Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(1):31-44. doi:10.7150/thno.21342 This issue Cite
Research Paper
A Novel DNA Aptamer for Dual Targeting of Polymorphonuclear Myeloid-derived Suppressor Cells and Tumor Cells
Haoran Liu1,2, Junhua Mai2, Jianliang Shen2, Joy Wolfram2,3, Zhaoqi Li2,4, Guodong Zhang2, Rong Xu5, Yan Li2, Chaofeng Mu2, Youli Zu6, Xin Li7, Ganesh L. Lokesh7, Varatharasa Thiviyanathan7, David E. Volk7, David G. Gorenstein7, Mauro Ferrari2,8, Zhongbo Hu1, Haifa Shen2,9 ![]()
1. College of Materials Science and Opto-Electronic Technology, University of Chinese Academy of Sciences, Beijing 100049, China
2. Department of Nanomedicine, Houston Methodist Hospital Research Institute, Houston, TX 77030, USA
3. Department of Transplantation, Mayo Clinic, Jacksonville, FL 32224, USA
4. Xiangya Hospital of Central South University, Changsha, Hunan 410013, China
5. Department of Pharmacology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China
6. Department of Pathology and Genomic Medicine, Houston Methodist Hospital, Houston, TX 77030, USA
7. Institute of Molecular Medicine and the Department of Nanomedicine and Biomedical Engineering, McGovern Medical School, The University of Texas Health Science Center at Houston, TX 77030, USA
8. Department of Medicine, Weill Cornell Medical College, New York, NY 10065, USA
9. Department of Cell and Developmental Biology, Weill Cornell Medicine, New York, NY 10065, USA
Received 2017-6-5; Accepted 2017-9-28; Published 2018-1-1
Abstract

Aptamers have the potential to be used as targeting ligands for cancer treatment as they form unique spatial structures.
Methods: In this study, a DNA aptamer (T1) that accumulates in the tumor microenvironment was identified through in vivo selection and validation in breast cancer models. The use of T1 as a targeting ligand was evaluated by conjugating the aptamer to liposomal doxorubicin.
Results: T1 exhibited a high affinity for both tumor cells and polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs). Treatment with T1 targeted doxorubicin liposomes triggered apoptosis of breast cancer cells and PMN-MDSCs. Suppression of PMN-MDSCs, which serve an immunosuppressive function, leads to increased intratumoral infiltration of cytotoxic T cells. Conclusion: The cytotoxic and immunomodulatory effects of T1-liposomes resulted in superior therapeutic efficacy compared to treatment with untargeted liposomes, highlighting the promise of T1 as a targeting ligand in cancer therapy.
Keywords: DNA aptamer, active targeting, tumor microenvironment, myeloid-derived suppressor cells (MDSCs), liposome
Introduction
The tumor microenvironment (TME), which consists of extracellular matrix components and various cell types, such as cancer-associated fibroblasts, infiltrating immune cells, and endothelial cells, plays a critical role in tumor progression [1]. Strategies that target the TME have the potential to overcome therapeutic challenges as surrounding cells often develop tumor-protective functions [2-5]. Among various infiltrating immune cells, myeloid-derived suppressor cells (MDSCs), a major population of immature myeloid cells, stimulate angiogenesis, promote metastasis, and serve an immunosuppressive role through inhibition of CD8+ T cell proliferation and function [6-8]. The dramatic expansion of MDSCs in blood, spleen, and bone marrow has been widely documented in many animal tumor models as well as in patients with various types of cancers [9]. Mouse MDSCs express two primary surface markers, CD11b and Gr-1, and can be further classified as polymorphonuclear (PMN-MDSCs, CD11b+Ly6G+Ly6Clow), which form the largest population of MDSCs, and monocytic (M-MDSCs, CD11b+Ly6G-Ly6Chigh) [10]. Recent studies have revealed that depletion [11-14] or inhibition [15-17] of MDSCs substantially augments antitumor responses, highlighting the promise of MDSC targeting strategies for cancer therapy.
Single-stranded oligonucleotide (DNA or RNA) aptamers can bind to biological targets with high affinity and specificity, owing to the unique 3D structure of these molecules. Aptamers can be selected from a random library using a procedure known as systematic evolution of ligands by exponential enrichment (SELEX) [18, 19]. Briefly, iterative selection rounds are applied to enrich for sequences with strong binding affinity for a specific target, eventually yielding several potential aptamer candidates. Both in vitro-based SELEX methods, such as cell-SELEX and protein-SELEX, and live-animal-based SELEX have been developed [20-22]. Aptamers selected with these techniques have shown promising potential for diagnostic and therapeutic applications. Recently, nanoparticle-based drug delivery systems have attracted attention as powerful tools to overcome biological barriers in the body in order to improve the therapeutic efficacy and safety of drugs [23-26]. However, there exists a need to increase site-specific delivery of systemically administered nanoparticles. Several studies have demonstrated that active targeting approaches involving nanoparticle functionalization with aptamers can dramatically improve drug delivery [27-36].
Here, a TME-targeting DNA thioaptamer candidate, T1, was selected and validated in an MDA-MB-231 breast cancer bone metastasis model. The targeting ability of T1 was further validated in MDA-MB-231 and 4T1 orthotopic breast cancer models. This study is the first to demonstrate the identification of an aptamer that displays high binding affinity for both PMN-MDSCs and tumor cells in vitro and in vivo. Furthermore, the conjugation of the T1 aptamer to a well-established liposomal delivery system for doxorubicin (Dox) extended the therapeutic potential of this drug beyond cancer cell cytotoxicity to immunomodulatory capacity. Anticancer efficacy was evaluated in the poorly immunogenic and highly metastatic 4T1 orthotopic breast cancer model, which is associated with PMN-MDSC expansion [15, 37]. T1-targeted liposomes induced transformation of the TME toward a more immunostimulatory state, leading to improved anticancer efficacy. In vivo results revealed that the combination of chemotherapy and immunotherapy effectively suppressed tumor growth. In summary, the successful identification of an aptamer with dual targeting capability for PMN-MDSCs and tumor cells was leveraged to improve cancer therapy.
Materials and Methods
Cell culture
The murine breast cancer cell line 4T1, human breast cancer cell lines MDA-MB-231, MDA-MB-468, and MCF-7, human lung cancer cell lines A549 and HCC827, the human Burkitt's lymphoma cell line Raji, and human acute monocytic leukemia cell lines (AMoL) MV4-11 were obtained from ATCC (American Type Culture Collection). H322 and H1299 human lung cancer cell lines were obtained from the National Cancer Institute (NCI). The TUBO mammary carcinoma cell line was generously provided by Dr. L. Pease (Mayo Clinic). The human chronic myeloid leukemia (CML) cell line 32Dp210 was derived from the interleukin 3 (IL-3)-dependent murine hematopoietic cell line [38]. The acute myeloid leukemia (AML) cell line Molm-13 was obtained from Leibniz-Institut DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH). Hematopoietic stem cells (HSCs) were isolated from bone marrow as previously described [39].
HSCs, Raji, MV4-11, and 32Dp210 cells were cultured in RPMI 1640 (Corning Inc., USA) medium supplemented with 10% fetal bovine serum (FBS) and penicillin (100 IU/mL)/streptomycin (100 μg/mL) (Cellgro, Corning, USA) at 37 °C with 5% CO2. Other cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Corning, USA) supplemented with 10% FBS and penicillin (100 IU/mL)/streptomycin (100 μg/mL) at 37 °C with 5% CO2.
Murine models
All animal studies were performed following protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the Houston Methodist Research Institute. Female athymic nude mice and BALB/c mice (6-8 weeks old) were purchased from Charles River Laboratories (Boston, MA, USA). To generate an MDA-MB-231 orthotopic breast cancer model, 3 × 106 cells were inoculated in the mammary fat pad of nude mice. An MDA-MB-231 murine bone metastatic model was established in nude mice through intracardiac inoculation of 1 × 105 cells transfected with a plasmid carrying the luciferase gene. Tumor growth in the bone was monitored through bioluminescence imaging using a Xenogen in vivo imaging system (IVIS)-200 imaging system (PerkinElmer, Inc., USA). A 4T1 orthotopic breast cancer model was generated by injecting 3 × 104 4T1 cells in the mammary fat pad of BALB/c mice.
In vivo aptamer selection
A DNA thioaptamer combinatorial library was synthesized as previously described [40]. The library consisted of a 21 base 5'-primer (5'-CGCTCGATAGATCGAGCTTCG-3'), a 23 base 3'-primer (5'-GTCGATCACGCTCTAGAGCACTG-3') in the flank, and a 30 base random region in the middle. The library (10 μg) was intravenously injected into mice bearing MDA-MB-231 breast cancer bone metastases. Mice were euthanized 4 h post-injection and tumor tissues were collected and homogenized. Bound thioaptamers were extracted and amplified with amplified polymerase chain reaction (PCR) using primers specific for the aptamer library. The amplified PCR products were reinjected into mice for a next round of screening. After ten iterative selection cycles, the amplified PCR products were subcloned into a plasmid vector for DNA sequencing, generating several candidates. The sequences with highest occurrence frequency was named T1 and used for further analysis. The full sequence of T1 is: (5'-CGCTCGATAGATCGAGCTTCGCTCGATGTGGTGTTGTGGGGGCTTGTATTGGTCGATCACGCTCTAGAGCACTG-3'). A random scrambled aptamer (Scr) sequence (5'-ATCCAGAGTGACGCAGCACTACTGGACTTCATCGGAGCTAGGTCATCGCTTGCATGCATGGACACGGTGGCTTA-3') was used as a control. T1 and Scr aptamers were synthesized (Integrated DNA Technologies, Inc., USA) and labeled with Cy5, as this dye is suitable for several applications, including flow cytometry, confocal microscopy, and IVIS imaging.
In vitro evaluation of T1
For cell viability studies, MDA-MB-231 cells were seeded in 96-well plates at a seeding density of 5 × 103 cells/well and incubated with Scr or T1 aptamers. Cell proliferation was measured 48 h later using a cell counting kit-8 (CCK8) viability assay (Dojindo Molecular Technologies, Inc. Japan) according to the manufacturer's instructions. For evaluation of cell migration using the scratch assay, MDA-MB-231 cells were seeded in 6-well plates (85% confluency). Cells were scratched with a pipette tip and then incubated with 500 nM Scr and T1 aptamer for 24 h. Microscopy images were taken immediately after scratching and 24 h later. To study the stability of the T1 aptamer, the aptamer was incubated with 2% FBS in phosphate buffered saline at 37 °C for 0 h, 1 h, 3 h, 5 h, 8 h, 12 h, 24 h, 48 h, and 72 h. Electrophoresis was performed on 2% agarose gels at a constant voltage of 115V for 25 min. To study the uptake of the T1 aptamer, MDA-MB-231 cells were seeded in 4-well chamber slides (LAB-TEK, Nagle Nunc, IL, USA) overnight at a density of 2 × 104 cells per well. Cells were then incubated with the Cy5-labeled T1 aptamer for 30 min. The cells were washed twice with pre-warmed PBS, fixed with 4% paraformaldehyde solution for 10 min, washed twice with PBS, and stained with 4′,6-diamidino-2-phenylindole (DAPI, Thermo Fisher scientific, USA) for 10 min. Finally, cells were washed twice with PBS and imaged with a confocal microscope (A1 Confocal Imaging System, Nikon). The binding buffer used in the aptamer binding studies was prepared by adding yeast tRNA (0.1 mg/mL) (Sigma) in the washing buffer (PBS/2% FBS). Cells were exposed to ethylenediaminetetraacetic acid (EDTA) and then washed, incubated with 100 μL of Cy5-labeled aptamer solution on ice for 30 min, and washed twice with washing buffer. Cells (1 × 105) were counted with flow cytometry (LSRII, BD Biosciences) and analyzed with Flowjo 7.6.1 software (Tree Star, USA). The apparent dissociation constants (Kd) of the aptamer-cell interaction was obtained by using the following equation Y = Bmax X/(Kd + X). To identify the cellular internalization pathway of T1, MDA-MB-231 cells were pretreated with various uptake inhibitors for 40 min and incubated with T1 for 30 min. Specifically, cells were exposed to amiloride (macropinocytosis; 50 μM, 100 μM and 200 μM), chlorpromazine (clathrin-dependent endocytosis; 7.5 μM, 15 μM and 30 μM), cytochalasin D (broad-spectrum inhibitor of actin-dependent endocytosis; 4 μM, 8 μM, and 16 μM), dynasore (dynamin inhibitor; 25 μM, 50 μM, and 100 μM), and genistein (caveolae-dependent endocytosis; 40 μM, 80 μM, and 160 μM). The inhibitor concentrations were selected based on maintaining cell viability above 80%. Cells were then detached with trypsin, washed twice with washing buffer, and analyzed with flow cytomerty using Flowjo software.
Ex vivo and in vivo biodistribution of T1
To investigate the biodistribution of the aptamers, Cy5-labeled Scr or T1 aptamers (0.5 nmol/mouse or 2 nmol/mouse) were intravenously injected in mice bearing MDA-MB-231 orthotopic breast cancer tumors, MDA-MB-231 bone metastases, or 4T1 orthotopic breast cancer tumors (n = 3). The biodistribution of the aptamers was assessed at various time points (5 min, 1 h, 2 h, 4 h, and 8 h) using an IVIS-200 imaging system. Animals were euthanized 4 h or 8 h post-injection, and major organs and bone samples were collected and visualized with the IVIS-200 imaging system. The signal-to-noise ratio (SNR) was obtained as previously described [41] using the following equation: SNR = ηA / ηB, where ηA and ηB are the total radiant efficiency of organs from aptamer-injected and control mice, respectively. Tumor samples were frozen and processed for confocal fluorescence microscopy to detect Cy5-labeled aptamers. Single-cell suspensions were prepared from blood, lung, spleen, and bone marrow samples to determine aptamer binding by flow cytometry (LSRII, BD Biosciences, USA). Specifically, tumor samples were minced with scalpels and incubated with 250 U/mL collagenase type III (Worthington Biochem, USA) for 2 h at 37 °C. Tumor samples were then homogenized by repeated pipetting and filtered through 70-µm nylon filters (BD Biosciences, USA). Single-cell suspensions from spleen samples were obtained by grinding the organ through 40-µm filters, while bone marrow cells were harvested from the tibia and femur by flushing with PBS. Red blood cell lysis with ACK Lysing Buffer (Lonza, USA) was performed as required and all samples were washed and re-suspended in washing buffer. Cells were stained with antibodies of cell specific markers on ice for 30 min (MDA-MB-231 cell, HLA-ABC+; PMN-MDSCs, CD45+CD11b+Ly6G+Ly6Clow; M-MDSCs, CD45+CD11b+Ly6G-Ly6Chigh; T cells, CD45+CD3+; B cells, CD45+B220+; Macrophages, CD45+CD11b+F4/80+). CD45, CD3, and CD11b antibodies were purchased from Tonbo Biosciences (USA), while all other antibodies were acquired from eBioscience (USA). The antibodies were diluted 1:100 in PBS containing 2% FBS.
Preparation and characterization of aptamer-Dox liposomes
Aptamer-lipid conjugates (Scr-lipid or T1-lipid) were prepared by conjugation of the aptamer to the distal end of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N- [maleimide (polyethylene glycol)-2000 (DSPE-PEG-Mal) (Avanti Polar Lipids, Inc. USA) through thiol-maleimide coupling. Briefly, aptamers with a 5' end thiol group (Integrated DNA Technologies, USA) were reduced by Tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Thermo Fisher Scientific, USA) to convert any disulfide aptamer dimers back into thiol monomers, and then reacted with DSPE-PEG-Mal micelles in PBS (pH = 7, 3 mM EDTA) at room temperature overnight with magnetic stirring. Excess DSPE-PEG-Mal was removed by dialysis (3,000-3500 molecular weight cut off (MWCO) dialysis tubing, Spectrum Laboratories, Inc., USA) in DMSO and phosphate buffer (50 mM; pH = 7). The aptamer-lipid conjugates were concentrated using centrifugal filter unit (MWCO 3000, Millipore, USA) and lyophilized for liposome preparation. Empty liposomes were prepared and Dox was loaded using the ammonium sulfate gradient method according to a previously reported protocol [42]. Briefly, PEGylated liposomes composed of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), cholesterol, and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000 (DSPE-PEG(2000)) (Avanti Polar Lipids, Inc. USA)) and aptamer-lipids were mixed at the molar ratio of 63:32:4.5:0.5. The lipid mixture was frozen, lyophilized, hydrated at 60 °C in 150 mM ammonium sulfate (pH = 4), and sonicated until transparent. Dialysis was performed to remove unencapsulated ammonium sulfate against 20 mM HEPES buffered saline for 4 h (MWCO 10,000, Thermo Fisher Scientific, USA). Dox was then loaded into liposomes at a drug-to-lipid ratio of 1:10 (w/w) and incubated at 60 °C for 1 h with gentle shaking. Finally, unencapsulated Dox was removed by dialysis as described above. Dox loading efficiency was determined using a spectrophotometric absorbance assay. Briefly, Triton X-100 (Sigma, USA) was added to liposomal Dox to produce a 1% (v/v) final detergent concentration and the absorbance at 490 nm was recorded on a microplate reader (iMarkTM, Bio-Rad Laboratories, Inc., USA). Absorbance values were compared to that of a Dox standard curve. The mean diameter and zeta potential of liposomes were determined with dynamic light scattering and laser Doppler electrophoresis, respectively, using a Zetasizer Nano ZS Zen 3600 (Malvern, UK) at 25 °C. Liposomes were diluted in Milli-Q water, PBS, or DMEM (1:100 dilution). To study the uptake of liposomes functionalized with Scr aptamer (Scr-Dox) or T1 aptamer (T1-Dox), 4T1 cells were seeded in 4-well chamber slides (LAB-TEK, Nagle Nunc, IL, USA) overnight at a density of 2 × 104 cells per well. Cells were then incubated with Scr-Dox and T1-Dox (500 μg/mL) for 30 min. The cells were washed twice with pre-warmed PBS, fixed with 4% paraformaldehyde solution for 10 min, washed twice with PBS, and stained with DAPI for 10 min. Finally, cells were washed twice with PBS and imaged with a confocal microscope.
Biodistribution of aptamer-Dox liposomes
To evaluate the biodistribution of aptamer-Dox liposomes, mice bearing 4T1 orthotopic breast cancer tumors were intravenously administered with 6 mg/kg Scr-Dox or T1-Dox (n = 3). Animals were sacrificed 24 h post-injection and major organs (heart, liver, spleen, lungs, kidneys, brain, tumor, bone, and spine) were collected. The drug concentration in tissues was measured by high performance liquid chromatography (HPLC) with a fluorescence detector (Hitachi LaChrom Elite, Japan) and a Zorbax 300SB-C18 reverse phase column (Agilent, USA) [43].
Evaluation of the therapeutic efficacy of aptamer-Dox liposomes
Mice bearing 4T1 orthotopic breast cancer tumors were intravenously administered with PBS, free Dox, Scr-Dox, or T1-Dox (3 mg/kg) on day 7, 14, 21, and 28 post-injection of cancer cells. Body weight and tumor measurements (length × width × height/2) were recorded every other day. On day 35, mice were euthanized and major organs (heart, liver, spleen, lungs, and kidneys) were harvested, fixed in formalin, and processed for histological evaluation with hematoxylin and eosin (H&E) staining. Tumor, spleen, and blood samples were collected for single cell isolation as described above. MDSCs were analyzed by flow cytometry. For tumor immunofluorescent staining, tissues were harvested and frozen. Frozen slides were fixed in ice-cold methanol for 10 min, blocked with 10% bovine serum albumin (BSA) in PBS, and permeabilized with 0.1% triton X-100 for 1 h at room temperature. Slides were the incubated with FITC-anti-Gr-1 (Affymetrix eBioscience, 1:100) and PE-anti-mouse CD11b (Affymetrix eBioscience, 1:100) antibodies at 4°C overnight. Nuclear counterstaining was performed using DAPI according to the manufacturer's instructions. Slides were mounted with fluorescence mounting medium (Thermo Fisher scientific, USA) and visualized using a confocal microscope.
Statistical analysis
Statistical analysis was performed with the Student's t-test when comparing two independent groups and with two-way ANOVA when comparing more than two groups.
Results
Evaluation of T1 aptamer accumulation in tumors
In vivo aptamer selection was applied to an animal model of MDA-MB-231 breast cancer bone metastasis. Specifically, luciferase-transfected MDA-MB-231 cells were intracardially injected to generate bone metastases and bioluminescence imaging was performed to confirm tumor growth (Fig. S1A). After intravenous injection of a random library, bones were harvested and homogenized. DNA molecules were extracted and amplified using aptamer library-specific primers. The resulting DNA pool was then reinjected into mice and the process was repeated ten times. The PCR products were sequenced and several candidates were identified (Fig. 1A). The sequence with highest occurrence frequency (T1) was selected for further evaluation. A database search with the Basic Local Alignment Search Tool (BLAST) of the National Center for Biotechnology Information (NCBI) revealed a lack of homology between the T1 sequence and existing genes.
Validation of T1 aptamer tumor accumulation in an orthotopic and metastatic MDA-MB-231 breast cancer model. (A) Table showing seven DNA aptamer sequences with more than 300 reads present in breast cancer bone metastases after ten rounds of in vivo selection. (B-D) Mice were intravenously administered with a Cy5-labeled T1 or scrambled (Scr) aptamer. Tissues were analyzed 4 h post-injection. (B, C) Fluorescent images of organs and bones obtained with an in vivo imaging system (IVIS- 200 Spectrum) in mice bearing breast cancer bone metastases (B) or orthotopic breast cancer tumors(C). Organs: Bn, bones; Br, brain; He, heart; Ki, kidney; Li, liver; Lu, lung; Sp, spleen; St, sternum; Tu, tumor. Scale bar, 1 cm. (D) Representative confocal microscopy images of orthotropic breast cancer tumor sections. Scr aptamer (left) and T1 aptamer (right). Scale bar, 100 μm.
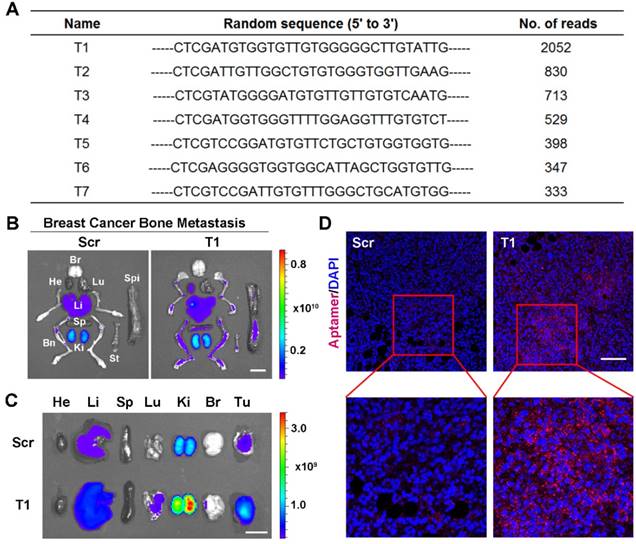
To confirm tumor-targeting capability of the T1 aptamer, Cy5-labeled T1 and Scr aptamers were intravenously injected into mice bearing MDA-MB-231 breast cancer bone metastases. Major organs and bones were collected for IVIS imaging. The results revealed that the T1 aptamer exhibited increased tumoritropic accumulation compared to the Scr aptamer (Fig. 1B and Fig. S1B). After confirming the targeting effect in the breast cancer bone metastasis model, biodistribution studies were performed in an MDA-MB-231 orthotopic breast cancer model to evaluate the utility of the aptamer for primary breast cancer. Consistent with the results obtained with the breast cancer bone metastasis model, T1 displayed increased tumor deposition compared to the Scr aptamer (Fig. 1C and Fig. S1C). Confocal microscopy analysis of tumor sections confirmed increased accumulation of T1 aptamers in tumors (Fig. 1D). Taken together, these results indicate that the T1 aptamer exhibits selectivity for the TME in both orthotopic and metastatic breast cancer models.
Identification of T1 aptamer binding targets
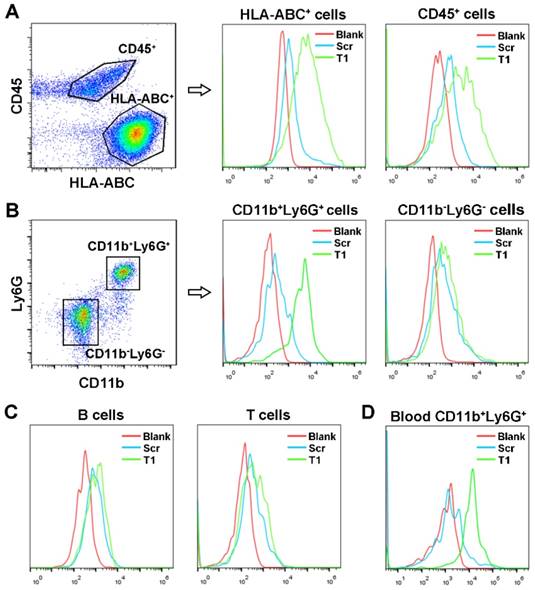
To identify cellular targets of the T1 aptamer, single cells from MDA-MB-231 orthotopic tumors were isolated, stained with antibodies, and incubated with T1 or Scr aptamers on ice. Due to the complexity of the TME, the two main cell populations, epithelial tumor cells (HLA-ABC+CD45-) and infiltrating immune cells (CD45+) were stained first. After gating on live singlet cells, flow cytometry analysis of the tumor specimens revealed that the T1 aptamer was bound to both MDA-MB-231 and immune cells (Fig. 2A). Further analysis of the infiltrating immune cell population demonstrated that T1 displayed increased binding to PMN-MDSCs (CD11b+Ly6G+) compared to Scr. On the contrary, the Scr and T1 aptamers exhibited similar binding to non-PMN-MDSCs (CD11b-Ly6G-) and (Fig. 2B). Further staining of B cells, T cells, and macrophage cells revealed no binding difference between Scr and T1 aptamer (Fig. 2C and Fig. S2). The ability of the T1 aptamer to preferentially bind to PMN-MDSCs in blood samples was also demonstrated (Fig. 2D). Together, these results indicate that that the T1 aptamer binds to PMN-MDSCs and MDA-MB-231 cells, both of which can be targeted for cancer therapy.
Identification of T1 aptamer binding targets in MDA-MB-231 orthotopic breast cancer model. (A-C) Representative flow cytometry graphs of Cy5-labeled Scr and T1 aptamer binding (ice incubation) to single cells isolated from orthotopic breast cancer tumors. (A) Tumor infiltrating-immune cells and MDA-MB-231 cells were defined as CD45+ and HLA-ABC+, respectively. (B) Tumor infiltrating polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) and non-PMN-MDSCs were defined as CD45+CD11b+Ly6G+, CD45+CD11b-Ly6G-, respectively. (C) Tumor infiltrating B cells and T cells were defined as CD45+B220+ and CD45+CD3+, respectively. (D) Representative flow cytometry graphs of Cy5-labeled aptamers incubated with PMN-MDSCs derived from the blood of mice bearing MDA-MB-231 orthotopic breast cancer.
Characterization of the T1 aptamer in vitro
Structure predictions for the aptamer were determined by Mfold [44]. The predicted secondary structure with lowest energy (∆G = -12.73 kcal/mol) for the T1 aptamer is shown in Fig. 3A. The characteristics of the T1 aptamer was studied in vitro using MDA-MB-231 cells, as these cells are easier to maintain in culture compared to PMN-MDSCs. The potential cytotoxicity of the T1 aptamer was determined, as safety is a major consideration for the development of therapeutic agents [45]. As shown in Fig. 3B, neither T1 nor Scr displayed any toxicity in MDA-MB-231 cells. Additionally, cell migration, determined with the scratch assay, was unaffected in response to aptamer treatment (Fig. S3A). Next, the stability of the T1 aptamer was assessed in serum solution. The deoxyadenosine (dA) residues on the aptamers were modified with a thiol group to improve in vivo stability [46]. Agarose gel electrophoresis results indicated that the T1 aptamer remained stable for at least 72 h (Fig. S3B).
Characterization of the T1 aptamer in vitro. (A) Predicted secondary structure of the T1 aptamer determined with Mfold. (B) Viability of MDA-MB-231 breast cancer cells treated with Scr or T1 aptamers for 48 h. (C) Flow cytometry of MDA-MB-231 cells treated with Cy5-labeled aptamers (25 nM). (D) Concentration-dependent binding of Cy5-labeled T1 in MDA-MB-231 cells. (E) The apparent equilibrium Kd of the T1 aptamer with MDA-MB-231 cancer cells. Data is presented as mean ± s.d. of three measurements. (F) Table showing cancer cell lines that display increased binding to T1 (Kd < 10 nM). (G) Table showing cancer cell lines that do not display increased binding to T1 (Kd > 500 nM). HSCs, Hematopoietic stem cells.
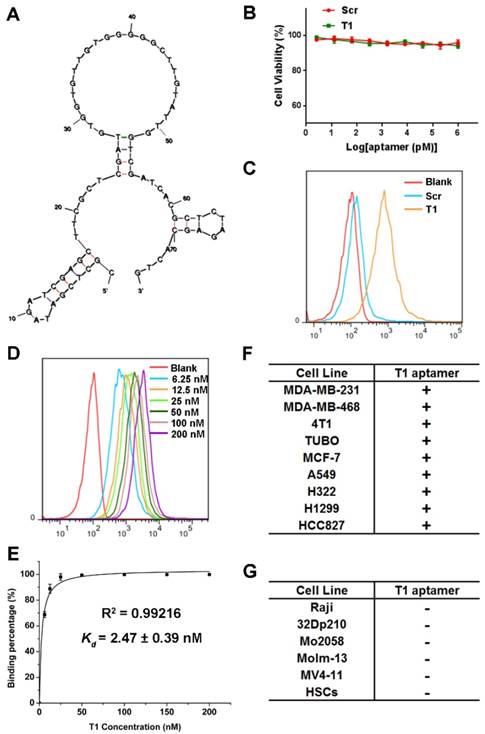
To verify that the T1 aptamer binds to MDA-MB-231 cells in culture, binding studies were performed using flow cytometry. The results reveal that the T1 aptamer exhibited increased binding to MDA-MB-231 cells compared to the Scr aptamer (Fig. 3C). The apparent dissociation constants (Kd) were measured by incubating cells with serial dilutions of the aptamers (Fig. 3D). As shown in Fig. 3E, the T1 aptamer binds to MDA-MB-231 cells with high affinity (Kd = 2.47 nM). To investigate cell-binding specificity, concentration gradient binding experiments were performed on other cell lines. Based on the Kd values, the cell lines were divided into two groups: binding cell lines (Kd < 10 nM) and non-binding cell lines (Kd > 500 nM). Interestingly, the T1 aptamer displayed increased binding to other solid tumor cell lines, such as those derived from murine and human breast cancer, lung cancer, and cervical cancer (Fig. 3F). However, the T1 aptamer did not bind to suspension cell lines, such as CML and AML cells (Fig. 3G). These results suggest that the T1 aptamer could have broader applicability beyond targeting the breast TME.
Uptake of the T1 aptamer in vitro
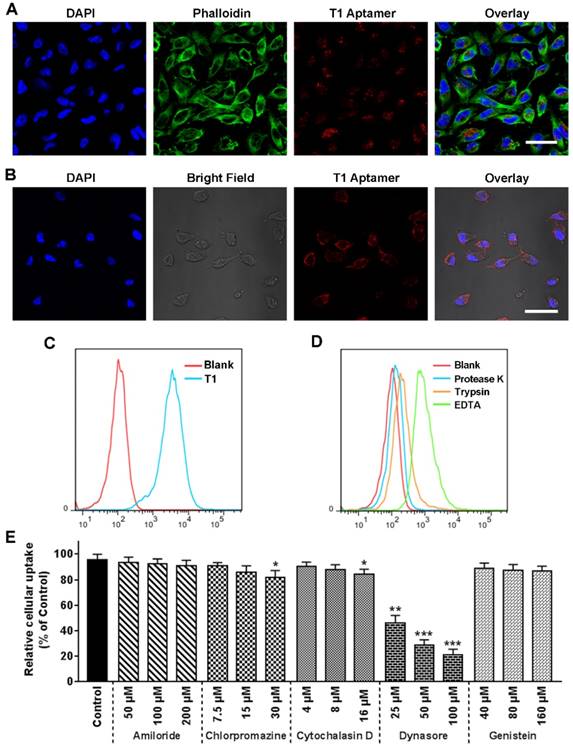
Previous reports have demonstrated that oligonucleotides longer than 25 bases do not readily penetrate the cell membrane [47]. Therefore, confocal microscopy studies were performed to determine whether the T1 aptamer, which consists of 74 bases, is internalized into cells. Confocal microscopy images revealed that Cy5-labeled T1 was present in large quantities inside the cells after a short period of time (Fig. 4A and Fig. S4A). Ice-incubation substantially reduced T1 cellular internalization (Fig. 4B), suggesting that the uptake process was energy-mediated. Flow cytometry results further confirmed that T1 was rapidly taken up by MDA-MB-231 cells (Fig. 4C). Cells were treated with proteinase K and trypsin to determine whether membrane proteins were involved in T1 uptake. As shown in Fig. 4D, treatment with trypsin or proteinase K lead to a substantial decrease in the fluorescent intensity of MDA-MB-231 cells, whereas exposure to EDTA had less of an effect on cellular uptake. Further studies were performed to identify T1 internalization pathways. As shown in Fig. 4E, T1 uptake was minimally affected by amiloride, chlorpromazine, cytochalasin D, and genistein, whereas dynasore dramatically reduced internalization in a dose-dependent manner. Confocal microscopy further confirmed that dynamin-dependent endocytosis plays an important role in T1 uptake (Fig. S4B).” These observations indicate that T1 is primarily internalized into cancer cells through interactions with membrane proteins that are involved in dynamin-dependent uptake.
Uptake of the T1 aptamer in vitro. MDA-MB-231 cells were treated with Cy5-labeled-T1 (25 nM) at 37 °C for 30 min unless otherwise indicated. (A) Confocal fluorescence microscopy images. T1 aptamer (red), phalloidin (green), and DAPI (blue). Scale bar, 20 μm. (B) Confocal fluorescence microscopy images. Cells were incubated with aptamers for 20 min on ice. Scale bar, 25 μm. (C) Flow cytometry graphs. (D) Flow cytometry graphs of cells treated with trypsin, proteinase K-treated, or ethylenediaminetetraacetic acid (EDTA) for 5 min. (E) Relative cellular uptake of T1 in response to uptake inhibitors. Data is presented as mean ± s.d. (n = 3). Statistics by Student's t-test. *, P < 0.05; ***, P < 0.001, ***, P < 0.0001, compared with control cells.
Revalidation of T1 aptamer binding targets in a 4T1 orthotopic breast cancer model
The ability of the T1 aptamer to bind to the TME of breast tumors was further validated in a 4T1 orthotropic breast cancer model. This model was selected, as 4T1 tumor development is associated with an expansion of MDSCs. Indeed, as shown in Fig. 5A, 4T1 tumor-bearing mice had a six- to eightfold increase in circulating MDSCs (CD11b+Gr-1+) compared to control mice. IVIS images revealed that the Cy5-labeled T1 aptamer displayed increased accumulation in the tumor in comparison to the Scr aptamer (Fig. 5B and Fig. S5A). Single cells from blood and tissue samples were analyzed by flow cytometry. Similar to the results obtained with MDA-MB-231 breast cancer models, the T1 aptamer displayed increased binding to PMN-MDSCs in the blood, bone, tumor, and spleen tissues compared to the Scr aptamer (Fig. 5C and Fig. S5B). In tumor samples, CD11b- cells were gated as 4T1 tumor cells, due to a lack of other makers to identify these cells. In accordance with previous results, the T1 aptamer exhibited increased binding to 4T1 cancer cells compared to the Scr aptamer (Fig. 5D). Moreover, neither Scr nor T1 showed a positive signal for M-MDSCs, B cells, T cells, or macrophages (Fig.S6A-D). Whole-body imaging time course experiments were performed with the Scr and T1 aptamers. The Scr aptamer was cleared from the body at a higher rate than T1. Notably, T1 was still present in high quantities in the body after 8 h (Fig. 5E). Taken together, these results confirm that the T1 aptamer binds to PMN-MDSCs and breast cancer cells, suggesting the potential use of the aptamer for tumor targeting applications.
Revalidation of T1 binding targets in a 4T1 orthotopic breast cancer model. (A) Percentage of myeloid-derived suppressor cells (MDSCs) in the blood following cancer cell implantation. (B-E) Cy5-labeled aptamers (0.5 nmol/mouse) were intravenously administered and analysis was performed 4 h post-injection unless otherwise indicated. (B) Fluorescent images of organs captured with the IVIS-200 imaging system. Organs: Tu, tumor; Sp, spleen; Bn, bones; Sp, spine. Scale bar, 1 cm. (C) Representative flow cytometry graphs of PMN-MDSCs from the blood, bone, tumors, and spleen. (D) Representative flow cytometry graphs of 4T1 cells (CD11b- cells) from tumor samples. (E) Whole body images of mice acquired with an IVIS-200 imaging system. Scale bar, 2 cm.
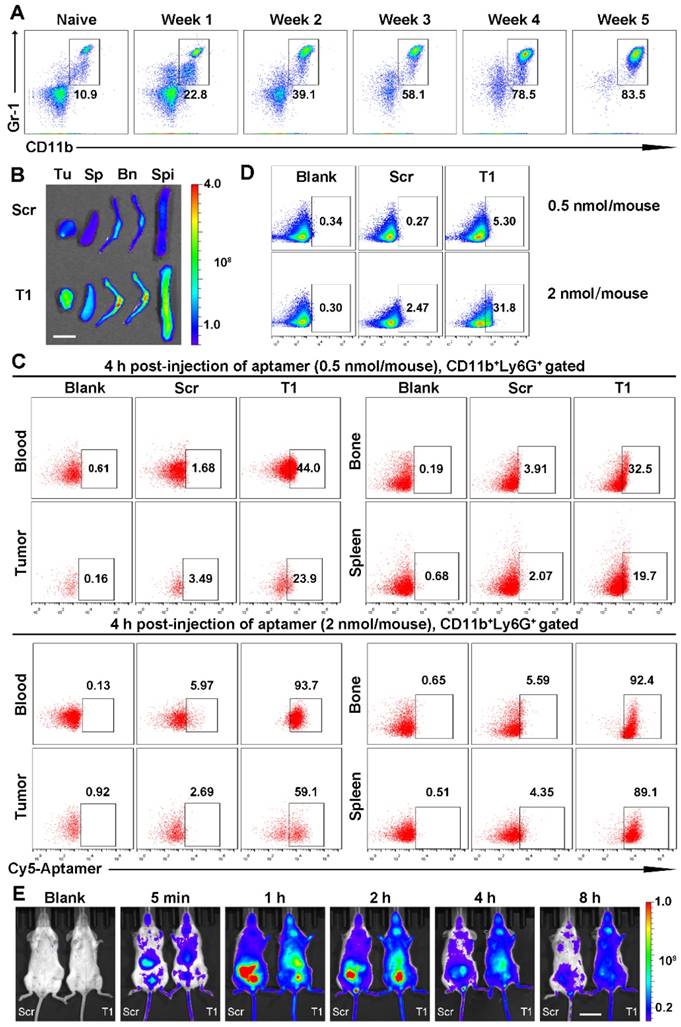
T1 aptamer conjugated liposomal Dox accumulation in tumors and bone tissue
Next, the potential use of T1 for targeted drug delivery was assessed. A liposomal-based system was utilized, as liposomes represent a major category of clinically approved delivery vehicles [48, 49]. The aptamer was conjugated to the outer lipid membrane and the liposomes were loaded with Dox (Fig. 6A) using an active loading procedure. The drug loading efficiency was >90% and was unaffected by aptamer conjugation. Liposomes functionalized with Scr or T1 were similar in size (~120 nm) (Fig. 6B). Tumor accumulation of liposomes in the nano-size range is usually facilitated through passive targeting due to the enhanced permeability and retention (EPR) effect. Size measurements revealed that the liposomes remained stable in various solutions (Fig. 6B). The targeting capability of the liposomes was assessed in 4T1 cancer cells using flow cytometry. Consistent with the results obtained with the free aptamers, the T1-functionalized liposomes displayed increased binding to 4T1 cells compared to liposomes conjugated to Scr (Fig. 6C). These results were further confirmed by confocal microscopy (Fig. 6D), which indicated that T1-liposomes delivered larger quantities of Dox inside cells. The biodistribution of the liposomes was evaluated in a 4T1 orthotopic breast cancer model using HPLC to detect Dox. Mice administered with T1-Dox exhibited higher intratumoral levels of Dox compared to those injected with Scr-Dox (Fig. 6E). These results suggest that the T1 aptamer could be used for targeted delivery of Dox liposomes to breast cancer tumors.
Tumor accumulation of aptamer-conjugated liposomal doxorubicin (Dox) in an orthotopic 4T1 breast cancer model. (A) Schematic illustration of aptamer-conjugated liposomal Dox. (B) Size and zeta potential of Scr aptamer-conjugated liposomal Dox (Scr-Dox) and T1 aptamer-conjugated liposomal Dox (T1-Dox) in phosphate buffered saline (PBS), water, or Dulbecco's Modified Eagle's Medium (DMEM). Three measurements were taken of each sample. (C) Flow cytometry analysis of 4T1 cells treated with Scr-Dox and T1-Dox at 37 °C for 30 min. (D) Confocal fluorescence microscopy images of 4T1 cells treated with Scr-Dox and T1-Dox for 30 min. Dox (red), phalloidin (green), and DAPI (blue). Scale bar, 25 μm. (E) Biodistribution of liposomes based on Dox content (6 mg/kg Scr-Dox or T1-Dox) 24 h post-injection. Data is presented as mean ± s.d. (n = 3). *, P < 0.05; **, P < 0.001; ***, P < 0.0001.
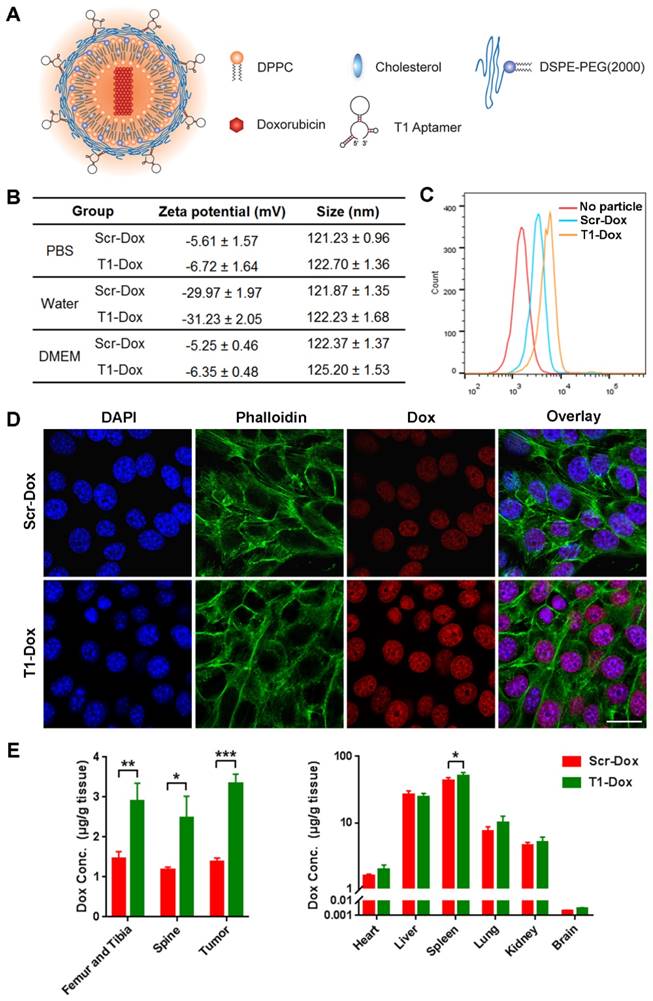
Therapeutic efficacy of aptamer-functionalized liposomal Dox in vivo
The therapeutic efficacy of aptamer-functionalized Dox liposomes was assessed in a 4T1 orthotopic breast cancer model (Fig. 7A). The 4T1 tumor model is highly aggressive and recalcitrant to most therapeutic agents, partially due to extreme expansion of MDSCs [15, 37]. The results revealed that T1-Dox liposomes outperformed Scr-Dox liposomes and free Dox (Fig. 7B). Treatment with T1-Dox resulted in an almost two- to three-fold decrease in tumor weight compared to the other groups (Fig. 7C). Moreover, flow cytometry analysis confirmed the T1-Dox selectively eliminated PMN-MDSCs in the tumor, spleen, and blood (Fig. 7D). Moreover, the percentage of CD8+ T cells was substantially higher in tumors of mice treated with T1-functionalized liposomes (Fig. S7), suggesting that PMN-MDSCs depletion promotes antitumor immunity. These results were further confirmed by confocal microscopy of tumor sections. As shown in Fig. 7E and F, treatment with T1-Dox liposomes dramatically decreased the intratumoral population of MDSCs and increased tumor infiltration of CD8+ T cells. Immunohistochemical analysis of Ki-67, a cell proliferation marker, showed a substantial decrease in this protein in response to T1-Dox (Fig. S8). Taken together, targeting and elimination of MDSCs facilitated a more immuneresponsive microenvironment, which likely contributed to increased cancer cell death. Notably, mice exhibited no clinical signs of distress, toxicity, or renal damage during the treatment period. Additionally, repeated injections of liposomes did not affect body weight (Fig. S9) or cause gross morphological changes in major organs (Fig. S10). In conclusion, the use of T1-Dox liposomes represents a safe and effective approach to target both PMN-MDSCs and breast cancer cells in order to improve conventional cancer therapy.
Therapeutic efficacy of T1-Dox in a 4T1 orthotopic breast cancer model. (A) Schematic of the experimental design. PBS, Dox, Scr-Dox, or T1-Dox (3 mg/kg) was administered intravenously on day 7, 14, 21, and 28 post-injection of cancer cells. Mice were sacrificed on day 35. (B) Tumor growth in response to treatment. Data is presented as mean ± s.d. (n = 8). (C) Tumor weight in response to treatment. Data is presented as mean ± s.d. (n = 8). (D) Left panel: Representative flow cytometry results of CD11b+Ly6G+ cells in tumor, blood, and spleen samples. Right panel: CD11b+Ly6G+ cells in tumor, blood, and spleen samples. Data is presented as mean ± s.d. (n = 5). (E) Representative confocal fluorescence microscopy images of CD11b+Gr-1+ cells in tumor samples. CD11b (green), Gr-1 (red), and DAPI (blue). Scale bar, 50 μm. (F) Representative confocal fluorescence microscopy images of CD8+ cells in tumor samples. CD8 (red) and DAPI (blue). Scale bar, 50 μm. *, P < 0.05; **, P < 0.001; ***, P < 0.0001.
Discussion
PMN-MDSCs are a key component of the TME and play an important role in the cellular networks that regulate immune responses in various cancers [4, 6]. These cells display high levels of reactive oxygen species and low nitric oxide production, which causes suppression of T cell function and subsequent promotion of tumor progression and metastasis [6, 7]. Furthermore, several studies have shown that suppression of MDSCs can overcome resistance to checkpoint blockade therapy, such as programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antibody therapies [15, 16]. Therefore, PMN-MDSCs present a promising target for improving anticancer immunity [5]. Indeed, several strategies have been explored to either eliminate or reduce the immunosuppressive function of MDSCs, resulting in restored T-cell antitumor activity [12-15]. Notably, conventional chemotherapeutic agents, such as gemcitabine and Dox, are able to cause cell death of MDSCs [13, 14]. However, the ability of these cytotoxic agents to eliminate MDSCs is highly dependent on targeted delivery, and there is a need to identify ligands that display preferential binding to these cells.
In this study, a DNA aptamer (T1) for dual targeting of PMN-MDSCs and tumor cells was identified and validated in multiple breast cancer models. To the best of our knowledge, this is the first example of an aptamer that can simultaneously target these two cell populations. An RNA aptamer that binds to MDSCs and tumor-associated macrophages has previously been described [50]. However, this aptamer is unable to serve as a combined targeting ligand for immunochemotherapy due to a lack of preferential binding to cancer cells. The T1 DNA aptamer identified in this study was conjugated to liposomal Dox. In a 4T1 orthotopic breast cancer model, treatment with T1-Dox caused cancer cell death and efficiently eliminated MDSCs in the blood, spleen, and tumor, increasing CD8+ T cell intratumoral infiltration. T1-Dox-induced transformation of the TME toward a more immunoactive state lead to improved anticancer efficacy compared to free Dox or non-targeted Dox liposomes. In addition to PMN-MDSCs and breast cancer cells, several other solid tumor cell lines were also shown to be targets for T1, indicating that this aptamer could have broader implications beyond breast cancer treatment. Taken together, this study highlights the promising potential of T1 to be used as a targeting ligand for cancer therapy.
Abbreviations
AML: acute myeloid leukemia; CML: chronic myeloid leukemia; DAPI: 4′,6-diamidino-2-phenylindole; EDTA: ethylenediaminetetraacetic acid; FBS: fetal bovine serum; HSCs: Hematopoietic stem cells; IVIS: in vivo imaging system; MDSCs: myeloid-derived suppressor cells; M-MDSCs: monocytic myeloid-derived suppressor cells; PCR: polymerase chain reaction; PMN-MDSCs: polymorphonuclear myeloid-derived suppressor cells; TME: The tumor microenvironment.
Acknowledgements
This work was supported by the National Institutes of Health grants 1R01CA193880-01A1 (HS), and U54CA210181 (MF), U. S. Department of Defense grant W81XWH-12-1-0414 (MF), and Huazhong University of Science and Technology funds 2016YXZD035 and 2017KFKJXX004 (RX). MF is an Ernest Cockrell Jr. Presidential Distinguished Chair at Houston Methodist Research Institute. HL is partially supported by the University of Chinese Academy of Sciences.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346-54
2. Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C. et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2015;518:107-10
3. Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148:135-46
4. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423-37
5. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263-74
6. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162-74
7. Lu T, Ramakrishnan R, Altiok S, Youn J-I, Cheng P, Celis E. et al. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. 2011;121:4015-29
8. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253-68
9. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y. et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409-21
10. Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF. et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150
11. Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7:513-20
12. Qin H, Lerman B, Sakamaki I, Wei G, Cha SC, Rao SS. et al. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat Med. 2014;20:676-81
13. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine Selectively Eliminates Splenic Gr-1+/CD11b+ Myeloid Suppressor Cells in Tumor-Bearing Animals and Enhances Antitumor Immune Activity. Clin Cancer Res. 2005;11:6713-21
14. Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B. et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014;74:104-18
15. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH. et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539:443-7
16. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL. et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014;111:11774-9
17. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E. et al. Disruption of CXCR2-Mediated MDSC Tumor Trafficking Enhances Anti-PD1 Efficacy. Sci Transl Med. 2014;6:237ra67-ra67
18. Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505-10
19. Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818-22
20. Zhou J, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov. 2017;16:181-202
21. Sefah K, Shangguan D, Xiong X, O'Donoghue MB, Tan W. Development of DNA aptamers using Cell-SELEX. Nat Protoc. 2010;5:1169-85
22. Mi J, Liu Y, Rabbani ZN, Yang Z, Urban JH, Sullenger BA. et al. In vivo selection of tumor-targeting RNA motifs. Nat Chem Biol. 2010;6:22-4
23. Cho K, Wang X, Nie S, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14:1310-6
24. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotech. 2015;33:941-51
25. Chen H, Zhang W, Zhu G, Xie J, Chen X. Rethinking cancer nanotheranostics. Nat Rev Mater. 2017;2:17024
26. Shen J, Liu H, Mu C, Wolfram J, Zhang W, Kim H-C. et al. Multi-step encapsulation of chemotherapy and gene silencing agents in functionalized mesoporous silica nanoparticles. Nanoscale. 2017;9:5329-41
27. Xiang D, Shigdar S, Qiao G, Wang T, Kouzani AZ, Zhou S-F. et al. Nucleic acid aptamer-guided cancer therapeutics and diagnostics: the next generation of cancer medicine. Theranostics. 2015;5:23-42
28. Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW. et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103:6315-20
29. Xing H, Tang L, Yang X, Hwang K, Wang W, Yin Q. et al. Selective delivery of an anticancer drug with aptamer-functionalized liposomes to breast cancer cells in vitro and in vivo. J Mater Chem B. 2013;1:5288-97
30. Mai J, Huang Y, Mu C, Zhang G, Xu R, Guo X. et al. Bone marrow endothelium-targeted therapeutics for metastatic breast cancer. J Control Release. 2014;187:22-9
31. Chu TC, Twu KY, Ellington AD, Levy M. Aptamer mediated siRNA delivery. Nucleic Acids Res. 2006;34:e73-e
32. Kang H, O'Donoghue MB, Liu H, Tan W. A liposome-based nanostructure for aptamer directed delivery. Chem Commun. 2010;46:249-51
33. McNamara JO, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E. et al. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol. 2006;24:1005-15
34. Zhu G, Niu G, Chen X. Aptamer-Drug Conjugates. Bioconjugate Chem. 2015;26:2186-97
35. Tang Y, Hu H, Zhang MG, Song J, Nie L, Wang S. et al. An aptamer-targeting photoresponsive drug delivery system using "off-on" graphene oxide wrapped mesoporous silica nanoparticles. Nanoscale. 2015;7:6304-10
36. Zeng Z, Parekh P, Li Z, Shi Z-Z, Tung C-H, Zu Y. Specific and sensitive tumor imaging using biostable oligonucleotide aptamer probes. Theranostics. 2014;4:945-52
37. Pulaski BA, Ostrand-Rosenberg S. Reduction of Established Spontaneous Mammary Carcinoma Metastases following Immunotherapy with Major Histocompatibility Complex Class II and B7.1 Cell-based Tumor Vaccines. Cancer Res. 1998;58:1486-93
38. Matulonis U, Salgia R, Okuda K, Druker B, Griffin J. Interleukin-3 and p210 BCR/ABL activate both unique and overlapping pathways of signal transduction in a factor-dependent myeloid cell line. Exp Hematol. 1993;21:1460-6
39. Weissman IL, Shizuru JA. The origins of the identification and isolation of hematopoietic stem cells, and their capability to induce donor-specific transplantation tolerance and treat autoimmune diseases. Blood. 2008;112:3543-53
40. King DJ, Ventura DA, Brasier AR, Gorenstein DG. Novel Combinatorial Selection of Phosphorothioate Oligonucleotide Aptamers. Biochemistry. 1998;37:16489-93
41. Du Y, Zhang Q, Jing L, Liang X, Chi C, Li Y. et al. GX1-conjugated poly (lactic acid) nanoparticles encapsulating Endostar for improved in vivo anticolorectal cancer treatment. International journal of nanomedicine. 2015;10:3791
42. Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. BBA-Biomembranes. 1993;1151:201-15
43. Xu R, Zhang G, Mai J, Deng X, Segura-Ibarra V, Wu S. et al. An injectable nanoparticle generator enhances delivery of cancer therapeutics. Nat Biotech. 2016;34:414-8
44. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406-15
45. Wolfram J, Zhu M, Yang Y, Shen J, Gentile E, Paolino D. et al. Safety of nanoparticles in medicine. Curr Drug Targets. 2015;16:1671-81
46. Volk DE, Yang X, Fennewald SM, King DJ, Bassett SE, Venkitachalam S. et al. Solution structure and design of dithiophosphate backbone aptamers targeting transcription factor NF-κB. Bioorg Chem. 2002;30:396-419
47. Patil SD, Rhodes DG, Burgess DJ. DNA-based therapeutics and DNA delivery systems: a comprehensive review. AAPS J. 2005;7:E61-E77
48. Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145-60
49. Gentile E, Cilurzo F, Di Marzio L, Carafa M, Anna Ventura C, Wolfram J. et al. Liposomal chemotherapeutics. Future Oncol. 2013;9:1849-59
50. Roth F, De La Fuente AC, Vella JL, Zoso A, Inverardi L, Serafini P. Aptamer-Mediated Blockade of IL4Rα Triggers Apoptosis of MDSCs and Limits Tumor Progression. Cancer Res. 2012;72:1373-83
Author contact
![]() Corresponding author: hshenorg
Corresponding author: hshenorg