Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(19):4777-4790. doi:10.7150/thno.21713 This issue Cite
Research Paper
miR34a/GOLPH3 Axis abrogates Urothelial Bladder Cancer Chemoresistance via Reduced Cancer Stemness
Qing Zhang1*, Junlong Zhuang1*, Yongming Deng1*, Lin Yang1*, Wenmin Cao1, Wei Chen1, Tingsheng Lin1, Xiaoyu Lv1, Hang Yu1, Yanshi Xue1, Hongqian Guo1 ![]()
1. Department of Urology, Drum Tower Hospital, Medical School of Nanjing University, Institute of Urology, Nanjing University, 321 Zhongshan Road, Nanjing 210008, Jiangsu, PR China
*These authors contributed equally to the work
Received 2017-6-30; Accepted 2017-9-12; Published 2017-10-17
Abstract

Rationale: Chemoresistance and subsequent recurrence of human urothelial bladder cancer (UBC) is partially driven by a subpopulation of tumor initiating cells, namely cancer stem cells (CSCs). However, the underlying molecular mechanism in chemotherapy-induced CSCs enrichment and following chemoresistance and recurrence remains largely unclear.
Methods: Gemcitabine and cisplatin (GC) chemoresistant cell lines (T24 GC 3rd and 5637 GC 3rd cells) and the chemo-sensitive UBC cell lines T24 and 5637 were established in vivo for the investigation of acquired resistance mechanisms. The role of miR34a/GOLPH3 axis in regulating UBC chemoresistance and recurrence was evaluated in cell and animal models. The expression levels of miR34a/GOLPH3 axis and CSCs markers were assayed in specimens of UBC. The association of GOLPH3 with clinicopathologic features and prognosis was analysed.
Results: RT-PCR and western blotting confirmed that the expression levels of miR34a were decreased and GOLPH3 were increased in GC chemoresistant UBC cell lines. Downregulation of miR34a resulted in the overexpression of GOLPH3, which is a target gene of miR34a confirmed by luciferase experiment. The ectopic expression of miR34a decreased the stem cell properties of chemoresistant UBC cells and re-sensitized these cells to GC treatment in vitro and in vivo. Moreover, miR34a/GOLPH3 axis has obvious clinical relevance with prognosis and recurrence in human UBC patients with standard GC chemotherapy.
Conclusion: Our results suggest that miR34a/GOLPH3 axis exert key role in CSCs involved UBC drug resistance and recurrence and warrant further development as a promising therapeutic approach in treating drug-resistant UBC.
Keywords: GOLPH3, miR-34a, Cancer stem cells, Urothelial bladder cancer, Drug-resistance
Introduction
Human urothelial bladder cancer (UBC), especially muscle-invasive bladder cancer (MIBC), is one of the most aggressive epithelial tumors and is characterized by a high rate of early systemic dissemination [1]. Patients with MIBC, advanced or metastatic UBC are routinely treated with cisplatin-based systemic cytotoxic chemotherapy [2], such as M-VAC (methotrexate, vinblastine, doxorubicin, cisplatin) or GC (gemcitabine, cisplatin) regimens [3, 4]. Although cisplatin-based regimens still constitute the gold standard, UBC ultimately acquires chemoresistance and no standard of care exists after cisplatin-based regimens [1].
Recent studies revealed that a small population of cancer stem cells (CSCs), which are enriched after therapy, may account for chemotherapy failure [5, 6]. CSCs have the ability to generate all types of differentiated cells to repopulate tumors and eventually lead to metastasis and are regarded as the sustaining force of a tumor [5, 7, 8]. It is reported that CSCs may actively proliferate in response to chemotherapy and induced UBC recurrence and metastasis [9]. Consequently, CSCs-targeting therapeutic approaches to prevent UBC recurrence and metastasis become promising regimens [6, 8].
Golgi phosphoprotein 3 (GOLPH3), is a newly identified membrane protein in the trans-Golgi matrix, which plays a role in anterograde and retrograde Golgi trafficking [10]. Previous studies have noted that a high proportion of human cancers harbor amplification and overexpression of GOLPH3, which correlates with a poor prognosis for several cancers [10-12], including UBC [13]. Strikingly, Suzette et al. recently found that overexpression of GOLPH3 confers resistance to killing by DNA-damaging agents [14, 15]. These findings indicate GOLPH3 may be important in cancer chemoresistance and may be responsible for the CSCs enrichment induced by DNA-damaging agents.
MicroRNAs are a conserved class of noncoding small RNAs that regulate the expression of certain key genes involved in self-renewal, survival, and tumor progression [16]. Some microRNAs, such as miR34a, are known as tumor suppressors, which are inactivated in various tumors, including UBC [5, 17, 18] and regulate cancer stemness and drug resistance [5, 19, 20]. Our small-scale miRNA screening using qRT-PCR showed that miR34a was greatly downregulated in GC chemoresistant UBC cells compared with their counterparts. Downregulation of miR34a resulted in GOLPH3 overexpression, which is a target gene of miR34a confirmed by luciferase experiment. The ectopic expression of miR34a decreased the stem cell properties of chemoresistant UBC cells and sensitized these cells to GC chemotherapy. These results provide new insight into miR34a/GOLPH3 axis function and open a new perspective for developing novel therapeutic strategies of drug-resistant UBC aimed at targeting CSCs.
Results
GC (gemcitabine and cisplatin) treatment enriches CSCs subpopulation and induces chemoresistance
In order to mimic the process of tumor chemoresistance and recurrence in patients treated with GC regimens, we first inoculated human UBC cell lines T24 or 5637 into nude mice subcutaneously and then 40 mg/kg of gemcitabine was administered to the mice on day 1, 5, 8 and 15, and 60 mg/kg cisplatin was administered to the mice on day 2 followed by a 15 day recovery period. The second round and third round treatment started on day 30 and day 60. As shown in Fig. 1A, overall the tumors derived from both cell lines grew much slower in the GC-treated mice than those in the control group. During the first 15 days, GC treatment almost completely inhibited the growth of tumors. This indicates that T24 and 5637 cell lines are GC chemotherapy sensitive cells. During the second 15 day recovery period, tumors grew much faster in control group, although the tumor growth rate was still lower in the GC group. Finally, GC treatment failed to inhibit the growth of T24 and 5637 xenografts in the third round of treatment (Fig. 1A).
Gemcitabine and cisplatin treatment enriches CSC subpopulation and induces chemoresistance in vivo. (A) In vivo gemcitabine and cisplatin chemotherapy simulates clinical regimen with multiple treatment cycles and gap periods. Tumor sizes of T24 (left) and 5637 (right) xenografts after PBS (red line, n=5) or gemcitabine and cisplatin treatment (green line, n=5). Dashed boxes indicate the time frame of each chemotherapy cycle. (B) Western blotting and (C) real-time Q-PCR of CSC markers (CD44, ALDH1A1, Sox2, and KLF4) and DNA damage and repair related protein GOLPH3 in T24 and 5637 xenografts of control (endpoint of red line) and chemotherapy groups (endpoint of green line). (D) Representative H&E and IHC data showing expression of CSC markers (CK5, CK14 and CD44) in T24 and 5637 xenografts of control and chemotherapy groups (n=5 per group). Unpaired 2-tailed Student's t test was used for analysis of statistical significance. The experiments were done in triplicate. CSC: cancer stem cell; GOLPH3: Golgi phosphoprotein 3; H&E: hematoxylin-eosin staining; IHC: immunohistochemistry; PCR: Polymerase Chain Reaction
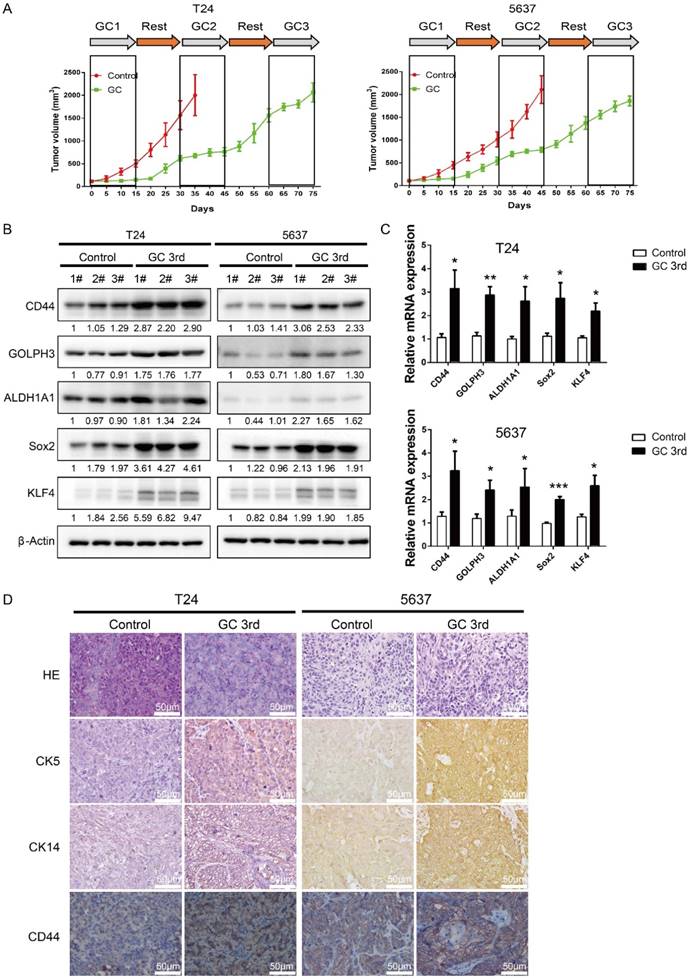
To explore the possibility that some of the tumor cells became resistant to GC treatment due to the existence of a subpopulation of CSCs, we sacrificed the mice and collected tumor samples at the end of the 3rd round of treatments (endpoint of green line in Fig. 1A), and collected the tumor tissue at endpoint of control group (endpoint of red line in Fig. 1A), which was treated with PBS for further analyses. Western blot and real-time Q-PCR results showed that four CSC markers (CD44, ALDH1A1, Sox2 and KLF4) and DNA damage and repair related protein GOLPH3 were elevated dramatically in the GC treated samples (Fig. 1B and C). Consistently, IHC analyses showed that three UBC stemness markers CK5, CK14 and CD44 were highly expressed in the xenografts treated with GC (Fig. 1D).
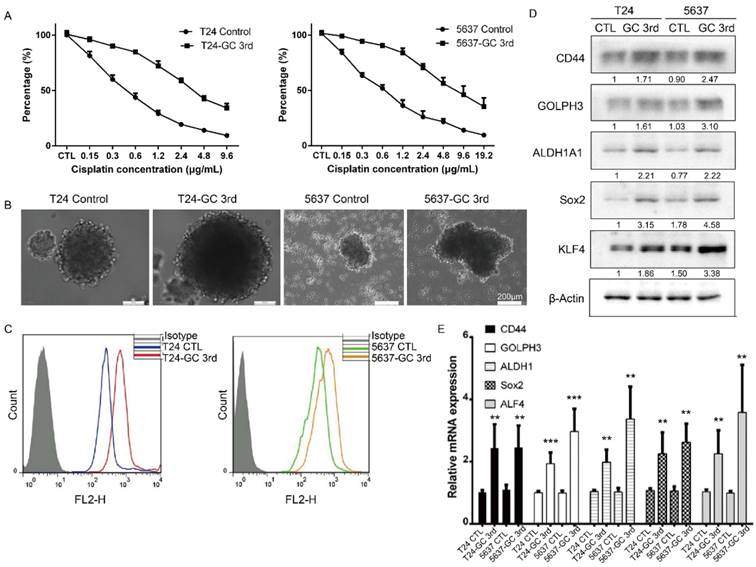
To further confirm that the GC chemotherapy-resistant UBC cell lines were successfully established, we isolated bladder cancer cells from xenografts of T24 GC 3rd, T24 control, 5637 GC 3rd and 5637 control. Then we treated T24 GC 3rd, T24 control, 5637 GC 3rd and 5637 control cells in vitro with various concentrations of cisplatin (0-19.2 µg/mL) combined with 1.5 µg/mL gemcitabine (Fig. 2A). We found that GC 3rd UBC cells had significantly higher viability compared to control cells, which indicated that the GC chemotherapy-resistant UBC cell lines were established. Moreover, GC treatment significantly increased the sphere formation ability (Fig. 2B) and the percentage of CD44+ subpopulation, a CSCs marker for UBC cells (Fig. 2C). Consistently, western blot and real-time Q-PCR showed that CSCs markers and GOLPH3 elevated dramatically in the GC-resistant UBC cell lines T24 GC 3rd and 5637 GC 3rd (Fig. 2D and 2E). Taken together, these data suggest that GC regimens treatment can enrich the CSCs subpopulation and cause chemoresistance in UBC.
Gemcitabine and cisplatin chemotherapy enhanced CSC subpopulation enrichment and chemoresistance in vitro. (A) T24 and 5637 cells isolated from xenografts of T24 GC 3rd, T24 control, 5637 GC 3rd and 5637 control cells were treated with various concentrations of cisplatin (0-19.2 µg/mL) and 1.5 µg/mL gemcitabine. (B) Sphere formation assay of control cells derived from xenografts of control and chemotherapy groups. (C) CD44+ subpopulation flow cytometry assay of control cells derived from xenografts of control group and chemotherapy groups. (D) Western blot and (E) real-time Q-PCR of CSC markers (CD44, ALDH1A1, Sox2, and KLF4) and DNA damage and repair related protein GOLPH3 in T24 and 5637 cells isolated from xenografts of control and chemotherapy groups. The experiments were done in triplicate. CSC: cancer stem cell; GOLPH3: Golgi phosphoprotein 3; PCR: Polymerase Chain Reaction
GC chemotherapy enhanced CSC subpopulation enrichment and chemoresistance is GOLPH3 dependent
A tumor sphere model, in which the stem cells have the capability to form spheres under suspension culture condition, was then generated. Using this model, we showed stem cell markers (CD44, ALDH1A1, Sox2, KLF4) and GOLPH3 level increased in T24 and 5637 spheres, compared to adherent T24 and 5637 cells, respectively (Fig. 3A) (all cells isolated from xenografts). These data demonstrated GOLPH3 may be associated with the stemness of UBC cells.
Gemcitabine and cisplatin chemotherapy enhanced CSC subpopulation enrichment and chemoresistance is GOLPH3 dependent. (A) CSC markers (CD44, ALDH1A1, Sox2, and KLF4) and DNA damage and repair related protein GOLPH3 protein level in T24 and 5637 tumor spheres (stem/progenitor cell population) and T24 and 5637 cells (all cells were isolated from control xenografts) under regular adherent culture conditions (CTL) were analyzed by western blot. (B) Sphere formation assay of T24 GC 3rd and 5637 GC 3rd cells with GOLPH3 knockdown and T24 and 5637 control cells (all cells were isolated from xenografts) with GOLPH3 elevated expression. (C) CD44+ subpopulation flow cytometry assay of T24 GC 3rd cell and 5637 GC 3rd cell with GOLPH3 knockdown and control cells T24 and 5637 (all cells were isolated from xenografts) with GOLPH3 elevated expression. (D) T24 GC 3rd cell and 5637 GC 3rd cell with GOLPH3 knockdown and control cells T24 and 5637 (all cells were isolated from xenografts) with GOLPH3 elevated expression were treated with various concentrations of cisplatin (0-19.2 µg/mL) and 1.5 µg/mL gemcitabine. (E) Western blot and (F) real-time Q-PCR of CSC markers (CD44, ALDH1A1, Sox2, and KLF4) and DNA damage and repair related protein GOLPH3 in T24 GC 3rd cell and 5637 GC 3rd cell with GOLPH3 knockdown and control cells T24 and 5637 (all cells were isolated from xenografts) with GOLPH3 elevated expression. The experiments were done in triplicate. CSC: cancer stem cell; GOLPH3: Golgi phosphoprotein 3; PCR: Polymerase Chain Reaction
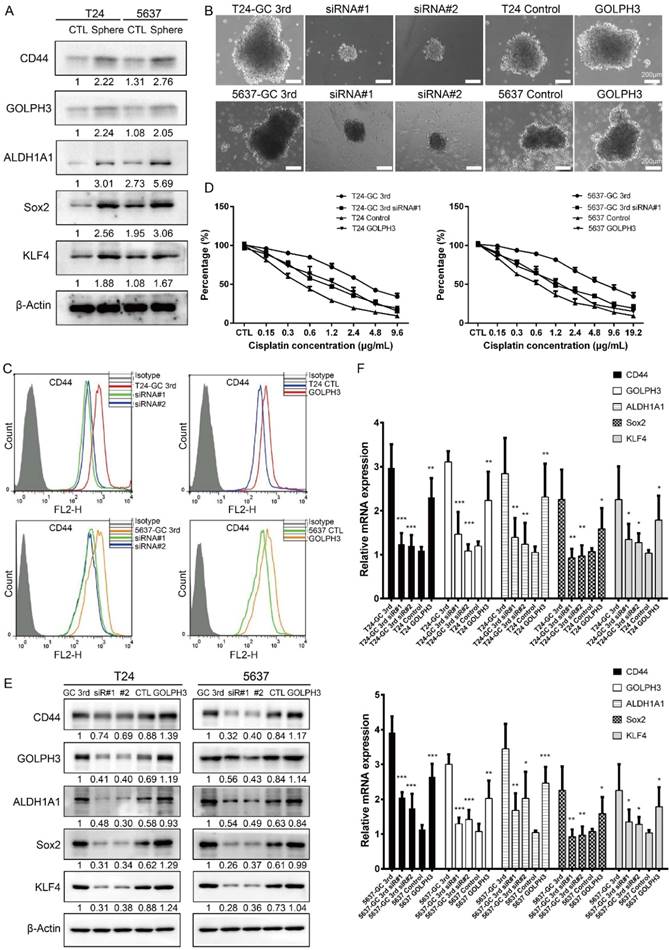
In order to test the role of GOLPH3 in UBC CSCs subpopulation enrichment and GC chemoresistance, we knocked down GOLPH3 by small interfering RNA and found its depletion reduced the sphere formation ability of GC chemoresistant UBC cells (T24 GC 3rd and 5637 GC 3rd). Consistently, overexpression of GOLPH3 increased the sphere formation ability of T24 and 5637 cells (Fig. 3B). Moreover, knockdown of GOLPH3 in GC chemoresistant UBC cells reduced the CD44+ cell population, compared with that in the vehicle-treated cells. Consistently, T24 and 5637 cells with high expression of GOLPH3 increased the CD44+ cell population (Fig. 3C). Furthermore, the results of CCK8 assay showed that inhibition of GOLPH3 expression contributed to GC sensitivity in T24 GC 3rd and 5637 GC 3rd cells. Increasing GOLPH3 expression contributed to T24 and 5637 cells' chemoresistance (Fig. 3D). Moreover, western blot and real-time Q-PCR showed that GOLPH3 ablation reduced the protein and mRNA expression of these CSCs markers and GOLPH3 overexpression increased the expression of these CSCs markers (Fig. 3E and F).
miR34a is directly regulating GOLPH3 and is responsible for CSCs enrichment and GC chemoresistance
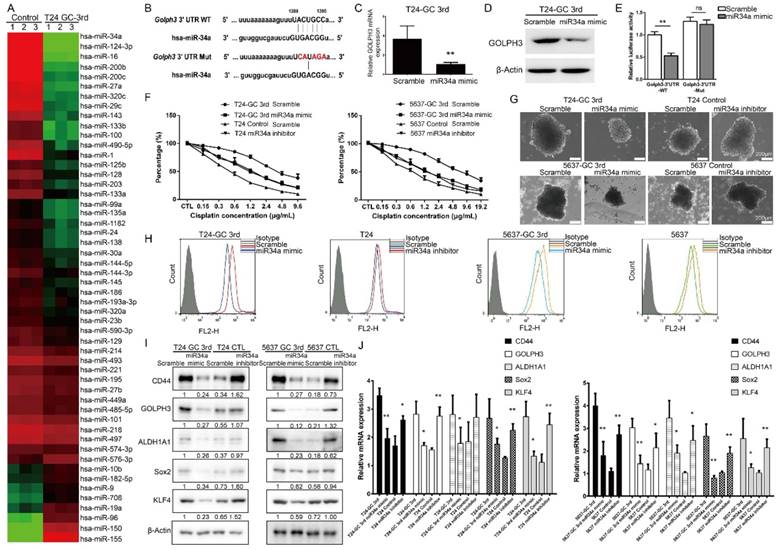
miRNAs have been implicated in various cellular processes, including drug response and cancer stemness. In this study, we first carried out a small scale qRT-PCR screening in 52 UBC-related miRNAs[21] to identify the miRNAs associated with resistance of T24 cells to GC 3rd treatment in vivo (Fig. 4A). Among them, miR34a was the one with the highest change. The expression of miR34a is decreased in many cancers because of inactivation and may play a crucial role in some CSCs survival and drug resistance [22, 23]. It is exciting to find that there is a seed sequence in the 3'UTR of GOLPH3, which suggests that GLOPH3 is a target of miR-34a (Fig. 4B). To examine whether miR-34a inhibits GOLPH3 expression, we transfected T24 GC 3rd cells with miR-34a mimic and scramble control and tested the GOLPH3 mRNA and protein levels. The results showed that GOLPH3 is inhibited at the mRNA and protein levels by miR-34a (Fig. 4C and D). To further examine whether the miR34a directly inhibits GOLPH3 expression, we cloned the wildtype 3'-UTR and mutant 3'UTR into luciferase reporter plasmids. Then we carried out luciferase assay and demonstrated that miR-34a mimics the inhibited activity of the reporter controlled by the wild-type, but not the mutant 3'UTR of GOLPH3 (Fig. 4E). Therefore, we focused on the functions of miR34a in subsequent experiments. First, we examined whether the changed expression of miR34a contributed to GC sensitivity in UBC cell lines, using the CCK8 assay. As expected, overexpression of miR34a in T24 3rd and 5637 3rd cells was more sensitive than scramble control to GC treatment (Fig. 4F). Furthermore, the sphere formation ability and the CD44+ cell population of T24 GC 3rd and 5637 GC 3rd cells, which indicated the CSC subpopulation enrichment, were decreased in miR34a mimic groups (Fig. 4G-H). Western blot and qRT-PCR results showed that the expression levels of four CSC markers and GOLPH3 decreased dramatically in miR34a mimic groups in T24 GC 3rd and 5637 GC 3rd cells and increased dramatically in the miR34a inhibitor groups in T24 and 5637 cells (Fig. 4I-J). Collectively, these results suggested that GOLPH3 is negatively regulated by miR34a, and takes part in stemness and GC chemoresistance of UBC.
miR34a directly regulates GOLPH3 and is responsible for CSC subpopulation enrichment and GC chemoresistance. (A) Small scale miRNA screening using qRT-PCR showed the altered folds of 52 UBC-related miRNAs in T24 GC 3rd xenograft tissue, compared with xenograft tissue from control group. (B) Sequences of wild type (GOLPH3-3'UTR-WT) and mutated 3'UTR Renilla luciferase reporters (GOLPH3-3'UTR-Mut) were listed. Expression of GOLPH3 was determined by real-time Q-PCR (C) and Western blotting (D) in scramble control and miR-34a overexpressed (miR-34a mimic) in T24-GC 3rd cell (n=3 per group). (E) Relative luciferase activity was measured in scrambled control miRNA and mimic miR-34a transfected T24-GC 3rd cells, simultaneously transfected also with GOLPH3-3'UTR-WT or GOLPH3-3'UTR-Mut expression vectors. (F) T24 GC 3rd cell and 5637 GC 3rd cell with miR34a elevated expression and control T24 and 5637 cells with miR34a knockdown were treated with various concentrations of cisplatin (0-19.2 µg/mL) and 1.5 µg/mL gemcitabine. (G) Sphere formation assay of T24 GC 3rd cell and 5637 GC 3rd cell with miR34a elevated expression and control cells T24 and 5637 with miR34a knockdown. (H) CD44+ subpopulation flow cytometry assay of T24 GC 3rd cell and 5637 GC 3rd cell with miR34a elevated expression and control cells T24 and 5637 with miR34a knockdown. (I) Western blot and (J) real-time Q-PCR of CSC markers (CD44, ALDH1A1, Sox2, and KLF4) and DNA damage and repair related protein GOLPH3 in T24 GC 3rd cell and 5637 GC 3rd cell with miR34a elevated expression and control cells T24 and 5637 with miR34a knockdown. Unpaired 2-tailed Student's t test was used for analysis of statistical significance. All cells were isolated from xenografts. The experiments were done in triplicate. CSC: cancer stem cell; GOLPH3: Golgi phosphoprotein 3; PCR: Polymerase Chain Reaction
Blocking miR34a/GOLPH3 axis inhibits UBC stemness and chemoresistance in vivo
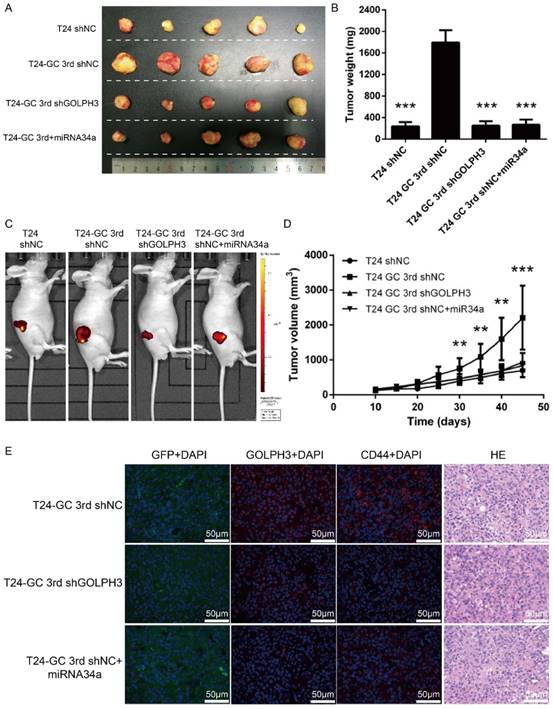
Next, we assessed the effect of restored miR34a expression or GOLPH3 ablation on tumor formation in an in vivo model with GC regimens treatment. We first established GOLPH3 stable knockdown T24 GC 3rd UBC cell lines (T24 GC 3rd shGOLPH3) and negative control T24 GC 3rd UBC cell lines (T24 GC 3rd shNC) by infecting T24 GC 3rd UBC cells with lentivirus containing shRNA targeting GLOPH3 and scramble negative control shRNA, respectively. As the context control, we also infected T24 cell with Lv-shRNA-NC (T24 shNC). Then, these cells were inoculated into nude mice subcutaneously and a nude mouse xenograft model was established (Fig. 5A). When the tumor size was ~100 mm3, 10 μg miR-34a mimic complex was injected into the tumor of T24 GC 3rd shNC, while scramble control mimic was injected into T24 shNC, T24 GC 3rd shNC and T24 GC 3rd shGOLPH3 groups. T24 GC 3rd shGOLPH3 with scramble control mimic group, T24 GC 3rd shNC with miR34a mimic group (intratumoral injection of miR34a mimic complexes) with GC regimens treatment resulted in a significant decrease in tumor volume and weight as measured at the end of the experiment at day 45 compared with control T24 GC 3rd shNC with scramble control mimic (Fig. 5B, 5C and 5D). Immunofluorescence staining revealed that the expression of GOLPH3 and CD44 was decreased significantly in T24 GC 3rd shGOLPH3 and T24 GC 3rd shNC with mimic miR34a group xenografts compared with control treated xenografts (Fig. 5E). Our findings suggest that the reduced expression of miR34a increases the expression of GOLPH3, and contributes to UBC stemness and GC chemoresistance in vivo.
Blocking miR34a/GOLPH3 axis inhibits UBC stemness and chemoresistance in vivo. (A) Photograph, (B) tumor weight, (C) tumor volume monitored using the IVIS Lumina II system and (D) tumor growths of T24 shNC, T24 GC 3rd shNC, T24 GC 3rd shGOLPH3 and T24 GC 3rd tumor xenografts injected with miR34a mimic complex groups and all groups subjected to gemcitabine and cisplatin treatment. Measurements were performed every five days for 35 days. (E) IF staining of GFP, GOLPH3 and CD44 on T24 GC3rd shNC, T24 GC3rd shGOLPH3, and T24 GC3rd tumor xenografts injected with miR34a mimic complex groups and all groups subjected to GC treatment. The experiments were done in triplicate.
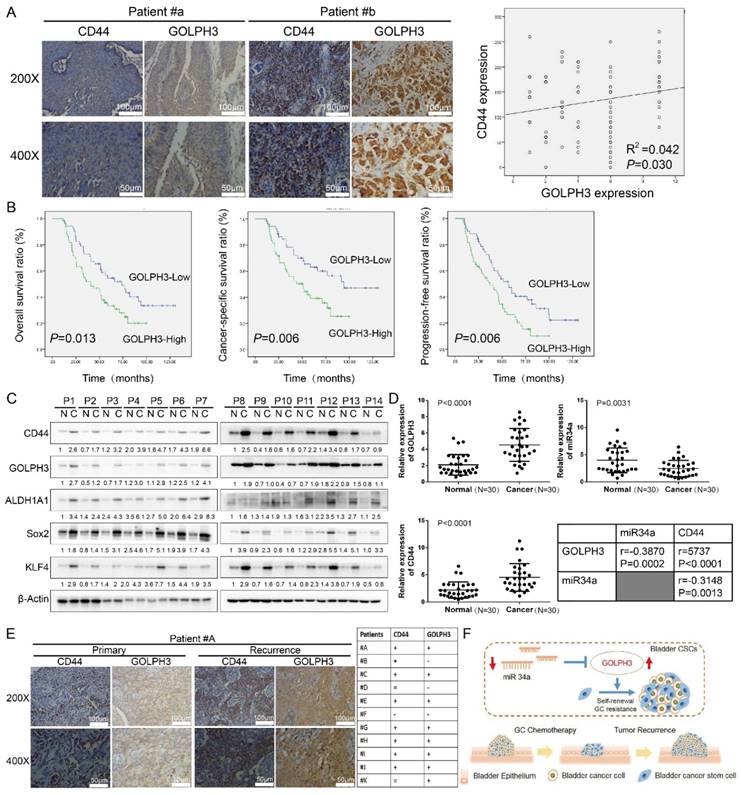
Clinical relevance of miR34a/GOLPH3 axis in human UBC specimens
To investigate the relationship between GOLPH3 expression and the clinicopathologic characteristics of UBC, GOLPH3 and CD44 expression was examined in 112 paraffin-embedded, archived bladder cancer tissues by IHC staining (all these patients received standard GC regimens chemotherapy) (Table 1). Statistical analysis revealed that the GOLPH3 levels were not associated with gender, size, N classification, or multiplicity classification (Table 1; all P > 0.05), but they are associated with age (≤ 65 y vs. > 65 y; P = 0.047) and pT status (T2 vs. T3 vs. T4; P = 0.033), which indicated that age and pT status might affect the function of GOLPH3 gene. Strikingly, we found that the areas in the UBC specimens displaying high levels of GOLPH3 staining also had strong CD44 staining signals, and areas with low GOLPH3 expression had weakly detectable CD44 expression (Fig. 6A). Statistical analysis indicated that the GOLPH3 expression was significantly associated with the CD44 expression level (P = 0.030; Fig. 6A, right panel), which indicated that GOLPH3 plays through CD44 a crucial role in UBC stemness and GC chemoresistance. Kaplan-Meier survival curves and log-rank test survival analysis were used to determine the prognostic value of GOLPH3 on survival of patients who received standard GC regimens chemotherapy. The results showed that patients with UBC exhibiting high GOLPH3 expression had substantially shorter overall survival (OS; P = 0.013), cancer-specific survival (CSS; P = 0.006) and progression-free survival (PFS; P = 0.006) than those with UBC exhibiting low GOLPH3 expression (Fig. 6B). These data suggested that high GOLPH3 expression is associated with poor prognosis in patients treated with GC adjuvant chemotherapy.
Association between GOLPH3 expression and the clinicopathologic characteristics of 112 patients treated with radical cystectomy followed by standard GC chemotherapy for urothelial carcinoma of the bladder
| Characteristic | Total cases | GOLPH3 expression | P | |||
|---|---|---|---|---|---|---|
| Low | High | |||||
| Total | 52 | 60 | ||||
| Gender | 0.322 | |||||
| Female | 24 | 9 | 15 | |||
| Male | 88 | 43 | 45 | |||
| Age | 0.047 | |||||
| ≤65y | 47 | 27 | 20 | |||
| >65y | 65 | 25 | 40 | |||
| Size | 0.288 | |||||
| ≤3cm | 62 | 26 | 36 | |||
| >3cm | 50 | 26 | 24 | |||
| pT status | 0.033 | |||||
| T2 | 46 | 27 | 19 | |||
| T3 | 49 | 16 | 33 | |||
| T4 | 17 | 9 | 8 | |||
| pN status | 0.064 | |||||
| N0 | 47 | 17 | 30 | |||
| N1-N2 | 65 | 35 | 30 | |||
| Multiplicity | 0.587 | |||||
| Unifocal | 74 | 33 | 41 | |||
| Multifocal | 38 | 19 | 19 | |||
Clinical relevance of miR34a/GOLPH3 axis in human UBC specimens. (A) IHC staining analysis of the association of GOLPH3 and CD44 protein expression in paraffin-embedded, archived urothelial bladder cancer tissues. (B) Kaplan-Meier survival analysis of overall survival (P = 0.013), cancer-specific survival (P = 0.006) and progression-free survival (P = 0.006) for all 112 UBC patients who underwent radical cystectomy with pelvic and iliac lymphadenectomy following standard GC (gemcitabine, cisplatin) chemotherapy regimens with low GOLPH3- expressing (n = 52) versus high GOLPH3-expressing tumors (n = 60). The log-rank test was used to calculate P-values. (C) Western blot (14 paired) analysis of CSC markers (CD44, ALDH1A1, Sox2, and KLF4) and DNA damage and repair-related GOLPH3 protein level in paired primary bladder cancer tissues (C) and normal bladder tissues (N). (D) Real-time PCR (30 paired) analysis of GOLPH3 mRNA, miR34a and CD44 mRNA levels, and the correlations among GOLPH3, miR34a and CD44 were determined by Pearson correlation analysis. (E) IHC analysis of GOLPH3 and CD44 in 11 patients with recurrence UBC and their corresponding primary cancer (CD44 enhanced in 8/11 patients; GOLPH3 enhanced in 8/11 patients; both CD44 and GOLPH3 were enhanced in 7/11 patients). (F) Schematic illustration of enhancement of cancer stem-like cell properties in gemcitabine and cisplatin chemotherapy-resistant UBC cells by the dysregulation of miR34a/GOLPH3 axis. CSC: cancer stem cell; GOLPH3: Golgi phosphoprotein 3; IHC: immunohistochemistry; PCR: Polymerase Chain Reaction
Furthermore, Cox proportional hazard regression analysis was performed to evaluate the prognostic significance of clinicopathological parameters (Table 2). Univariate analysis showed that high GOLPH3 expression was significantly associated with shorter OS (HR, 1.805; 95%CI, 1.122-2.902; P = 0.015) in patients treated with GC adjuvant chemotherapy. Moreover, pT (HR, 1.637; 95%CI, 1.004-2.671; P = 0.048) and pN (HR, 1.654; 95%CI, 1.019-2.686; P = 0.042) status was also significantly associated with shorter OS. All factors that showed prognostic significance in the univariate analysis were included in the multivariate analysis. Multivariate analysis showed that GOLPH3 appeared to be an independent prognostic factor for short OS (HR, 1.808; 95% CI, 1.085-3.013; P = 0.023, Table 2). Moreover, pT (HR, 1.843; 95% CI, 1.063-3.196; P = 0.030) and pN (HR, 2.360; 95% CI, 1.386-4.019; P = 0.002) status also appeared to be an independent prognostic factor for short OS (Table 2) in patients treated with GC adjuvant chemotherapy, respectively.
(a) Univariate and (b) multivariate analyses for the effects of GOLPH3 expression on the overall survival of 112 patients treated with radical cystectomy followed by standard GC chemotherapy for urothelial carcinoma of the bladder
| Univariate analysis | Multivariate analysis | |||||||
|---|---|---|---|---|---|---|---|---|
| Variable | HR | 95%CI | P | HR | 95%CI | P | ||
| Gender | ||||||||
| Male vs Female | 0.854 | 0.509-1.432 | 0.549 | 0.792 | 0.460-1.365 | 0.401 | ||
| Age | ||||||||
| >65 y vs ≤65 y | 1.186 | 0738-1.905 | 0.482 | 1.068 | 0.649-1.759 | 0.795 | ||
| pT status | ||||||||
| T3-T4 vs T2 | 1.637 | 1.004-2.671 | 0.048 | 1.843 | 1.063-3.196 | 0.030 | ||
| Size | ||||||||
| >3 cm vs ≤3 cm | 1.319 | 0.833-2.088 | 0.238 | 1.224 | 0.733-2.045 | 0.440 | ||
| pN status | ||||||||
| N1-N2 vs N0 | 1.654 | 1.019-2.686 | 0.042 | 2.360 | 1.386-4.019 | 0.002 | ||
| Multiplicity | ||||||||
| Multifocal vs Unifocal | 1.231 | 0.765-1.983 | 0.392 | 1.112 | 0.661-1.870 | 0.689 | ||
| GOLPH3 expression | ||||||||
| High vs Low | 1.805 | 1.122-2.902 | 0.015 | 1.808 | 1.085-3.013 | 0.023 | ||
Western blot and real-time q-PCR analyses showed that the GOLPH3 expression level in most human UBC tissues (C) was higher than that in paired normal bladder tissues (N) (Fig. 6C). Moreover, the CSCs markers CD44, Sox2, KLF4 and ALDH1A1 were increased in most human UBC tissues. These results suggested that GOLPH3 and CSCs markers expressions were increased in most human UBC. To determine if there was any correlation among GOLPH3, CD44 and miR-34a in human UBC specimens, we analyzed the mRNA levels of the GOLPH3, CD44 and miR-34a in 30 paired fresh UBC samples and normal tissues. More importantly, the level of miR34a in tumor tissues was negatively correlated with GOLPH3 (r = -0.3870; P < 0.0002) and CD44 (r = -0.3148; P = 0.0013). Next, we further assessed the protein expressions of GOLPH3 and CD44 in archived recurrence UBC tissues. The results showed that the GOLPH3 and CD44 expressions in recurrence UBC were higher than in primary cancer from patients who received standard GC chemotherapy (CD44 enhanced in 8/11 patients; GOLPH3 enhanced in 8/11 patients; both CD44 and GOLPH3 were enhanced in 7/11 patients) (Fig. 6E). This indicates that GOLPH3 and CD44 may play a pivotal role in UBC GC recurrence. Collectively, our findings suggested that the reduced expression of miR34a contributes to UBC stemness, chemoresistance and recurrence by regulating the expression of GOLPH3 (Fig. 6F).
Discussion
CSCs have intrinsic chemoresistant properties and can be selectively enriched during chemotherapy and ultimately cause chemotherapy failure and cancer recurrence. In this study, we found that UBC CSC-like subpopulation enriched during gemcitabine and cisplatin three round chemotherapy in vivo, which was confirmed by the increased levels of CSC markers, such as CK14, CK5 and CD44. Then we found that the expression of GOLPH3 was significantly increased in GC chemotherapy-resistant human UBC tissues, xenograft, isolated cells and sphere cells. Moreover, we revealed the mechanism by which UBC cells escape chemotherapeutic drug (gemcitabine and cisplatin) is mediated by UBC stem-like cells enrichment via inhibition of the miR34a/GOLPH3 axis. Notably, our in vivo data implied that a miR34a mimic was a potential therapeutic agent for treating GC chemoresistant UBCs.
The prognosis for patients with advanced or metastatic UBC remains poor. The vast majority of patients treated with cisplatin-based regimens develop progressive disease within 8 months of treatment, and the median survival was reported to be only 13-15 months [24]. CSCs may generate other CSCs and populations of cells forming the bulk of the tumor [9, 25]. Hidden CSCs contribute to treatment resistance and relapse [9]. This is consistent with the observation in our study that cytotoxic chemotherapy (gemcitabine and cisplatin) induced CK5+, CK14+, CD44+ UBC cells enrichment despite reducing tumor size and ultimately led to UBC chemotherapy failure. Therefore, targeting CSCs has some advantages by eliminating the root cause of tumors, improving therapeutic sensitivity and minimizing side effects [5].
GOLPH3 has been implicated in protein trafficking, receptor recycling, and protein glycosylation and is a potential link between these cellular processes and tumorigenesis [26]. It has been reported previously that GOLPH3 is amplified at the 5p13 region, and high expression of GOLPH3 has been suggested to be a predictor of poor prognosis in several types of cancer, including UBC [13]. Strikingly, Suzette et al. found that overexpression of GOLPH3 confers resistance to killing by DNA-damaging agents [14, 15]. Since DNA-damaging agents like cisplatin and gemcitabine remain the mainstay of UBC treatment regimens, this raises the possibility that GOLPH3 may be a clinically relevant marker of responsiveness to UBC chemotherapy. In this study, we found that expression of GOLPH3 is dramatically increased in GC chemotherapy-resistant UBC cell lines and xenografts. Consistently, the CSC markers CK5, CK14, CD44, Sox2, KLF4 or ALDH1A1 are highly expressed in GC chemotherapy-resistant human UBC tissues, xenografts, and isolated cells. Upon changing the expression of GOLPH3 in UBC cells, the level of CSC markers and the sensitivity to GC chemotherapy changed accordingly. All these results indicated that GOLPH3 plays a key role in UBC stem cell enrichment and GC chemoresistance.
In conclusion, miR34a-mediated regulation of GOLPH3 is involved in UBC CSCs self-renewal and GC chemoresistance. Restoration of miR34a and inhibition of GOLPH3 reduced UBC CSCs properties and GC chemoresistance in vivo and in vitro. In addition, miR34a is one of the master tumor suppressor miRNAs and the miR34a-GOLPH3 axis plays a crucial role in UBC CSCs survival. Thus, modulating miR34a-GOLPH3 axis may serve as a novel therapeutic strategy for drug-resistant UBC by targeting CSCs and shifting UBC therapy from conventional therapies to targeted therapies.
Materials and Methods
Patient information and tissue specimens
112 consecutive patients with UBC who underwent radical cystectomy with pelvic and iliac lymphadenectomy following standard GC (gemcitabine, cisplatin) chemotherapy regimens at Drum Tower Hospital, Medical School of Nanjing University, between January 2006 and December 2012 were included in this study. No patients had distant metastases at the time of cystectomy and GC chemotherapy. Human UBC paraffin embedded tissues of the 112 patients were analyzed for IHC study. Tumor stage and grade were defined according to Unio Internationale Contra Cancrum and WHO classification as previously described [13]. The study protocol was approved by the Ethics Committee of Drum Tower Hospital, Medical School of Nanjing University (Nanjing, China), and written informed consent was obtained from each patient.
Cell lines and culture
Two invasive human UBC cell lines, parental T24 and 5637, were obtained from the American Type Culture Collection (Rockville, MD, USA). All cells were routinely maintained in RPMI-1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Dainippon Pharmaceutical, Tokyo, Japan), at 37 °C in a humidified 5% CO2 atmosphere.
Cell growth assay
Cell growth was measured with the CCK8 assay (cell counting kit-8, Vazyme, Piscataway, NJ, USA). All cell lines were seeded at a density of 1×104 per well into 96-well culture plates. Following 24 h incubation in RPMI 1640 medium with 10% fetal bovine serum, the cells were incubated for appropriate times with various concentrations of cisplatin (0-19.2 µg/mL) and 1.5 µg/mL gemcitabine, to investigate the sensitivity of each cell line. At the end of the incubation period, the cell monolayer was washed three times with phosphate-buffered saline (PBS), and then CCK8 solution diluted 1:10 in RPMI 1640 was added and the cells were incubated for 2 h at 37 °C. The results were measured with a microplate reader at 450 nm and expressed as a percentage of control values (obtained for cells treated with vehicle).
Western blotting
Tissues or cells were lysed in lysis buffer containing protease inhibitor cocktail. Following quantification, the protein lysates were resolved in an SDS-PAGE gel, transferred to a PVDF membrane (Millipore, Billerica, MA, USA), and immunoblotted with different antibodies such as GOLPH3, Sox2, ALDH1A1, CD44 (Proteintech Inc., Chicago, IL, USA), KLF4, β-actin (Cell Signaling Technology Inc., Danvers, MA, USA) and then a horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology Inc., Danvers, MA, USA). Bound antibody was visualized using standard chemical luminescence methodology. β-actin was used as a loading control. The quantification was performed through optical density methods using the ImageJ software (National Institutes of Health). The results are presented as relative density to β-actin, normalized to the mean value of the control group. The experiment was done in triplicate (Supplementary Figure S1).
Real-time quantitative PCR
Total RNA was isolated from tissue specimens or cells using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. First-strand cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) with special stem-loop primer for miRNA and oligo-dT or random primer for mRNA. Quantitative real-time PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) with the 7900 Real-Time PCR System (Applied Biosystems). U6 (RNU6B) snRNA and β-actin (ACTB) mRNA were used as references for miRNA and mRNA, respectively. The Ct values of the samples were calculated, and the relative levels of mRNA were analyzed by the 2-△△ Ct method. Primer sequences used in the study were listed in Supplementary Table S1.
PCR array analysis
We used the home-made PCR array by analyzing 52 cancer related miRNAs in T24 control and T24 GC-3rd xenograft tissue. The RNA of T24 control and T24 GC-3rd tissue were isolated, reverse transcribed and used for qRT-PCR by 52 miRNA primers as described in Supplementary Table S1. The relative amounts of mature miRNAs were normalized by U6 transcript. Three independent experiments were performed. The normalized, individual data point is identical to the 2-ΔΔCt. The values were used for Hierarchical clustering through Cluster and TreeView. The median adjusted values were used to create expression files. Primer sequences used in our study are listed in Supplementary Table S1.
Immunohistochemistry
Paraffin-embedded UBC specimens were cut into 5 μm sections. Immunohistochemical staining was performed as described previously [13]. GOLPH3 expression was assessed by evaluating the proportion and intensity of positively stained carcinoma cells. A score was assigned to represent the estimated percentage of positively stained carcinoma cells as follows: 0: none, 1: ≤ 50%, 2: 50-75%, and 3: ≥ 75%. An intensity score was assigned to represent the average estimated intensity of staining in positive carcinoma cells as follows: 0: none, 1: weak, 2: intermediate, 3: strong. The proportion score and intensity score were multiplied to obtain a total score ranging from 0 to 9. The IHC results were classified based on total scores with 0 to 4 classified as low expression and 5 to 9 indicating high expression. Scoring for the expression of CD44 was determined using light microscopy, by multiplying the intensity times the area (%) where staining was observed in urothelium; the possible scores ranged from 0 to 300. The IHC results were independently judged by two well-trained uropathologists. Differences in evaluations were discussed using a double headed microscope until a consensus was reached.
Immunofluorescence (IF)
5 µm thick OCT-Freeze Medium-embedded sections were used for IF staining. Sections were deparaffinized, ethanol hydrated and then incubated with primary antibodies against GOPLH3, CD44 overnight at 4 °C. After washing, the following secondary antibody was used: Alexa Fluor 594 goat anti-mouse (Invitrogen). The dilution of the second antibody was 1:500. DAPI was used with a dilution ratio of 1:1000 at last. Sections were scanned using a Leica TCS SP5 laser confocal microscope.
Sphere formation assay
Cells were trypsinized by TrypLE (Life Technologies) and washed in PBS. ~1.0×104 cells per well were seeded in 6-well ultra-low attachment plates (Corning, Steuben County, NY, USA) in DMEM/F12 culture medium supplemented with 10 ng/mL human recombinant bFGF and 10 ng/mL EGF (PeproTech, Rocky Hill, NJ, USA). After culturing for 9-12 days, photos were taken and spheres with diameters greater than 50 µm were counted.
Flow cytometry
CD44 (BD Biosciences, San Jose, CA, USA, 1:25) was added to cell suspension in PBS / 0.5% BSA for 15 min at 4 °C, followed by analysis on a BD FACScan flow cytometer.
Plasmid construction and cell transfection
Pri-miR-34a-WT and pri-miR-34a-MUT were cloned into pcDNA3.1. GOLPH3-3'UTR, GOLPH3-3'UTR mutant were constructed in psiCHECK2 at XhoI and NotI sites. UBC cells were plated in 6 cm dish overnight, transfected with 20 nM of GOLPH3 siRNA#1 (5′-GUUAAGAAAUGUACGGGAATT-3′), GOLPH3 siRNA#2 (5′-GGUGAGACAUGGAAUCCAUTT-3′) or scramble control siRNA (5'-UUCUCCGAACGUGUCACGUTT-3'), 50 nM miR-34a mimic, miR-34a inhibitor and corresponding scramble control (Genepharma, Shanghai, China) by Lipofectamine® 3000 Reagent (Life Technologies) for 48 h. For forced expression of GOLPH3, pcDNA3-GOLPH3 and empty vector (pcDNA3) were transfected into UBC cells by Lipofectamine 3000, respectively. Plasmid construction, retroviral production and infection were conducted as previously described. Briefly GOLPH3-targeting siRNA#1 sequences (5′-GUUAAGAAAUGUACGGGAATT-3′) were cloned into the pSuper-retro-puro vector to generate pSuper-retro-GOLPH3-RNAi. Stable T24 GC 3rd UBC cell lines expressing GOLPH3 (Lv-shRNA-GOLPH3 with GFP) and negative control (Lv-shRNA-NC with GFP) short hairpin RNAs were selected for 10 days with 0.5 mg/mL puromycin.
Luciferase assay
For miRNA targeting assay, the wild type GOLPH3 3'UTR target sequence containing miR-34a binding site was cloned downstream of the luciferase gene in the psiCheck2 luciferase vector. Mutant GOLPH3 3'UTR sequence (represented in Fig. 4B) was cloned in the same vector. Control/WT GOLPH3/mutant GOLPH3 3'-UTR constructs (0.2 μg) were transfected into T24 GC 3rd cells cultured in 24-well plates along with miR-34a or scramble control mimics using Lipofectamine 3000. 48 h later, cell lysates were harvested and measured by the dual luciferase reporter assay system (E1910, Promega) according to the manufacturer's protocol. Firefly luciferase was normalized to renilla luciferase activity. Experiments were performed in triplicate. At least three biological repeats were carried out. The results are shown as mean ± SD.
Xenograft tumor model
The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC), Drum Tower Hospital, Medical School of Nanjing University. Six-week-old male BALB/c nude mice (Experimental Animal Center of Nanjing Medical University, Nanjing, China) were housed in a specific pathogen free (SPF) environment at the Animal Laboratory Center, Drum Tower Hospital, Medical School of Nanjing University. For the part of establishment of GC chemotherapy-resistant UBC cell lines. ~5.0×106 cells were suspended in 100 μL PBS and subcutaneously injected in the right flank region of nude mice. Gemcitabine (40 mg/kg body weight) were administered to the mice on day 1, 5, 8 and 15, and cisplatin (60 mg/kg body weight) were administered to the mice on day 2 via intraperitoneal injection followed by a 15 day recovery period. The second round and third round treatments started on day 30 and day 60. The establishment of control cell lines T24 and 5637 were the same as GC chemotherapy-resistant UBC cell lines, but without chemotherapy treatment (PBS treatment). The tumors were harvested 24 h after the last treatment and snap frozen or fixed in 10% formalin, followed by embedding in paraffin. Xenografts were rinsed in PBS and mechanically minced with sterile blades in RPMI1640 / 5% FBS with antibiotics/antimycotics and digested with 0.5% trypsin for 5-10 min at 37 °C. The dissociated cells from xenografts were strained through strainer (BD) and cultured as GC chemoresistant UBC cell lines (T24 GC 3rd and 5637 GC 3rd) and control cell lines (T24 and 5637).
For the xenograft model, we first designed shRNA targeting GOLPH3 and cloned it into pLKO.1, which is used for lentivirus generation. The sequences for GOLPH3 shRNAs were: sh GOLPH3, 5′-GUUAAGAAAUGUACGGGAATT-3′; shNC, 5'-UUCUCCGAACGUGUCACGUTT-3'. Lentiviral particles were produced in 293FT cells co-transfected with a shGOLPH3 expressing plasmid, an envelope plasmid (VSVG) and a packing plasmid (gag-pol). The supernatant with virus particles was collected 48 h after transfection for transduction of UBC cells. The infected cells were selected by 0.5 mg/mL puromycin for 10 days. We first established GOLPH3 stable knockdown T24 GC 3rd UBC cell lines (T24 GC 3rd shGOLPH3) and control T24 GC 3rd UBC cell lines (T24 GC 3rd shNC) by infecting T24 GC 3rd UBC cells with lentivirus containing shRNA targeting GLOPH3 and negative control shRNA, respectively. We also infected T24 control cell with Lv-shRNA-NC (T24 shNC) as the context control. Then, 5.0×106 cells suspended in 100 μL PBS were inoculated into nude mice subcutaneously. When tumor size was 100 mm2 (~2 weeks after cells inoculation), miR34a mimic complex (each complex contained 10 μg of miR-34a mimic and 7.5 μL Lipofectamine® 3000 reagent in PBS, total volume 15 μL) was injected into tumor of T24 GC 3rd shNC, while scramble control complex (each complex contained 10 μg of scramble control mimic and 7.5 μL Lipofectamine® 3000 reagent in PBS, total volume 15 μL) was injected into T24 shNC group, T24 GC 3rd shNC and T24 GC 3rd shGOLPH3 group. Therefore, four groups were set: T24shNC + scramble mimic, T24 GC 3rd shNC + scramble mimic, T24 GC 3rd shGOLPH3 + scramble mimic and T24 GC 3rd shNC + miR34a mimic (n = 5 for each group). Microliter syringes (Gaoge, Shanghai, China) were used for the injection of miR34a mimic complex and scramble mimic complex. The nude mice received an intratumoral injection every 5 days. Tumor volume was calculated by the formula: tumor volume [mm3] = (length [mm]) × (width [mm])2/2. We monitored the tumor volume using the IVIS Lumina II system (Caliper Life Sciences, Hopkinton, MA, USA) at the end point. At the endpoint, tumors were weighed and processed for further histological analysis.
Statistical analysis
All statistical analyses were conducted using SPSS version 17.0 statistical software (SPSS Inc., Chicago, IL, USA). Comparisons between groups for statistical significance were performed with an unpaired 2-tailed Student's t test. The relationship between GOLPH3 expression and clinicopathologic characteristics was analyzed by the χ2 test. Survival curves were plotted using the Kaplan-Meier method and compared using the log-rank test. Survival data were evaluated by univariate and multivariate cox regression analyses. Values were the mean ± SD of 3 independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001.
Abbreviations
CSCs: cancer stem cells; CSS: cancer-specific survival; GC: gemcitabine and cisplatin; GOLPH3: Golgi phosphoprotein 3; H&E: hematoxylin-eosin staining; IHC: immunohistochemistry; MIBC: muscle-invasive bladder cancer; M-VAC: methotrexate, vinblastine, doxorubicin and cisplatin; OS: overall survival; PFS: progression-free survival; RT-qPCR: Real-time quantitative Polymerase Chain Reaction; UBC: urothelial bladder cancer.
Supplementary Material
Table S1 and figure S1.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81572519, 81772710, 81502203, 81602221), the Project of Invigorating Health Care through Science, Technology and Education Jiangsu Provincial Key Medical Discipline, the Social Development Project of Nanjing (201503014), the National Natural Science Foundation of Jiangsu Province (BK20160117), the “Summit of the Six Top Talents” Program of Jiangsu Province (SWYY-084) and the Fundamental Research Funds for the Central Universities (021414380041).
Author contributions
QZ and HQG designed the study. JLZ, YMD, LY, WMC, TSL, WC, XYL, YSX, HY performed experiments and JLZ, YMD, LY, and WC analyzed data. YMD and JLZ, HY provided clinical samples and clinical information. QZ, JLZ, YMD, and LY wrote the manuscript. HQG and WC supervised research.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tanaka N, Miyajima A, Kosaka T, Miyazaki Y, Shirotake S, Shirakawa H. et al. Acquired platinum resistance enhances tumour angiogenesis through angiotensin II type 1 receptor in bladder cancer. Br J Cancer. 2011;105:1331-7
2. Bambury RM, Rosenberg JE. Advanced Urothelial Carcinoma: Overcoming Treatment Resistance through Novel Treatment Approaches. Front Pharmacol. 2013;4:3
3. Herr HW, Dotan Z, Donat SM, Bajorin DF. Defining optimal therapy for muscle invasive bladder cancer. J Urol. 2007;177:437-43
4. Luo T, Yu J, Nguyen J, Wang CR, Bristow RG, Jaffray DA. et al. Electron transfer-based combination therapy of cisplatin with tetramethyl-p-phenylenediamine for ovarian, cervical, and lung cancers. Proc Natl Acad Sci U S A. 2012;109:10175-80
5. Park EY, Chang E, Lee EJ, Lee HW, Kang HG, Chun KH. et al. Targeting of miR34a-NOTCH1 axis reduced breast cancer stemness and chemoresistance. Cancer Res. 2014;74:7573-82
6. Zhang Q, Zhao W, Ye C, Zhuang J, Chang C, Li Y. et al. Honokiol inhibits bladder tumor growth by suppressing EZH2/miR-143 axis. Oncotarget. 2015;6:37335-48
7. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275-84
8. Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727-38
9. Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP. et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2015;517:209-13
10. Scott KL, Kabbarah O, Liang MC, Ivanova E, Anagnostou V, Wu J. et al. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature. 2009;459:1085-90
11. Gorringe KL, Boussioutas A, Bowtell DD. Novel regions of chromosomal amplification at 6p21, 5p13, and 12q14 in gastric cancer identified by array comparative genomic hybridization. Genes Chromosomes Cancer. 2005;42:247-59
12. Zeng Z, Lin H, Zhao X, Liu G, Wang X, Xu R. et al. Overexpression of GOLPH3 promotes proliferation and tumorigenicity in breast cancer via suppression of the FOXO1 transcription factor. Clin Cancer Res. 2012;18:4059-69
13. Zhang Q, Zhuang J, Deng Y, Zhao X, Tang B, Yao D. et al. GOLPH3 is a potential therapeutic target and a prognostic indicator of poor survival in bladder cancer treated by cystectomy. Oncotarget. 2015;6:32177-92
14. Farber-Katz SE, Dippold HC, Buschman MD, Peterman MC, Xing M, Noakes CJ. et al. DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3. Cell. 2014;156:413-27
15. Foiani M, Bartek J. Golgi feels DNA's pain. Cell. 2014;156:392-3
16. Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW. et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317-23
17. Corney DC, Hwang CI, Matoso A, Vogt M, Flesken-Nikitin A, Godwin AK. et al. Frequent downregulation of miR-34 family in human ovarian cancers. Clin Cancer Res. 2010;16:1119-28
18. Ji Q, Hao X, Zhang M, Tang W, Yang M, Li L. et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One. 2009;4:e6816
19. Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C. et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109-23
20. Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE. et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol. 2011;195:417-33
21. Enokida H, Yoshino H, Matsushita R, Nakagawa M. The role of microRNAs in bladder cancer. Investig Clin Urol. 2016;57(Suppl 1):S60-76
22. Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193-9
23. Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H. et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211-5
24. Niedersüss-Beke D, Puntus T, Kunit T, Grünberger B, Lamche M, Loidl W. et al. Neoadjuvant Chemotherapy with Gemcitabine plus Cisplatin in Patients with Locally Advanced Bladder Cancer. Oncology. 2017
25. Chakraborty C, Chin KY, Das S. miRNA-regulated cancer stem cells: understanding the property and the role of miRNA in carcinogenesis. Tumour Biol. 2016;37:13039-48
26. Scott KL, Chin L. Signaling from the Golgi: mechanisms and models for Golgi phosphoprotein 3-mediated oncogenesis. Clin Cancer Res. 2010;16:2229-34
Author contact
![]() Corresponding author: Hongqian Guo, Department of Urology, Drum Tower Hospital, Medical School of Nanjing University, Institute of Urology, Nanjing University, 321 Zhongshan Rd., Nanjing 210008, Jiangsu, People's Republic of China; E-mail: dr.ghqedu.cn
Corresponding author: Hongqian Guo, Department of Urology, Drum Tower Hospital, Medical School of Nanjing University, Institute of Urology, Nanjing University, 321 Zhongshan Rd., Nanjing 210008, Jiangsu, People's Republic of China; E-mail: dr.ghqedu.cn