Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(18):4566-4576. doi:10.7150/thno.19840 This issue Cite
Research Paper
In-Tether Chiral Center Induced Helical Peptide Modulators Target p53-MDM2/MDMX and Inhibit Tumor Growth in Stem-Like Cancer Cell
Kuan Hu1*, Feng Yin1*, Mengyin Yu1, Chengjie Sun1, Jingxu Li1, Yujie Liang1, Wenjun Li1, Mingsheng Xie1, Yuanzhi Lao3, Wei Liang2 ![]() , Zi-gang Li1
, Zi-gang Li1 ![]()
1. School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen, 518055, China
2. Department of Radiation Oncology, the First Affiliated Hospital, Anhui Medical University, Hefei, 230022, China
3. School of Pharmacy, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, China
* These authors contributed equally to this work.
Received 2017-2-27; Accepted 2017-9-8; Published 2017-10-13
Abstract

Inhibition of the interaction between p53 and MDM2/MDMX has attracted significant attention in anticancer therapy development. We designed a series of in-tether chiral center-induced helical stabilized peptides, among which MeR/PhR effectively reactivated p53. The activation of p53 inhibits cell proliferation and induces apoptosis in both the MCF-7 normal tumor cell line and the PA-1 pluripotent cancer cell line with only minimal cellular toxicity towards normal cells or cancer cell lines with p53 mutations. The in vivo bioactivity study of the peptide in the ovarian teratocarcinoma (PA-1) xenograft model showed a tumor growth rate inhibition of 70% with a dosage of 10 mg/kg (one injection every other day). This is the first application of a stabilized peptide modulator targeting stem-like cancer cell both in vitro and in vivo and provides references to cancer stem cell therapy.
Keywords: Chirality-induced helical peptide, stem-like cancer cell, Cancer, p53-MDM2/MDMX.
Introduction
Cancer is one of the most formidable diseases to combat. The cancer stem cell (CSC) hypothesis provides a compelling cellular mechanism to account for the therapeutic refractoriness and dormancy in cancer development [1-4]. CSCs are a subset of stem-like cells that exhibit a unique spectrum of biologic, biochemical, and molecular features and possess the ability to efficiently propagate the bulk of tumors [5]. Their unlimited self-renewal and multipotency make CSCs more resistant to conventional and targeted therapies [1, 2]. CSCs are believed to be the primary cause of tumor recurrence and metastases. In clinical practice, CSCs need to be eradicated for long-term disease-free survival. While drug resistance and issues in drug delivery are the main obstacles in developing CSC therapy, progress is compromised by the lack of compounds with suitable biological functions and pharmacological properties [6, 7].
Peptide stabilization is a technique for constraining short peptides into a fixed secondary conformation, typically an α-helix [8, 9]. As protein-protein interactions (PPIs) were previously thought to be 'undruggable' by small molecules due to their limited interacting surface area, stabilized peptides have become a promising drug modality to target PPIs [10]. We precisely added a chiral center into the peptide tether to constrain peptides into an α-helical conformation as shown in Fig. 1A. Compared to the S epimer, the R epimer peptide showed significantly enhanced helical content, cellular uptake and target binding affinity [11-13]. One major advantage of our chirality-induced helicity (CIH) strategy is that we are able to fine-tune the peptides' biophysical properties, including target binding affinity and cellular uptake, by switching the substitution group at the chiral center. While stabilized peptides have previously been used to target various PPIs [8, 9, 14-16], to our knowledge, there has been no report of using stabilized peptides to regulate PPIs in CSCs or stem-like cancer cells. In our initial trials, we found that short peptides constructed via our strategy could successfully penetrate CSCs, and we hypothesized that stabilized peptide modulators could be developed to interrupt PPIs in CSCs and inhibit CSC growth and differentiation.
(A) A chirality-induced helicity system was used to construct stabilized peptides. The non-natural amino acids used were synthesized following previously reported methodology, [11] and the tether was cross-linked by a UV-initiated thiol-ene reaction. (B) The p53/MDM2 or p53/MDMX inter-action could be disrupted by CIH peptide modulators.
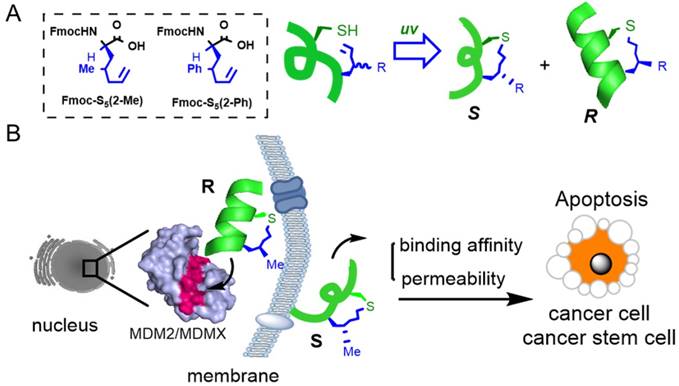
The human transcription factor protein p53 plays a pivotal role in protecting cells from malignant transformation by inducing cell-cycle arrest and apoptosis in response to DNA damage and cellular stress [17-19]. Disruption of the interaction between wild-type p53 and (murine double minute 2) MDM2/(murine double minute X) MDMX may release and reactivate p53 as a promising approach in cancer therapy [20-23]. Extensive research has been done on small molecular leads and stapled peptides, such as nutlin-3a [24, 25] and RG7112 [26], and SAH-P53-8 [27] or ATSP-7041 [28]. Although great successes in the modulation of p53-MDM2/X interaction by peptide-based ligands have been achieved, only limited in vivo studies were reported and more research is required to tackle this important PPI.
Ovarian teratocarcinoma (OVTC) arises from germ cells and comprises pluripotent cells [21, 22, 29]. The PA-1 cell line, derived from human ovarian teratocarcinoma cells, is a well-accepted model for studying cancer cell stemness and expresses endogenous, nonfunctional wild-type p53 (Fig. S1 and S2) [23-25]. Here we designed CIH peptide modulators that target p53-MDM2/MDMX PPIs in CSCs with suitable cell permeability and binding affinity to reactivate the p53 apoptosis pathway and eradicate malignant CSCs (Fig. 1B). In assessing the bioactivity of peptide drug leads, we thought that conducting in vivo animal model experiments in addition to in vitro cell-based experiments would provide more convincing results than the latter alone. In this study, for the first time, we assessed the peptide drug lead efficiency both in vitro and in vivo in CSCs.
Materials and Methods
Fluorescence Polarization Assay
Fluorescein isothiocyanate (FITC)-labeled peptides (10 or 20 nM) were incubated with MDM2 or MDMX protein in binding assay buffer (140 mM NaCl, 50 mM, Tris pH 8.0) at room temperature for 1 h. Fluorescence polarization experiments were performed in 96-well plates (Perkin Elmer Optiplate-96F) on a plate reader (Perkin Elmer, Envision, 2104 multilabel reader). Concentrations of the peptides were determined by 494 nm absorption of FITC. Kd values were determined by nonlinear regression analysis of dose response curves using Origin pro 9.0.
Confocal Microscopy and Co-localization Assay
PA-1 cells (or MCF-7 cells) were cultured with DMEM with 10% fetal bovine serum (FBS) (v/v) in imaging dishes (50000 cells/well) in a 37°C, 5% CO2 incubator for one day until they were ~80% adherent. Peptide were first dissolved in dimethyl sulfoxide (DMSO) to make a 1 mM stock and then added to cells to a final concentration of 5 μM. The cells were incubated with peptides for 1 h at 37°C. After incubation, cells were washed 3 times with phosphate buffered saline (PBS) and then fixed with 4% (wt/vol) formaldehyde (Alfa Aesar, MA) in PBS for 10 min. They were then washed 3 times with PBS and stained with 1 μg/mL 4, 6-diamidino-2-phenylindole (DAPI) (Invitrogen, CA) in PBS for 5 min. Images of peptide localization in cells were taken via PerkinElmer confocal microscopy. Image processing was done using the Volocity software package (Zeiss Imaging).
Cell Viability Assay
Cell viability was measured by the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenylt-etrazolium bromide (MTT) (Sigma) assay. Cells were seeded in a 96-well plate at a density of 5 × 103 cells/well and incubated with p53 peptides and nutlin-3a in serum-free media for 4 h, followed by serum replacement and additional incubation for 44 h. MTT (5 mg/mL, 20 μL) in PBS was added and the cells were incubated for 4 h at 37 °C with 5% CO2. DMSO (150 μL, Sigma) was then added to solubilize the precipitate with 5 min of gentle shaking. Absorbance was measured with a microplate reader (Bio-Rad) at a wavelength of 490 nm.
Immunoprecipitation
Exponentially growing PA-1 cells were treated with 40 μM nutlin-3a and equivalent volume of DMSO. Whole-cell extracts were generated using lysis buffer [50 mM Tris (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40]. Protein extracts (500 μg) were precleared for 2 h with 40 μL protein G Sepharose beads (50%, Sigma) before addition of the indicated antibodies. For immunoprecipitation, rabbit monoclonal antibody anti-p53 (Cell Signaling Technology, 2 mg/mL) and mouse monoclonal anti-MDM2 (abcam, 1 mg/mL) were used. Immune complexes were then collected on protein G Sepharose beads at 4°C overnight, and beads were washed five times with cold lysis buffer. Precipitated proteins were subjected to Western blotting with rabbit monoclonal antibody anti-p53 (Cell Signaling Technology), mouse monoclonal anti-MDM2(abcam, 1 mg/mL), polyclonal antibodies pan-actin (Cell Signaling Technology, 2 mg/mL).
Western Blot Analysis
For western blot analysis, cells were seeded in 6-well plates and treated for 48 h with p53 peptides and nutlin-3a. To isolate the protein, cells were washed with PBS and harvested using lysis buffer (50 mM Tris·Cl pH 6.8, 2% (wt/vol) SDS, 6% (wt/vol) Glycerol, 1% (wt/vol) β-mercaptoethanol, 0.004% (wt/vol) bromophenol blue). Total cellular protein concentrations were determined by a spectrophotometer (Nano-Drop ND-2000). 20 μg of denatured cellular extracts were resolved using 10% SDS-PAGE gels. Protein bands in the gel were then transferred to nitrocellulose blotting membranes and incubated with the appropriate primary antibody. The antibody dilutions were used as follows: 1:500 for mouse monoclonal anti-MDM2 (abcam, 1 mg/mL), mouse monoclonal anti-MDMX (abcam, 1 mg/mL), rabbit monoclonal antibody anti-p53 (Cell Signaling Technology, 2 mg/mL) and 1:1000 for polyclonal antibodies pan-actin (Cell Signaling Technology, 2 mg/mL), anti-H3 (Sigma, 2 mg/mL). Membranes were incubated overnight at 4 °C and washed the next day with buffer (1×PBS, 0.05% (wt/vol) Tween 20). Goat anti-rabbit or anti-mouse secondary antibodies were used for secondary incubation for 1 h at room temperature. Proteins were then visualized with chemiluminescent substrates.
RNA Extraction and RT-PCR
For real-time quantitative PCR (rt-qPCR) analysis, cells were seeded in 6-well plates and treated for 48 h with P53 peptides and nutlin-3a as described for the western blot assay. Then total RNA was extracted from cells using TRIzol reagent (Invitrogen) and the amount of RNA was quantified by a spectrophotometer (Nano-Drop ND-2000). Total RNA (2 μg) was reverse transcribed to cDNA using the reverse transcriptase kit from Promega according to the manufacturer's instructions. The mRNA levels of the target genes were quantified by real time PCR using SYBR green (Promega) in an ABI Prism 7500 real-time PCR system (Applied Biosystems). The primers used are listed in Table S3.
Ubiquitination Analysis
For the cytoplasmic translocation assay, HCT116 cells were plated on Costar six-well-containing glass coverslips until they reached 80-90% confluence. The next day, the cells were transfected with GFP-p53 (1 μg) using HD transfection reagent (Roche, USA) according to the manufacturer's protocol (for low levels of Mdm2 expression, 0.5 μg of pCMV-MMD2 was used). At 4 h after transfection, cells were treated with peptide. 24 h after transfection, cells on the coverslips were washed three times with PBS and then fixed in 4% paraformaldehyde/PBS for 10 min at room temperature. After 3 washes with ice-cold PBS, cells were perforated in ice-cold PBS containing 0.2% (v/v) Triton X-100 for 10 min. Cells were blocked in PBS containing 1% (v/v) bovine serum albumin and 1 μg/mL of DAPI (Sigma) at room temperature for 30 min in a dark environment. Cells were washed three times with PBS, and the stained cells were mounted with mounting medium and the cover slips were sealed with nail polish. Fluorescence was recorded using a confocal microscope (Zeiss, ×100, oil lens).
Antitumor efficacy in human xenograft model using PhR peptide
Athymic nude mice (BALB/c ASlac-nu) were obtained from Vital River Laboratory Animal Technology Co. Ltd. of Beijing, People's Republic of China and allowed an acclimation period of 1 week. Mice were maintained in an isolated biosafety facility for specific pathogen free (SPF) animals with bedding, food and water. All operations were carried out in accordance with the National Standard of Animal Care and Use Procedures at the Laboratory Animal Center of Shenzhen University, Guangdong Province, People's Republic of China (the permit number is SZU-HC-2014-02). For tumor suppression assay, athymic nude mice (female; 6 weeks old) were inoculated with 1 × 107 PA-1 cells (PA-1 cells were trypsinized, harvested and re-suspended in DMEM, with 100 mL volume of each) propagated in vitro subcutaneously in the lower flank of mice. After 10-15 days, mice with tumors exceeding 100-150 mm3 in volume were randomly divided into 3 groups of 5-6 mice per treatment group. Mice bearing PA-1 tumors were injected with PhR (10 mg/kg) or nutlin-3a (10 mg/kg) using PBS as negative control. Mice were injected once every 2 days starting on day 0. Tumor volumes were measured by calipers (accuracy of 0.02 mm) every other day and calculated using the following formula: V= L×W2/2 (W, the shortest dimension; L, the longest dimension). Each tumor was independently measured and calculated by changes in volume (folds) relative to day 0. Statistical significances between groups were tested by one-way analysis of variance.
Results
Design and Synthesis of MDM2/X modulators-CIH peptide MeR/PhR
A p53 mimetic-stabilized peptide library was designed based on a recently reported dual inhibitor peptide (pDI) of MDM2/MDMX with the initial sequence of LTFEHYWAQLTS discovered via phage display named as PDI-1 [30] (Table S1). Notably, the Glu-to-Gln mutation at position 4 was made to facilitate permeation by removal of one negative charge. Previous structural elucidation of MDM2 and its stapled peptide ligands suggested that the peptide tether also interacts with the hydrophobic cleft of MDM2/X [28, 31]. We performed iterative optimization and synthesized a library of stapled i, i+4 peptides (entry 2-12) with the tether at different positions, different substitution groups (Methyl- or Phenyl-) at the chiral center, and amino acid mutations at selected positions (Table S1). For each peptide, it has two epimers, S and R, for which are different at the in-tether chiral center configurations[incomplete sentence, please revise]. The R epimer is helical and the S epimer is mainly random coil [11]. All peptides retained the core binding triad of Phe19, Trp23 and Leu26 referring to the crystal complex of p53-MDM2 [22, 32]. However, unique contributions also exist, including (i) the staple moiety of peptides, which binds to the hydrophobic cleft of MDM2, (ii) the Tyr of PDI, which has been identified as a fourth key amino acid by several phage display studies [33, 34]. The characterization of all peptides is summarized in Table S2. Fluorescence polarization assays were used to measure the binding affinity between the FITC-labeled peptide and GST-labeled MDM2/MDMX proteins (Fig. S3). MeR and PhR exhibited the highest binding abilities toward both MDM2 and MDMX. Notably, all the R epimers have a higher binding affinity than their respective S epimers, which unambiguously showed the importance of maintaining peptides' secondary structures (Fig. S3).
Permeability and cell killing ability of MeR/PhR peptide
To investigate the cellular uptake of MeR/PhR, FITC-labeled TAT (structure shown in Supplementary data - the appendix section), a commonly used cell-penetrating peptide derived from HIV integrase [35], was used as a cellular uptake control for MeR and PhR. PA-1 cells were treated with 5 µM peptides and imaged at 2 h post-treatment with confocal microscopy. Fluorescence activated cell sorting (FACS) was used to quantify the fluorescence intensity in cells. The results are summarized in Fig. 2A and 2B. MeR and PhR showed diffused intracellular localization, while TAT-treated cells showed almost no detectable intracellular fluorescence. PhR showed higher cellular uptake than MeR in both PA-1 and MCF-7 cells (PA-1 in Fig. 2A-B; MCF-7 in Fig. S4-a). In order to exclude the probability that cell fixation led to artifactual internalization/location of peptides, we further performed the live cell confocal imaging assay, and the results suggested that PhR peptide could be internalized into the cells both in the cytoplasm and nucleus (Fig. S4-b). Immunofluorescence was also used to study the co-localization of the peptides and MDM2. The MDM2 protein was stained with an Alexa-647-labeled antibody and was mainly located in the nucleus. The results showed that PhR can accumulate in the nucleus and bind to MDM2/X (Fig. S4-c).
(A) Confocal microscopy images of PA-1 cells treated with 5 µM FITC-labelled peptides at 37°C for 2 h. Scale bar, 20 µm. DAPI: blue; peptide: green. (B) Flow cytometry analysis of PA-1 cells treated with 5 µM FITC-labelled peptides at 37°C for 2 h (blank-red, TAT-cyan, MeR-green, PhR-purple). (C, D) Viability of two p53 mutant cancer cells (MDA-231 and skov3) and two normal cells (HEK-293 and QSG-7701). Skov-3, MDA-MB-231, HEK-293 and QSG-7701 were incubated with 3.12, 6.25, 12.5, 25 and 50 µM MeR or PhR for 48 h. Both MeR and PhR had little effect on the cell growth and proliferation. Error bars represent SEMs of at least three independent measurements. (E, F) Viability of two p53wt cancer cells (MCF-7 and PA-1). PA-1 and MCF-7 cells were incubated with 5, 10, 20, 40 and 50 µM MeR, PhR or their epimers-MeS and PhS for 48 h. Error bars represent SEMs of at least three independent measurements. (G) PhR inhibits p53-MDM2 binding in cancer cells. PA-1 cells were incubated with 40 μM PhR or equivalent volume of DMSO for 24 h, and the levels of p53, MDM2 were determined in protein complexes immunoprecipitated with anti-MDMX or anti-p53 antibodies by Western blotting.
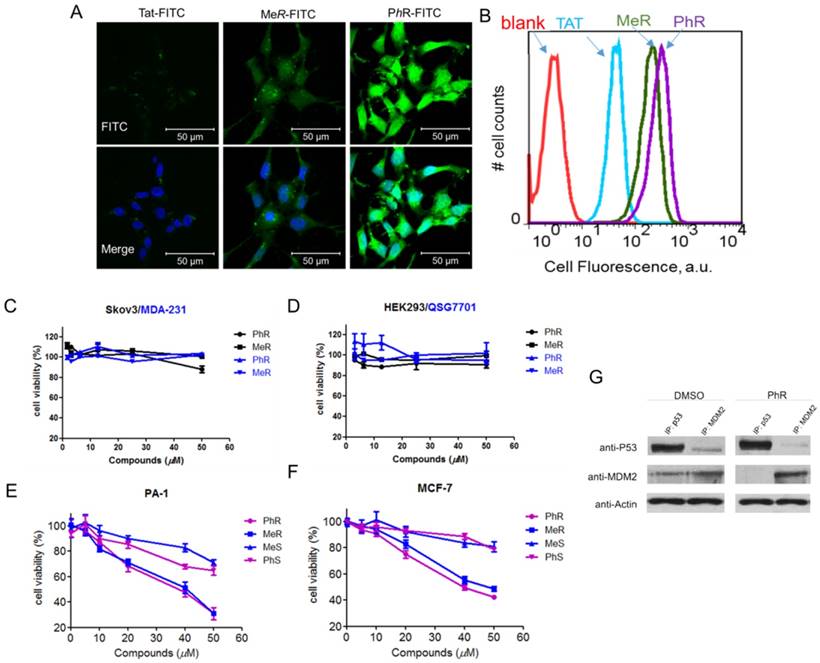
Inhibition of p53 binding to its negative regulators, MDM2 and MDMX, could possibly stabilize p53 and activate the pathway. Activation of the p53 pathway only occurs in cells that express wild-type p53 but not in cells that express the mutant form of the p53. To assess the activity of MeR/PhR, we chose cancer cells lines representing two clinically relevant populations of tumors that overproduce either MDM2 (PA-1 ovarian teratocarcinoma) or MDMX (MCF-7 breast cancer). First, we measured the IC50 of our peptides using cell viability assays. Nutlin-3a was chosen as a positive control. Two mutant p53 cell lines (Skov-3 and MDA-231) were chosen to exclude the nonspecific toxicity of MeR and PhR. Negligible effects were detected for these two cell lines' viability up to 50 μM of either MeR and PhR (Fig. 2C). To exclude the potential that the minimal cytotoxicity was caused by the lack of internalization in these cell lines, we performed cellular uptake assays and proved that the level of cellular uptake in these cells is similar to that of PA-1 (Fig. S4-d). Two normal cell lines (HEK-293 and QSG-7701) were also tested and negligible cytotoxic effects were detected with peptide treatment as shown in Fig. 2D. In contrast, treatment of MCF-7 and PA-1 cells with MeR and PhR led to significant cell growth inhibition in a strictly dose-dependent manner (IC50=40 μM) (Fig. 2E-F). From the bright field images of cells treated with MeR/PhR, we observed apparent cell proliferation inhibition in a time- and dose- dependent manner (Fig. S5a-b). These results proved the target specificity of the peptides MeR and PhR in the p53wt stem-like cancer cell line and normal cancer cell lines. To assess the ability of PhR to inhibit p53 binding to MDM2 and MDMX, we incubated with PA-1 cells with PhR at 40 μM for 4 h and determined the levels of MDM2 and MDMX associated with p53 by immunoprecipitation (IP) from cell lysates followed by western blotting. Our results revealed substantial reduction of MDM2 protein bound to p53 in lysates from cells treated with PhR compared with DMSO control. These studies confirmed the ability of PhR to penetrate living cancer cells in the presence of 10% FBS and inhibit the binding of its target proteins MDM2 to p53.
PhR activates p53 signaling in PA-1 cancer cells
The protein and mRNA expression levels were measured to assess the biological activity of our peptides. PA-1 and MCF-7 cells were treated with MeR, PhR and nutlin-3a for 48 h, and then p53, MDM2 and MDMX expression levels were monitored via western blot analysis (Fig. 3A-B). Notably, p53 expression was upregulated with MeR and PhR treatment. The peptides' S epimers showed negligible effects. The activation of p53 by peptide MeR and PhR was also demonstrated with the induction of the mRNA levels of three p53 target genes, MDM2, MDMX, and MIC-1 in both PA-1 (Fig. 3C) and MCF-7 (Fig. 3D). Direct evidence of CSC inhibition by PhR came from the significant downregulation of stemness genes sox2 and upregulation of differentiation genes foxA2 in PA-1 cells after treatment, which further suggested that the deactivation of p53 could play an essential role in the proliferation of PA-1 cells. Similar changes were observed in the expression of sox2 and foxA2 in MCF-7 cells treated with PhR [36]. In summary, PhR served as an efficient modulator that reactivated p53 to regulate pluripotency related genes. Notably, changes in gene regulation caused by peptide treatment were observed only in p53wt cancer cells (PA-1 and MCF-7) and not in normal (QSG-7701) cells (Fig. S5c-d).
(A, B) MeR/PhR stabilize p53 and elevate protein levels of p53 and its targets MDM2 and MDMX. Log-phase PA-1 and MCF-7 cells were incubated with 40 µM peptides (MeR/S, PhR/S) or 1 µM nutlin-3a, and cell lysates were analyzed by Western blotting. (C, D) PhR shows induction of p53 and p53 target genes in p53wt cell lines. Exponentially growing p53wt cancer cell lines were incubated with 40 µM peptides or 1 µM nutlin-3a for 48 h, and mRNA level of p53 and p53 targets MDM2, MDMX, and MIC or stemness-related genes were analyzed by quantitative PCR and expressed as fold increase. Error bars represent SEMs for triplicates of three independent experiments, **, P<0.01. *, P<0.05.
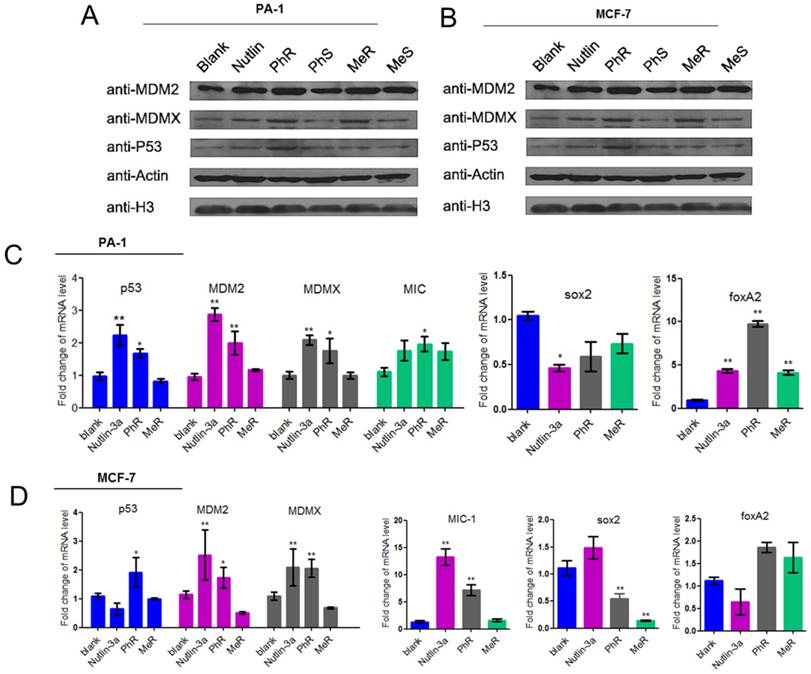
The S epimers of MeR and PhR, MeS and PhS were used as control peptides, a practice that circumvented the use of scrambled or mutated peptides as negative controls. Their effects were shown in Fig. 2E-F and Fig. S6, labeled as MeS and PhS. The functional difference between the peptide epimers clearly indicated the importance of the tether structure.
PhR reactivates major p53 cellular functions in PA-1 cancer cells
The tumor suppressor p53 possesses many cellular functions, the most important of which is the induction of apoptosis and cell-cycle arrest [33, 37, 38]. PA-1 cells were exposed to 40 μM peptides or 5 μM nutlin-3a for 48 h, the latter of which was known to induce apoptosis by inhibiting MDM2/MDMX [32, 34, 39]. Annexin-V/PI assays were used to quantify the apoptotic effect of MeR and PhR. The apoptotic effect of PhR was more pronounced in PA-1 than those of MeR and nutlin-3a (Fig. 4A and Fig. S7a); this was consistent with the viability results. Notably, PhR was more effective in inducing cell-cycle arrest in the G2/M phase than MeR and nutlin-3a in PA-1 cells (Fig. 4B and Fig. S7b). It is worth noting that PhR did not induce cell cycle arrest in the normal cell line QSG-7701 (Fig. S7c-d), suggesting that PhR is more specific and less toxic than nutlin-3a. The caspase-3 assay indicated that PhR induced dose-dependent apoptosis in PA-1 cells (Fig. 4C). Nutlin-3a only induced minimal apoptosis at a low dose, while at higher doses (>5 μM), no caspase-3 activity was detected (Fig. S7e). These results further indicated nutlin-3a killed PA-1 cells partly through its nonspecific toxicity rather than only through p53 pathway activation.
(A) A column graph was used to show the ratio of apoptotic cells in total cells. The apoptotic cells were calculated by Q1+Q2+Q3 in Fig. S9. (B) Column graph to show the distribution of cell-cycle phase for PA-1 after treatment with MeR, PhR or nutlin-3a. The original cell cycle analysis data of PA-1 are shown in Fig. S12. (C) The extent of apoptosis was measured through the detection of caspase-3 activity by exposing the cells to a caspase-3-specific substrate. (D) PhR inhibited the ubiquitination of p53 by MDM2. PhR effectively inhibited the translocation process in 24 h. GFP-p53: green; DAPI: blue.
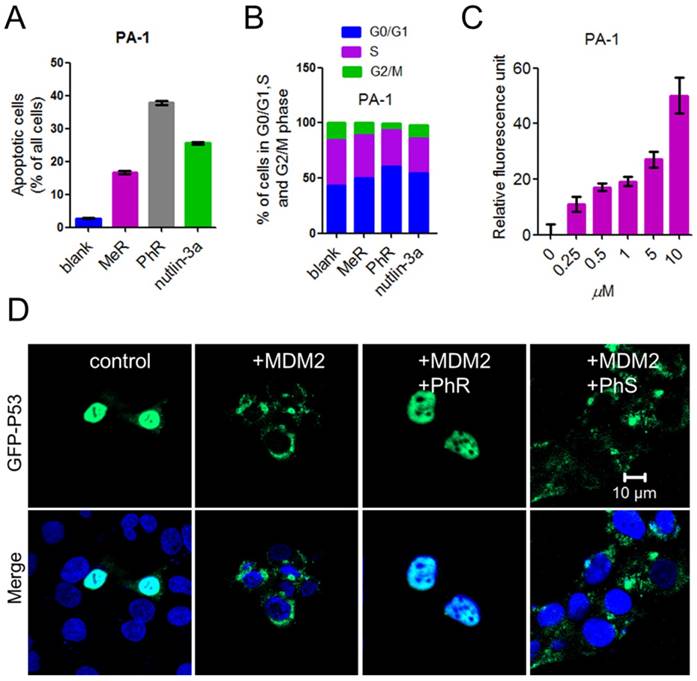
Reactivating p53 by inhibiting MDM2 inhibits the ubiquitination of p53 and blocks its export from the nucleus for degradation [40]. GFP (green fluorescent protein)-labeled p53 and MDM2 plasmids were co-transfected into HCT-116 cells for 24 h; the cells were then treated with peptides for 24 h. The confocal imaging results showed that p53 ubiquitination was inhibited when treated with PhR, as no green fluorescence was observed in the cell plasma (Fig. 4D), while PhS had little effect on this process. Here we further confirmed our peptides' capacity for nucleus penetration.
PhR activated the p53 pathway and induced cell apoptosis. PA-1 cells were treated with a dose ladder of PhR from 0 μM to 40 μM with 10% serum. After 2 days, a dose-dependent increase in the level of p53 and a concomitant elevation of the p53 transcriptional targets MDM2 and MDMX was observed. The activation of p53 by PhR was demonstrated by the dose-dependent induction of the mRNA of p53 and three p53 target genes - p21, MDM2 and MDMX (Fig. S8a). The response time of p53 activation after treatment with PhR was tested by using a fixed peptide concentration of 40 μM from 0 to 48 h. Apparent p53 and MDM2 accumulation both in protein and mRNA levels was detected, with the highest mRNA level observed slightly before the highest protein level (Fig. S8b). Recently, Lahav et al. reported that cell death depends on the accumulation kinetics of p53 [41]. With HCT-116 Venus knock-in cell line (p53-VKI) (a kind gift from the Lahav laboratory (Yale University)), we detected a more rapid accumulation of p53 protein in PhR treated cells than in nutlin-3a treated cells. For PhR, the highest levels of p53 and p21 were reached in 12 h, while that for nutlin-3a was 48 h (Fig. S9), which clearly indicated a delay as compared with PhR. The faster accumulation of p53 in PhR-treated cells was further confirmed via measurements of the mRNA levels of p53 and related transcriptional genes (Fig. S9). This phenomenon may help to explain PhR's working mechanism and its greater efficacy in vivo (in the following section) compared with nutlin-3a.
PhR suppresses tumor growth in vivo in PA-1 xenograft model via reactivation of the p53 pathway
PhR was examined for its efficacy in a human cancer xenograft model carrying p53wt and elevated levels of MDM2 proteins, namely the PA-1 human xenograft model. As shown in Fig. 5, PhR resulted in statistically significant tumor growth inhibition (TGI) in the MDM2-amplified human teratocarcinoma xenograft model PA-1. When dosed for 3 weeks at 10 mg/kg every other day with intratumoral injections, PhR induced a TGI of 70% (Fig. 5A). In comparison, the selective MDM2 small-molecule inhibitor nutlin-3a (administrated by intratumoral injection every other day at doses of 10 mg/kg) resulted in a TGI of 30%, which is much less effective compared to PhR. Actually, the drug concentration in the tumor section is much higher for nutlin-3a than PhR for this dosage [M.W.: PhR (1640) vs. nutlin-3a (581)]. Significantly, the mice treated with PhS peptides at the same dose as PhR displayed negligible inhibition effect on tumor growth. These results are consistent with the in vitro tests, which is a direct proof that peptides' secondary structures are critical for bioactivity. The inhibition effects are confirmed by tumor weight and tumor volume measurements (Fig. 5B, C). At the same time, the weight of the mice showed no significant change (Fig. S10a), suggesting that our peptides have little side effect with respect to mouse growth. Furthermore, the mortality of the mice during the survival study are shown as survival curves (Fig. 5D), which indicate that PhR was able to prolong the lives of the mice. Especially, no mice died during the treatment period in the PhR group while three in ten mice died in the nutlin-3a group. To assess the pharmacokinetics and duration time of PhR within the tumor, Cy3-labeled PhR was treated by intratumoral injection and imaged at different time points (0 min, 5 min, 30 min, 2 h, 6 h and 24 h) using the IVIS Lumina II small animal in vivo optical imaging system. The optical signal intensity of the PhR-Cy3 peptide remained consistent without apparent diminishment for 24 h (Fig. 5E, Fig. S10b). Furthermore, the ex vivo fluorescence results from tumors and major organs were consistent with previous reports on the biodistribution of peptides, and indicate that the majority of the peptide accumulated in the tumor and liver/kidney (Fig. S10c).
(A) Tumor tissue image of mice treated with (1) PBS, (2) nutlin-3a, (3) PhR, (4) PhS. (B) Mice treated with PhR peptides exhibited on average the smallest tumors. Data are plotted as mean ± SEM. (C) Relative changes in tumor volume versus time. Error bars represent SEMs for triplicate data. Mean tumor volumes were analyzed using one-way ANOVA. Values are means ± SEM, n=4-6 tumors. (D) Kaplan-Meier survival curves for epidermal graft tumor nude mice treated with PhR, PhS and Nutlin-3a for 21 days and raised for another 24 days post treatment. The nutlin-3a caused 20% mice death in the first week of treatment. (E) The body distribution of PhR-Cy3 at different time points after intratumoral injection.
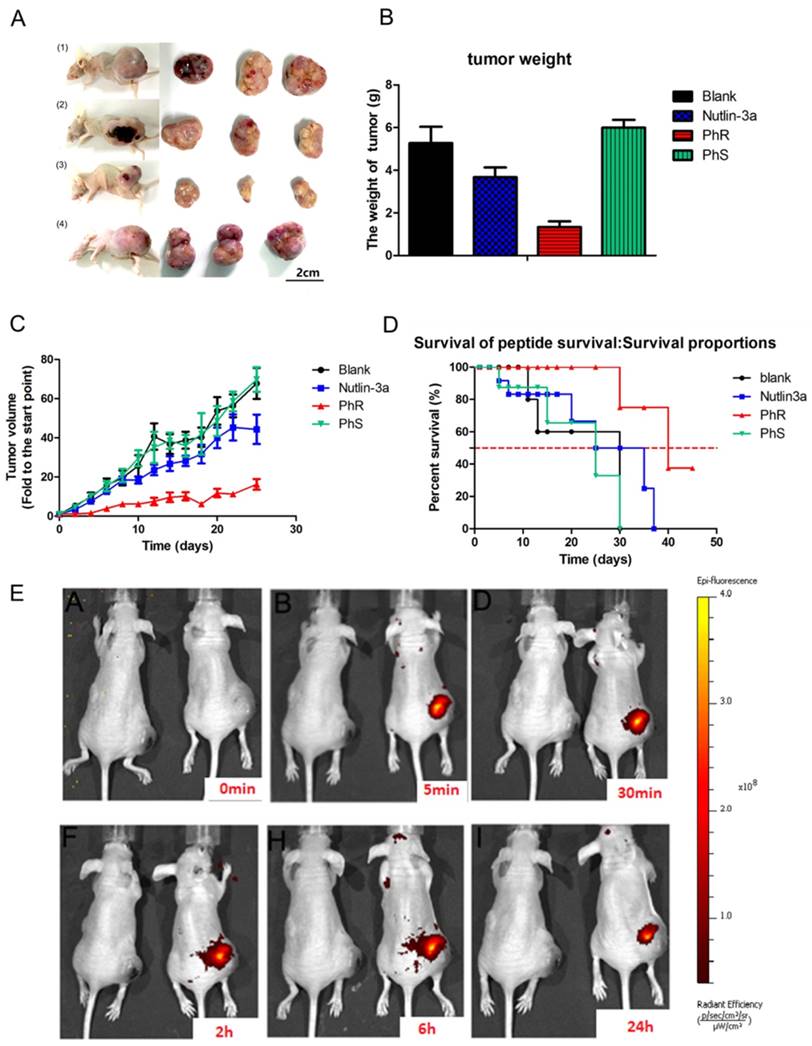
Immunohistochemistry assay was used to check the p53 and target protein expression levels. Notably, higher expression levels of p53 and p21 in PhR-treated tumor tissues were detected via immunohistochemistry (Fig. S11a-b); these results were consistent with the in vitro results. Caspase-3 and proliferating cell nuclear antigen (PCNA) detection via immunohistochemistry was used to determine the apoptosis level (Fig. S11c-d). Although nutlin-3a displayed a lower IC50 than PhR in vitro, when the mice were treated with higher doses (10 mg/kg) in vivo it induced a lower level of apoptosis and displayed less tumor inhibition than peptide (10 mg/kg).
According to the in vitro and in vivo experiments, PhR efficiently inhibited cancer growth in p53 function-deficient mice. However, low-toxicity and harmless elimination from the body within a reasonable timeframe are as important to consider as efficacy. A mouse voluntary cage-wheel exercise assay was designed to assess the toxicity, and histological studies were performed for the biodistribution of PhR in vivo. In the mouse voluntary cage-wheel exercise assay, BALB/c mice were randomly divided into two groups and were subcutaneously injected with PBS or PhR (10 mg/kg). Over a period of 20 days after injection, the voluntary running cycles increased steadily and no significant differences were found between these two groups, which indicates no obvious harmful effect to the motor learning ability of mice treated with PhR peptide (Fig. S12). Histological analysis was performed to further evaluate the toxicity of PhR in vivo. Major organs including the tumor, heart, liver, spleen, lung, kidney, and brain tissue sections were harvested for hematoxylin and eosin (H&E) staining to observe histological changes at 21 days after injection. The PhR group had a more apparent tumor inhibition effect compared to other groups (Fig. S13a). Compared with the PBS-treated mice, no signs of organ lesions were observed in the mice treated with PhR (Fig. S13b). Therefore, these in vivo toxicity assessments strongly indicate PhR as a low-toxic and biocompatible modulator for cancer therapy.
Discussion
In summary, a potent dual peptide inhibitor based on the CIH concept was designed to specifically target MDM2/X and could further induce p53-dependent apoptosis and inhibit cell proliferation. Notably, the CIH strategy provides an ideal way to obtain peptides with identical chemical compositions as controls instead of mutated or scrambled control peptides used in all previous studies. The S-PDI epimers were used as controls for target binding affinity, cellular uptake, cell viability, and protein/mRNA regulation. The results clearly emphasize the decisive correlation between the peptides' secondary structure and their functions.
To our knowledge, this is the first report of stabilized peptides showing inhibitory effects on CSCs. Despite its relatively moderate in vitro activity, the superior in vivo efficacy and minimal toxicity of the CIH peptide PhR compared with nutlin-3a shows that it is a promising candidate for further development. From this study, we learned that binding affinity, cellular uptake and function should be evaluated synergetically to provide a suitable candidate, and that in vivo efficacy and toxicity should be examined to obtain a more comprehensive evaluation of a candidate's reactivity.
Abbreviations
CSC: cancer stem cell; PPIs: protein-protein interactions; CIH: chirality-induced helicity; MDM2: murine double minute 2; MDMX: murine double minute X; OVTC: ovarian teratocarcinoma; FITC: fluorescein isothiocyanate; FBS: fetal bovine serum; PBS: phosphate buffered saline; DAPI: 4, 6-diamidino-2-phenylindole; DMSO: dimethyl sulfoxide; MTT: 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenylt-etrazolium bromide; rt-qPCR: real-time quantitative PCR; FACS: fluorescence activated cell sorting; IP: immunoprecipitation; TGI: tumor growth inhibition; PCNA: proliferating cell nuclear antigen.
Supplementary Material
Experimental methods and procedures for peptide synthesis, sphere formation assay, protein production, flow cytometry, IHC, Mice voluntary cage-wheel exercise and in vivo imaging were provided. Supplementary figures about fluorescence polarization assay data, confocal image, the original data for cell cycle analysis and apoptosis, the RT-PCR analysis, the immunohistochemistry analysis, the H&E stained images and other data; tables about peptide sequences, peptide mass statistics, primer sequence of genes for RT-PCR analysis, schemes, and the chemical structure of peptides were showed. The full gel image of western blot also included.
Acknowledgements
We appreciate professor Lahav (Harvard Medical School) for kindly help and serve us HCT-116 p53 Venus knock-in cell line (p53-VKI) cells. Thank professor Olaf (University of Notre Dame) for the kind help of discussing the manuscript. We acknowledge financial support from the Natural Science Foundation of China Grants 21372023 and 81572198; MOST 2015DFA31590, the Shenzhen Science and Technology Innovation Committee, JCYJ20170412150719814, JCYJ20170412150609690, JCYJ20150403101146313, JCYJ20160301111338144, JCYJ20160331115853521, JSGG20160301095829250 and GJHS201703100931223 65; China Postdoctoral Science Foundation 2017M610704and the Fundamental Research Funds for the Central Universities (2682017CX092) and the Provincial Natural Science Research Project of Anhui Province (Grant KJ2013A142 to W.L.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest. 2010;120:41-50
2. Li Y, Laterra J. Cancer Stem Cells: Distinct Entities or Dynamically Regulated Phenotypes? Cancer Res. 2012;72:576-80
3. Chen K, Huang Y-h, Chen J-l. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34:732-40
4. Francescangeli F, Contavalli P, De Angelis ML, Baiocchi M, Gambara G, Pagliuca A. et al. Dynamic regulation of the cancer stem cell compartment by Cripto-1 in colorectal cancer. Cell Death Differ. 2015;22:1700-13
5. Marhold M, Tomasich E, El-Gazzar A, Heller G, Spittler A, Horvat R. et al. HIF1α Regulates mTOR Signaling and Viability of Prostate Cancer Stem Cells. Mol Cancer Res. 2015;13:556-64
6. Liu J, Kopečková P, Bühler P, Wolf P, Pan H, Bauer H. et al. Biorecognition and Subcellular Trafficking of HPMA Copolymer-Anti-PSMA Antibody Conjugates by Prostate Cancer Cells. Mol Pharm. 2009;6:959-70
7. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167-74
8. Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC. et al. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462:182-188
9. Bird GH, Madani N, Perry AF, Princiotto AM, Supko JG, He XY. et al. Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic. P Natl Acad Sci U S A. 2010;107:14093-98
10. Milroy L-G, Grossmann TN, Hennig S, Brunsveld L, Ottmann C. Modulators of Protein-Protein Interactions. Chem Rev. 2014;114:4695-748
11. Hu K, Geng H, Zhang Q, Liu Q, Xie M, Sun C. et al. An In-tether Chiral Center Modulates the Helicity, Cell Permeability, and Target Binding Affinity of a Peptide. Angew Chem Int Ed. 2016;55:8013-17
12. Hu K, Li W, Yu M, Sun C, Li Z. Investigation of Cellular Uptakes of the In-Tether Chiral-Center-Induced Helical Pentapeptides. Bioconjugate Chem. 2016;27:2824-27
13. Jiang Y, Hu K, Shi X, Tang Q, Wang Z, Ye X. et al. Switching substitution groups on the in-tether chiral centre influences backbone peptides' permeability and target binding affinity. Org Biomol Chem. 2017;15:541-44
14. Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD. et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466-70
15. Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J Am Chem Soc. 2007;129:2456-57
16. Grossmann TN, Yeh JTH, Bowman BR, Chu Q, Moellering RE, Verdine GL. Inhibition of oncogenic Wnt signaling through direct targeting of beta-catenin. P Natl Acad Sci U S A. 2012;109:17942-47
17. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307-10
18. Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis - the p53 network. J Cell Sci. 2003;116:4077-85
19. Cheok CF, Verma CS, Baselga J, Lane DP. Translating p53 into the clinic. Nat Rev Clin Oncol. 2011;8:25-37
20. Mogi A, Kuwano H. TP53 Mutations in Nonsmall Cell Lung Cancer. J Biomed Biotechnol. 2011;2011:583929
21. Chung W-M, Chang W-C, Chen L, Lin T-Y, Chen L-C, Hung Y-C. et al. Ligand-independent androgen receptors promote ovarian teratocarcinoma cell growth by stimulating self-renewal of cancer stem/progenitor cells. Stem Cell Research. 2014;13:24-35
22. Sekar D, Krishnan R, Panagal M, Sivakumar P, Gopinath V, Basam V. Deciphering the role of microRNA 21 in cancer stem cells (CSCs). Genes & Diseases. 2016;4:277-81
23. Yaginuma Y, Westphal H. Abnormal Structure and Expression of the p53 Gene in Human Ovarian Carcinoma Cell Lines. Cancer Res. 1992;52:4196-99
24. Reich NC, Oren M, Levine AJ. Two distinct mechanisms regulate the levels of a cellular tumor antigen, p53. Mol Cell Biol. 1983;3:2143-50
25. Wang L, Wu Q, Qiu P, Mirza A, McGuirk M, Kirschmeier P. et al. Analyses of p53 Target Genes in the Human Genome by Bioinformatic and Microarray Approaches. J Bio Chem. 2001;276:43604-10
26. Tovar C, Graves B, Packman K, Filipovic Z, Xia BHM, Tardell C. et al. MDM2 Small-Molecule Antagonist RG7112 Activates p53 Signaling and Regresses Human Tumors in Preclinical Cancer Models. Cancer Res. 2013;73:2587-97
27. Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the p53 Tumor Suppressor Pathway by a Stapled p53 Peptide. J Am Chem Soc. 2007;129:2456-57
28. Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH. et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A. 2013;110:E3445-54
29. Chung W-M, Chang W-C, Chen L, Chang Y-Y, Shyr C-R, Hung Y-C. et al. MicroRNA-21 promotes the ovarian teratocarcinoma PA1 cell line by sustaining cancer stem/progenitor populations in vitro. Stem Cell Res & Ther. 2013;4:1-10
30. Hu B, Gilkes DM, Chen J. Efficient p53 activation and apoptosis by simultaneous disruption of binding to MDM2 and MDMX. Cancer Res. 2007;67:8810-17
31. Baek S, Kutchukian PS, Verdine GL, Huber R, Holak TA, Lee KW. et al. Structure of the stapled p53 peptide bound to Mdm2. J Am Chem Soc. 2012;134:103-106
32. Chen L, Lu W, Agrawal S, Zhou W, Zhang R, Chen J. Ubiquitous induction of p53 in tumor cells by antisense inhibition of MDM2 expression. Mol Med. 1999;5:21-34
33. Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L. et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661-665
34. Wang W, Takimoto R, Rastinejad F, El-Deiry WS. Stabilization of p53 by CP-31398 Inhibits Ubiquitination without Altering Phosphorylation at Serine 15 or 20 or MDM2 Binding. Mol Cell Biol. 2003;23:2171-81
35. Brooks H, Lebleu B, Vivès E. Tat peptide-mediated cellular delivery: back to basics. Adv Drug Deliver Rev. 2005;57:559-77
36. Zhang X, Lu F, Wang J, Yin F, Xu Z, Qi D. et al. Pluripotent Stem Cell Protein Sox2 Confers Sensitivity to LSD1 Inhibition in Cancer Cells. Cell Rep. 2013;5:445-57
37. Wahl AF, Donaldson KL, Fairchild C, Lee FY, Foster SA, Demers GW. et al. Loss of normal p53 function confers sensitization to Taxol by increasing G2/M arrest and apoptosis. Nat Med. 1996;2:72-79
38. Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol. 2001;13:332-37
39. Hsieh J-K, Chan FSG, O'Connor DJ, Mittnacht S, Zhong S, Lu X. Regulates the Stability and the Apoptotic Function of p53 via MDM2. Mol Cell. 1999;3:181-93
40. Brooks CL, Gu W. p53 Ubiquitination: Mdm2 and Beyond. Mol Cell. 2010;21:307-15
41. Paek AL, Liu JC, Loewer A, Forrester WC, Lahav G. Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell. 2016;165:631-42
Author contact
![]() Corresponding authors: Zi-gang Li, email: lizgedu.cn Wei Liang, email: williamleungcom
Corresponding authors: Zi-gang Li, email: lizgedu.cn Wei Liang, email: williamleungcom