Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(17):4204-4216. doi:10.7150/thno.21516 This issue Cite
Research Paper
pH-Sensitive Nano-Complexes Overcome Drug Resistance and Inhibit Metastasis of Breast Cancer by Silencing Akt Expression
Jieying Yin1,2*, Tianqun Lang1,3*, Dongmei Cun2, Zhong Zheng1, Yan Huang1,2, Qi Yin1 ![]() , Haijun Yu1, Yaping Li1
, Haijun Yu1, Yaping Li1 ![]()
1. State Key Laboratory of Drug Research & Center of Pharmaceutics, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;
2. School of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, China;
3. University of Chinese Academy of Sciences, Beijing 100049, China.
* These authors contributed equally to this work.
Received 2017-6-19; Accepted 2017-8-17; Published 2017-9-26
Abstract

The therapy of breast cancer is encumbered by drug resistance and metastasis, which can be due to a defective PI3K/AKT/mTOR signaling pathway. This study was aimed at improving the anti-cancer effect of the chemotherapeutic agent paclitaxel (PTX) on the drug resistant and metastatic breast cancer by co-delivering PTX and a siRNA, siAkt, directed at silencing the Akt expression.
Methods: The pH-sensitive amphiphilic polymer, poly [(1,4-butanediol)-diacrylate-β-N, N-diisopropylethylenediamine]-polyethyleneimine (BDP) was synthesized. The PTX-loaded BDP micelle/siAkt nano-complex (PMA) was prepared and characterized. The cellular uptake, cytotoxicity, RNA interference efficiency, biodistribution, pharmacokinetics, pharmacodynamics, and biocompatibility of PMA in the murine metastatic breast cancer 4T1 cells and the 4T1 tumor-bearing mice were evaluated.
Results: PMA was stable at the neutral as well as tumor extracellular pH and released the drugs in the intra-endo/lysosome acidic environment. In 4T1 cells, the RNA interference against the Akt gene down-regulated the expression of Akt and P-glycoprotein and up-regulated the expression of Caspase-3. The down-regulated P-gp inhibits the efflux of PTX thereby increasing its intracellular concentration, improving the cytotoxicity, and inhibiting the migration and invasion of 4T1 cells. In the 4T1 tumor-bearing mice, co-delivery of PTX and siAkt by PMA achieved a tumor inhibiting rate of 94.1% and suppressed 96.8% lung metastases. PMA did not cause pathological lesions in normal organs.
Conclusion: PMA, by virtue of overcoming drug resistance and simultaneously restraining lung metastasis, might be an efficient drug delivery system for the therapy of breast cancer.
Introduction
Breast cancer is the cause of high morbidity and mortality in women world-wide [1]. Two major obstacles, drug resistance and metastasis, ultimately lead to the failure of clinical treatment of breast cancer [2-4]. Especially, metastasis accounts for 90% of death in breast cancer patients [5]. Drug resistance and metastasis result from the mutations in the genes that regulate cell proliferation, and angiogenesis [6, 7]. An interesting theory describes cancer-related signaling pathways as the 'trunk' and 'limb' of a cancer evolutionary tree, where the 'trunk' fuels initial tumorigenesis and the 'limb' induces or accelerates acquired resistance to therapy and metastasis [8]. Consequently, drug resistance is always a concomitant of metastasis in patients; therefore, both issues must be resolved for a successful treatment.
Akt, the central node of the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway, plays a key role in the regulation of cell survival, proliferation, and angiogenesis [9, 10]. The deregulation of Akt in human cancer often underlies the development of drug resistance [11, 12] and metastasis [13, 14]. Thus, Akt is a key target in cancer therapy for tackling both drug resistance and metastasis [15]. The silencing of Akt expression can greatly substantiate the efficacy of chemotherapy. In this respect, the siRNA targeting Akt (siAkt) and chemotherapeutic agents can be co-delivered by nano-vectors, which are popular tools in the field of anti-cancer drugs delivery [16]. pH-sensitive nanoparticles are good candidates with their passive tumor-targeting ability due to the enhanced permeability and retention effect of solid tumors [17] and site-specific drug release exploiting the pH gradient between the blood circulation and the tumor sites [18]. The endosomal pH-responsive nanoparticles have previously been used for enhanced cytosolic siRNA delivery [19-21]. The N, N-diisopropylethylenediamine (DPA) group has a pKa value of 6.3 [22], which is below the extracellular pH in the tumor tissues (6.8-7.0) [23, 24] and above the pH inside the endo/lysosomes (4.5-6) [25]. It is therefore an appropriate candidate to be employed in the drug delivery system for intracellular drug release.
In this study, we synthesized a pH-sensitive amphiphilic polymer, in which the hydrophobic block contained repeating units of DPA groups and the hydrophilic block was the low molecular weight polyethyleneimine (PEI) forming micelles via self-assembly. The anti-cancer agent paclitaxel (PTX) was loaded at the neutral pH and the siAkt was condensed in the micelle by the cationic charges of PEI. The resulting PM/siAkt nano-complex (PMA) was characterized and the inhibitory effect of PMA on drug resistance and metastasis of breast cancer was evaluated in the metastatic murine mammary cancer 4T1 cells and the 4T1-tumor bearing mice.
Materials and Methods
Materials
Polyethyleneimine 2000 Da branched (PEI2000), 1,4-butanediol diacrylate (BDA), and DPA were purchased from J&K Scientific Ltd. (Shanghai, China). Sulforhodamine B-based in vitro toxicology assay kit was obtained from Sigma-Aldrich (St. Louis, USA). RPMI 1640 medium, fetal bovine serum (FBS), YOYO-1, Hoechst 33342, LysoTracker Red DND-99, Oregon Green® 488 Conjugate paclitaxel (Oregon Green-PTX), Dead Cell Apoptosis Kit with Annexin V Alexa Fluor™ 488 & Propidium Iodide, and 1,1'-dioctadecyl-3,3,3',3'-tetramethylindotricarbocyanine iodide (DiR) were purchased from Thermo Fisher Scientific Inc (Waltham, USA). PTX was obtained from Melun Biotechnology Co., Ltd. (Dalian, China). Anti-P glycoprotein (P-gp) antibody, Anti-β actin antibody, and Goat anti-rabbit IgG H&L (HRP) for Western blotting were purchased from Abcam Trading Company Ltd. (Cambridge, UK). Anti-Akt2 polyclonal antibody (HRP) was purchased from Bioss Antibodies Inc. (Woburn, U.S.A.). Anti-Akt2 antibody for immunohistochemistry (IHC) was acquired from R&D Systems, Inc. (Minneapolis, U.S.A.). Anti-Caspase-3 antibody for IHC was purchased from Cell Signaling Technology, Inc. (Danvers, U.S.A.). Matrigel was obtained from BD Biosciences (San Jose, U.S.A.). Crystal violet was purchased from Beyotime Biotechnology (Shanghai, China). Small interfering RNA duplexes against Akt2 (siAkt) [5'-CGCCAUGGAUUACAAGUGUTT-3' (sense)], negative control RNA nonspecific to any human gene (siNC) [5'-UUCUCCGAACGUGUCACGUTT-3' (sense)], FAM labeled siAkt (FAM-siAkt), and Cy3 labeled siAkt (Cy3-siAkt) were ordered from GenePharm Co. Ltd (Shanghai, China). All other reagents were purchased from Sinopharm Chemical Reagent Co. Ltd and of analytical grade.
Cell culture
The murine mammary carcinoma cell line 4T1 was obtained from Cell Bank of Shanghai, Chinese Academy of Sciences (Shanghai, China) and was cultured in RPMI 1640 medium containing 10% FBS, 1.5 g/L sodium bicarbonate, 2.5 g/L glucose and 0.11 g/L sodium pyruvate. The cells were cultured in a humidified atmosphere containing 5% CO2 at 37℃.
Animals
BALB/c nude mice (female, 4-6 weeks, 16-20 g) were purchased from Shanghai Experimental Animal Center (Shanghai, China) and kept in the SPF grade environment of the Animal Care Facility. All experiments utilizing animals were supervised by the Institutional Animal Care and Use Committee (IACUC) of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
Synthesis of the polymers
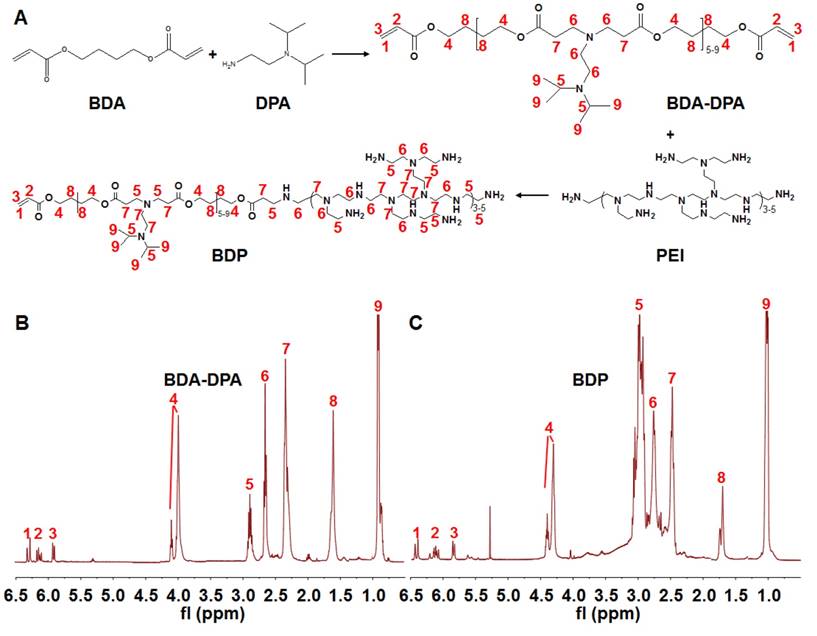
Synthesis of poly[(1,4-butanediol)-diacrylate-β-N, N-diisopropylethylenediamine] (BDA-DPA) was carried out by mixing BDA (1.0 g) and DPA (0.48 g) at 80℃ for 24 h (Figure 1a). The product was dialyzed (MWCO 3.5 kDa) in ethanol for 24 h. After ethanol had been removed, PEI2000 (0.11 g) dissolved in dimethyl sulfoxide (DMSO, 3 mL) was added dropwise into the DMSO solution (3 mL) of BDA-DPA followed by stirring at 50℃ for 4 h. The product in DMSO was dialyzed in water (MWCO 3.5 kDa) for 48 h to remove the redundant reactants. The final products (BDA-DPA-PEI, BDP) were lyophilized and stored at -80℃ until use. The structures of BDA-DPA and BDP were confirmed by 1H NMR spectra (Mercury Plus-400, Varian, USA) at 400 MHz. The mean molecular weight (Mw) was calculated according to its retention time and the standard curve tested by the gel permeation chromatograph (GPC) (Waters 2695, USA). The pKa of BDP was measured by the potentiometric titration method.
Synthesis of the polymers. (A) The flowchart of the synthesis of BDA-DPA and BDP. (B, C) The 1H NMR spectra of BDA-DPA (B) and BDP (C).
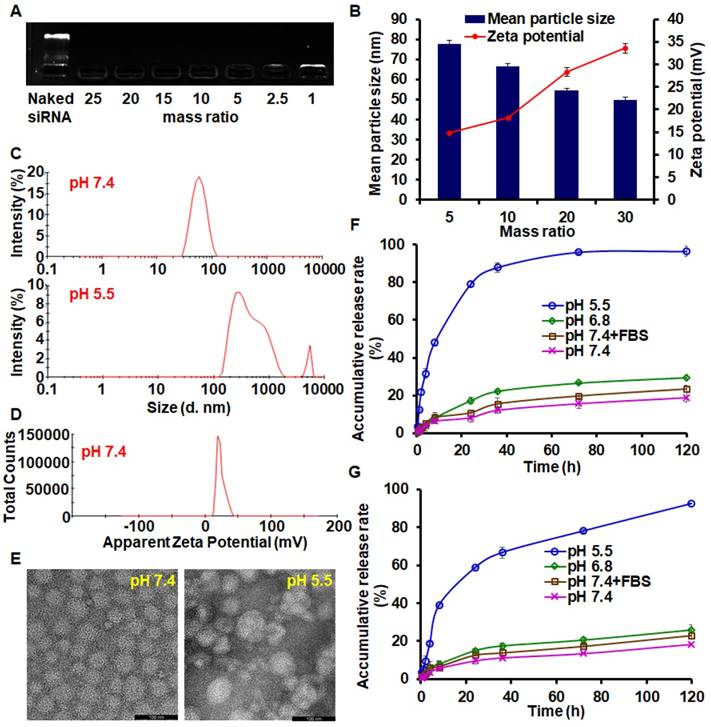
Preparation and characterization of PTX-loaded BDP/siAkt nano-complex (PMA)
BDP (30 mg) and PTX (2 mg) were dissolved in 5 mL methanol, and the PTX-loaded BDP micelle (PM) was formed by thin film hydration method. The blank BDP micelle without PTX (BM) was prepared by a similar method. siAkt or siNC (negative control) solution in water was mixed with PM at different BDP/siAkt mass ratios and vortexed to form PMA or PM/siNC nano-complex (PMN). The siRNA-condensing capability of PMA was determined by agarose gel electrophoresis. The mean particle sizes and ζ-potential were measured by dynamic light scattering (DLS) on Zetasizer Nano ZS90 (Malven, USA). The appearance of PMA was visualized by a transmission electron microscope (TEM). The concentration of PTX in PMA was measured with the high-performance liquid chromatography (HPLC) method, and the drug loading (DL) and encapsulation efficiency (EE) were calculated according to the following formula:
DL% = (weight of the drug in PMA)/(weight of the feeding polymer and drug)×100% (1)
EE% = (weight of the drug in PMA)/(weight of the feeding drug)×100% (2)
The releasing profile of PTX from PMA in different environments was investigated through the dialysis method. PMA (BDP/siAkt mass ratio: 20) containing 1 mg/mL PTX was added into the dialysis bags, which were incubated in different releasing media: phosphate-citric acid buffered saline (pH 5.5), phosphate buffered saline (PBS) (pH 6.8), PBS (pH 7.4) and PBS containing 10% FBS (pH 7.4). All media contained 1 M sodium salicylate. After incubation at 37℃ with shaking for different times, the releasing media were taken out to measure the drug concentration by the HPLC method and fresh media were supplemented. Since siRNA has a large molecular weight, the siRNA release behavior from PMA was tested by the ultracentrifugation method. PMA containing 0.5 mg/mL siAkt in 10 mL media was shaken at 37℃. At preset time points, 0.5 mL medium was taken and stained with YOYO-1. The fluorescent intensity was immediately measured on a microplate reader at 491 nm excitation and 509 nm emitted light. The cumulative releasing rates of PTX and siAkt were calculated.
Cellular uptake and intracellular localization
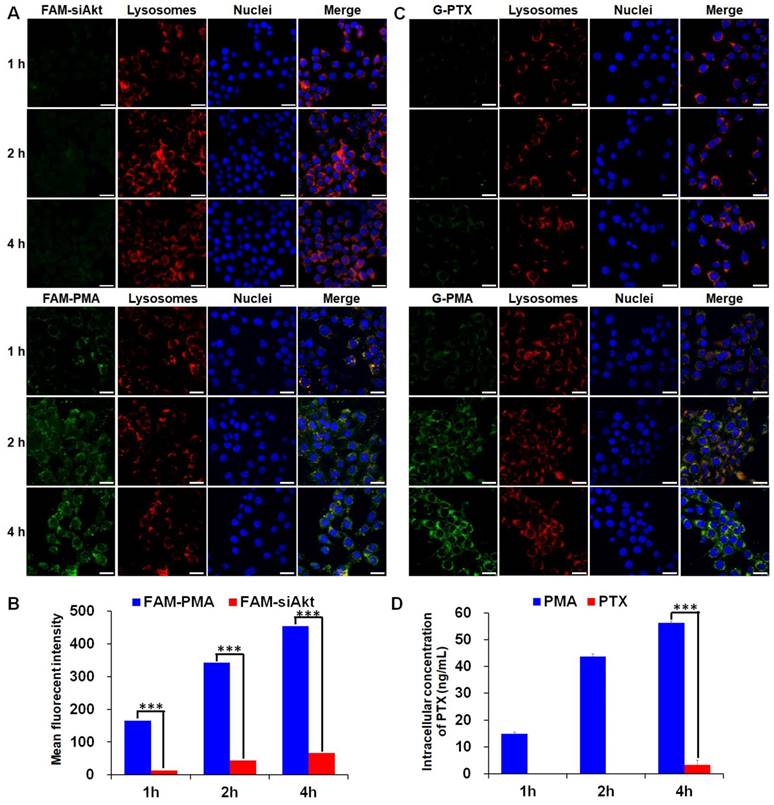
FAM-labeled PMA was formed by mixing PM and FAM-siAkt, and Oregon Green® 488-labeled PMA (G-PMA) was prepared by replacing PTX with Oregon Green-PTX (G-PTX). Murine mammary carcinoma 4T1 cells were seeded on the round glass coverslips (Ø10 mm) in 24-well plates at the density of 5×104 cells per well. After cells had attached, FAM-siAkt, FAM-PMA, G-PTX (DMSO solution) or G-PMA containing 50 nM siRNA and 0.1 μg/mL PTX was added into each well. Following incubation for 40, 100, and 220 min, Hoechst 33342 and Lyso Tracker Red were added to stain the nuclei and lysosomes for another 20 min. Subsequently, the cells were washed, fixed with paraformaldehyde (4 %), and photographed under a laser scanning confocal microscope (LSCM, FluoView TM FV1000, Olympus). For quantitative analysis, cells were incubated with FAM-siAkt, FAM-PMA, free PTX (DMSO solution) or PMA for 1, 2, and 4 h. For FAM-siAkt and FAM-PAM groups, the relative fluorescent intensity was determined by the FACS Calibur flow cytometer (Becton Dickinson, USA). For PTX and PMA groups, the cells were shattered by ultrasonication and the intracellular concentration of PTX was measured by HPLC.
RNA interference efficiency
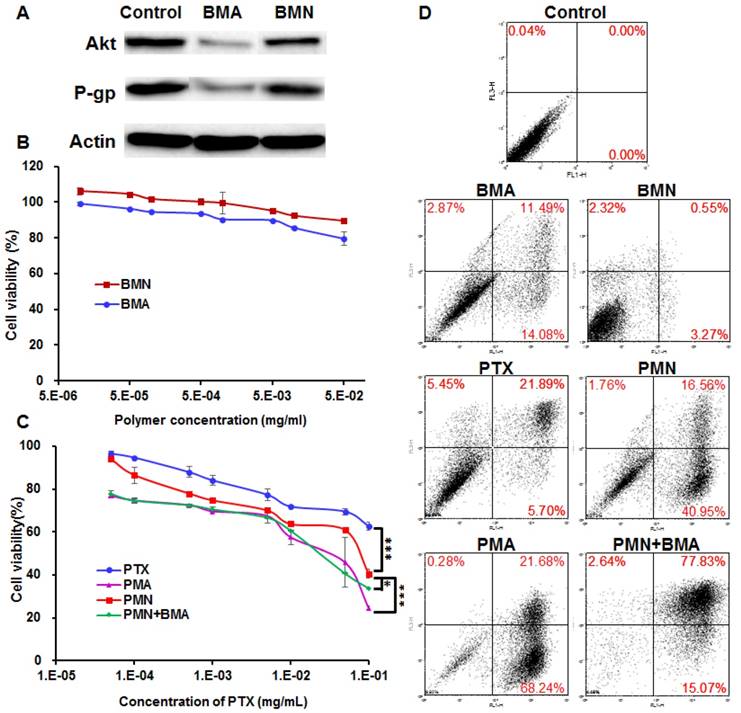
To investigate the siRNA delivery efficiency of the BDP micelle/siAkt nano-complex, BM/siAkt nano-complex (BMA) and BM/siNC nano-complex (BMN) at BDP/siRNA mass ratio of 20 were prepared and added to 4T1 cells at the siRNA concentration of 50 pmol/mL. PTX was not loaded in the nanoparticles because its cytotoxicity could interfere with the experimental results. The expression level of Akt and P-gp was determined by Western blotting after incubation for 48 h.
Cytotoxicity
BMA, BMN, free PTX (DMSO solution), PMA, PMN, and the mixture of PMN and BMA (PMN+BMA) were added onto 4T1 cells that were seeded in 96-well plates. The medium was replaced with the fresh one 4 h later, followed by another 44 h-incubation. Cell viability was tested using the Sulforhodamine B-based in vitro toxicology assay kit. The IC50 values were fitted by the GraphPad Prism software. The cells were stained with the Cell Apoptosis Kit and the cell apoptosis rate was determined by flow cytometry.
Cell migration and invasion
The influence of Akt silencing on the mobility of 4T1 cells in vitro was evaluated. PTX was not incorporated in the nanoparticles to avoid cytotoxicity, which would reduce the cell activity.
Wound healing assay
4T1 cells were incubated with BMN or BMA (BDP/siAkt mass ratio 20, 50 pmol/mL siRNA) for 24 h and scratched with a pipette tip. The cicatrization of the wound was observed under a microscope and the percentage of the wound area was calculated.
Migration and invasion assays
4T1 cells pre-treated with BMN or BMA (50 pmol/mL siRNA) for 24 h were added into the transwell in the serum-free medium at the density of 1 × 105 cells per well and 2 × 105 cells per well for the migration assay and invasion assay, respectively. In the invasion assay, the transwell top chambers were pre-coated with the Matrigel. 10% FBS-containing medium was used as the chemoattractant in the lower chambers. 24 h later, cells passing onto the other side of the transwell membrane were stained with crystal violet, photographed, and counted.
Biodistribution and pharmacokinetics
siAkt was labeled with Cy3 and the PM/Cy3-siAkt nano-complex (PMC) and the DiR-loaded BDP/siAkt nano-complex (DMA) were prepared. The 4T1 breast cancer-bearing mice model was generated by injecting 1 × 105 4T1 cells on the right mammary gland of the female nude mice. The mice were randomly grouped when the tumor volume reached approximately 100 mm3 and administrated with Cy3-siAkt solution, PMC, DiR solution, DMA, Taxol (the solution of PTX dissolved in the ethanol/Cremophor EL mixture, which is the commercial injection of PTX) and PMA (1.5 mg/kg siRNA, 2 mg/kg DiR and 10 mg/kg PTX). The formulation of free PTX in vivo is different from that used in vitro as DMSO cannot be used in living animals due to its toxicity. At 2, 4, and 8 h post injection, mice were sacrificed and the tumor, heart, liver, spleen, lung, and kidney were removed. The tissues of the Cy3-siAkt, PMC, DiR and DMA groups were photographed using an in vivo imaging system (Carestream Health, USA). For quantitative analysis, the tissues of the Cy3-siAkt, PMC, Taxol and PMA groups were homogenized. The concentration of Cy3-siAkt and PTX was measured by the microplate reader (Perkin Elmer, USA) and HPLC, respectively.
SD rats were randomly grouped and administrated with Taxol, PMA, Cy3-siAkt, and PMC (2.8 mg/kg PTX and 0.4 mg/kg siRNA). At 0, 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24 and 48 h post injection, about 0.5 mL per mouse blood were taken to harvest the plasma. The concentration of PTX and siAkt in the plasma was measured by HPLC and the microplate reader, respectively.
In vivo anti-tumor effect
Nude mice bearing 4T1 tumors were divided into 6 groups randomly and intravenously injected with saline, Taxol, BMA, PMN, BMA+PMN, and PMA twice a week for 3 weeks (1.5 mg/kg siRNA and 10 mg/kg PTX). The tumor volumes and body weights were monitored in the following 25 days after the first administration. At day 25, the mice were sacrificed and the tumors and lungs were collected. The tumors were weighed and sectioned. To test the in vivo gene silencing efficiency, the expression of Akt and Caspase-3 in the tumors was examined by IHC. Cell apoptosis was detected by TdT-mediated dUTP Nick-End Labeling (TUNEL). The metastasis foci on the lungs were counted, then the lungs were sectioned and stained with hematoxylin and eosin (H&E).
Pathological evaluation
Healthy BALB/c mice were divided into 6 groups randomly and intravenously injected with saline, Taxol, BMA, PMN, BMA+PMN, and PMA (1.5 mg/kg siRNA and 10 mg/kg PTX) twice a week for three weeks. On day 25 after the first administration, the mice were sacrificed and the organs (heart, liver, spleen, lung and kidney) were removed and the H&E staining was performed.
Statistical analysis
All experiments were done with at least three replications and the results were shown as the mean values ± SD. The difference between two groups was evaluated by the Student's t-test and considered significant when p<0.05, and very significant when p<0.01 and p<0.001.
Results and Discussion
Synthesis of the polymers
Figure 1A displays the diagram for the synthesis of the polymers. BDA-DPA was hydrophobic and converted to amphiphilic after conjugating with PEI. The three peaks in the 1H NMR spectrum at ppm 5.80, 6.09 and 6.48 represented the acrylate terminal group of BDA-DPA, and the sharp peak at ppm 0.99 was the signal of the methyl group of DPA on the side chain (Figure 1B). Hence, DPA and BDA were successfully polymerized via the Michael addition reaction. The peak areas at ppm 2.32-2.48, 2.56-2.78 and 2.80-3.06, which belonged to the methylene groups next to the amines, remarkably increased after BDA-DPA reacted with PEI, proving that PEI was successfully conjugated onto BDA-DPA. According to GPC analysis, the molecular weights of BDA-DPA and BDP were 4150 Da and 6393 Da, respectively. Thus, the molar ratio of BDA-DPA to PEI in BDP was 1:1. The pKa value of BDP was 6.29.
Characterization of PTX-loaded BDP/siAkt nano-complex (PMA)
The migration of siAkt bands on the electrophoresis strips was completely retarded by BDP when the BDP/siAkt mass ratios were no less than 1 (Figure 2A). Although the low molecular weight PEI had poor nucleic acid-condensing ability [26], the PEI block on BDP had stronger condensing ability because the amphiphilic BDP assembled to form a micelle structure and the cationic charges of PEI aggregated. At pH 7.4, the complexes of PM and siAkt at the BDP/siAkt mass ratios ranging from 5 to 30 formed nanoparticles with the mean particle sizes of 50 - 80 nm and the zeta potential of +14 - +35 mV (Figure 2B). The particle sizes decreased with the increasing BDP/siAkt mass ratio, but the variation of the particle size was minimal when the BDP/siAkt mass ratio was over 20. The PMA at BDP/siAkt mass ratio of 20 was therefore chosen in the subsequent experiments to avoid the cytotoxicity caused by the high positive charge [27]. At pH 5.5, the particle size enlarged and lost the homogeneity (Figure 2C). The signal/noise ratio was too low for the detection of zeta-potential which might be because PMA dissociated and no dispersed particle existed at low pH (Figure 2D); the amorphous structure of PMA was also affected by the low pH. PMA appeared as spheres with homogeneous size at pH 7.4 under TEM but showed irregular shape and size at pH 5.5 (Figure 2E). These phenomena indicated that PMA underwent structure changes at the acidic pH. The DL and EE of PMA measured by HPLC were 4.45% and 90.85%, respectively. As shown in Figure 2F, PTX release behavior from PMA was environment-dependent. Different release media were used to mimic the environments of the blood circulation (pH 7.4+FBS), the extracellular space of the tumor tissues (pH 6.8), and the endosomal/lysosomal vesicles (pH 5.5). At the neutral and weak acidic pH (pH 7.4, pH 7.4+FBS and pH 6.8), less than 30% drug could be released in 120 h, indicating that PMA could be stable in the blood circulation and in the extracellular environment of tumor tissues. While in the stronger acidic pH in endosomes/lysosomes (pH 5.5), the cumulative release rate reached 79% and 96% in 24 h and 120 h, respectively. This was due to the protonation of the DPA group in the backbone of BDP at the pH value less than 6.3, leading to the conversion of the BDA-DPA block from hydrophobic to hydrophilic resulting in the dissociation of PMA. The release behavior of siAkt was similar but slightly slower than that of PTX, suggesting that the dissociation of the core of PMA led to the decrease of the cation density on the shell and the siRNA condensing ability was greatly weakened (Figure 2G). Thus, PTX and siAkt would be rapidly released inside the tumor cells.
Characterization of PMA. (A) Agarose gel electrophoresis of PMA. (B) Mean particle sizes and zeta potential of PMA at different BDP/siAkt mass ratio determined by DLS. (C) Size distribution of PMA at pH 7.4 and 5.5 at the BDP/siAkt mass ratio of 20. (D) Zeta potential distribution of PMA at pH 7.4 at the BDP/siAkt mass ratio of 20. (E) TEM images of PMA at pH 7.4 and 5.5 at the BDP/siAkt mass ratio of 20 (Scale bar: 100 nm). (F, G) The PTX (F) and siAkt (G) release profiles from PMA in media at different pH values. Data are shown as mean ± SD (n=3).
Cellular uptake and intracellular localization of PTX and siAkt
The cellular uptake of free siRNA and PTX by 4T1 cells was inefficient as the green fluorescence of FAM-siAkt and G-PTX was hardly detected by LSCM (Figure 3A, 3C). After cells had been incubated with free PTX for 1 h and 2h, the intracellular PTX concentration was below the detection limit of HPLC. Even at 4 h, the concentration was less than 5 ng/mL. The low concentration of free PTX, which can easily localize in the cells, might result from the rapid drug efflux by the overexpressed P-gp. The concentrations of siAkt and PTX mediated by PMA, on the other hand, were 6-13 fold and 16 fold higher, respectively, than that in the free state (Figure 3B, 3D), suggesting that PMA helped retain PTX inside 4T1 cells. The co-localization of siRNA and PTX in PMA with lysosomes appeared (yellow fluorescence) at 1 h. The green fluorescence did not always co-localize with the red one at 2 h, and the merged yellow fluorescence almost disappeared at 4 h, suggesting that siRNA and PTX in PMA could escape out of the lysosomes and reach the cytoplasm.
Cellular uptake of PMA. (A, C) Confocal microscopy images of 4T1 cells incubated with FAM-siAkt, FAM-PMA, G-PTX, or G-PMA for 1 h, 2 h, and 4 h (Scale bar: 20 μm). (B, D) Quantitative analysis of siAkt (B) and PTX (D) in 4T1 cells incubated with FAM-siAkt, FAM-PMA, PMA, or free PTX for 1 h, 2 h and 4 h. Data are shown as mean ± SD (n=3). ***p< 0.001.
RNA interference efficiency
The downregulation of both Akt and P-gp expression was observed in the 4T1 cells treated with BMA but not BMN (Figure 4A). It is obvious that BMA efficiently mediated RNA interference against the Akt gene. Furthermore, P-gp expression was also reduced by knocking down Akt which might account for the increased intracellular PTX concentration.
Anti-proliferative effect of PMA on 4T1 cells. (A) Immunoblotting of Akt and P-gp in 4T1 cells pre-incubated with BMA and BMN. (B, C) Viability of 4T1 cells after incubating for 48 h with BMN, BMA, free PTX, PMN, PMN+BMA, and PMA at different concentrations. (D) Flow cytometry analysis of cell apoptosis induced by BMN, BMA, free PTX, PMN, PMN+BMA and PMA. Data are shown as mean ± SD (n=3). ***p< 0.001.
Cytotoxicity
The cell viability following BMN treatment was above 90% at various polymer concentrations, suggesting that the polymer was safe for the cells (Figure 4B). The siNC neutralized partial positive charges of BM, which helped in avoiding a high zeta potential and not interrupting the cell proliferation. BMA with the same polymer concentration was slightly more effective than BMN, which might be because the downregulation of Akt gene relieved the apoptosis suppression by Akt. PTX dissolved in DMSO inhibited less than 40% cell proliferation even at a high concentration up to 100 μg/mL, reflecting resistance of 4T1 cells to PTX (Figure 4C). The poor solubility of PTX also limited its anti-tumor effect, since crystallization of PTX was evident microscopically at the concentrations over 10 μg/mL. When encapsulated in PMN, the IC50 value decreased by 8.2-fold to 7.04×10-2 mg/mL, which probably resulted from enhanced cellular uptake of PTX. PMA and the mixture of PMN and BMA had similar cytotoxicity with the IC50 values of 1.58×10-2 and 2.06×10-2 mg/mL, respectively, suggesting that proliferation was further inhibited by siAkt. Similar results were observed for cell apoptosis with the SRB assay (Figure 4D). BMN barely induced apoptosis while BMA and free PTX had weak effect on cell apoptosis. PMN, PMA and PMN+BMA induced 59%, I 90% and 95% apoptotic cells, respectively, indicating that the cytotoxicity of PTX was enhanced when it was loaded in the BDP-based nanoparticles. Furthermore, the combination of PTX and siAkt showed stronger in vitro anti-proliferative activity than the individual drugs.
Cell migration and invasion
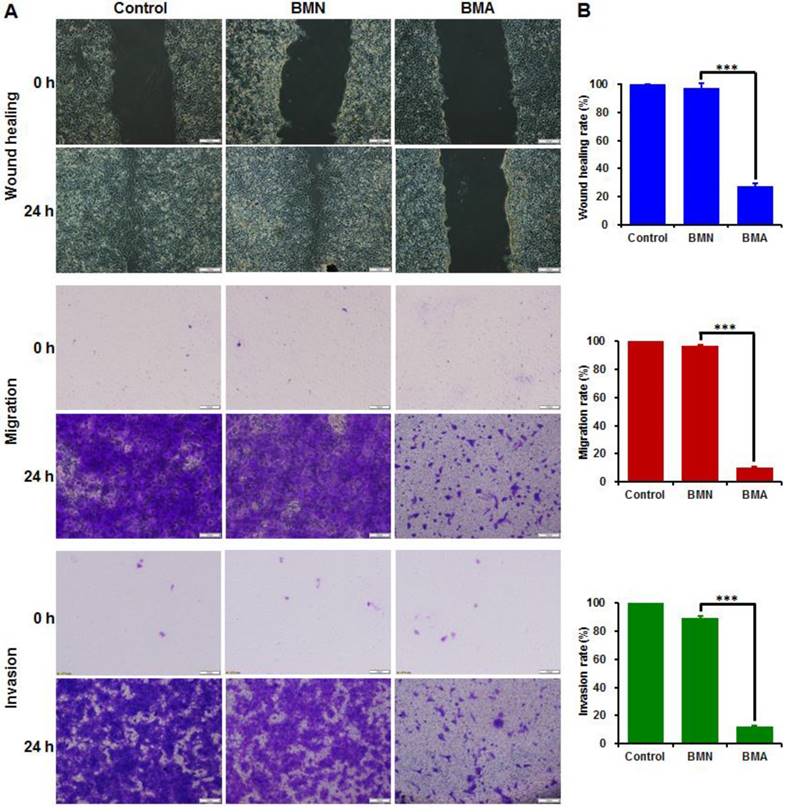
The wound healing, cell migration and invasion assays had consistent results (Figure 5). The wounds were almost completely healed in the control and BMN groups and nearly all the cells crossed the transwell membrane. The BMA-treated cells, on the other hand, showed large wound areas with few cells crossing to the other side of the transwell membrane. The wound healing, migration, and invasion rates of the BMA group were 27.4%, 10.1% and 11.9%, respectively. These results indicated that the interference against the Akt gene could restrain the in vitro metastatic behavior of 4T1 cells.
Anti-metastasis effect of BMA and BMN on 4T1 cells. Microscopy images (A) and quantitative analysis (B) of 4T1 cells pre-incubated with BMA and BMN in the wound healing, migration, and invasion assays. Cells without treatment were used as controls. Data are shown as mean ± SD (n=3). ***p< 0.001. (Scale bar: 100 μm)
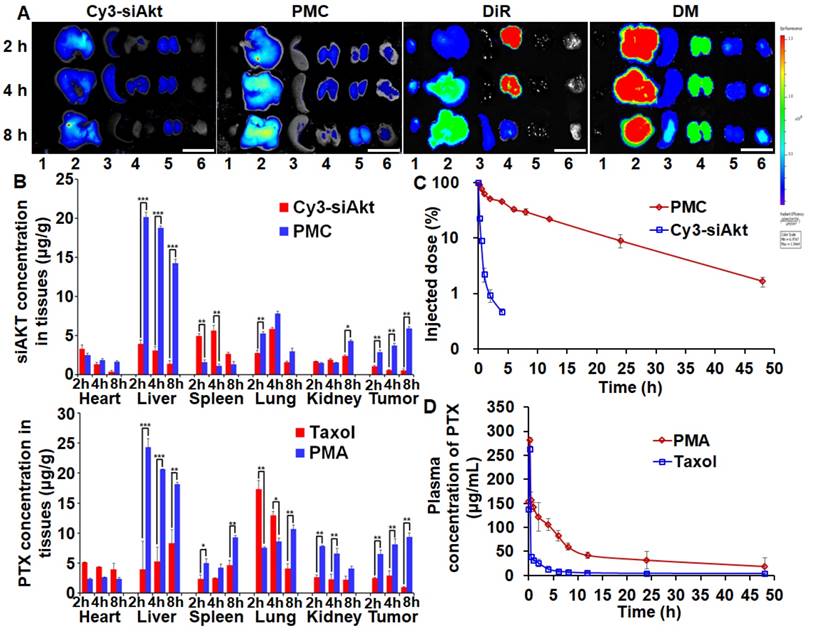
Biodistribution and pharmacokinetics
PTX and siAkt in different formulations displayed discrepant distribution patterns after systemic administration in the 4T1-tumor bearing mice (Figure 6A, 6B). Taking advantage of the enhanced permeability and retention effect of solid tumors, PMA could improve the siAkt and PTX concentrations in tumors by 11.5- and 10-fold, respectively, compared with free siAkt and Taxol, respectively. Moreover, their accumulation in the tumor increased with time up to 8 h post injection. PMA increased the accumulation of both siAkt and PTX in the liver and kidney and PTX also in the spleen, while free drugs had more accumulation in the heart and lung. It might be attributed to the cationic charge on the surface of the nanoparticles by PEI. In the plasma, free siAkt and PTX were both cleared rapidly upon injection with t1/2 less than 10 min and 1 h, respectively. When delivered through the nanoparticles, the clearance was slowed down. The t1/2 of siAkt was more than 4 h and the area under the curve (AUC(0-t)) of PTX was increased by 3.75 folds. The results indicated that PMA could protect PTX and siRNA and enhance their accumulation in the tumor.
Biodistribution and pharmacokinetics of PMA in 4T1 tumor-bearing mice. Fluorescence imaging (A) and quantitative analysis (B) of tumors and organs at 2, 4, and 8 h after intravenous administration of Cy3-siAkt, PMC, DiR, DMA, Taxol and PMA. Lane 1-6 in A represent heart, liver, spleen, lung, kidney and tumor, respectively. (Scale bar: 2 cm) (C) The percentage of injection dose of siAkt in plasma at different time points after intravenous administration of PMC and Cy3-siAkt. (D) The plasma concentration-time curve of PTX after intravenous administration of PMA and Taxol. Data are shown as mean ± SD (n=3). *p<0.05, **p<0.01, and ***p< 0.001.
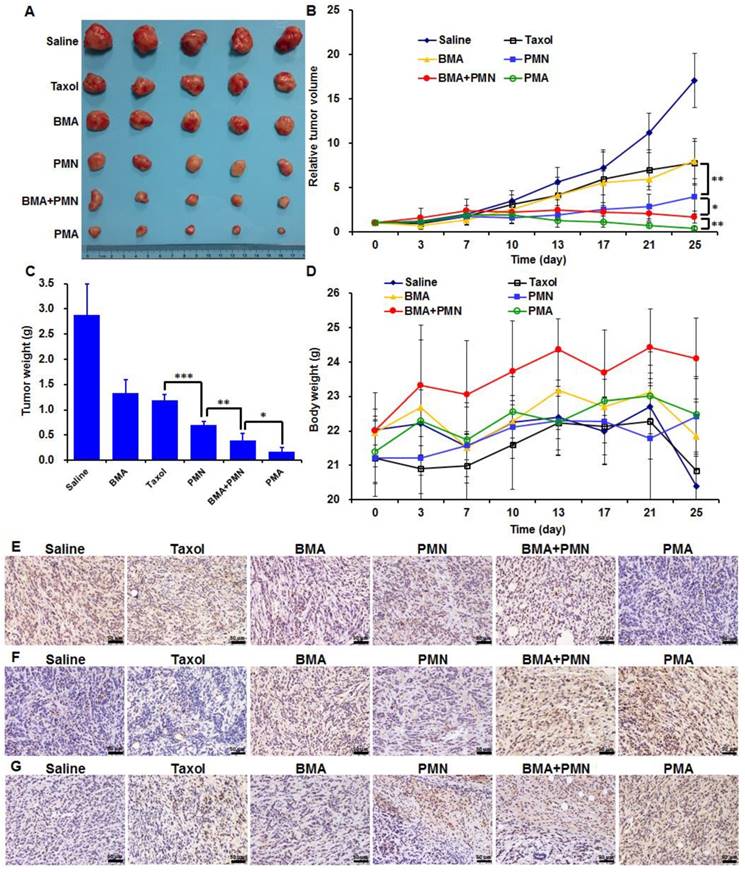
Anti-tumor and anti-metastasis effect
Both Taxol and BMA showed weak inhibitory effects on 4T1 tumors with the tumor inhibiting rate (TIR) of 58.7% and 53.5%, respectively (Figure 7A-C). PMN enhanced the anti-tumor efficiency by 1.3-fold compared with Taxol, since more drug accumulated in tumor cells. When combined PTX and siAkt, which were encapsulated in nanoparticles, the tumor growth was further suppressed as the TIR of PMN+BMA and PMA groups was 86.4% and 94.1%, respectively. The stronger anti-tumor effect of PMA compared with PMN+BMA indicated the superiority of the strategy of co-delivering different drugs with a single vector over the one that delivered the mixture of separate drugs in vivo. The animal body weights of all groups showed a growing trend, suggesting that PMA had no severe toxic effect (Figure 7D). The expression of Akt and Caspase-3 examined by IHC revealed that PMA led to significant down-regulation of Akt and up-regulation of Caspase-3, which is a key protein in the cell apoptosis signaling pathway and was inhibited by Akt (Figure 7E, 7F). This result indicated that PMA possessed good in vivo gene silencing ability. TUNEL assay revealed efficient cell apoptosis-inducing efficacy by PMA, which was consistent with the tumor volumes variation (Figure 7G). The suppression of tumor growth by PMA could be due to the synergistic effect between the cytotoxicity of PTX and the apoptosis-inducing ability of siAkt.
Anti-tumor efficacy of various formulations in 4T1 tumor-bearing mice treated with multiple doses (described in M&M). (A) The images of tumors at day 25. (B) The profiles of tumor volumes variation. (C) The weights of tumors at day 25. (D) The profiles of body weights variation. (E-G) The Akt expression (E), Caspase-3 expression (F) and TUNEL assay (G) of tumor sections analyzed by IHC. Data are shown as mean ± SD (n=5). *p<0.05, **p<0.01, and ***p< 0.001. (Scale bar: 50 μm)
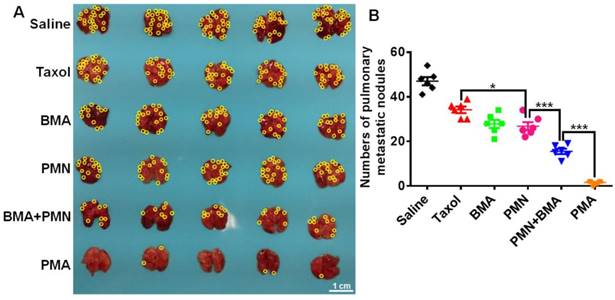
The 4T1 tumor-bearing mice showed severe lung metastasis with numerous metastatic nodules in the lungs (Figure 8A). Silencing of the Akt gene impaired the metastatic ability of 4T1 cells since mice treated with the nanoparticles containing siAkt presented varying degrees of decreased lung metastasis. Also, PMN had a metastasis-inhibiting level comparable with BMA, suggesting that the killing of the primary tumor cells also contributed to fewer metastatic lesions in the lungs. The co-delivery of PTX and siAkt displayed synergism of anti-metastatic activity. The PMA group had clean lungs with a 96.8% decrease in the number of metastasis foci compared with the saline group (Figure 8B).
Anti-metastasis effect of different formulations in 4T1 tumor-bearing mice. (A) Images of the lungs at day 25. (B) Quantitative analysis of the pulmonary metastatic nodules at day 25. The yellow circles indicate metastatic nodules on the lungs. (Scale bar: 1 cm) Data are shown as mean ± SD (n=5). *p<0.05 and ***p< 0.001.
Pathological evaluation
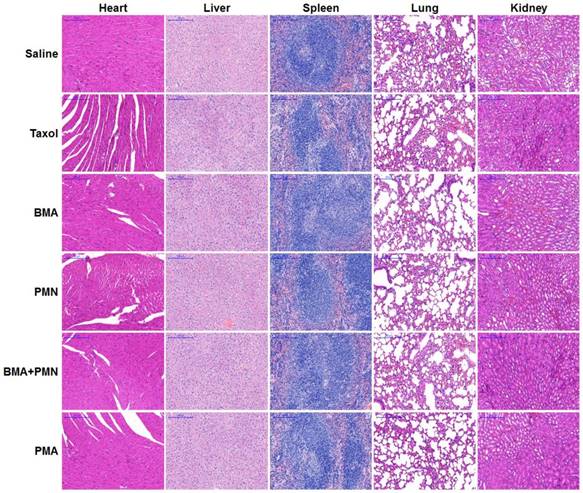
The histopathological analysis of organs was performed in mice receiving free drugs or other formulations. The H&E staining images showed no pathological variations in all organs, suggesting that all nanoparticles had good biocompatibility (Figure 9).
Histopathological analysis in healthy mice following treatment with multiple doses of various formulations. H&E staining images of tissue sections from the mice treated with Saline, Taxol, BMA, PMN, BMA+PMN, and PMA. (Scale bar: 200 μm)
Conclusion
The BDP copolymer containing a pH-sensitive hydrophobic block and a cationic hydrophilic block was synthesized which provided the basis for PMA preparation. PMA was stable at neutral pH and tumor extracellular pH while releasing the drugs in the intra-endosomal/lysosomal environment of acidic pH. In the 4T1 breast cancer cells, PMA increased the intracellular concentration of PTX, enhanced its anti- proliferative effect, and inhibited cell migration and invasion by down-regulating the expression of Akt and P-gp. In 4T1 tumor-bearing mice, the TIR of PMA reached 94.1% and the number of the lung metastasis foci was reduced by 96.8%. In summary, PMA is a promising drug delivery system to simultaneously overcome drug resistance and suppress metastasis for treating breast cancer.
Abbreviations
PI3K: phosphatidylinositol 3-kinase; mTOR: mammalian target of rapamycin; siAkt: siRNA targeting Akt; DPA: N,N-diisopropylethylenediamine; PEI: polyethyleneimine; PTX: paclitaxel; PM: PTX-loaded micelle; PMA: PTX-loaded micelle/siAkt nano-complex; BDA: 1,4-butanediol diacrylate; FBS: fetal bovine serum; G-PTX: Oregon Green® 488 Conjugate paclitaxel; DiR: 1,1'-dioctadecyl-3,3,3',3'-tetramethylindotricarbocyanine iodide; P-gp: P glycoprotein; IHC: immunohistochemistry; siNC: negative control RNA nonspecific to any human gene; FAM-siAkt: FAM labeled siAkt; Cy3-siAkt: Cy3 labeled siAkt; BDA-DPA: poly[(1,4-butanediol)-diacrylate-β-N,N-diisopropylethylenediamine]; DMSO: dimethyl sulfoxide; BDP: BDA-DPA conjugated with PEI; Mw: molecular weight; GPC: gel permeation chromatograph; BM: blank BDP micelle without PTX; PMN: PM/siNC nano-complex; DLS: dynamic light scattering; TEM: transmission electron microscopy; HPLC: high performance liquid chromatography; DL: drug loading; EE: encapsulation efficiency; PBS: phosphate buffered saline; G-PMA: Oregon Green® 488-labeled PMA; BMA: BM/siAkt nano-complex; BMN: BM/siNC nano-complex; PMC: PM/Cy3-siAkt nano-complex; DMA: DiR-loaded BDP/siAkt nano-complex; TUNEL: TdT-mediated dUTP Nick-End Labeling; H&E: hematoxylin and eosin; AUC: area under the curve; TIR: tumor inhibiting rate.
Acknowledgements
The National Basic Research Program of China (2014CB931900), the National Natural Science Foundation of China (81690265, 81521005), and the Youth Innovation Promotion Association of CAS (2015226) are gratefully acknowledged for financial support.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global Cancer Statistics, 2012. Ca-Cancer J Clin. 2015;65:87-108
2. Bock C, Lengauer T. Managing drug resistance in cancer: lessons from HIV therapy. Nat Rev Cancer. 2012;12:494-501
3. Brown R, Curry E, Magnani L, Wilhelm-Benartzi CS, Borley J. Poised epigenetic states and acquired drug resistance in cancer. Nat Rev Cancer. 2014;14:747-53
4. Wan LL, Pantel K, Kang YB. Tumor metastasis: moving new biological insights into the clinic. Nat Med. 2013;19:1450-64
5. Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer. 2014;14:430-9
6. Altrock PM, Liu LL, Michor F. The mathematics of cancer: integrating quantitative models. Nat Rev Cancer. 2015;15:730-45
7. Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323-34
8. Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16:201-18
9. Keane NA, Glavey SV, Krawczyk J, O'Dwyer M. AKT as a therapeutic target in multiple myeloma. Expert Opin Ther Targets. 2014;18:897-915
10. Burris HA. Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemoth Pharm. 2013;71:829-42
11. Gdowski A, Panchoo M, Van Treuren T, Basu A. Emerging therapeutics for targeting Akt in cancer. Front Biosci (Landmark Ed). 2016;21:757-68
12. Cassinelli G, Zuco V, Gatti L, Lanzi C, Zaffaroni N, Colombo D. et al. Targeting the Akt Kinase to Modulate Survival, Invasiveness and Drug Resistance of Cancer Cells. Curr Med Chem. 2013;20:1923-45
13. Li H, Huang F, Fan L, Jiang Y, Wang X, Li J. et al. Phosphatidylethanolamine-binding protein 4 is associated with breast cancer metastasis through Src-mediated Akt tyrosine phosphorylation. Oncogene. 2014;33:4589-98
14. Jiang WG, Sanders AJ, Katoh M, Ungefroren H, Gieseler F, Prince M. et al. Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Semin Cancer Biol. 2015;35:S244-S75
15. Wang C, Jin HJ, Wang N, Fan SH, Wang YY, Zhang YR. et al. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3 beta/beta-catenin Signaling. Theranostics. 2016;6:1205-19
16. Xu R, Zhang GD, Mai JH, Deng XY, Segura-Ibarra V, Wu SH. et al. An injectable nanoparticle generator enhances delivery of cancer therapeutics. Nat Biotechnol. 2016;34:414-8
17. Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Delivery Rev. 2015;91:3-6
18. Wei H, Zhuo RX, Zhang XZ. Design and development of polymeric micelles with cleavable links for intracellular drug delivery. Prog Polym Sci. 2013;38:503-35
19. Yu HJ, Zou YL, Wang YG, Huang XN, Huang G, Sumer BD. et al. Overcoming Endosomal Barrier by Amphotericin B-Loaded Dual pH-Responsive PDMA-b-PDPA Nano-complexes for siRNA Delivery. ACS Nano. 2011;5:9246-55
20. Xu XD, Wu J, Liu YL, Yu MY, Zhao LL, Zhu X. et al. Ultra-pH-Responsive and Tumor-Penetrating Nanoplatform for Targeted siRNA Delivery with Robust Anti-Cancer Efficacy. Angew Chem Int Ed. 2016;55:7091-4
21. Xu XD, Wu J, Liu YL, Saw PE, Tao W, Yu M. et al. Multifunctional Envelope-Type siRNA Delivery Nanoparticle Platform for Prostate Cancer Therapy. ACS Nano. 2017;11:2618-27
22. Hu JM, Zhang GY, Ge ZS, Liu SY. Stimuli-responsive tertiary amine methacrylate-based block copolymers: Synthesis, supramolecular self-assembly and functional applications. Prog Polym Sci. 2014;39:1096-143
23. Du JZ, Mao CQ, Yuan YY, Yang XZ, Wang J. Tumor extracellular acidity-activated nanoparticles as drug delivery systems for enhanced cancer therapy. Biotechnol Adv. 2014;32:789-803
24. Zhou Q, Hou YL, Zhang L, Wang JL, Qiao YB, Guo SY. et al. Dual-pH Sensitive Charge-reversal Nanocomplex for Tumor-targeted Drug Delivery with Enhanced Anticancer Activity. Theranostics. 2017;7:1806-19
25. Pang X, Jiang Y, Xiao QC, Leung AW, Hua HY, Xu CS. pH-responsive polymer-drug conjugates: Design and progress. J Controlled Release. 2016;222:116-29
26. Sun YX, Zeng X, Meng QF, Zhang XZ, Cheng SX, Zhuo RX. The influence of RGD addition on the gene transfer characteristics of disulfide-containing polyethyleneimine/DNA complexes. Biomaterials. 2008;29:4356-65
27. Chen CK, Jones CH, Mistriotis P, Yu Y, Ma XN, Ravikrishnan A. et al. Poly(ethylene glycol)-block-cationic polylactide nanocomplexes of differing charge density for gene delivery. Biomaterials. 2013;34:9688-99
Author contact
![]() Corresponding authors: Dr Qi Yin (qyinac.cn) and Prof. Yaping Li (ypliac.cn), 501 Haike Road, Shanghai 201203, China Tel/Fax: +86-21-2023-1979
Corresponding authors: Dr Qi Yin (qyinac.cn) and Prof. Yaping Li (ypliac.cn), 501 Haike Road, Shanghai 201203, China Tel/Fax: +86-21-2023-1979