel: 82-2-2220-2323, Fax: 82-2-2220-5943, E-mail: hyooac.kr
Corresponding authors: Jin Won Kim, M.D., Ph.D., F.A.C.C. Address: Cardiovascular Center, Korea University Guro Hospital, 80, Guro-dong, Guro-gu, Seoul 152-703, Republic of Korea. Tel: 82-2-2626-3021, Fax: 82-2-863-1109, E-mail: kjwmmac.kr and Kyeongsoon Park, Ph.D. Address: Department of Systems Biotechnology, College of Biotechnology and Natural Resources, Chung-Ang University, Anseong-si, Gyeonggi-do, 17546, Korea, Tel: +82-31-670-3357. Fax: 82-31-675-1381. E-mail: kspark1223ac.kr and Hongki Yoo, Ph.D. Address: Department of Biomedical Engineering, Hanyang University, 222 Wangsimni-ro, Seongdong-gu, Seoul 133-791, Republic of Korea. Tel: 82-2-2220-2323, Fax: 82-2-2220-5943, E-mail: hyooac.kr