Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(19):4671-4688. doi:10.7150/thno.21216 This issue Cite
Research Paper
Follicular Stimulating Hormone Accelerates Atherogenesis by Increasing Endothelial VCAM-1 Expression
Xiaosa Li1*, Weiyu Chen2*, Ping Li1*, Jinzhi Wei1, Yang Cheng3, Pei Liu4, Qing Yan1, Xingyan Xu1, Yuhong Cui1, Zhengtian Gu3, Tommaso Simoncini5, Xiaodong Fu1 ![]()
1. Guangzhou Institute of Cardiovascular Disease, The Second Affiliated Hospital; Key Laboratory of Cardiovascular Diseases, School of Basic Medical Sciences, Guangzhou Medical University, Guangzhou 511436, China;
2. School of Medical Sciences, Faculty of Medicine, University of New South Wales, Sydney, NSW 2052, Australia;
3. Department of Gynecology and Obstetrics, the Municipal First People' s Hospital of Guangzhou, Guangzhou, 510180, Guangdong Province, China;
4. Department of Anesthesiology, the Third Affiliated Hospital of Southern Medical University, Guangzhou 510630, Guangdong Province, China;
5. Molecular and Cellular Gynecological Endocrinology Laboratory (MCGEL), Department of Reproductive Medicine and Child Development, University of Pisa, Pisa 56100, Italy.
* These authors contributed equally to this work.
Received 2017-5-28; Accepted 2017-9-8; Published 2017-10-17
Abstract

Rationale: Postmenopausal atherosclerosis (AS) has for decades been attributed to estrogen deficiency. Although the follicular stimulating hormone (FSH) levels rise sharply in parallel, the direct effect of FSH on AS has never been investigated. In this study, we explored the possible role of FSH in the development of AS.
Methods: This was a prospective cohort study of 48 healthy premenopausal and 15 postmenopausal women. ApoE knockout mice were used as atherosclerosis model and human umbilical vascular endothelial cells (HUVECs) were cultured as cell model. Serum hormones and vascular cell adhesion molecule-1 (VCAM-1) levels were measured. Real-time PCR, histology for atherosclerotic lesions, immunofluorescence, luciferase assay, transfection experiments, flow chamber adhesion assay and western blot were performed.
Results: In ApoE knockout mice, administration of FSH increased the atherosclerotic lesions and serum VCAM-1 concentration. Importantly, in blood samples of postmenopausal women, we detected significantly higher levels of FSH and VCAM-1 compared with those from premenopausal women, and there was a positive correlation between these two molecules. In cultured HUVECs, FSH receptor (FSHR) mRNA and protein expression were detected and FSH enhanced VCAM-1 expression. This effect was mediated by the activation of nuclear factor κB (NF-κB), which was sequentially enhanced by the activation of PI3K/Akt/mTOR cascade. FSH first enhanced GαS activity resulting in elevated cAMP level and PKA activity, which relayed the signals from FSHR to the PI3K/Akt/mTOR cascade. Furthermore, FSHR was detected in endothelial caveolae fraction and interacted with caveolin-1 and GαS. The disruption of caveolae or the silencing of caveolin-1 blocked FSH effects on signaling activation and VCAM-1 expression, suggesting the existence of a functional signaling module in membrane caveolae. Finally, FSH increased human monocyte adhesion to HUVECs which was reversed by the VCAM-1 neutralizing antibody.
Conclusion: FSHR was located in the membrane caveolae of HUVECs and FSH promoted VCAM-1 expression via FSHR/GαS /cAMP/PKA and PI3K/Akt/mTOR/NF-κB pathway. This may contribute to the deleterious role of FSH in the development of AS in postmenopausal women.
Keywords: Follicular Stimulating Hormone, Vascular Cell Adhesion Molecule-1, Atherosclerosis, Vascular Endothelial Cells, Caveolae.
Introduction
Cardiovascular disease (CVD) is the leading cause of death among postmenopausal women [1]. It is generally believed that estrogen deficiency after menopause is the critical risk factor for increased incidence of CVD and estrogen replacement therapy would provide beneficial cardiovascular effects [2]. However, currently, the overall effect of postmenopausal estrogen therapy on the risk of CVD remains controversial and the estrogen deficiency alone does not fully explain the increased incidence of CVD [3, 4]. Therefore, new mechanisms should be investigated and novel strategies should be developed to reduce the CVD incidence in postmenopausal women.
Follicle-stimulating hormone (FSH) plays a critical role in the control of follicular growth in females and its secretion is negatively regulated by estrogenic feedback. Together with estrogen deficiency, a high circulating FSH concentration is regarded as a marker for the onset of menopause. Serum FSH rises from 19.5 mIU/mL to 109.7 mIU/mL across the early menopausal transition, remaining at this high concentration for a decade [5]. This period is considered as the “window of opportunity” for efficient interventions to prevent CVD in postmenopausal women [4]. Until now there was no evidence for a direct relationship between FSH and CVD since FSH receptor (FSHR) has long been believed to be exclusively located in sertoli and granulosa cells [6]. However, in recent years we and other groups found that FSHR was expressed and played an important role in extragonadal tissues such as cholangiocytes, osteoclasts, and breast cancer cells [7-9]. For example, FSHR activation enhanced osteoclast formation and function, which accounts for postmenopausal osteoporosis independent of estrogen deficiency [7]. In vascular system, it has been reported that FSHR was selectively expressed on the surface of the blood vessels of a wide range of tumors [10]. Recently, it was reported that FSHR was also expressed in the human vascular endothelial cells [11]. Thus, the vascular effects of FSHR remain a topic of interest.
Endothelial dysfunction is the first step towards CVD such as atherosclerosis (AS) [12]. A variety of stimulators increase expression of adhesion molecules in vascular endothelial cells where they regulate leukocyte adhesion to the endothelium [13]. Among these molecules, increased expression of vascular cellular adhesion molecule-1 (VCAM-1) was reported on the endothelial cell surface in lesion-prone areas [14] and the blockade of VCAM-1 greatly reduced monocyte adhesion and diminished atherosclerotic lesions in a mouse model [15]. In this study, we therefore chose to investigate the effects of FSH on the expression of VCAM-1 in human vascular endothelial cells and explored the underlying signaling mechanisms. First, we measured the concentrations of plasma FSH and VCAM-1 in pre- and postmenopausal women and detected a positive relationship between them. Subsequently, we used ApoE-/- female mice as atherosclerosis model to examine the role of FSH in atherogenesis. Finally, we used human vein umbilical endothelial cells (HUVECs) as a model system and found that FSHR was located in the caveolae of human vascular endothelial cell and by coupling GαS protein it activated cAMP/PKA and PI3K/Akt/mTOR/NF-κB pathways, resulting in increased expression of VCAM-1 and monocyte adhesion. Our findings indicate that FSH may contribute to the development of AS in postmenopausal women.
Results
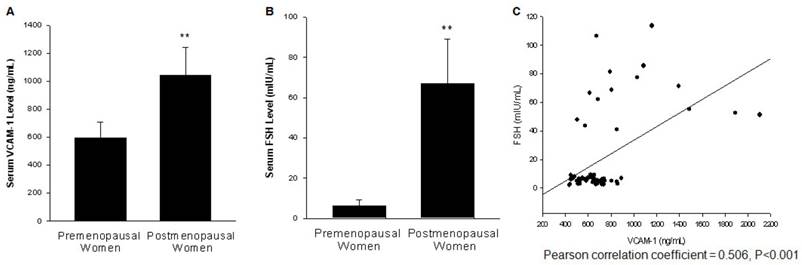
The serum level of FSH was positively associated with VCAM-1 level
Blood samples were collected from 48 premenopausal and 15 postmenopausal women. The serum level of FSH and VCAM-1 were measured. A complete table with the parameters including height, weight, FSH, LH, E2 and progesterone levels of each participants is shown in Table S1. Both FSH and VCAM-1 level were significantly higher in postmenopausal women when compared to premenopausal women (Fig. 1A, 1B). As shown in Fig. 1C, the serum level of FSH was positively associated with serum VCAM-1 level.
The serum level of FSH was positively associated with serum VCAM-1 level. (A). Serum VCAM-1 from 48 premenopausal women at follicular phase and 15 postmenopausal women were assessed by ELISA. ** = P<0.01 vs. premenopausal women group. (B). Serum FSH levels from these samples were determined by immunoassay. ** = P<0.01 vs. premenopausal women group. (C). Scatter diagram shows the association between the serum FSH and VCAM-1 levels.
FSH promoted aortic atherosclerotic lesion formation
The levels of 17β-estradiol (E2) and FSH were measured in different groups of mouse models and are displayed in Table 2. Aorta sinus plaques were found in sham-operated ApoE-/- mice. As expected, OVX aggravated the lesion formation, which was partly reversed by the supplementation of E2. However, the size of plaque was significantly increased when OVX mice were both supplemented with E2 and injected with FSH (Fig. 2A, 2B).
Serum levels of 17β-estradiol and FSH in mice groups.
| SHAM (n=8) | OVX (n=6) | OVX+E2 (n=9) | OVX+E2+FSH (n=6) | |
|---|---|---|---|---|
| E2 (pg/mL) | 52.95±7.08 | 7.84±2.55*** | 42.93±11.22 | 45.03±9.74 |
| FSH (mIU/mL) | 6.41±1.21 | 13.07±2.52*** | 8.73±3.02 | 12.14±4.37# |
*** P<0.001 VS SHAM group; # P<0.05 VS OVX+E2 group.
FSH promoted aortic atherosclerotic lesion formation. (A). Oil-red O-stained aortic root lesion in ApoE-/- mice with different treatments as indicated. Scale bar = 100 μm. (B). Statistical analysis of the size of atherosclerotic plaque in the aortic root. One-way ANOVA analysis was used. n=6-9, *** = P < 0.001 vs. SHAM group; ### = P < 0.001 vs. OVX or OVX+E2+FSH group. (C-D). Immunofluorescent staining of frozen sections of aorta sinus was performed with VCAM-1 antibody (green) (C) or Specific marker antibodies for macrophages (anti-CD68) (green) (D). Nuclei were counterstained with and DAPI (blue). Scale bar = 250 μm. All experiments were repeated at least six times with consistent results and the representative images are shown. (E). Serum VCAM-1 from ApoE-/- mice with different treatments were detected by ELISA. n=6-9, ** = P<0.01, *** = P < 0.001 vs. SHAM group; ### = P<0.001 vs. OVX or OVX+E2+FSH group.
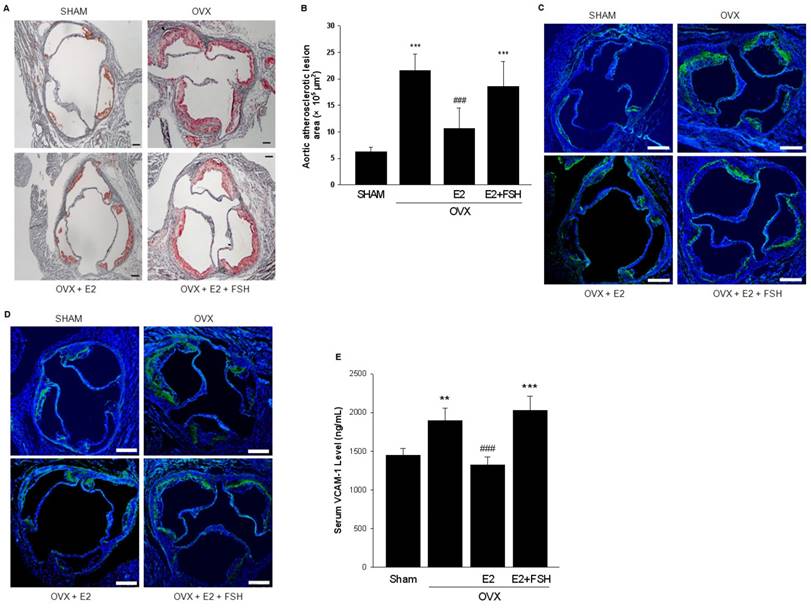
The staining of the histopathology slides of aortic roots showed that in OVX group, VCAM-1 expression and presence of macrophages were extensively detected in plaque areas while both were reduced in the OVX+E2 group. In the OVX+E2+FSH group, VCAM-1 expression and the number of infiltrated macrophages were increased (Fig. 2C, 2D). In line with this, serum level of VCAM-1 was significantly increased in OVX+E2+FSH group as compared to OVX+E2 group (Fig. 2E).
FSHR was expressed in HUVECs and FSH upregulated expression of adhesive molecules
To confirm the existence of FSHR in HUVECs, we performed RT-PCR to detect its mRNA expression. As expected, a 631 bp FSHR mRNA fragment was found both in HUVECs and in the positive control, the human granulosa cells (Fig. 3A). A semi-quantitative comparison suggested a much lower amount of FSHR mRNA in HUVECs than in granulosa cells. Also, FSHR protein expression was detected in HUVECs by Western blotting (Fig. 3B).
FSHR was expressed in HUVECs and FSH upregulated expression of adhesive molecules. (A). FSHR mRNA fragment was assessed by RT-PCR in HUVECs and in the positive control, the human granulosa cells (GCs). (B). FSHR protein expression was detected by Western blotting. (C-D). HUVECs were treated with different concentration of FSH for 24 h (C), or treated with FSH (50 mIU/mL) for different time as indicated (D). VCAM-1, ICAM-1 and E-selectin protein expressions were detected. * = P<0.05, ** = P<0.01, *** = P < 0.001 vs. corresponding control. All experiments were repeated at least three times with consistent results, and the representative images are shown. (E). HUVECs were treated with control vehicle (CON), FSH (50 mIU/mL) or the positive control TNF-α (0.1 ng/mL) for 24 h. VCAM-1, ICAM-1 and E-selectin mRNA were assessed by Quantitative Real-time PCR. n=5, * = P < 0.05, ** = P < 0.01 vs. corresponding control.
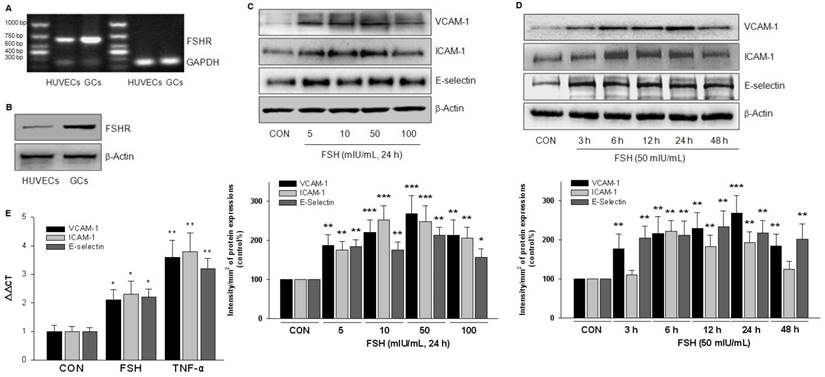
To explore the functional role of FSHR, HUVECs were treated with different doses of FSH for 24 h. Increased expression of adhesive molecules, including VCAM-1, ICAM-1 and E-selectin, was detected with the maximal effect achieved at 50 mIU/mL FSH (Fig. 3C). At this dose of FSH, increased expression of adhesive molecules was observed at 3 h, which reached a peak value at 24 h and then began to decline (Fig. 3D). Also, FSH (50 mIU/mL) increased the mRNA of these molecules, but the potency was much less than that observed with TNF-α, the well-known up-regulator of adhesive molecules (Fig. 3E).
PI3K/Akt and NF-κB signaling pathways were implicated in FSH-enhanced VCAM-1 expression
To clarify the signaling pathways involved, we used several inhibitors that impede the signaling relevant to G-coupled transmembrane receptors. FSH-enhanced VCAM-1 expression was not altered by ERK1/2 inhibitor PD98059, c-Src inhibitor PP2, or the Gi protein inhibitor PTX, but was blocked by PI3K inhibitor LY294002 or the NF-κB inhibitor PDTC (Fig. 4A, 4B, 4C) but 17β-estradiol (E2) antagonized FSH effect (Fig. 4A). When TNF-α was used as a positive control, it increased VCAM-1 expression much stronger than FSH, which was blocked by PDTC (Fig. 4C).
PI3K/Akt and NF-κB signaling were implicated in FSH-enhanced VCAM-1 expression. (A-C). HUVECs were treated with control vehicle (CON) or FSH (50 mIU/mL) for 24 h in the presence or absence of PI3K inhibitor LY294002 (5 μM), ERK1/2 inhibitor PD98059 (2.5 μM), 17β-estradiol (E2, 10 nM), c-Src inhibitor PP2 (5 μM), Gαi protein inhibitor PTX (50 ng/mL), NF-κB inhibitor PDTC (10 μM). VCAM-1 was detected by Western blotting. β-actin was used as an internal control. (D-E). HUVECs were treated with different concentrations of FSH for 24 h, or treated with 50 mIU/mL of FSH over 48 h. Total Akt, phosphorylated Akt (p-Akt), total mTOR, phosphorylated mTOR (p-mTOR), total p65, phosphorylated p65 (p-p65) and β-actin were detected by Western blotting. * = P<0.05, ** = P<0.01, *** = P < 0.001 vs. corresponding control. All experiments were repeated at least three times with consistent results, and the representative images are shown.
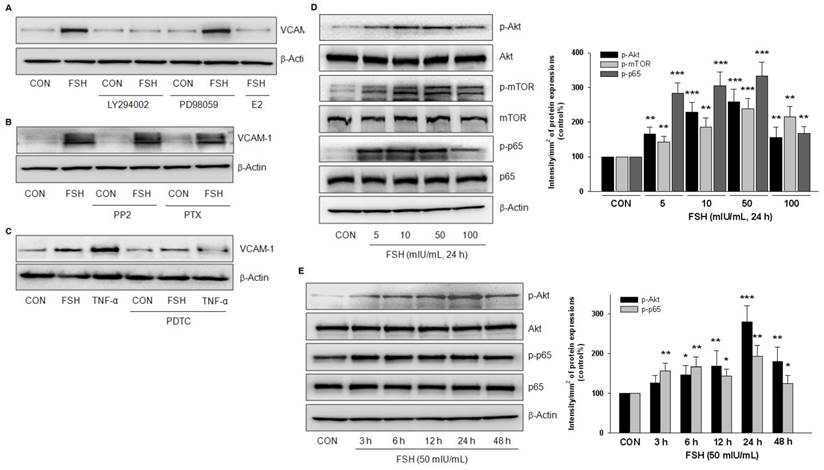
Treatment with different concentrations of FSH for 24 h increased the phosphorylation of Akt, mTOR and p65 but had no effect on the total protein expressions of these molecules (Fig. 4D). Furthermore, FSH (50 mIU/mL) increased Akt and p65 phosphorylation levels from 3 h to 48 h (Fig. 4E). Taken together, these results suggested that PI3K/Akt and NF-κB signaling pathways were implicated in FSH-enhanced VCAM-1 expression.
PI3K/Akt/mTORC1 pathway was upstream of NF-κB signaling
FSH failed to activate Akt and p65 in the presence of PI3K inhibitor LY294002, but it was still able to activate Akt in the presence of NF-κB inhibitor PDTC (Fig 5A), suggesting that PI3K signaling was upstream of NF-κB. To further confirm the role of PI3K, HUVECs were transfected with wild-type p85α (WT p85α) or dominant-negative p85α (△p85α) constructs. Fig. 5b shows that both constructs were efficiently transfected and expressed. Transfection with WT p85α increased VCAM-1 expression with or without FSH treatment (Fig. 5B). On the contrary, transfection with △p85α abrogated FSH effect (Fig 5B).
PI3K/Akt/mTORC1 pathway was upstream of NF-κB signaling. (A). HUVECs were treated with control vehicle (CON) or FSH (50 mIU/mL) for 24 h in the presence or absence of PI3K inhibitor LY294002 (5 μM) and NF-κB inhibitor PDTC (10 μM). Phosphorylated Akt (p-Akt), phosphorylated p65 (p-p65) and β-actin were detected by Western blotting. (B). HUVECs were transiently transfected with constitutively active p85α (WT p85α) or dominant-negative p85α (△p85α) plasmid (both 15 μg) for 48 h. Next, cells were treated with vehicle or FSH (50 mIU/mL) for another 24 h. Cell contents of p85α, VCAM-1 and β-actin were detected by Western blotting. (C). HUVECs were transfected with scrambled siRNA (100 nM) or Akt siRNA (100 nM) for 48 h followed by treatment with vehicle or FSH (50 mIU/mL) for another 24 h. Western blot of proteins is shown. (D). HUVECs were treated with vehicle or 50 mIU/mL FSH for 24 h in the presence or absence of mTOR inhibitor KU 0063794 (KU, 1 μM). Western blot of proteins is shown. (E). HUVECs were treated with control vehicle (CON) or FSH (50 mIU/mL) for 24 h in the presence or absence of PI3K inhibitor LY294002 (5 μM). Total rictor or raptor, phosphorylated rictor (p-rictor) or phosphorylated raptor (p-raptor) and β-actin were detected by Western blotting. (F). HUVECs were transfected with scrambled siRNA, raptor siRNA or rictor siRNA (all 100 nM) for 48 h. Cells were then treated with vehicle or FSH (50 mIU/mL) for another 24 h and the proteins were detected by Western blotting.
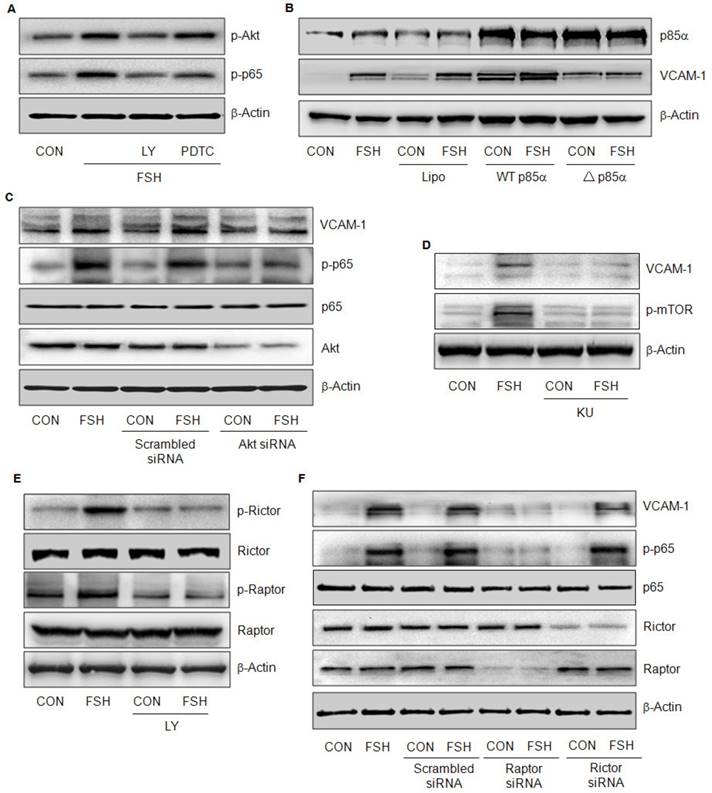
Next, we investigated the role of Akt. Transfection of Akt siRNA greatly reduced Akt expression (Fig 5C). When Akt was knock-down, FSH failed to increase VCAM-1 expression (Fig. 5C) and was also unable to trigger p65 phosphorylation (Fig 5C), further implying that Akt was upstream of NF-κB in FSH signaling cascades.
Mammalian target of rapamycin (mTOR) pathway is known to be downstream of Akt. In the presence of mTOR inhibitor KU 0063794 (KU), FSH failed to increase mTOR phosphorylation and VCAM-1 expression (Fig. 5D), indicating that mTOR also played an important role in this signaling pathway. mTOR forms an enzyme complex with raptor (mTORC1) or rictor (mTORC2). Apparently, FSH activated both mTORC1 and mTORC2 via Akt pathway as indicated by rictor and raptor phosphorylation levels (Fig. 5E). However, FSH failed to activate p65 and increase VCAM-1 when raptor but not rictor, was knocked down by siRNA (Fig. 5F). These results suggested that PI3K/Akt/mTORC1 was responsible for NF-κB activation and VCAM-1 expression induced by FSH.
FSH activated NF-κB signaling through IKK/IkBα pathway
TNF-α is known to activate NF-κB and increase its transcriptional activity of up-regulating VCAM-1 expression. Similar but weaker than TNF-α, FSH caused translocation of p65 protein to the nucleus and increased NF-κB transcriptional activity (Fig. 6A, 6B). Also, the effect of FSH was antagonized by E2 (Fig 6A, 6B). When p65 was efficiently knocked down, FSH-enhanced VCAM-1 expression was abolished, while the activation of Akt and mTOR by FSH was not affected (Fig. 6C). These data further confirmed that p65 relayed the signals from PI3K/Akt/mTOR to enhance VCAM-1 expression.
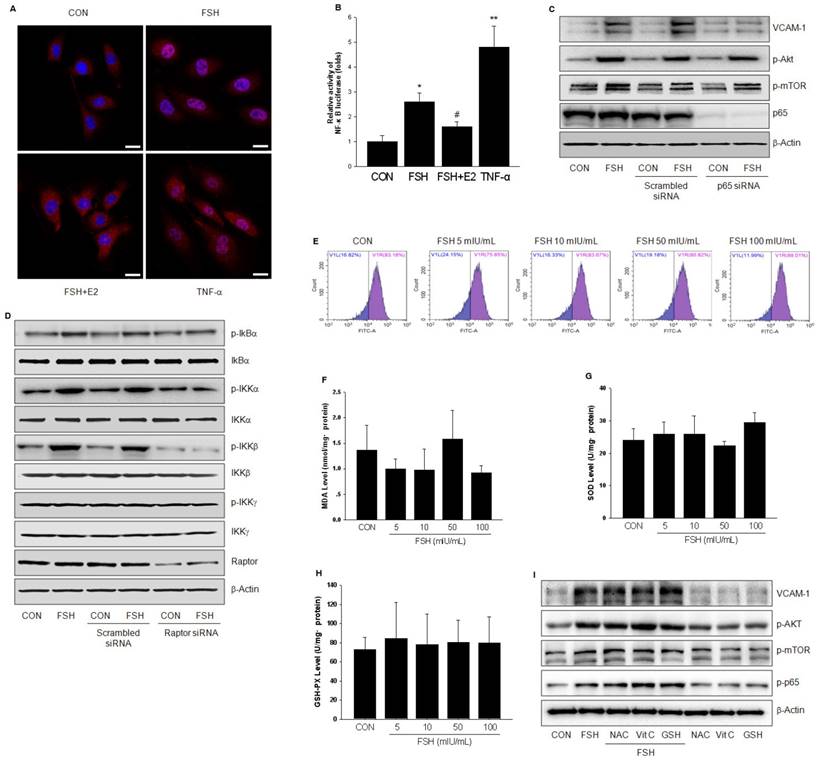
FSH activated NF-κB signaling through IKK/IkBα pathway. (A). HUVECs were treated with control vehicle (CON), FSH (50 mIU/mL) or TNF-α (0.1 ng/mL) for 24 h in the presence or absence of 17β-estradiol (E2, 10 nM). Subcellular localization of the p65 protein was assayed by immunofluorescence (red staining). Scale bar = 250 μm. (B). HUVECs were transiently transfected with 750 ng of NF-κB-luciferase reporter and 250 ng pRL-TK vector expressing renella luciferase. Subsequently, cells were treated with control vehicle (CON), FSH (50 mIU/mL) or TNF-α (0.1 ng/mL) for 24 h in the presence or absence of 17β-estradiol (E2, 10 nM). Relative luminescent units (RLU) were examined and were normalized to fold change from control. * = P < 0.05, ** = P<0.01 vs. control; # = P<0.05 vs. FSH. (C-D). HUVECs were transfected with scrambled siRNA (100 nM), p65 siRNA (100 nM) (C) or raptor siRNA (100 nM) (D) for 48 h. Cells were then treated with vehicle or FSH (50 mIU/mL) for another 24 h and analyzed by Western blotting. (E). HUVECs were treated with control vehicle (CON) or different concentrations of FSH for 24 h. Then cells were washed and loaded with H2DCFDA and subjected to fluorescence measurement by flow cytometry. V1R gate (right gate) showed the percentage of positive cells loaded with probe. No significance was found between these groups. (F-H). HUVECs were treated as indicated for 24 h and the levels of MDA, SOD, GSH-PX were measured. No significance was found between these groups. (I). HUVECs were treated with control vehicle (CON) or FSH (50 mIU/mL) for 24 h, in the presence or absence of antioxidants (including N-Acetyl-L-cysteine (NAC, 1 mM), Vitamin C (Vit C, 200 μM) and glutathione (GSH, 1 mM)). Proteins were analyzed by Western blotting. All experiments were repeated at least three times with consistent results and the representative images are shown.
Activation of IkB kinase (IKK) is believed to induce phosphorylation and rapid degradation of IkBα, thus resulting in activation of p65. In HUVECs, FSH promoted IkBα phosphorylation, which was due to the activation of IKKα and IKKβ but not IKKγ (Fig. 6D). Once raptor was silenced, FSH was unable to activate these molecules, further indicating that PI3K/Akt/mTORC2 activates p65 via IKK/IkBα pathway.
It is well known that reactive oxygen species mediate NF-κB activation. To observe the effect of FSH on oxidative stress status, the levels of several indicators, including reactive oxygen species (ROS), malonaldehyde (MDA), superoxide dismutase (SOD) and glutathione peroxidase (GSH-PX), were measured. As shown in Fig. 6E-6H, different concentrations of FSH didn't alter the levels of these indicators. Furthermore, FSH still led to Akt, mTOR, p65 activation and increased VCAM-1 expression in HUVECs in the presence of antioxidants (including N-Acetyl-L-cysteine (NAC), Vitamin C (Vit C) or glutathione (GSH)) (Fig. 6I). These data suggested that FSH activates NF-κB independent of oxidative stress status.
FSHR coupled to Gαs/AC/cAMP/PKA cascade to activate PI3K/Akt/mTOR
FSHR is a 7-transmembrane receptor that transduces extracellular signals by coupling to heterotrimeric G proteins. To confirm the role of GαS, HUVECs were treated with GαS antagonist NF499 which abolished the phosphorylation of Akt/mTORC2 and IKKα/β/IkBα/p65 induced by FSH (Fig. 7A). On the contrary, treatment with FSH for 2 h and 6 h increased activity of Gαs (Fig. 7B). When HUVECs were treated with FSH or Forskolin (FSK), an adenylyl cyclase activator, cAMP level was increased. On the other hand, GαS antagonist NF499 reduced cAMP level (Fig. 7C). Furthermore, FSH enhanced PKA activity, which was blocked by NF499 and adenylate cyclase inhibitor MDL12330A (Fig. 7D).
FSHR coupled to GαS /AC/cAMP/PKA cascade to activate PI3K/Akt/mTOR. (A). HUVECs were treated with vehicle or FSH (50 mIU/mL) for 24 h in the presence or absence of GαS antagonist NF499 (10 μM) and analyzed by Western blotting. (B). HUVECs were treated with FSH (50 mIU/mL) for over 6 h. Cell lysates were subjected to immunoprecipitation using anti-active GαS monoclonal antibody. Immunoprecipitates were analyzed by Western blotting with anti- GαS antibody. (C). HUVECs were treated with vehicle, FSH, or adenylyl cyclase activator Forskolin (FSK, 10 μM) in the presence or absence of GαS antagonist NF499. cAMP concentration was determined by ELISA. ** = P < 0.01, *** = P < 0.001 vs. control; ## = P<0.01, ### = P<0.001 vs. FSH or FSK. (D). HUVECs were treated with vehicle or FSH in the presence or absence of NF499 or adenylate cyclase inhibitor MDL12330A (MDL, 10 μM). PKA activity was determined by ELISA. ** = P < 0.01 vs. control; ## = P<0.01 vs. FSH. (E). Western blots of HUVECs treated with vehicle, FSH or FSK in the presence or absence of PKA inhibitor H89 (10 μM) pretreatment. (F). HUVECs were transfected with scrambled siRNA (100 nM) or PKA Cα siRNA (100 nM) for 48 h. Then cells were treated with vehicle or FSH (50 mIU/mL) for another 24 h and analyzed by Western blotting. All experiments were repeated at least three times with consistent results and the representative images are shown.
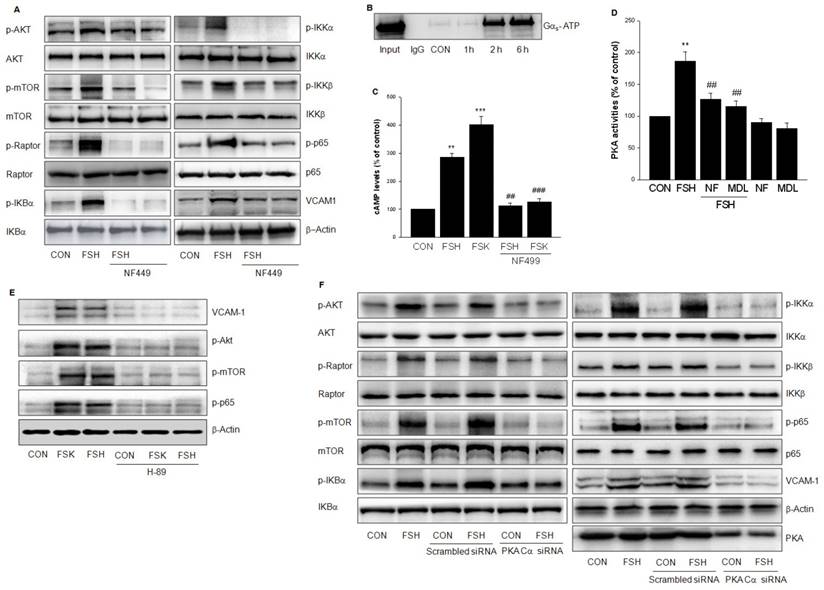
To confirm the role of PKA, H89, a PKA inhibitor was used and the PKA catalytic subunit (PKA Cα) was silenced to block FSH- or FSK-enhanced VCAM-1 expression and the phosphorylation of Akt/mTOR and p65 (Fig. 7E, 7F). Taken together, above results suggested that FSHR signaling occurs via GαS /AC/cAMP/PKA cascade to activate PI3K/Akt/mTOR.
FSHR was in caveolae and its effects were caveolin-1-dependent
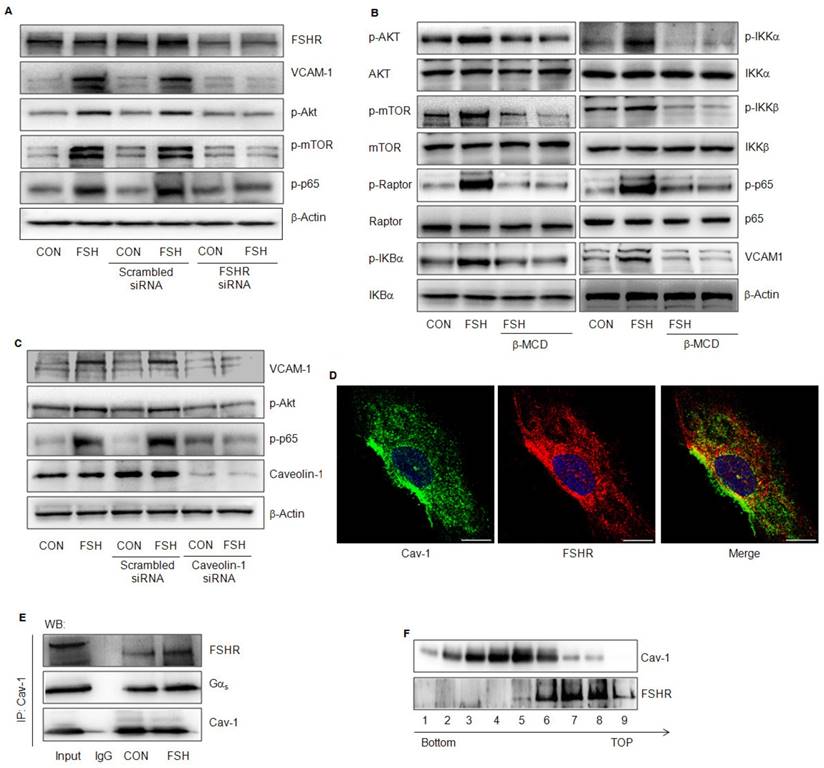
FSH exerts its biological effect by binding to FSHR; therefore, when FSHR was knocked down, FSH failed to activate Akt/mTOR, p65 or to enhance VCAM-1 expression (Fig. 8A). Also, caveolae represent the signaling centers that are abundant in vascular endothelial cells. The lipid raft disruptor Methyl-β-cyclodextrin (β-MCD), or the silencing of caveolin-1, blocked FSH-induced activation of the downstream signaling and VCAM-1 expression (Fig. 8B, 8C). Interaction between Cav-1 and FSHR or GαS was detected in HUVECs in a quiescent state by immunofluorescence or immunoprecipitation assays (Fig 8D, 8E). FSH enhanced the interactions between these molecules (Fig 8D). This was further supported when caveolae were extracted and FSHR was detected in caveolin-1 positive fractions of caveolae (#6) (Fig. 8F).
FSHR was located in caveolae and its effects were caveolin-1-dependent. (A). HUVECs were transfected with scrambled siRNA (100 nM) or FSHR siRNA (100 nM) for 48 h, treated with vehicle or FSH (50 mIU/mL) for another 24 h and analyzed by Western blotting. (B). HUVECs were treated with vehicle or FSH for 24 h in presence or absence of lipid raft disruptor Methyl-β-cyclodextrin (β-MCD, 0.1 mM) and analyzed by Western blotting. (C). HUVECs were transfected with scrambled siRNA (100 nM) or caveolin-1 siRNA (100 nM) for 48 h, treated with vehicle or FSH (50 mIU/mL) for another 24 h and analyzed by Western blotting. (D). Subcellular colocalization of the Cav-1 and FSHR proteins was determined with laser scanning confocal microscopy. Cav-1 (green staining), FSHR (red staining), DAPI-stained nuclei (blue staining). Merged image (yellow staining) is shown. Scale bar = 50 μm. (E). HUVECs were treated with vehicle or FSH for 24 h. Cells were then lysed and cell lysates were subjected to immunoprecipitation using anti-caveolin-1 antibody. Immunoprecipitates were analyzed by Western blotting with anti- GαS and anti-FSHR antibodies. (F). Cell fractionation was performed by OptiPrep density gradient centrifugation as described. Distribution of FSHR in Cav-1-enriched caveolae membrane domain fractions was detected by Western blotting. Representative immunoblotting of cell fractionation from the tubes top fraction 1 to the bottom fraction 9 are shown. All experiments were performed in triplicates and representative images are shown.
FSH promoted monocyte adhesion to vascular endothelial cells
HUVECs were pretreated with different concentrations of FSH for 48 h or treated with TNF-α as a positive control and monocyte adhesion to cells was measured. As shown in Fig. 9A and 9B, FSH increased the number of monocytes adhering to endothelial cells in a dose- and time-dependent manner, which was antagonized by VCAM-1 neutralizing antibody. Moreover, treatment with FSH for 6 h to 48 h enhanced adhesion of monocyte to endothelial cells (Fig. 9A). PI3K inhibitor LY294002, NF-κB inhibitor PDTC, or mTOR inhibitor Rapamycin all inhibited the FSH effect on monocyte-endothelial adhesion (Fig. 9C).
FSH promoted monocyte adhesion to vascular endothelial cells. (A). Representative images of monocyte cell adhesion to HUVECs after treatment with positive control TNF-α (0.1 ng/mL) for 24 h, or with different concentrations of FSH for 24 h in the presence or absence of VCAM-1 neutralizing antibody (Ab). Total cell number of monocyte cell adhesion to HUVECs was observed and calculated by fluorescence microscopy. n=5, * = P < 0.05, ** = P < 0.01, *** = P < 0.001 vs. control; ### = P<0.001 vs. FSH at 50 mIU/mL. Scale bar = 500 μm. (B). Representative images of monocyte cell adhesion to HUVECs after various treatments as indicated. n=5, ** = P < 0.01, *** = P < 0.001 vs. control; ### = P<0.001 vs. FSH treatment for 24 h. (C). HUVECs were treated with FSH (50 mIU/mL) or treated with TNF-α as positive control for 24 h in the presence or absence of PI3K inhibitor LY294002 (LY, 5 μM), NF-κB inhibitor PDTC (10 μM), or mTOR inhibitor KU (1 μM). n=5, *** = P < 0.001 vs. control; # = P<0.05, ## = P<0.01 vs. FSH. (D). The rolling velocity of monocytes adhered to HUVECs were calculated. n= 10-20, ** = P < 0.01 vs. control.
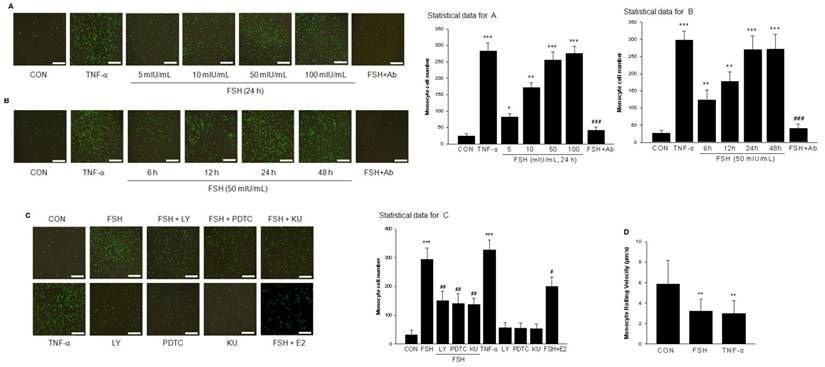
To mimic physiological conditions, parallel-plate flow chamber experiments were performed. HUVECs were treated with vehicle (CON), FSH (50 mIU/mL), TNF-α (0.1 ng/mL) for 24 h, and then U937 monocytoid cell suspension was perfused. The rolling velocity of monocytes in FSH or TNF-α treatment group was significantly slower than that in control group (Fig. 9D, Supplementary video 1(CON), 2 (FSH), 3 (TNF-α)).
Discussion
Currently, direct evidence on the relationship between FSH and cardiovascular diseases (CVD) remains to be established. Our present study provides the first experimental evidence that FSH acts on vascular endothelial cells and alters their function to promote AS development. This suggests that the interference with the FSHR signaling cascades is an alternative way for the prevention of CVD in postmenopausal women.
Vascular endothelial cells express adhesion molecules such as VCAM-1, ICAM-1, and E-selectin, which regulate leukocyte adhesion to and transmigration through endothelium [13]. VCAM-1 is the initial molecule, the expression of which was abnormally increased on the endothelial cell surface in lesion-prone areas [14] and perfusion of specific VCAM-1 antibody greatly reduced monocyte adhesion and diminished atherosclerotic lesions in a mouse model [15]. In this study, we detected a positive relationship between serum FSH and VCAM-1 level in participating women consistent with the data reported in another clinical study of patients undergoing in-vitro fertilization (IVF) [16]. Indeed, one complication associated with IVF is venous thromboembolism [17] in which VCAM-1 plays an important role [18, 19]. Also, elevated serum VCAM-1 levels and increase in blood viscosity were reported in ovulatory phase (FSH peak) in healthy women [20, 21]. Consistent with these observations, our data suggests the possibility that high levels of FSH change the hemorheology by upregulating VCAM-1 and accounts for these physiological and pathophysiological phenomena.
Generally, E2 level decreases and FSH level rises in parallel due to negative feedback, making it difficult to investigate the independent role of FSH in cardiovascular system. To circumvent this, we used OVX mice administrated with E2 to maintain its normal level, and then injected FSH to raise its level while avoiding the E2 production from hyper-stimulated ovaries. Consequently, there was no difference in E2 levels between OVX+E2 and OVX+E2+FSH groups. Under these circumstances, the size of plaque was significantly increased in OVX+E2+FSH group compared to OVX+E2 group, suggesting that FSH acts independently to accelerate atherogenesis. Consistent with the data from human samples, VCAM-1 expression was enhanced in the serum and lesion area in the mouse model, possibly due to the increased number of macrophages found in aortic roots by histopathology.
FSH exerts its function via binding to FHSR. However, controversy exists about the location of FSHR in the vascular system [10, 11]. In this study, we detected a much lower level of FSHR mRNA and protein as compared to granulose cells. FSHR was visible on the surface of HUVECs by immunofluorescence. Also, we have clearly shown, for the first time, that FSH up-regulated VCAM-1 mRNA and protein expressions as well as those of two other adhesion molecules, ICAM-1 and E-selectin. These findings clearly demonstrated that FSH is capable of directly regulating vascular endothelial functions.
VCAM-1 gene expression is regulated by the transcription factors NF-κB, SP-1, Ap-1, and interferon regulatory factor-1 [13]. For instance, we have previously reported that sex steroids inhibited VCAM-1 expression by interfering with NF-κB signaling [22, 23]. In this study, we have shown that FSH caused translocation of p65 to the nucleus and increased its transcriptional activity, resulting in an increased VCAM-1 level. It has been reported that NF-κB was activated by FSH in sertoli cells and osteoclasts [7, 24]. Together, these observations imply that NF-κB signaling is an important pathway for FSH actions in various organs and tissues.
Oxidative stress, a state characterized by an imbalance between pro-oxidant molecules and antioxidant defenses, plays a crucial role in the activation of NF-κB and endothelial dysfunction [25]. Here we found that FSH didn't change the levels of ROS, MDA, SOD and GSH-PX. In the presence of several antioxidants, FSH still activated NF-κB. These data indicate that FSH activates NF-κB independent of oxidative stress. Interestingly, a latest report showed that FSH protects granulosa cells from oxidative injury by repressing autophagy via PI3K-Akt axis [26]. Therefore, the regulatory effect of FSH on oxidative stress may be different dependent on different tissues or organs.
In cardiovascular cells, Akt/mTOR pathway may serve upstream of NF-κB [27-29], which is consistent with our results that FSH activated NF-κB via the activation of PI3K/Akt/mTOR. Indeed, Akt/mTOR signaling was indispensable for FSH-stimulated proliferation of Sertoli or granulosa cells [30, 31]. Similar to the signaling involved in thrombin-induced ICAM-1 expression [32], our findings further indicated that Akt/mTORC2 stimulated NF-κB via the activation of IKKα/β, possibly due to direct interaction between rictor and IKK [33]. TNF-α, which is known to upregulate VCAM-1, was used as the positive control in our study and increased VCAM-1 expression via mTORC2 [34]. However, in that experimental setting, mTORC2 promoted VCAM-1 expression by repressing ERK1/2 activity and the subsequent stimulation of transcription factor IRF-1 [34]. Thus, the implicated signaling mechanism between Akt/mTOR and VCAM-1 depends, at least in part, upon the stimulus used.
FSHR is the member of GPCRs that are known to activate PI3K [35]. The predominant mechanisms of PI3K activation involve the binding of active Ras to the p110 catalytic subunit of PI3K [36], the phosphorylation of the p85 regulatory subunit of PI3K by PKA [37], the regulation of p85 and p110, or by Gβγ [38, 39]. It is well recognized that FSHR activates PKA through coupling to GαS subunit and the subsequent elevation of cAMP level [6]. This was confirmed by our data that FSH increased GαS activity and cAMP levels to stimulate PKA, which was then responsible for Akt/mTOR activation. The underlying mechanism of how PKA activates PI3K/Akt remains unclear. In this respect, a recent study showed that in granulosa cells, FSH activated PKA, which in turn phosphorylated protein phosphatase 1/insulin-like growth factor 1 receptor cascade to activate PI3K/Akt signaling [40]. Also, it has been reported that PKA directly regulates Akt activity via physical interaction in vascular endothelial cells [41]. Thus, PKA may serve as the important intermediate of FSH to activate PI3K/Akt via direct or indirect pathways.
Caveolae are the specialized lipid domains prevalent on the plasma membrane of vascular endothelial cells, where Caveolin-1 is the principal scaffold protein serving as the switch for many signal transduction pathways [42]. In the present study, we have shown the localization of FSHR in caveolae of vascular endothelial cells as it was detected from Caveolin-1 enriched membrane fractions and was colocalized with Caveolin-1 by immunostaining. Furthermore, under FSH stimulation, an increased interaction between Caveolin-1 and FSHR was observed. The disruption of caveolae or the silencing of Caveolin-1 abolished FSH effects on cascade activation and VCAM-1 expression. These results suggested that Caveolin-1 may function as a co-activator of FSHR in caveolae to facilitate its downstream activation.
A corroborative finding of an interaction between FSHR and Caveolin-1 was reported in human embryonic kidney 293 (HEK-293) cells [43]. Indeed, a number of GPCRs (such as adrenergic receptor, thyrotropin receptor) have been found to be enriched in caveolae [44]. These GPCRs contain caveolin-binding motifs (ФXФXXXXФ and ФXXXXФXXФ, where Ф = Trp, Phe or Tyr, and X = any amino acid) to facilitate their interaction with caveolin and reside within caveolae membranes [45]. Similar with a previous report [46], by using protein motif analysis we detected a sequence of caveolin binding motif (FAFAAALFPIF) between amino acids 503-510 in human FSHR. However, the exact role of this motif remains to be confirmed in the future. Collectively, these findings may expand our current understanding of molecular mechanisms of FSH not only in the cardiovascular system but also in the female reproductive system, since Caveolin-1 is abundantly expressed in ovaries [47]. Indeed, it was shown that Caveolin-1 mRNA expression in granulosa cells varies during different follicular developmental stages [48]. We, therefore, suggest that the increased Caveolin-1 expression is important for FSHR signaling in controlling follicular development.
Although the present study provided the first experimental evidence that FSH acted directly on vascular endothelial cells and altered their function to promote atherosclerosis development, several limitations should be addressed. First, endothelial cell-specific FSHR/ApoE double knockout mouse should be generated and would be the better animal model to observe the development and progression of atherotic plaque in the absence of endothelial FSHR. Secondly, the relationship between serum FSH and the level of VCAM-1 or other atherosclerotic markers should be investigated in postmenopausal women with established atherosclerosis. Finally, although a direct interaction of FSHR and caveolin-1 was found, the exact binding site or motif and molecular binding dynamics between these two molecules should be investigated to expand our understanding of the cellular mechanism of FSH in a variety of cells or tissues.
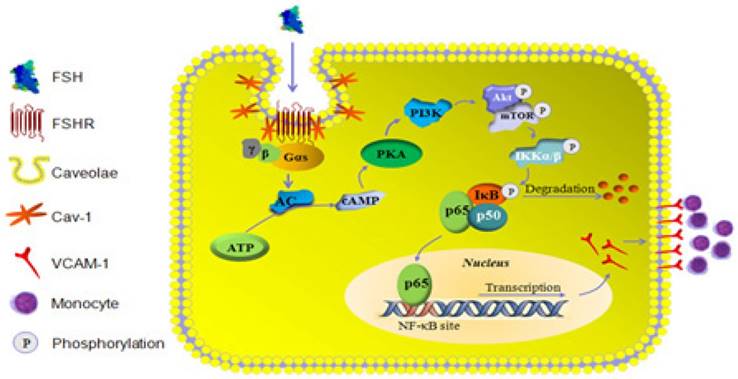
In conclusion, the present study revealed that FSHR was located within caveolae of HUVECs and interacted with caveolin-1. This interaction facilitated activation of GαS by FSHR and the downstream AC/cAMP/PAK cascade, which then enhanced PI3K/Akt/mTORC2 activity to stimulate NF-κB nuclear translocation and VCAM-1 gene transcription. Increased levels of VCAM-1 promoted monocyte adhesion to HUVECs, which is likely the reason for the development of AS (Schema Fig. 10). Thus, our findings provide the first experimental evidence that FSH directly influences CVD and interference in FSHR or its signaling may be an alternative way for the prevention of CVD in postmenopausal women. These observations suggest new avenues to explore the effects and molecular mechanisms of FSH in different cell types in cardiovascular system.
Schema illustration of the signalling involved in FSH-induced signalling activation and VCAM-1 expression.
Materials and Methods
Participants
Participants in this study included 48 healthy young women, aged 23-26 years with regular menstrual cycle (cycle length 28-30 days, menses length 3-5 days) recruited from Guangzhou Medical University. Fasting serum samples were collected at follicular phase (Day 8-10). Fifteen healthy postmenopausal women aged 52-59 years (at least 3 years since menopause, without hormone replacement therapy at least 3 months) were recruited from the Municipal First People's Hospital of Guangzhou. Exclusion criteria included current use of oral contraceptives or other medications. Further details on exclusion criteria have been reported [49]. The Guangzhou Medical University Review Board approved the study. FSH concentration was measured by an independent Laboratory of Kingmed Medical Diagnostic Center (Guangzhou, China) using the DPC Immulite2000 analyzer (Siemens Medical Solutions Diagnostics, IL, USA).
Animals
Six-week-old homozygous ApoE-/- female mice in C57BL/6 background were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and raised in the Experimental Animal Center of Guangzhou Medical University. Mice were kept under specific pathogen-free and temperature-controlled conditions on a 12 h light/dark cycle and fed ad libitum an atherogenic diet containing 1.25% (w/w) cholesterol (Guangdong Medical Animal Experimental Center, Guangzhou, China) and drinking water. At 8 weeks of age, female mice were anesthetized and sham-operated (SHAM) or bilaterally ovariectomized (OVX) through a 1 cm dorsal incision. After surgery, mice were allowed to recover for 1 week, at which point pellets containing 17β-estradiol (E2, 0.25 mg, 60 day release, Innovative Research of America, FL, USA) were implanted subcutaneously using a trochar. The control pellets (Innovative Research of America) were implanted into those without E2 treatment. Mice were randomly divided into 4 groups (6 to 9 mice per group) as follows: Sham, OVX, OVX + E2, and OVX + E2 + FSH. Recombinant FSH (Gonal-F, Serono, Sweden) was dissolved in physiologic saline and administerd into mice by subcutaneous injection daily. The dose of FSH (3 IU/day) was established according to several reports [50-52]. Mice without FSH treatment were injected daily with saline. After treatment mice were killed by exsanguination under anesthesia and blood samples were collected. FSH and E2 concentrations were measured by the Laboratory of Kingmed Medical Diagnostic Center. All procedures were performed in accordance with the guidelines for animal welfare of Guangzhou Medical University.
Assessment of aortic atherosclerotic lesions
Aortic atherosclerotic lesions were accessed as previously described [53, 54]. In brief, after 8 weeks of treatment, mice were killed by exsanguination under anesthesia. The heart and arterial tree were dissected carefully and were incubated with phosphate-buffered saline (PBS, 0.01 mol/L, pH 7.4) and fixed in 4% formalin. Tissues were embedded in ornithine carbamoyltransferase and serially sectioned at 6 μm using a Leica cryostat (Leica Microsystems, Wetzlar, Germany). To quantify the dimensions of atherosclerotic lesions, 10-12 sections with tricuspid valves were stained with oil-red O and counterstained with hematoxylin. Images were captured and lesion areas were quantified with Leica Qwin imaging software (Leica Microsystems).
Materials
FSH (#F4021), 17β-estradiol (E2, #E2758), TNF-α(#T0157), pertussis toxin (PTX, #3097), β-methyl cyclodextrin (β-MCD, #C-4555), and KU0063794 (#SML0382) were purchased from Sigma Aldrich (St. Louis, MO, USA). 4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo (3,4-d) pyrimidine (PP2, #1407), pyrrolidine dithiocarbamate (PDTC, #0727), PD98059 (#1213), NF499 (#1391), forskolin (#1099), MDL12330A(#1436) were obtained from Tocris Bioscience (Minneapolis, MN, USA). LY294002 (LY, #9901s) and H89 (#9844s) were acquired from Cell Signaling Technologies (CST, Beverly, MA, USA). Endothelial Basal Medium-2 (EBM-2) (#190860) was purchased from Lonza (Walkersville, MD, USA). Opti-MEM (#31985) and fetal bovine serum (FBS, #12484) were obtained from Invitrogen (Carlsbad, CA, USA). All other chemicals were of analytical grade and purchased from Guangzhou Chemical Reagents (Guangzhou, China).
Elisa for VCAM-1
The concentration of VCAM-1 in the plasma from participants was estimated by ELISA according to the manufacturer's protocol (Sigma Aldrich, # ab201278). Briefly, 50 μL of standard cytokine or serum and 50 μL of Antibody Cocktail were added to each well and incubated for 60 min at room temperature. The wells were then incubated for 10 min with 100 μL of TMB Substrate. After incubation, 100 μL of stop solution was added to terminate the reaction. The optical density was determined using a microplate reader at 450 nm within 30 min. A standard curve for each cytokine was generated. Based on this standard curve, linear regression analysis was performed using the Microcal Origin 6.0 software.
Cell cultures and treatments
Human umbilical vein endothelial cells (HUVECs) were cultured as previously described [55]. Human KGN granulosa cell line cells were obtained from China Center for Type Culture Collection. Before various treatments, HUVECs were kept for 24 h in EBM-2 containing steroid-deprived FBS. Whenever an inhibitor was used, the compound was added 30 min before starting the treatments. The control cells received the same volume of phosphate buffer saline (solvent for FSH).
cAMP level measurement
The level of cAMP was estimated by ELISA (ENZO Life Sciences, NY, USA. #ADI-900-066). Briefly, 50 μL of Neutralizing Reagent was added to each well before 100 μL of standard cytokine or culture supernatant was added. After 2 h incubation, each well was incubated for 60 min with 200 μL of pNpp Substrate solution at room temperature. Finally, 50 μL of stop solution was added to terminate the reaction. The optical density was determined using a microplate reader at 450 nm. The standard curve was generated and linear regression analysis was performed.
PKA activity assay
The PKA Kinase Activity Assay is based on ELISA that utilizes a specific synthetic peptide as a substrate for PKA and a polyclonal antibody that recognizes the phosphorylated form of the substrate (ENZO Life Sciences, #ADI-EKS-390A). Briefly, each well of PKA Substrate Microtiter Plate was soaked with 50 μL Kinase Assay Dilution Buffer. Subsequently, 10 μL of ATP solution was added followed by 30 μL of standard cytokine or culture supernatant and incubated for 90 min at 37 °C. Each well was then sequentially incubated with 40 μL of the Phosphospecific Substrate Antibody, 40 μL of the Anti-Rabbit IgG-HRP Conjugate and 60 μL of the TMB Substrate. Finally, 20 μL of stop solution was added to each well to terminate the reaction. The optical density was determined using a microplate reader at 450 nm. The standard curve was generated and linear regression analysis was performed.
Transfection experiments
Scrambled control siRNA (#SR30004) and siRNAs for caveolin-1 (#SR300603), raptor (#SR311531), rictor (#SR316744), Akt (#SR300143), p65 (#SR304030), and FSHR (#SR301661) were purchased from Origene (Rockville, MD, USA). Wide type p85 α (WT p85α) or dominant-negative p85α (△p85α) constructs were obtained from the Guthrie cDNA Resource Center (www.cdna.org) and were transfected into HUVECs using Lipofectamine (Invitrogen, #10349361) according to the manufacturer's protocol. Cells (40% confluent) were serum-starved for 1 h followed by incubation with siRNA or plasmid for 6 h in serum-free media. The serum-containing media was then added (2% serum final concentration) for 42 h before experiments and/or functional assays were conducted. Target protein silencing or overexpression was assessed through protein analysis up to 48 h after transfection.
Luciferase assay
HUVECs were transiently transfected with 750 ng of NF-κB-responsive reporter plasmid (#219078, Stratagene, La Jolla, CA) and 250 ng pRL-TK vector (#E2241, Promega, Madison, WI, USA) expressing renella luciferase using Lipofectamine LTX reagent (Thermo Fisher Scientific, Waltham, MA) for 6 h at 37°C. After transfection, cells were incubated (> 80% confluent) and treated as indicated and luciferase activity was measured using the dual-luciferase assay kit (Promega) according to the provided protocol. Relative luminescent units (RLU) were examined by using the GloMax 96 microplate luminometer (Promega). Data were normalized to fold change from control cells treated with solvent.
Real-time PCR and Quantitative Real-time PCR Analysis
Total RNA was isolated from HUVECs or granulosa cells by using RNeasy mini kit. Real-time PCR was performed using the RT system according to the manufacturer's instructions. The PCR program was as follows: hold at 95 °C for 5 min followed by at 95°C for 45 s, at 60°C for 45 s and at 72 °C for 1 min for 40 cycles. The specific sense and antisense primers for human FSHR included 5'-tcaggctaggggtcagagat-3' and 5'-gctcaccttcatgtagctgc-3'; the expected size of FSHR fragments was 631 bp. The specific sense and antisense primers for human GAPDH included 5'-gtcagtggtggacctgacct-3' and 5'-tgctgtagccaaattcgttg-3'; the expected size of GAPDH fragments was 245 bp. A 20 μL aliquot of RT-PCR product was loaded into 1% agarose gel.
The primers of human VCAM-1, ICAM-1, E-Selectin genes are listed in Table 1. Quantitative Real-time one-step RT-PCR was carried out on an ABI 7500 (Applied Biosystems) using standard 25 μL Universal PCR Master Mix on 1-2 μg total RNA. Reaction conditions were 95 °C for 90 s followed by 38 cycles at 95 °C for 10 s and at 60 °C for 20 s with an annealing temperature of 55 °C for 45 s. No-template control was included in each assay. β-actin was used as an endogenous control and vehicle control was used as a calibrator. Each sample was run in triplicate. The comparative threshold cycle method was used to calculate the relative changes in the expression of these genes. The relative change of gene expression was calculated using the following formula: fold change in gene expression, 2-△△Ct = 2-[△Ct (treatment samples) -△Ct (vehicle control)], where △Ct = Ct (specific gene) - Ct (β-actin) and Ct represents the threshold cycle number.
The primers of human VCAM-1, ICAM-1, E-Selectin genes
| Gene | Accession Number | Primers | Size(bp) |
|---|---|---|---|
| VCAM-1 | NM_001078.3 | F:5'-CCCTTGACCGGCTGGAGATT-3' R:5'-CTGGGGGCAACATTGACATAAAGTG-3' | 241 |
| ICAM-1 | NM_000201.2 | F: 5'-TCTTCCTCGGCCTTCCCATA-3' R: 5'-AGGTACCATGGCCCCAAATG-3' | 152 |
| E-selectin | NM_000450.2 | F: 5'-GGCAGTTCCGGGAAAGATCA-3' R: 5'-CCTTCAGGACAGGCGAACTT-3' | 81 |
| β-Actin | NM_001101.3 | F:5'-AGCACAGAGCCTCGCCTTTG-3' R:5'-ACATGCCGGAGCCGTTGT-3' | 107 |
Immunoblottings
After various treatments, HUVECs were washed once with ice-cold PBS before addition of the lysis buffer (100 mM Tris-HCl, pH=6.8, 4% SDS, 20% glycerol, 1 mM sodium orthovanadate, 1 mM NaF and 1 mM phenylmethylsulfonylfluoride, PMSF) to cell-culture dish on ice. Subsequently, cell lysates were scraped, boiled, and centrifuged for 2 min at 13000 rpm and separated by SDS-PAGE. Antibodies used were: FSHR (Abcam, #ab75200), VCAM-1 (Abcam, #ab134047), ICAM-1 (CST, #4915s), E-selectin (Sigma, #S9555), P-Akt (CST, #4060s), Akt (CST, #4691s), P-p65 (CST, #3033s), p65 (CST, #8242s), P-mTOR (CST, #5536p), mTOR (CST, #3983p), P-rictor (CST, #3806s), rictor (CST, #2114p), P-raptor (CST, #2083s), raptor (CST, #2280p), IKKα (Abcam, #ab32041), IKKβ (CST, #8943), IKKγ (Abcam, #ab178872), P-IKKα (Abcam, #ab38515), P-IKKβ (Abcam, #ab59195), P-IKKγ (CST, #2689), IκBα (CST, #4814), P-IκBα (CST, #2859), caveolin-1 (CST, #3267s), β-actin (Santa Cruz, #sc-81178). Primary and secondary antibodies were incubated with the membranes using the standard technique. Immunodetection was accomplished using enhanced chemiluminescence.
Immunoprecipitation assay
HUVECs were harvested in 100 mM Tris-HCl (pH 6.8), 4% SDS, 20% glycerol, 1 mM Na3VO4, 1 mM NaF, and 1 mM PMSF. Equal amounts (700 μg) of cell lysates were incubated with 1 μg of precipitating anti-caveolin-1 antibody for 1 h at 4°C under gentle agitation. 25 μL of protein A-agarose slurry (Cell Signaling) was added, and the samples were rotated and incubated at 4°C for another hour. The samples were then pelleted, washed, and resuspended in 50 μL of 2×Laemmli buffer for immunoblotting.
GαS activity assay
GαS Activation Assay Kit (NewEast Biosciences, PA, USA, #80801) is based on the monoclonal antibody specifically recognizing the active GTP-bound GαS proteins. Briefly, HUVECs were harvested in assay/lysis buffer and cell lysates were incubated with 1 μL anti-active GαS monoclonal antibody and 20 μL of protein A-agarose slurry. The samples were then pelleted, washed, and resuspended in 50 μL of 2×Laemmli buffer for immunoblotting by using anti- GαS monoclonal antibody.
Reactive oxygen species production measurement
2′,7′-Dichlorodihydrofluorescein diacetate (H2DCFDA, #C6827, C6827, Invitrogen, Carlsbad, CA) was used to detect the production of reactive oxygen species. Briefly, cells were seeded in 35 mm dishes and cultured. After treatment, cells were washed and loaded with H2DCFDA (10 μM in phenol-red-free DMEM medium) at 37 °C for 30 min in the dark. Cells were then washed and subjected to fluorescence measurement by flow cytometry on a FACSCalibur flow cytometer using a 488 nm argon excitation laser.
MDA, SOD, GSH-PX measurement
The levels of MDA (malonaldehyde), SOD (superoxide dismutase) and GSH-PX (glutathione peroxidase) were measured according to the manufacturer instructions. MDA kit (#A003-2), SOD kit (#A001-3) and GSH-PX kit (#A005) were all purchased from Nanjing Jiancheng Bioengineering institute (Nanjing, China) .
Immunofluorescence
Histopathologic slides of aortic roots or cultured HUVECs were incubated with antibody against VCAM-1 (Invitrogen, #MA5-11447), FSHR (Abcam, #ab75200), caveolin-1 (CST, #3267s), p65 (CST, #8242s), specific marker antibodies for macrophages (anti-CD68, Abcam, #ab955) and FITC-conjugated secondary antibody. Nuclei were counterstained with 4'-6-diamidino-2-phenylindole (DAPI) (Sigma). The coverslips were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Immunofluorescence was visualized by using an Olympus BX41 microscope (Olympus Corporation, Japan) or laser scanning confocal microscopy (ZEISS LSM 710, ZEISS, Germany) and pictures were photographed.
Cell adhesion assay
Cell adhesion assay was performed according to the previously described procedure [56]. HUVECs were seeded on 0.1% gelatin-coated cover slides and were subjected to the indicated treatments. VCAM-1 neutralizing antibody (Abcam, #ab134047, 1:200) was added 30 min before FSH treatment. Human monocytic U937 cells (ATCC CRL-1593.2; Manassas, Virginia, USA) were incubated with CellTracker Orange CMTMR fluorescent dye (0.25 μL/mL; Molecular Probes; Life Technologies) in RPMI-1640 for 30 min, centrifuged and resuspended in RPMI-1640 containing 10% FBS culture medium. Subsequently, fluorescently-labelled U937 cells (1000 cells/mL) were seeded over HUVECs and allowed to adhere for 1 h. The non-adherent cells were washed away and the adherent fluorescent cells were counted under a fluorescence microscope and expressed relative to the total number of cells per field.
Flow chamber adhesion assay
Flow Chamber Adhesion Assay was performed as previously described [57, 58]. Briefly, the chamber consisted of a glass slide with a confluent HUVEC monolayer that was attached to a polycarbonate base. After treatment as indicated, flow across the monolayer was controlled with a syringe pump. U937 monocytoid cell suspension (2×106 cell/mL) in HBSS was perfused through the flow chamber at the speed of 8 µL/min (which equals a shear stress of 0.2 dynes/cm2). Experiments were videotaped using a color camera mounted on an inverted microscope. The velocity of rolling U937 cells adhensive to HUVECs were recorded and calculated by Image-pro-plus software.
Caveolae/Rafts isolation
Caveolae/Rafts isolation kit (#CS0750) was purchased from Sigma and was used according to manul of the kit and protocols reported by our group and others [59, 60]. Cells were homogenized in 1 mL of lysis buffer (10 mM Tris-HCl, pH=7.4, 1 mM EDTA, 200 mM sucrose and proteases inhibitors mix). Nuclei and cellular debris were removed by centrifugation and discarded. The resulting supernatant was centrifuged at 110,000 × g for 75 min at 4 °C to obtain crude membrane pellet which was solubilized in 1 mL of Lysis buffer containing 1% Triton X-100 and agitated end to end for 30 min at 4 °C. The sample was mixed with OptiPrep to a final concentration of 35%. The density gradient was made of 5 layers of OptiPrep with 35%, 30%, 25%, 20%, and 0%. The volume of each layer was 2 mL except for 1 mL of 0% OptiPrep. Samples were centrifuged at 200,000 × g for 4 h at 4 °C. After centrifugation, 9 fractions of 1 ml from the top to bottom of the tube were collected. Caveolin-1 highly concentrated fractions were the major caveolae-like plasma membrane microdomains under our experimental conditions. Equal volumes (30 μL) of each fraction were analyzed directly by Western blotting.
Statistical Analysis
All statistical analyses were performed with the Statistical Package for Social Sciences version 11.0 (SPSS Inc., Chicago, IL, USA). Data are presented as mean ± SD, representing at least 3 independent experiments. Statistical comparisons were made using Student's t test or one-way analysis of variance followed by a post hoc analysis (Tukey test) where applicable to find means that are significantly different from each other and the significance level was set at P < 0.05. Correlation analysis was also performed between variables and Spearman rank correlation coefficients were calculated.
Abbreviations
AS: atherosclerosis; FSH: follicular stimulating hormone; HUVECs: human umbilical vascular endothelial cells; VCAM-1: vascular cell adhesion molecule-1; FSHR: FSH receptor; TNF-α: tumor necrosis factor; NF-κB: nuclear factor κB; PI3K: phosphatidylinositol 3-kinase; mTOR: mammalian target of rapamycin; PKA: protein kinase A; CVD: cardiovascular disease; E2: 17β-estradiol; OVX: ovariectomized; IKK: IkB kinase; ROS: reactive oxygen species; MDA: malonaldehyde; SOD: superoxide dismutase; GSH-PX: glutathione peroxidase; NAC: N-Acetyl-L-cysteine; Vit C: Vitamin C (Vit C); GSH: glutathione; cAMP: cyclic adenosine monophosphate; β-MCD: methyl-β-cyclodextrin; Cav-1: caveolin-1; GPCR: G protein-coupled receptor.
Supplementary Material
Table S1 shows a complete table with the parameters including height, weight, FSH, LH, E2 and progesterone levels of each participant.
Supplementary videos show the monocyte rolling and adhesion onto HUVECs in real time by a flow chamber microscope. Video 1: control group. HUVECs were treated with vehicle.
Video 2: FSH group. HUVECs were treated with FSH (50 mIU/mL) for 24 h.
Video 3: TNF-α group. HUVECs were treated with TNF-α (0.1 ng/mL) for 24 h. After treatment, U937 monocytoid cell suspension was perfused and videotaped.
Acknowledgements
This work was supported by the National Natural Science Foundation (To X.D. Fu., No. 81270669; No: 81471426), the Project of Department of Education of Guangdong Province (To X.D. Fu., No. 2015KTSCX109), the National Funds of Developing Local Colleges and Universities (To. X.D. Fu., No. B16056001), the Science and Technology Planning Project of Guangdong Province (To Z.T. Gu., No. 2014J4100113), and the Medical Scientific Research Foundation of Guangdong Province (To. Y.C., No. A2015058). We thank Prof. Jianhua Wu (Institute of Biomechanics, South China University of Technology, Guangzhou, China) for providing us the flow chamber equipment.
Author Contributions
X.D.F. initiated and supervised the study and wrote the manuscript. X.S.L, W.Y.C., and P. L. performed all the experiments and collected the data. J.Z.W. performed some Western blotting experiments. Y.C. provided plasma samples, cultured HUVECs and contributed reagents. P.L., Q.Y., and X.Y.X. performed animal experiments. Y.H.C. performed PCR and RT-PCR experiments. Z.T.G. contributed reagents and analyzed the data. S.T revised the manuscript. All coauthors read and commented on successive drafts of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M. et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38-360
2. Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801-11
3. Harman SM. Menopausal hormone treatment cardiovascular disease: another look at an unresolved conundrum. Fertil Steril. 2014;101:887-97
4. Genazzani AR, Simoncini T. Pharmacotherapy: Benefits of menopausal hormone therapy-timing is key. Nat Rev Endocrinol. 2013;9:5-6
5. Randolph JF Jr, Sowers M, Bondarenko IV, Harlow SD, Luborsky JL, Little RJ. Change in estradiol and follicle-stimulating hormone across the early menopausal transition: effects of ethnicity and age. J Clin Endocrinol Metab. 2004;89:1555-61
6. Meduri G, Bachelot A, Cocca MP, Vasseur C, Rodien P, Kuttenn F. et al. Molecular pathology of the FSH receptor: new insights into FSH physiology. Mol Cell Endocrinol. 2008;282:130-42
7. Sun L, Peng Y, Sharrow AC, Iqbal J, Zhang Z, Papachristou DJ. et al. FSH directly regulates bone mass. Cell. 2006;125:247-60
8. Mancinelli R, Onori P, Gaudio E, DeMorrow S, Franchitto A, Francis H. et al. Follicle-stimulating hormone increases cholangiocyte proliferation by an autocrine mechanism via cAMP-dependent phosphorylation of ERK1/2 and Elk-1. Am J Physiol Gastrointest Liver Physiol. 2009;297:G11-26
9. Sanchez AM, Flamini MI, Russo E, Casarosa E, Pacini S, Petrini M. et al. LH and FSH promote migration and invasion properties of a breast cancer cell line through regulatory actions on the actin cytoskeleton. Mol Cell Endocrinol. 2016;437:22-34
10. Radu A, Pichon C, Camparo P, Antoine M, Allory Y, Couvelard A. et al. Expression of follicle-stimulating hormone receptor in tumor blood vessels. N Engl J Med. 2010;363:1621-30
11. Stilley JA, Guan R, Duffy DM, Segaloff DL. Signaling through FSH receptors on human umbilical vein endothelial cells promotes angiogenesis. J Clin Endocrinol Metab. 2013;99:E813-20
12. Vanhoutte PM. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circ J. 2009;73:595-601
13. Cook-Mills JM, Marchese ME, Abdala-Valencia H. Vascular cell adhesion molecule-1 expression and signaling during disease: regulation by reactive oxygen species and antioxidants. Antioxid Redox Signal. 2011;15:1607-38
14. Iiyama K, Hajra L, Iiyama M, Li H, DiChiara M, Medoff BD. et al. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ Res. 1999;85:199-207
15. Huo Y, Hafezi-Moghadam A, Ley K. Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circ Res. 2000;87:153-9
16. Souter I, Huang A, Martinez-Maza O, Breen EC, Decherney AH, Chaudhuri G. et al. Serum levels of soluble vascular cell adhesion molecule-1, tumor necrosis factor-alpha, and interleukin-6 in in vitro fertilization cycles. Fertil Steril. 2009;91:2012-9
17. Kasum M, Danolic D, Oreskovic S, Jezek D, Beketic-Oreskovic L, Pekez M. Thrombosis following ovarian hyperstimulation syndrome. Gynecol Endocrinol. 2014;30:764-8
18. Mosevoll KA, Lindas R, Tvedt TH, Bruserud O, Reikvam H. Altered plasma levels of cytokines, soluble adhesion molecules and matrix metalloproteases in venous thrombosis. Thromb Res. 2015;136:30-9
19. Chiu GN, Bally MB, Mayer LD. Targeting of antibody conjugated, phosphatidylserine-containing liposomes to vascular cell adhesion molecule 1 for controlled thrombogenesis. Biochim Biophys Acta. 2003;1613:115-21
20. Bonello N, Norman RJ. Soluble adhesion molecules in serum throughout the menstrual cycle. Hum Reprod. 2002;17:2272-8
21. Solerte SB, Fioravanti M, Spinillo A, Ferrari E, Guaschino S. Association between hormonal and haemorheological changes during the menstrual cycle in healthy women. Br J Obstet Gynaecol. 1988;95:1305-8
22. Simoncini T, Maffei S, Basta G, Barsacchi G, Genazzani AR, Liao JK. et al. Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ Res. 2000;87:19-25
23. Simoncini T, Mannella P, Fornari L, Caruso A, Willis MY, Garibaldi S. et al. Differential signal transduction of progesterone and medroxyprogesterone acetate in human endothelial cells. Endocrinology. 2004;145:5745-56
24. Lukas-Croisier C, Lasala C, Nicaud J, Bedecarras P, Kumar TR, Dutertre M. et al. Follicle-stimulating hormone increases testicular Anti-Mullerian hormone (AMH) production through sertoli cell proliferation and a nonclassical cyclic adenosine 5'-monophosphate-mediated activation of the AMH Gene. Mol Endocrinol. 2003;17:550-61
25. Chaudhari N, Talwar P, Parimisetty A, Lefebvre d'Hellencourt C, Ravanan P. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci. 2014;8:213
26. Shen M, Jiang Y, Guan Z, Cao Y, Li L, Liu H. et al. Protective mechanism of FSH against oxidative damage in mouse ovarian granulosa cells by repressing autophagy. Autophagy. 2017:1-22
27. Hassanian SM, Dinarvand P, Smith SA, Rezaie AR. Inorganic polyphosphate elicits pro-inflammatory responses through activation of the mammalian target of rapamycin complexes 1 and 2 in vascular endothelial cells. J Thromb Haemost. 2015;13:860-71
28. Parra V, Verdejo HE, Iglewski M, Del Campo A, Troncoso R, Jones D. et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFkappaB-Opa-1 signaling pathway. Diabetes. 2014;63:75-88
29. Furgeson SB, Simpson PA, Park I, Vanputten V, Horita H, Kontos CD. et al. Inactivation of the tumour suppressor, PTEN, in smooth muscle promotes a pro-inflammatory phenotype and enhances neointima formation. Cardiovasc Res. 2010;86:274-82
30. Riera MF, Regueira M, Galardo MN, Pellizzari EH, Meroni SB, Cigorraga SB. Signal transduction pathways in FSH regulation of rat Sertoli cell proliferation. Am J Physiol Endocrinol Metab. 2012;302:E914-23
31. Alam H, Weck J, Maizels E, Park Y, Lee EJ, Ashcroft M. et al. Role of the phosphatidylinositol-3-kinase and extracellular regulated kinase pathways in the induction of hypoxia-inducible factor (HIF)-1 activity and the HIF-1 target vascular endothelial growth factor in ovarian granulosa cells in response to follicle-stimulating hormone. Endocrinology. 2009;150:915-28
32. Minhajuddin M, Bijli KM, Fazal F, Sassano A, Nakayama KI, Hay N. et al. Protein kinase C-delta and phosphatidylinositol 3-kinase/Akt activate mammalian target of rapamycin to modulate NF-kappaB activation and intercellular adhesion molecule-1 (ICAM-1) expression in endothelial cells. J Biol Chem. 2009;284:4052-61
33. Xu Y, Lai E, Liu J, Lin J, Yang C, Jia C. et al. IKK interacts with rictor and regulates mTORC2. Cell Signal. 2013;25:2239-45
34. Wang C, Qin L, Manes TD, Kirkiles-Smith NC, Tellides G, Pober JS. Rapamycin antagonizes TNF induction of VCAM-1 on endothelial cells by inhibiting mTORC2. J Exp Med. 2014;211:395-404
35. Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195-203
36. Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ. et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527-32
37. Torella D, Gasparri C, Ellison GM, Curcio A, Leone A, Vicinanza C. et al. Differential regulation of vascular smooth muscle and endothelial cell proliferation in vitro and in vivo by cAMP/PKA-activated p85alphaPI3K. Am J Physiol Heart Circ Physiol. 2009;297:H2015-25
38. Xiong Y, Zhou Y, Jarrett HW. Dystrophin glycoprotein complex-associated Gbetagamma subunits activate phosphatidylinositol-3-kinase/Akt signaling in skeletal muscle in a laminin-dependent manner. J Cell Physiol. 2009;219:402-14
39. Kurosu H, Maehama T, Okada T, Yamamoto T, Hoshino S, Fukui Y. et al. Heterodimeric phosphoinositide 3-kinase consisting of p85 and p110beta is synergistically activated by the betagamma subunits of G proteins and phosphotyrosyl peptide. J Biol Chem. 1997;272:24252-6
40. Law NC, White MF, Hunzicker-Dunn ME. G protein-coupled receptors (GPCRs) That Signal via Protein Kinase A (PKA) Cross-talk at Insulin Receptor Substrate 1 (IRS1) to Activate the phosphatidylinositol 3-kinase (PI3K)/AKT Pathway. J Biol Chem. 2016;291:27160-9
41. Bellis A, Castaldo D, Trimarco V, Monti MG, Chivasso P, Sadoshima J. et al. Cross-talk between PKA and Akt protects endothelial cells from apoptosis in the late ischemic preconditioning. Arterioscler Thromb Vasc Biol. 2009;29:1207-12
42. Mineo C, Shaul PW. Regulation of eNOS in caveolae. Adv Exp Med Biol. 2012;729:51-62
43. Miralles G, Cohen BD. Analyzing the Role of Caveolin in Human Follicle Stimulating Hormone Receptor Regulation. GPCRs, Growth Factors, Tyrosine Kinases, Inhibits, Activins, and TGF Beta Superfamily (posters): Endocrine Society. 2014 p. SAT-112-SAT-
44. Chini B, Parenti M. G-protein coupled receptors in lipid rafts and caveolae: how, when and why do they go there? J Mol Endocrinol. 2004;32:325-38
45. Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem. 1997;272:6525-33
46. Pereira J, Cohen BD. Interaction of Human Follicle Stimulating Hormone Receptor with Caveolin-1. Female Reproductive Endocrinology: Endocrine Society. 2016 p. SAT-0007-SAT-
47. Bullejos M, Bowles J, Koopman P. Extensive vascularization of developing mouse ovaries revealed by caveolin-1 expression. Dev Dyn. 2002;225:95-9
48. Diouf MN, Lefebvre R, Silversides DW, Sirois J, Lussier JG. Induction of alpha-caveolin-1 (alphaCAV1) expression in bovine granulosa cells in response to an ovulatory dose of human chorionic gonadotropin. Mol Reprod Dev. 2006;73:1353-60
49. Wactawski-Wende J, Schisterman EF, Hovey KM, Howards PP, Browne RW, Hediger M. et al. BioCycle study: design of the longitudinal study of the oxidative stress and hormone variation during the menstrual cycle. Paediatr Perinat Epidemiol. 2009;23:171-84
50. Singh J, Handelsman DJ. The effects of recombinant FSH on testosterone-induced spermatogenesis in gonadotrophin-deficient (hpg) mice. J Androl. 1996;17:382-93
51. Shaikh A, Bhartiya D, Kapoor S, Nimkar H. Delineating the effects of 5-fluorouracil and follicle-stimulating hormone on mouse bone marrow stem/progenitor cells. Stem Cell Res Ther. 2016;7:59
52. Ritter V, Thuering B, Saint Mezard P, Luong-Nguyen NH, Seltenmeyer Y, Junker U. et al. Follicle-stimulating hormone does not impact male bone mass in vivo or human male osteoclasts in vitro. Calcif Tissue Int. 2008;82:383-91
53. Zhou K, Gao Q, Zheng S, Pan S, Li P, Suo K. et al. 17beta-estradiol induces vasorelaxation by stimulating endothelial hydrogen sulfide release. Mol Hum Reprod. 2013;19:169-76
54. Li P, Wei J, Li X, Cheng Y, Chen W, Cui Y. et al. 17beta-estradiol enhances vascular endothelial Ets-1/miR-126-3p expression: the possible mechanism for attenuation of atherosclerosis. J Clin Endocrinol Metab. 2016 jc20162974
55. Fu XD, Flamini M, Sanchez AM, Goglia L, Giretti MS, Genazzani AR. et al. Progestogens regulate endothelial actin cytoskeleton and cell movement via the actin-binding protein moesin. Mol Hum Reprod. 2008;14:225-34
56. Zhang Y, Li P, Gao Q, Simoncini T, Fu X. 2-Methoxyestradiol prevents monocyte adhesion to vascular endothelial cells via downregulation of VCAM-1 expression. Gynecol Endocrinol. 2016:1-6
57. Ito F, Mori T, Takaoka O, Tanaka Y, Koshiba A, Tatsumi H. et al. Effects of drospirenone on adhesion molecule expression and monocyte adherence in human endothelial cells. Eur J Obstet Gynecol Reprod Biol. 2016;201:113-7
58. Li Q, Wayman A, Lin J, Fang Y, Zhu C, Wu J. Flow-Enhanced Stability of Rolling Adhesion through E-Selectin. Biophys J. 2016;111:686-99
59. Liu P, Li X, Song F, Li P, Wei J, Yan Q. et al. Testosterone promotes tube formation of endothelial cells isolated from veins via activation of Smad1 protein. Mol Cell Endocrinol. 2017
60. Sen S, Roy K, Mukherjee S, Mukhopadhyay R, Roy S. Restoration of IFNgammaR subunit assembly, IFNgamma signaling and parasite clearance in Leishmania donovani infected macrophages: role of membrane cholesterol. PLoS Pathog. 2011;7:e1002229
Author contact
![]() Corresponding author: Xiaodong Fu., M.D., Ph.D., Tel: +86.020.83412002, E-mail: fuxiaodgzhmu.edu.cn
Corresponding author: Xiaodong Fu., M.D., Ph.D., Tel: +86.020.83412002, E-mail: fuxiaodgzhmu.edu.cn