Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(19):4658-4670. doi:10.7150/thno.20582 This issue Cite
Research Paper
Material-induced Senescence (MIS): Fluidity Induces Senescent Type Cell Death of Lung Cancer Cells via Insulin-Like Growth Factor Binding Protein 5
Sharmy Saimon Mano1, Koichiro Uto2, Mitsuhiro Ebara1, 3, 4 ![]()
1. International Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan;
2. International Center for Young Scientist (ICYS), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan;
3. Graduate School of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8577, Japan;
4. Graduate School of Tokyo University of Science, 6-3-1 Niijuku, Katsushika-ku, Tokyo 125-8585, Japan.
Received 2017-4-15; Accepted 2017-8-11; Published 2017-10-17
Abstract

Objective: We propose here material-induced senescence (MIS) as a new therapeutic concept that limits cancer progression by stable cell cycle arrest. This study examined for the first time the effect of material fluidity on cellular senescence in lung carcinoma using poly(ε-caprolactone-co-D, L-lactide) (P(CL-co-DLLA)) with tunable elasticity and fluidity.
Methods: The fluidity was varied by chemically crosslinking the polymer networks: the crosslinked P(CL-co-DLLA) shows solid-like properties with a stiffness of 260 kPa, while the non-crosslinked polymer exists in a quasi-liquid state with loss and storage moduli of 33 kPa and 11 kPa, respectively.
Results: We found that cancer cells growing on the non-crosslinked, fluidic substrate undergo a non-apoptotic form of cell death and the cell cycle was accumulated in a G0/G1 phase. Next, we investigated the expression of biomarkers that are associated with cancer pathways. The cancer cells on the fluidic substrate expressed several biomarkers associated with senescence such as insulin-like growth factor binding protein 5 (IGFBP5). This result indicates that when cancer cells sense fluidity in their surroundings, the cells express IGFBP5, which in turn triggers the expression of tumor suppressor protein 53 and initiates cell cycle arrest at the G1 phase followed by cellular senescence. Furthermore, the cancer cells on the fluidic substrate maintained their epithelial phenotype, suggesting that the cancer cells do not undergo epithelial to mesenchymal transition.
Conclusion: By considering these results as the fundamental information for MIS, our system could be applied to induce senescence in treatment-resistant cancers such as metastatic cancer or cancer stem cells.
Keywords: fluidity, p(ε-caprolactone), senescence, insulin-like growth factor binding protein 5, tumor suppressor protein 53.
Introduction
The dynamic nature of cancer and its complexity and diversity pose many challenges in its study and treatment. To overcome this, many tools and techniques were established and employed to detect and treat cancer in its early stage [1]. Due to these facts, the number of long-term cancer survivors is increasing. Yet, cancer metastasis or recurrence is the major obstacle of successful cancer therapy. Cancer metastasis constitutes the emergence of secondary tumors at distant sites from the primary tumors [2]. One mechanism contributing to the first phase of metastasis is the epithelial to mesenchymal transition (EMT). EMT is well known for its primary roles in embryogenesis during development. In EMT, the epithelial cells acquire properties of mesenchymal cells. The mesenchymal cells exhibit greater resistance to traditional therapeutics and the ability to establish secondary tumors after treatment [3]. Another reason why cancer often develops resistance to treatment is that a few cancerous cells act as stem cells. Like stem cell, they acquire self-renewal, heterogeneity, and apoptosis resistance [4]. If the therapies are not assassinating the cancer stem cells (CSCs), the tumor will shortly grow back.
Current research suggests that therapy-induced senescence (TIS) is an attractive therapeutic concept that limits cancer progression by a stable cell cycle arrest [5]. Cellular senescence is induced by a variety of stimuli including alterations of the telomere, DNA damage, oxidative stress, and activation of certain oncogenes. Recent evidence suggests that senescence may differ qualitatively depending on the stimulus that leads to its establishment. For example, radiation-induced senescence abolishes the protective immunoregulatory effect of human mesenchymal stem cells (hMSCs) in a sepsis murine model [6]. Tumor suppressor protein 53 (p53) plays a vital role in senescence, which has several anti-proliferative activities including stimulation and expression of p21 and cyclin-dependent kinase inhibitor (CDKI) [7]. A report says that p53 is an essential protein in cellular senescence to suppress tumorigenesis in the prostate cancer mouse model [8]. Abegglen et. al. reported that an elephant's cancer risk is about 2-5x lower than a human's because they have multiple copies of the p53 gene [9]. On the other hand, BRAF(V600E) expression in human melanocytes induces senescence by cell cycle arrest followed by the induction of both p16(INK4a) and beta-galactosidase (SA-beta-Gal) [10]. In recent years, therapeutic approaches such as p53 activation [11] or treatment with cyclin-dependent kinase 2 (CDK 2) inhibitors [12] have shown an effective senescence response. Therefore, senescence is considered a relevant technique for the treatment and diagnosis of treatment-resistant cancer [13].
In this study, we propose a novel strategy to induce senescence in human lung epithelial adenocarcinoma cells by mechanical stimulus of materials; hereinafter called material-induced senescence (MIS). Towards this goal, we designed a fluidic cell culture platform using poly(ε-caprolactone-co-D,L-lactide) (P(CL-co-DLLA)), which can dynamically alter the cancer cells' surroundings. The fluidity was varied by chemically crosslinking the functionalized end chains. We found that cells growing on the non-crosslinked (fluidic) P(CL-co-DLLA) substrate undergo a non-apoptotic form of cell death and the cells were accumulated in a G0/G1 phase of cell cycle. Next, we investigated the non-apoptotic form of cell death on non-crosslinked P(CL-co-DLLA) substrate. To do this, cancer cells grown on crosslinked and non-crosslinked P(CL-co-DLLA) substrates were analyzed for several biomarkers associated with the regulation of cellular processes like apoptosis, cell cycle, DNA damage and response, metabolism, epithelial to mesenchymal transition and senescence. We believe that these investigations will give crucial evidence on MIS for the next generation of cancer therapy.
Materials and Methods
Preparation of fluidic substrate
Four-branched copolymers poly(ε-caprolactone-co-D,L,lactide) (P(CL-co-DLLA) were synthesized as described in our earlier reports [14, 15]. The structure and molecular weights were determined by 1H NMR spectroscopy (JEOL, Tokyo, Japan) and gel permeation chromatography (GPC; JASCO International, Tokyo, Japan) respectively. The viscoelastic spectrum (storage modulus, G' and loss modulus, G'') of the substrate was tested as a function of frequency and temperature using a rheometer (MCR 301, Anton Paar, Tokyo, Japan). The non-crosslinked P(CL-co-DLLA) substrate for cell culture was prepared by a spin-coating technique as described in our previous report [14]. Crosslinked substrate was prepared by thermal crosslinking P(CL-co-DLLA) macromonomers as mentioned in our previous reports [14,15]. The mechanical property of the crosslinked substrate was characterized by a tensile test (EZ-S 500N; Shimadzu, Kyoto, Japan). Scanning electron microscope (SEM) images of crosslinked and non-crosslinked P(CL-co-DLLA) substrates were examined with SU-8000 (Hitachi, Japan).
Cell culture
Human lung epithelial adenocarcinoma cells (NCI-H23; CRL-10317TM, ATCC, University Boulevard, Manassas VA, USA) were cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS; ATCC), 1% antibiotic- antimycotic (anti-anti, Gibco, Grand Island, NY, USA), MEM non-essential amino acids (Gibco, Grand Island, NY, USA) and sodium pyruvate (Gibco, Grand Island, NY, USA). Human breast epithelial cells (MCF 10A; ATCC, University Boulevard, Manassas VA, USA) were cultured in DMEM/F12 medium (Invitrogen, Carlsbad, CA, USA (supplemented with 5% horse serum ((Invitrogen) along with 20 ng/mL EGF (Sigma-Aldrich, St. Louis, MO, USA), 1% hydrocortisone (Sigma-Aldrich), 100 ng/mL cholera toxin (Sigma-Aldrich), 0.02% insulin (Sigma-Aldrich) and 1% antibiotic- antimycotic. CSCs were prepared from NCI-H23 cells by culture the cells on 6 well ultralow cell adhesion plate with CSC medium (PromoCell, Sickingenstr, Heidelberg, Germany) according to the manufacturer's instruction. CSC tumor spheres were collected after 9 days of culture. All cells were maintained under humidified atmosphere of 5% CO2 at 37°C.
Immunofluorescent staining and confocal microscopy
Cells were seeded on glass coverslip or P(CL-co-DLLA) (crosslinked and non-crosslinked) substrates at a density of 1×104 cells/cm2 and incubated for required time periods. The cells were fixed in 4% paraformaldehyde (PFA; Wako Pure Chemical Industries, Tokyo, Japan) and blocked with 1% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO, USA) (in PBS) for 30 min. The IGFBP 5, E-cadherin and vimentin were stained independently with anti-IGFBP5 antibody, anti-E-cadherin and anti-vimentin respectively (Proteintech, Chicago, IL, USA), and the corresponding secondary antibody conjugated with Alexa Fluor® 488 fluorescent dye (Invitrogen, Carlsbad, CA, USA) for 1 h each. F-actin and nuclei were counterstained with tetramethylrhodamine B isothiocyanate-conjugated phalloidin (Sigma-Aldrich, St. Louis, MO, USA) and Hoechst 33258 (Sigma-Aldrich, St. Louis, MO, USA) respectively. The images were taken by Leica SP5 confocal laser scanning microscope (Leica, Wetzlar, Germany).
Cell viability assay
NCI-H23, MCF 10A and CSC cells were seeded on glass coverslip or P(CL-co-DLLA) (crosslinked and non-crosslinked) substrates of 24-well plate inserts at a density of 1×104 cells/cm2 independently. The number of cells was determined using after 48 h and 72 h culture period by cell counting kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's instructions. The percentage of cell viability was calculated from the control group (glass). For CSC, the viability of suspended cells (CSC) was calculated from the total cell population. For IGFBP5 proliferation inhibition assay, after one day incubation on glass or P(CL-co-DLLA) (crosslinked and non-crosslinked) substrates, the cells were exposed with rhIGFBP5 protein (R&D System, Minneapolis, MN, USA) at a concentration ranges from 0 ng/mL, 50 ng/mL, 100 ng/mL, and 200 ng/mL independently and continued the incubation for 1 more day. For MCF 10A cells, 200 ng/mL of rhIGFBP5 protein was used. The number of cells was determined using CCK8 and the percentage of cells were calculated from the control groups (without rhIGFBP5) in the corresponding glass or P(CL-co-DLLA) substrates.
Cell death analysis
5×105 cells/well were seeded on glass or P(CL-co-DLLA) (crosslinked and non-crosslinked) substrates in 6-well inserts and incubated. After 3 days, cells were stained by FITC Annexin V/Dead Cell Apoptosis Kit with FITC annexin V and PI for Flow Cytometry (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The percentage of apoptotic and dead cells were measured by analyzing the cells with spectral cell analyzer (LE-SP6800A, Sony, Japan).
Cell cycle analysis
The cells were seeded at a density of 5×105 cells/mL on glass or P(CL-co-DLLA) (crosslinked and non-crosslinked) substrates in 6-well plate inserts. Three days later, the cells were harvested, fixed by 1 mL of 70% ethanol (in PBS) overnight at 4°C and stained by FxCycleTM PI/RNase staining solution (Invitrogen, Carlsbad, CA, USA) for 30 min according to the manufacturer's instruction. The cells were analyzed by spectral cell analyzer (LE-SP6800A, Sony, Tokyo, Japan) and the percentage in each phase of the cell cycle was analyzed by ModFit LT4.1 software (Verity software).
Gene expression analysis
Polymerase chain reaction (PCR) array
For a polymerase chain reaction (PCR) array, 5×105 cells/mL NCI-H23 cells were seeded on crosslinked and non-crosslinked P(CL-co-DLLA) substrates for 3 days. Total cellular RNA was extracted using an RNeasy Kit (Qiagen, Germany) according to the manufacturer's instructions. Extracted RNA was purified with DNaseI (Takara, Bio Inc. Tokyo, Japan) to remove DNA contamination. 4 μg of total RNA was reverse transcribed into cDNA with a random hexamer primer using the PrimeScriptTM 1st strand cDNA synthesis kit (Takara, Bio Inc. Tokyo, Japan) according to the manufacturer's instruction. Human cancer pathway finder PCR array (PAHS-0334Z; SA Bioscience, Qiagen, Germany) was performed using an ABI PRISM 7500 Sequence Detection System (Applied Biosystem, CA, USA). The cDNA was diluted 5 times with distilled water and mixed with 2xPCR master mixes according to the manufacture's protocol. 25 μL of the reaction mixture containing the test cDNA was added to the 96 wells of the reaction plate. The thermocycling conditions were 95ºC for 10 s, followed by 40 cycles at 95ºC for 15 s, and 60ºC for 1 min.
Quantitative real-time (qRT) PCR
5×105 cells/mL of NCI-H23 cells were seeded on crosslinked and non-crosslinked P(CL-co-DLLA) substrates for mRNA expression analysis. The expression levels of marker genes were determined by quantitative real-time PCR (qRT-PCR) technique. The total cellular RNA was extracted and purified as mentioned above. 2 μg of total RNA was reverse-transcribed into cDNA using the PrimeScriptTM 1st strand cDNA Synthesis kit (Takara, Bio Inc. Tokyo, Japan). Real-time PCR was performed using an ABI PRISM 7500 Sequence Detection System (Applied Biosystem, CA, USA). The reaction mixture was composed of 10 μL of SYBR Premix Ex Taq II (Takara, Japan), 10 pmol each of the forward and reverse primers, 2 μL of cDNA, and distilled water to a final volume of 20 μL. The thermocycling conditions were 95ºC for 30 s, followed by 40 cycles at 95ºC for 5 s, and 60ºC for 34 s. The relative change in gene expression was quantified using the threshold cycle (Ct), 2-ΔΔCt method. Data were normalized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a housekeeping gene and endogenous control in the same reaction as the gene of interest. The primers used in this study were as follows (Table 1).
List of oligomers used in qRT-PCR
| Genes | Forward primer (5'-3') | Reverse primer (5'-3') |
|---|---|---|
| GAPDH | CCCCCACCACACTGAATCTC | GCCCCTCCCCTCTTCAAG |
| GPD2 | CCCCACACTCTACATTAGGCTTGT | TTAGAAAGGCCAGGCGAGTA |
| LPL | GGAGATGTG GACCAGCTAGTGAA | AGCCCTTTCTCAAAGGCTTC |
| IGFBP5 | AACTGAGGACCTCGGAATCTCTAG | ATCCTGGGAAACCCCAATAG |
| IGFBP7 | GTGCCATGCATCCAATTTC | TATAGCTCGGCACCTTCACC |
| MAP2K3 | GATGGAGCACCCCTTCT TCA | CAGTAGCTTATGGTGTGGGTGA |
Reverse transcriptase (RT) PCR
For Reverse Transcriptase (RT) PCR, the total cellular RNA was extracted from NCI-H23 and MCF 10A cells cultured on crosslinked and non-crosslinked P(CL-co-DLLA) substrates as mentioned previously. RT-PCR was performed to analyze the expression of p53 or p21 with a OneStep RT-PCR kit (Qiagen, Germany) according to the manufacturer's instruction. GAPDH was used as an endogenous control. The RNA was also extracted from NCI-H23 and MCF 10A cells grew on crosslinked and non-crosslinked P(CL-co-DLLA) substrates treated with or without rhIGFBP5 protein (200 ng/mL) at day one and day three. The primers used for p53 are: forward primer = 5'-3' CCCAAGCAATGGATGATTTGA; reverse primer = 5'-3' GGCATTCTGGGAGCTTCATCT. The primers used for p21 are: forward primer = 5'-3' TGAGCCGCGACTGTGATG; reverse primer = 5'-3' GTCTCGGTGACAAAGTCGAAGTT. The products were electrophoresed using 2% (w/v) agarose gel and the images were documented.
Statistical analysis
All results are represented as mean ± standard deviation (SD) obtained from three independent experiments. The degree of significance of each data for non-crosslinked P(CL-co-DLLA) substrates compared with crosslinked P(CL-co-DLLA) substrates was analyzed by Student's t-test, where p < 0.05 is considered as statistically significant.
Results
Characterization of the fluidic substrate
In our previous reports, a novel technique to control the crystallinity of CL copolymers was reported by adjusting the melting temperature (Tm) [15-18]. In this study, we used a four-branched copolymer with CL/DLLA of 60/40 and 100 units per branch because this has a Tm below body temperature. The polymer with desired branched chain and structure was confirmed by 1H NMR (Supplementary Figure S1). The weight-average molecular weight (Mw) and number-average molecular weight (Mn) of the copolymers are 59,600 and 40,900 respectively (Mw/Mn = 1.46) as determined by GPC (Supplementary Table S1). The viscoelastic properties of the non-crosslinked substrate are: storage modulus G' = 10.8 kPa (represents the elastic or solid-like component); loss modulus G'' = 33 kPa (represents the liquid-like component); and, tan δ (=G''/G') = 3.06 (>1 means that the substrate is viscous dominant). These data were measured with an angular frequency of 10 s-1 at 37°C. The complex modulus (G*) of the non-crosslinked P(CL-co-DLLA) substrate was 34.7 kPa calculated from the formula √ (G')2+ (G”)2. The stiffness of the crosslinked P(CL-co-DLLA) substrate is 261 kPa measured by the tensile test [14]. Of particular importance is that the surface roughness and water contact angle of crosslinked and non-crosslinked P(CL-co-DLLA) substrates remain unchanged (Supplementary Figure S2).
Effects of the fluidic substrate on cancer cell viability and cell death
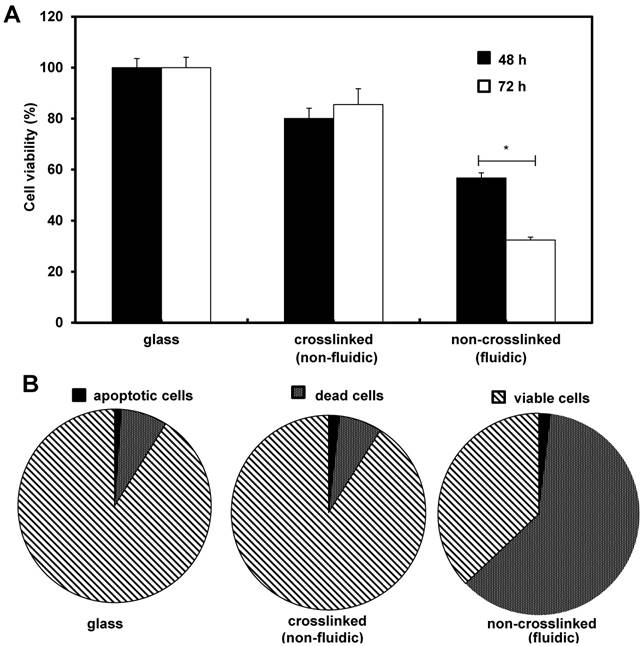
Initially we investigated the effects of crosslinked and non-crosslinked P(CL-co-DLLA) substrates on NCI-H23 viability. After the desired incubation periods, cell viability was performed independently for 48 h and 72 h. The viability of the cancer cells on non-crosslinked P(CL-co-DLLA) substrates was significantly decreased to 57% and 32% for 48 h and 72 h respectively (Figure 1A). To understand the decrease in the cell viability of NCI-H23 cells on non-crosslinked P(CL-co-DLLA) substrates, probable reasons such as cells apoptosis or cell death were examined. To quantitatively monitor the apoptotic and dead cells, apoptosis and dead cell assays were performed after 72 h of growth of NCI-H23 cells on glass, crosslinked or non-crosslinked P(CL-co-DLLA) substrates (Figure 1B). Cancer cells on non-crosslinked P(CL-co-DLLA) showed an enormous dead cell population while the cells did not exhibit significant apoptosis. We also investigated the effect of crosslinked and non-crosslinked P(CL-co-DLLA) substrates on a normal cell model such as MCF 10A. Interestingly, we did not detect any significance in the reduction of cell viability on non-crosslinked P(CL-co-DLLA) substrates (Supplementary Figure S3).
Effects of crosslinked and non-crosslinked P(CL-co-DLLA) substrates on cancer cell viability. NCI-H23 cells were cultured on glass, crosslinked (non-fluidic) or non-crosslinked (fluidic) P(CL-co-DLLA) substrates. (A) The percentage of viable cells was calculated from the control group (glass). (B) Apoptotic and dead cells were counted after 3 days of cell culture and the percentages were determined in comparison with viable cells. n = 3 ± SD and *p < 0.05.
Next, we checked the effect of crosslinked and non-crosslinked P(CL-co-DLLA) substrates on CSC cytotoxicity. Firstly, we cultured NCI-H23 cells in stem cell culture medium for a period of 9 days. Then the tumor spheres were collected and cultured on crosslinked and non-crosslinked P(CL-co-DLLA) substrates and the incubation was continued. As the CSCs are suspended cells by nature, we determined the viability of suspended cells on crosslinked and non-crosslinked P(CL-co-DLLA) substrates. A significant reduction in the number of viable cells on the non-crosslinked P(CL-co-DLLA) substrate was observed (Supplementary Figure S4).
Analysis of mRNA expression in cells cultured on fluidic substrate
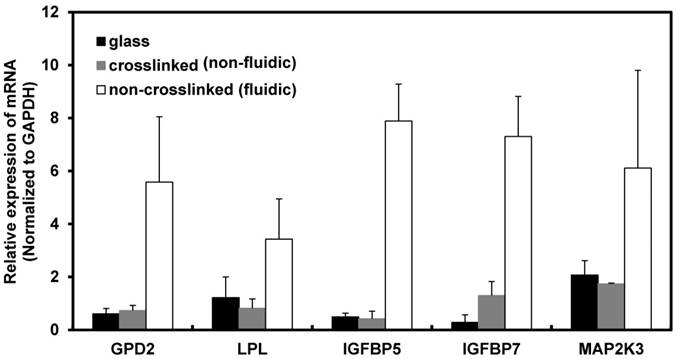
Based on the above result, the maximum time period for the gene expression study was fixed to 72 h because the cells on the non-crosslinked P(CL-co-DLLA) substrate underwent a non-apoptotic form of cell death at this time period. To understand the molecular mechanism of cancer cell behavior on crosslinked and non-crosslinked P(CL-co-DLLA) substrates, PCR array analysis was carried out to investigate the expression of biomarkers. The candidate biomarkers selected based on the PCR array results are GPD2 and LPL, which are responsible for cell metabolism, and IGFBP5, IGFBP7 and MAP2K3, which are responsible for cellular senescence (Table 2). On the other hand, several other biomarkers such as CASP7, CCND3, DDIT3, GADD45G and SLC2A1 (Table 3) were downregulated in NCI-H23 cells cultured on non-crosslinked P(CL-co-CLLA) substrate compared to the crosslinked P(CL-co-DLLA) substrate.
List of up-regulated genes in NCI-H23 cells cultured on non-crosslinked (fluidic) P(CL-co-DLLA) substrate compared to the crosslinked (non-fluidic) P(CL-co-DLLA) substrate.
| Role | Symbol | Description | Fold Change |
|---|---|---|---|
| Cell metabolism | GPD2 | Glycerol-3-phospahte dehydrogenase 2 (mitochondrial) | 6.25 |
| LPL | Lipoprotein lipase | 4.38 | |
| Cell senescence | IGFBP5 | Insulin-like growth factor binding protein 5 | 13.6 |
| IGFBP7 | Insulin-like growth factor binding protein 7 | 4.66 | |
| MAP2K3 | Mitogen-activated protein kinase 3 | 5.62 |
List of down-regulated genes in NCI-H23 cells cultured on non-crosslinked (fluidic) P(CL-co-DLLA) substrate compared to the crosslinked (non-fluidic) P(CL-co-DLLA) substrate.
| Role | Symbol | Description | Fold Change |
|---|---|---|---|
| Apoptosis | CASP7 | Caspase 7, apoptosis- related cysteine peptidase | -5.63 |
| Cell cycle | CCND3 | Cyclin D3 | 0.04 |
| E2F4 | E2F transcription factor 4, p107/p130 binding | 0.03 | |
| DNA damage and repair | DDIT3 | DNA-damage-inducible transcript 3 | 0.02 |
| GADD45G | Growth arrest and DNA-damage-inducible, gamma | 0 | |
| Hypoxia | SLC2A1 | Solute carrier family 2, member 1 | -11.23 |
The up-regulated biomarkers were selected as the candidate biomarkers and their expression level was further analyzed by a quantitative real-time (qRT) PCR technique. The fold induction of metabolic biomarkers such as GPD2 and LPL in cells grown on non-crosslinked P(CL-co-DLLA) substrate were 7.5 and 4 respectively compared to crosslinked P(CL-co-DLLA) substrate. Expression of senescence biomarkers such as IGFBP5, IGFBP 7 and MAP2K3 in cells grown on the non-crosslinked P(CL-co-DLLA) substrate were 19, 5 and 3 times higher respectively than on the crosslinked P(CL-co-DLLA) substrate (Figure 2). Among these up-regulated biomarkers, the mechanism of IGFBP5 involvement in senescence is well studied and its expression level is significantly higher than the other biomarkers. For these reasons IGFBP5 was selected for the further time-dependent study.
Induction of mRNA in NCI-H23 cells cultured on glass (black bars), crosslinked (gray bars) and non-crosslinked (white bars) P(CL-co-DLLA) substrates. Each data was normalized with glyceraldehide-3-phosphate dehydrogenase (GAPDH) as an endogenous control in the same sample. Each bar represents mean ±SD, n=3 for each, p<0.05 for all samples of non-crosslinked P(CL-co-DLLA) substrates compared to crosslinked P(CL-co-DLLA) substrate.
Time-dependent expression of IGFBP5 senescence biomarker in NCI-H23 cells on fluidic substrate
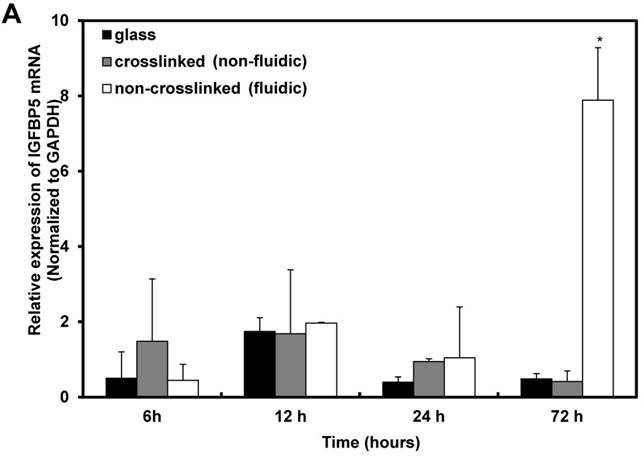
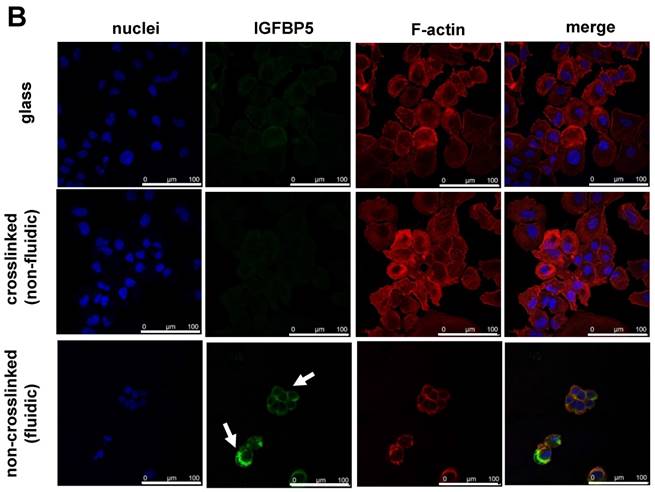
Furthermore, the time-dependent expression of IGFBP5 was analyzed to optimize the expression time of IGFBP5. IGFBP5 mRNA expression was analyzed at 6 h, 12 h, 24 h and 72 h after culturing the cells on crosslinked or non-crosslinked P(CL-co-DLLA) substrates. From 6 h to 24 h, there was no change in the expression of IGFBP5 in cells cultured on crosslinked or non-crosslinked P(CL-co-DLLA) substrates (Supplementary Figure S5). We previously reported that the non-crosslinked P(CL-co-DLLA) substrate did not affect the proliferation of cells until 48 h and later at 72 h cell proliferation was reduced [14]. We correlate the reduction in the proliferation of the cells with the expression of IGFBP5. At 72 h, cells on the non-crosslinked P(CL-co-DLLA) substrate showed a significant induction of IGFBP5 mRNA (Figure 3A). This was confirmed by immunostaining with the IGFBP5 antibody. Figure 3B shows that IGFBP5 is expressed in the cytoplasm of the cells grown on non-crosslinked P(CL-co-DLLA) substrates (Figure 3B, bottom images, white arrows).
(A) Time-dependent expression of IGFBP5 mRNA in NCI-H23 cells on glass (black bars), crosslinked (gray bars) and non-crosslinked P(CL-co-DLLA) substrates (white bars). Each data was normalized with glyceraldehide-3-phosphate dehydrogenase (GAPDH) as an endogenous control in the same sample. Each bar represents mean ± SD, n=3 for each, p<0.05 for non-crosslinked P(CL-co-DLLA) substrate compared to the crosslinked P(CL-co-DLLA) substrate. (B) Confocal microscopy images of IGFBP5 protein expressed in NCI-H23 cells grown on the glass or P(CL-co-DLLA) substrates. The white arrow indicates the expression of IGFBP5 protein in the cytoplasm of NCI-H23 cells (bottom image), scale bars are 100 µm.
IGFBP5 accelerates cellular senescence via induction of p53
Cell cycle arrest in the G1 phase is the hallmark of cellular senescence. To understand the effect of our non-crosslinked P(CL-co-DLLA) substrate on cell cycle, the distribution of cells in different phases of cell cycle was also measured during 3 days of cell culture (Figure 4A). Cells on crosslinked P(CL-co-DLLA) substrate showed a reduced G0/G1 phase (13.87%) and increased S phase (75.55%). However, cells on non-crosslinked P(CL-co-DLLA) substrate showed an accumulation of G0/G1 phase (66.3%) and reduction of S phase (13.92%). This suggests that there is a block in the G1 phase that limits the transition to S phase in cells grown on non-crosslinked P(CL-co-DLLA) substrate.
(A) Cell cycle of NCI-H23 cells cultured on crosslinked (non-fluidic) or non-crosslinked (fluidic) P(CL-co-DLLA) substrates. (B) Growth behavior of NCI-H23 cells in rhIGFBP5 protein-supplemented medium. The cells were exposed to different concentrations (50-200 ng/mL) of rIGFBP5 protein and the cell numbers were determined after 1 day of culturing on glass or crosslinked or non-crosslinked P(CL-co-DLLA) substrates. Cell numbers were calculated and the data were compared to the control group (without rhIGFBP5 exposure) of the corresponding samples. Each data represents mean ± SD, n=3 for each, p<0.05 with respect to control. (C) Expression of p53 at 1 day or 3 days after the cells were treated with 200 ng/mL of rhIGFBP5 protein (+) or without rhIGFBP5 protein (-) by reverse transcriptase (RT) PCR.
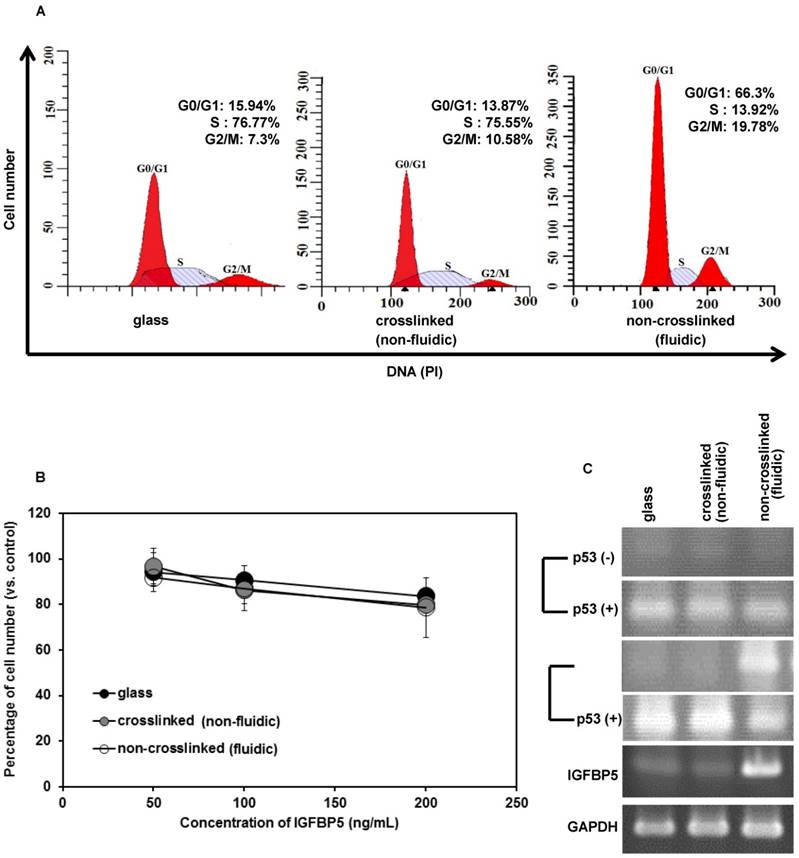
IGFBP5 has a multifunctional role in various cellular phenomena, such as promotion or inhibition of proliferation, apoptosis as well as senescence. To confirm the role of IGFBP5 in proliferation inhibition and senescence, the cells cultured on crosslinked or non-crosslinked P(CL-co-DLLA) substrates were treated with rhIGFBP5 protein and the cell numbers were determined. The cell number decreased in all cultures (glass or crosslinked or non-crosslinked P(CL-co-DLLA) substrates) when they were exposed to rhIGFBP5 protein (Figure 4B). This suggests that IGFBP5 has a growth inhibitory effect on NCI-H23 cancer cells. Meanwhile, we also confirmed the effect of IGFBP5 in MCF 10 A cells. The result indicates that IGFBP5 is not involved in the proliferation or inhibition of MCF 10A cells on any of the substrates (Supplementary Figure S6A).
To know whether IGFBP5 drives p53 expression, we checked the expression of p53 by RT-PCR (Figure 4C). On day 1, the expression of p53 was not observed in any of the samples (Figure 5C, top image). However, at day 3, p53 bands were significant only for cells grown on the non-crosslinked P(CL-co-DLLA) substrate. Additionally, p53 expression in MCF 10 A cells was also assessed (Supplementary Figure S6B). To confirm whether IGFBP5 is actually involved in the induction of p53, the expression of p53 was monitored in rhIGFBP5 protein-exposed NCI-H23 cancer cells grown on crosslinked and non-crosslinked P(CL-co-DLLA) substrates at day 1 and day 3. The expression of p53 was observed in all the samples treated with rhIGFBP5 proteins. This also suggests that IGFBP5 is involved in the induction of p53. Interestingly, we could not see any significant expression of p53 in rhIGFBP5 protein-exposed MCF 10A cells (Supplementary Figure S6C). Some reports state that inhibition of cell proliferation is also associated with the expression of p21 independently of p53 [19,20]. To confirm this phenomenon, we also checked the expression of p21 in NCI-H23 cells grown on crosslinked and non-crosslinked P(CL-co-DLLA) substrates (Supplementary Figure S6D). The expression of p21 was not observed for any of the substrates. This also suggests that p53 is involved in senescence but p21 is not.
Epithelial to mesenchymal transition (EMT) behavior of NCI-H23 cells on crosslinked or non-crosslinked P(CL-co-DLLA) substrates. NCI-H23 cells were stained for epithelial (E-cadherin) and mesenchymal (vimentin) markers after 3 days of culturing on the substrates.
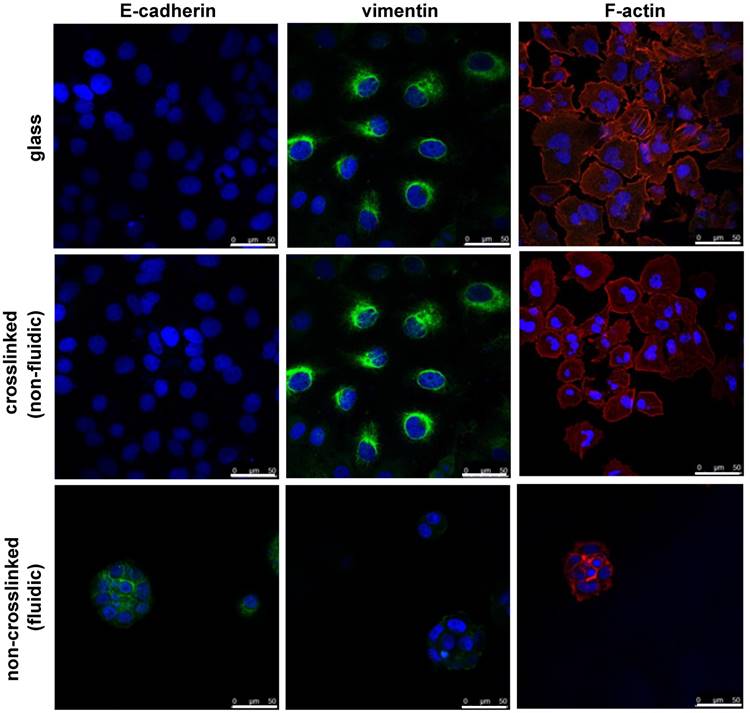
Epithelial to mesenchymal transition (EMT) behavior of NCI-H23 cells on the fluidic substrate
Finally, the effect of material fluidity on EMT was investigated. EMT is a biological process contributing to cancer metastasis during cancer progression. The efficacy of cancer therapy not only depends on the inhibition of the growth of cancer cells but also on the avoidance of metastasis. To understand the effect of the fluidic substrate on the prevention of metastasis in cancer cells, EMT immunostaining is an ideal choice. For this, we stained NCI-H23 cells cultured on crosslinked or non-crosslinked P(CL-co-DLLA) substrates for epithelial and mesenchymal specific proteins. The cells were cultured on the substrates for 3 days followed by staining with E-cadherin as an epithelial marker and vimentin as a mesenchymal marker. The cells on the crosslinked P(CL-co-DLLA) substrate underwent an EMT phenomena by losing the epithelial phenotype and gaining the mesenchymal phenotype (Figure 5, middle image and Supplementary Figure S7). No significant differences in the EMT were observed between the glass and crosslinked P(CL-co-DLLA) substrates. Interestingly, cells on the non-crosslinked, fluidic P(CL-co-DLLA) substrates maintained their epithelial phenotype (Figure 6, bottom image), suggesting that the cells did not undergo EMT.
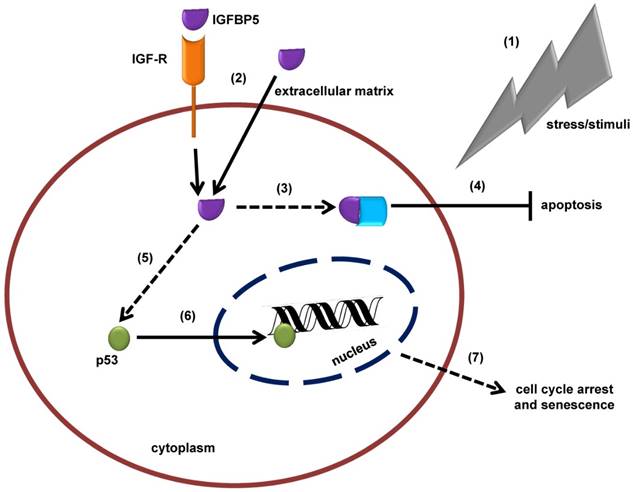
Schematic illustration of the insulin-like growth factor binding protein 5 (IGFBP5)-mediated, tumor suppressor protein 53 (p53)-dependent mechanism of cellular senescence. (1) In response to stress or external stimuli, cells secrete IGFBP5 to the extracellular matrix (ECM). (2) IGFBP5 in ECM is incorporated into the cells via insulin-like growth factor receptor (IGF-R) dependent or independent pathways and accumulates in the cytoplasm of the cells. (3) IGFBP5 in the cytoplasm may interact with other proteins and (4) stimulate an anti-apoptotic effect. (5) On the other hand, IGFBP5 activates p53 expression and (6) p53 translocates to the nuclear region where it interacts with the DNA and alters normal cellular mechanisms, which is followed by (7) cell cycle arrest and senescence.
Discussion
Cells have been known to sense mechanical signals from the surrounding matrix to regulate their functions [21, 22]. These mechanical signals are crucial to determining cellular fate and proliferative or inhibitory effects. In addition, cells possess an intracellular mechanical memory from past physical environments that influence their fate [23]. Furthermore, natural [24] and synthetic polymer-based scaffold materials [25] that modulate cellular behaviors are tremendously attractive in the biomedical area. In view of this, material stiffness-based studies for the regulation of cell behavior have been well known. On the other hand, the role of material fluidity to regulate cell behavior and functions is poorly understood. Recently, Mooney et al. [26] reported that ECM stress-relaxation influences cell behavior, determined both by computational modeling and cellular experiments. Here we proposed MIS therapy for cancer treatment using a biodegradable, fluidic PCL-based material. We have prepared crosslinked (non-fluidic) and non-crosslinked (fluidic) copolymers of CL and DLLA. Our previous report [14] states that cancer cells undergo a non-apoptotic form of cell death when they are grown on non-crosslinked P(CL-co-DLLA) substrate. This is a key factor to investigate the type and molecular mechanism of the non-apoptotic form of cell death exhibited by cancer cells on the non-crosslinked P(CL-co-DLLA) substrate. At first, we investigated the up and down regulation of biomarkers associated with cancer pathways by PCR array for cells grown on crosslinked and non-crosslinked P(CL-co-DLLA) substrates. The mRNA of the biomarkers associated with cellular metabolism (GPD and LPL) and senescence (IGFBP5, IGFBP7 and MAP2K3) were up-regulated by the cells cultured on non-crosslinked P(CL-co-DLLA) substrate compared to the cells on crosslinked P(CL-co-DLLA) substrate. GPD and LPL are the metabolic enzymes involved in mitochondrial intermediary metabolism [27] and the metabolism of fatty acids and triglycerides [28] respectively. No further evidence reported the regulation of GPD and LPL in cancer cell growth and behavior. Hence, we focused only on the senescent biomarkers.
Cellular senescence is a complex process in which cells lose their proliferation efficiency by an arrest in the G1 phase in response to external stress [29]. Growth arrest of the senescent cell is initiated with the activation of p53 [30]. Cancer cells grown on non-crosslinked P(CL-co-DLLA) substrate showed a cell cycle arrest at the G0/G1 phase and an overexpression of mRNA of senescence biomarkers such as IGFBP5, IGFBP7 and MAP2K3. Among these senescent biomarkers, IGFBP5 is well studied. Some evidence has shown that mitogen-activated protein kinase (MAPK) may activate both p53 and p16 via Ras-induced senescence [31]. IGFBP are a group of proteins that bind to insulin-like growth factor (IGF) receptors (IGF-I and IGF-II) with high affinity and modulate the biological functions of IGFs. IGF signaling pathway plays a crucial role in regulating cell proliferation, differentiation, and apoptosis [32]. Based on conservation of gene organization, structural similarity and binding affinity for IGF receptors, the IGFBP family has been categorized into seven distinct subgroups (IGFBP1- IGFBP7). Among these, IGFBP5 is the most conserved member of high-affinity IGF binding protein. IGFBP7 supports cell adhesion and stimulates the proliferation of fibroblasts [33], which is similar to tumor-derived adhesion factor (TAF) identified in human bladder carcinoma cell lines as a cell adhesion molecule [34]. However, in response to external cues, cells regulate the function of IGFBP7 by proteolytic cleavage [33], which abolishes its function as a growth enhancer [35]. IGFBP7 promotes premature senescence in MSCs, and extrinsic factors secreted by senescent MSCs affect neighboring cells [36]. However, the actual mechanism involved in IGFBP7-based cellular senescence is unclear. IGFBP5 plays a multifunctional role [37], possessing growth promoting as well as growth inhibitory effects. IGFBP5 expressed in the cytoplasmic region could promote cell growth in breast cancer [38]. On the other hand, IGFBP5 overexpression in mice retarded growth and reduced litter size [39]. The previous report states that IGFBP5 directly regulates apoptosis by interfering with the IGF signaling cascade [40]. Moreover, cytoplasmic accumulation of IGFBP5 interacted with sphingosine kinase (SK) and protein kinase C (PKC) and stimulated anti-apoptotic effects [41, 42]. The up-regulation of IGFBP5 accelerates p53-dependent cellular senescence in dermal fibroblast and endothelial cells [43]. Here, IGFBP5 induced growth inhibition in cells grown on non-crosslinked P(CL-co-DLLA) (fluidic) substrate. A reduced viability of cancer cells grown on the non-crosslinked P(CL-co-DLLA) substrate was observed. This may be due to cells feeling the fluidic system to be a stressful condition for their growth and survival and so the cells become senescent. IGFBP5 induces cellular senescence by a p53-dependent pathway [44]. To confirm whether IGFBP5 is involved in the induction of p53, we exposed the cells to rhIGFBP5 protein and the cell proliferation was assessed. The rhIGFBP5 protein induces the expression of p53 regardless of the cell culture substrate. This also suggests that IGFBP5 has a vital role in the induction of p53, which accelerated cell cycle arrest and was followed by cell death (Figure 6). Based on previous reports [19,20] describing the role of p53 in the induction of p21 to inhibit cell proliferation we also checked the expression of p21 in NCI-H23 cells grown on crosslinked and non-crosslinked P(CL-co-DLLA) substrates. The expression of p21 was not observed for any of the substrates. This also suggests that p53 is not induced by the expression of p21 and that p53 is involved in cellular senescence regardless of p21 expression.
EMT is a biochemical process in which epithelial cells acquire properties of mesenchymal cells and is well known for its important roles in embryogenesis and development. EMT is accompanied by the loss of cell-cell adhesion and the acquisition of migratory and motile properties. Because of these reasons, EMT can have adverse effects on the organism, leading to pathological conditions such as fibrosis and cancer [45]. The transcription factor Twist acts as a regulator of EMT and is involved in cancer metastasis [46]. A report suggests that high nitric oxide concentration is involved in the inhibition of EMT in cancer cells [47]. On the other hand, the bioactive compound curcumin has an inhibitory effect on EMT in lung cancer cells [48]. Interestingly, cells grown on the non-crosslinked P(CL-co-DLLA) substrate maintained their epithelial phenotype, suggesting that the cells did not undergo EMT. This result implies the possibility of material-induced EMT without any biochemical factor by regulating the fluidic nature of the substrate. Although a better understanding of the molecular mechanism accounting for the activation of senescence and inhibition of EMT is crucial, the fluidity of the material is considered to be a crucial factor in determining the cellular fate.
We also investigated the effect of crosslinked and non-crosslinked P(CL-co-DLLA) substrates on a normal cell model in order to distinguish the specificity of cytotoxicity of the non-crosslinked P(CL-co-DLLA) substrate on cancer cells. Interestingly, we didn't detect any significant reduction in cell viability for non-crosslinked P(CL-co-DLLA) substrates. Next, we checked the effect of crosslinked and non-crosslinked P(CL-co-DLLA) substrates on CSC cytotoxicity. Tumor sphere size reduction on non-crosslinked P(CL-co-DLLA) substrate suggests that non-crosslinked P(CL-co-DLLA) is effective for CSC therapy.
Conclusion
Senescence growth arrest is an attractive concept to suppress the development of cancer. No known physiological stimuli can stimulate senescent cells to reenter the cell cycle. Thus, senescence arrest is considered to be irreversible. We investigated the effects of substrate fluidity on cancer cell senescence, and explored the molecular mechanism. Cancer cells showed diverse behaviors on crosslinked and non-crosslinked P(CL-co-DLLA) substrates. Our findings state that cancer cells grown on the crosslinked P(CL-co-DLLA) substrate showed an increased proliferation rate with an active S phase. However, cells grown on non-crosslinked P(CL-co-DLLA) substrate showed a decreased proliferation with an accumulated G0/G1 phase. Here we also showed that the cancer cells grown on the non-crosslinked P(CL-co-DLLA) substrate expressed a relevant biomarker, IGFBP5, associated with cellular senescence. We also investigated the molecular mechanism of cellular senescence induced by IGFBP5. We found that IGFBP5 expression is also associated with the expression of p53 and is responsible for the cell cycle arrest that leads to cellular senescence. Cellular senescence has become an attractive therapeutic concept because it mimics the process of programmed cell death by suspending the cells from active progression by cell cycle arrest. Thus, our non-crosslinked P(CL-co-DLLA) substrate is considered to be a valuable system for cancer therapy. The cellular senescence observed in NCI-H23 carcinoma cells by non-crosslinked P(CL-co-DLLA) substrate might constitute a good model to further understanding of the significance of senescence in vivo. By considering these evidences as fundamental information for MIS for cancer therapy, our system could be applied for CSC therapy. It is recommended that senescence therapy combined with conventional cancer therapies might entirely abolish cancer.
Abbreviations
MIS: material-induced senescence; P(CL-co-DLLA): poly(ε-caprolactone-co-D,L-lactide); IGFBP5: insulin-like growth factor binding protein 5; EMT: epithelial to mesenchymal transition; CSCs: cancer stem cells; TIS: therapy-induced senescence; hMSCs: human mesenchymal stem cells; CDKI: cyclin-dependent kinase inhibitor; NMR: nuclear magnetic resonance; SEM: scanning electron microscopy; PBS: phosphate buffered saline; PFA: paraformaldehyde; BSA: bovine serum albumin; CCK-8: cell counting kit-8; PCR: polymerase chain reaction; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GPC: gel permeation chromatography; Mw: weight-average molecular weight; Mn: number-average molecular weight; G': storage modulus; G'': loss modulus; SK: sphingosine kinase; PKC: protein kinase C; ECM: extracellular matrix.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gu L, Mooney DJ. Biomaterials and emerging anticancer therapeutics: engineering the microenvironment. Nat Rev Cancer. 2016;16:56-66
2. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420-28
3. Smith MA, Peeper DS. Epithelial-mesenchymal transition and senescence: two cancer related processes are crossing path. Aging. 2010;2:735-41
4. Gil J, Stembalska A, Pesz KA. et al. Cancer stem cells: the theory and perspectives in cancer therapy. J Appl Genet. 2008;49:193-99
5. Acosta JC, Gi J. Senescence: a new weapon for cancer therapy. Trends Cell Biol. 2012;22:211-19
6. Sepúlveda JC, Tomé M, Fernández ME. et al. Cell senescence abrogates the therapeutic potential of human mesenchymal stem cells in the lethal endotoxemia model. Stem Cells. 2014;32:1865-77
7. Sharpless NE, DePinho RA. Telomeres. Stem cells, senescence, and cancer. J Clin Invest. 2004;113:160-68
8. Chen Z, Trotman LC, Shaffer D. et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725-30
9. Abegglen LM, Caulin AF, Chan A. et al. Potential mechanisms for cancer resistance in elephants and comparative cellular response to DNA damage in humans. JAMA. 2015;314:1850-60
10. Michaloglou C, Verdeveld LC, Soengas MS. et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720-24
11. Kim RH, Kang MK, Kim T. et al. Regulation of p53 during senescence in normal human keratinocytes. Aging Cell. 2015;14:838-46
12. Riggelen JV, Felsher DW. Myc and a Cdk2 senescence switch. Nat Cell Biol. 2010;12:7-9
13. Collado M, Gil J, Efeyan A. et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642
14. Mano SS, Uto K, Aoyagi T. et al. Fluidity of biodegradable substrate regulates carcinoma cell behavior: A novel approach to cancer therapy. AIMS Mater Sci. 2016;3:66-82
15. Uto K, Yamamoto K, Hirase S. et al. Temperature-responsive cross-linked poly(ε-caprolactone) membrane that functions near body temperature. J Control Release. 2006;110:408-13
16. Ebara M, Uto K, Idota N. et al. Shape-memory surface with dynamically tunable nano-geometry activated by body heat. Adv Mater. 2012;24:273-78
17. Uto K, Ebara M, Aoyagi T. Temperature-responsive poly(ε-caprolactone) cell culture platform with dynamically tunable nano-roughness and elasticity for control of myoblast morphology. Int J Mol Sci. 2014;15:1511-24
18. Uto K, Mano SS, Aoyagi T. et al. Substrate fluidity regulates cell adhesion and morphology on poly(ε-caprolactone)-based materials. ACS Biomater Sci Eng. 2016;2:446-53
19. Tarek A, Anindya D. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400-14
20. Maria TP, Stefania C. The dual role played by p21 may influence the apoptotic or anti-apoptotic fate in cancer. J Can Res Updates. 2012;1:189-202
21. Benjamin G, Joachim PS, Alexander DB. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10:21-33
22. Koohestani F, Braundmeier AG, Mahdian A. et al. Extracellular matrix collagen alters cell proliferation and cell cycle progression of human uterine leiomyoma smooth muscle cells. PLoS One. 2013;8:e75844
23. Chang Y, Mark WT, Lena B. et al. Mechanical memory and dosing influence stem cell fate. Nat Mater. 2014;13:645-52
24. Sell SA, Wolfe PS, Garg K. The use of natural polymers in tissue engineering: a focus on electrospun extracellular matrix analogues. Polymers. 2010;2:522-53
25. Gunatillake PA, Adhikari R. Biodegradable synthetic polymers for tissue engineering. Eur Cell Mater. 2003;5:1-16
26. Chaudhuri O, Gu L, Darnell M. et al. Substrate stress relaxation regulates cell spreading. Nat Commun. 2015;6:6364
27. Mracek T, Drahota Z, Houstek J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim Biophys Acta. 2013;1827:401-10
28. Sha H, Sun S, Francisco AB. et al. The ER-associated degradation adaptor protein Sel1L regulates LPL secretion and lipid metabolism. Cell Metab. 2014;2:458-70
29. Chang B-D, Xuan Y, Broude EV, Zhu H. et al. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999;18:4808-18
30. Rufini A, Tucci P, Celardo I. et al. Senescence and aging: the critical roles of p53. Oncogene. 2013;24:5129-43
31. Lin AW, Barradas M, Stone JC. et al. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12:3008-19
32. Ferbeyre G, Stanchina Ed, Lin AW. et al. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol. 2002;22:3497-508
33. Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505-18
34. Sie CG, Hesler S, Maas S. et al. IGFBP7's susceptibility to proteolysis is altered by A-to-I RNA editing of its transcript. FEBS Lett. 2012;586:2313-17
35. Akaogi K, Okabe Y, Funahashi K. et al. Cell adhesion activity of a 30-kDa major secreted protein from human bladder carcinoma cells. Biochem Biophys Res Commun. 1994;198:1046-53
36. Ahmed S, Yamamoto K, Sato Y. et al. Proteolytic processing of IGFBP-related protein-1 (TAF/angiomodulin/mac25) modulates its biological activity. Biochem Biophys Res Commun. 2003;310:612-18
37. Severino V, Alessio N, Farina A. et al. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis. 2013;4:e911
38. Akkiprik M, Hu L, Sahin A. et al. The subcellular localization of IGFBP5 affects its cell growth and migration functions in breast cancer. BMC Cancer. 2009;9:103
39. Salih DAM, Tripathi G, Holding C. et al. Insulin-like growth factor-binding protein 5 (Igfbp5) compromises survival, growth, muscle development, and fertility in mice. PNAS. 2004;101:4314-19
40. Marshman E, Streuli CH. Insulin-like growth factors and insulin-like growth factor binding proteins in mammary gland function. Breast Cancer Res. 2002;6:231-39
41. Akkiprik M, Feng Y, Wang H. et al. Multifunctional roles of insulin-like growth factor binding protein 5 in breast cancer. Breast Cancer Res. 2008;10:212
42. McCaig C, Perks CM, Holly JM. Signaling pathways involved in the direct effects of IGFBP-5 on breast epithelial cell attachment and survival. J Cell Biochem. 2002;84:784-94
43. Rufini A, Tucci P, Celardo I. et al. Senescence and aging: the critical roles of p53. Oncogene. 2013;24:5129-43
44. Kim KS, Seu YB, Baek S-H. et al. Induction of cellular senescence by insulin-like growth factor binding protein-5 through a p53-dependent mechanism. Mol Biol Cell. 2007;18:4543-52
45. Lamouille S, Xu J, Derynck R. Molecular mechanism of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178-96
46. Wang Y, Zhou BP. Epithelial-mesenchymal transition in breast cancer progression and metastasis. Chin J Cancer. 2011;30:603-11
47. Baritaki S, Yepez SH, Sahakyan A. et al. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: Inhibition of the metastasis-inducer Snail and induction of the metastasis-suppressor RKIP. Cell Cycle. 2010;9:4931-40
48. Jiao D, Wang J, Lu W. et al. Curcumin inhibited HGF-induced EMT and angiogenesis through regulating c-Met dependent PI3K/Akt/mTOR signaling pathways in lung cancer. Mol Ther Oncolytics. 2016;3:16018
Author contact
![]() Corresponding author: EBARA.Mitsuhirogo.jp; Tel: +81-29-860-4775; Fax: +81-29-860-4708
Corresponding author: EBARA.Mitsuhirogo.jp; Tel: +81-29-860-4775; Fax: +81-29-860-4708