Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Conclusion
Materials and Methods
Abbreviation
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(17):4135-4148. doi:10.7150/thno.20955 This issue Cite
Research Paper
Tethering Interleukin-22 to Apolipoprotein A-I Ameliorates Mice from Acetaminophen-induced Liver Injury
Wei Chen*, Xuyao Zhang*, Jiajun Fan*, Wenjing Zai, Jingyun Luan, Yubin Li, Shaofei Wang, Qicheng Chen, Yichen Wang, Yanxu Liang, Dianwen Ju ![]()
Department of Microbiological and Biochemical Pharmacy & The Key Laboratory of Smart Drug Delivery, Ministry of Education, School of Pharmacy, Fudan University, 826 Zhangheng Road, Shanghai, 201203, P. R. China.
* These authors contributed equally to this work.
Received 2017-5-10; Accepted 2017-8-14; Published 2017-9-26
Abstract

Increasing evidence indicates that interleukin-22 (IL-22) holds tremendous potential as a protective agent in preventing liver injury, but its pleiotropic effects and pathogenic role in carcinogenesis, rheumatoid arthritis and psoriasis restrict its systemic application. Here, we first developed a nanoparticle (liposIA) as a liver-targeted agent through IL-22 tethered to apolipoprotein A-I (ApoA-I) in a gene therapy vector. LiposIA was prepared using thin film dispersion method and the complexes exhibited desirable nanoparticle size, fine polydisperse index, highly efficient transfection, and excellent serum and storage stability. Biodistribution and hepatic STAT3 phosphorylation studies revealed that IL-22 tethered to ApoA-I led to highly efficient liver targeting. More importantly, our studies showed that a single-dose of liposIA was able to protect mice against acetaminophen-induced liver injury and did not initiate inflammatory response or systemic toxicity in vivo. During this process, activated STAT3/Erk and Akt/mTOR signaling transductions were observed, as well as inhibition of reactive oxygen species (ROS) generation, which prevented mitochondrial dysfunction. These studies demonstrated that IL-22 tethered to apolipoprotein A-I could target and ameliorate acetaminophen-induced acute liver injury, which highlighted that a targeted strategy for IL-22 delivery might have broad utility for the protection of hepatocellular damage.
Keywords: apolipoprotein A-I, interleukin-22, liver targeting, acetaminophen-induced acute liver injury.
Introduction
Interleukin-22 (IL-22), which was found as a member of the IL-10 cytokine family in 2000 [1-2], not only induces the production of selected chemokines and antibacterial proteins, but also promotes tissue repair via upregulating a great number of proliferative genes, antiapoptotic factors and antioxidants in various types of cells, including hepatocytes [3-6]. There is growing evidence that IL-22 plays a beneficial role in preventing acute liver injury induced by concanavalin A, carbon tetrachloride (CCl4), FAS ligand, or alcohol in vivo [7-10]. In addition, although it does not block virus replication, IL-22 may play a protective role in chronic hepatitis B (HB) or hepatitis C (HC) by means of promoting proliferation of progenitor cells in patients [11-12]. Therefore, application of IL-22 could be a promising approach in preventing hepatocellular damage, including acute or viral hepatitis diseases.
Despite the enormous progress that has been made in the protective potential of IL-22, significant drawbacks remain. Without considering the unclear mechanism of IL-22 as a liver-protective cytokine, its binding protein (IL-22BP) is widely identified in tissues of the respiratory and digestive systems, as well as cells of the placenta, skin, lung, liver, appendix, pancreas, breast, lymph nodes, bladder, kidney, and joints, which is enough to make us feel a headache [13-16]. Although IL-22 appears ideally suitable for liver-protective therapy in diseases that damage hepatocytes, systemic application of the cytokine may constitute a menace through its pleiotropic effects and inherent capacity to promote rheumatoid arthritis, psoriasis and carcinogenesis [17-20]. Thus, further evaluation of the method to assemble IL-22 in liver is urgently needed, which could help us to overcome its side effects.
In recent years, apolipoprotein A-I (ApoA-I), the main protein component of high-density lipoprotein (HDL), has been confirmed to be available for liver targeting [21-23]. These findings suggested that ApoA-I, which guides the transport of cholesterol from peripheral tissues to the liver, could help the cargos be predominantly absorbed to hepatocytes via scavenger receptor class B type I (SR-BI), which is primarily expressed in the liver. Moreover, as a targeting moiety, ApoA-I is an endogenous product and does not trigger immunological side effects [21]. According to the attractive features mentioned above, we expect that IL-22 tethered to ApoA-I may provide liver tropism and increase its potential.
With the rapid development of nanotechnology, cationic liposomes for gene-based therapy have emerged as a potential option. Most of all, this approach may provide obvious advantages with regard to localized gene expression, safety profile and rapid customization. The present work developed a liposome-based nanoparticle (liposIA) that co-expresses IL-22 and ApoA-I fusion protein for liver-targeted application. Furthermore, the hepatic targeting efficiency of IL-22 by the nanoparticle was demonstrated in vivo. A key finding of this study was that the nanoparticle, which had an excellent protective effect in a mouse model of acetaminophen (APAP)-induced acute liver injury, did not induce prominent side effects in mice. In addition, we also investigated its underlying molecular mechanisms. To our best knowledge, this is the first study to report a liver-targeted approach through interleukin-22 tethered to apolipoprotein A-I, which may be a promising strategy against hepatocellular damage.
Results
Development and characterization of interleukin-22/ApoA-I fusion protein-expressing plasmid
The DNA nucleotide encoding IL-22 and ApoA-I was generated after analysis of the genomic sequences in the GenBank-NCBI (National Center for Biotechnology Information) database. The DNA sequence of IL-22 was subcloned into the Kpn I and BamH I sites of pVAX1 to construct pVAX1-IL22 (pIL-22). Subsequently, the DNA of ApoA-I was subcloned into the BamH I and Xho I sites of pIL-22 to construct pVAX1-IA (pIA) (Figure 1A). The GGATCC sequence (restriction site for BamH I) was translated to a Gly-Ser small linker peptide for flexibility.
Construction and characterization of pIA. (A) These DNA sequences were subcloned into the Kpn I, BamH I and Xho I sites of pVAX1 to construct pVAX1-IL22 (pIL-22) and pVAX1-IA (pIA) respectively. (B) Expression of the IL-22 and ApoA-I fusion protein was detected by western blot from the indicated amount in HEK-293T cells after transfection with pIA, pVAX1 vector, or PBS (control group). (C) Densitometric values were quantified and normalized to control (n = 3; mean ± SD; **P < 0.01). The values of control were set to 1. (D) Immunofluorescence assay of HEK-293T cells transfected with pIA. IL-22 and ApoA-I fusion protein expression was indicated by Alexa Fluor 488 (AF488) staining, and Hoechst 33342 staining shows cell nuclei. (E) The time course of pIA intramuscular injection and blood sample collection. (F) IL-22 serum levels in C57BL/6 mice after intramuscular injection with 40 µg of pIA, pVAX1 vector, or PBS (control group) (n = 4; mean ± SD; **P < 0.01). (G) Western blot analysis of the presence of STAT3 phosphorylation after intramuscular injection with a single dose of pIA or the pVAX1 vector (40 µg) in liver after 3 days. (H) Quantitative analysis showing that phosphorylation of STAT3 increased significantly (n = 4; mean ± SD; **P < 0.01). The values of control were set to 1. FITC, fluorescein isothiocyanate.
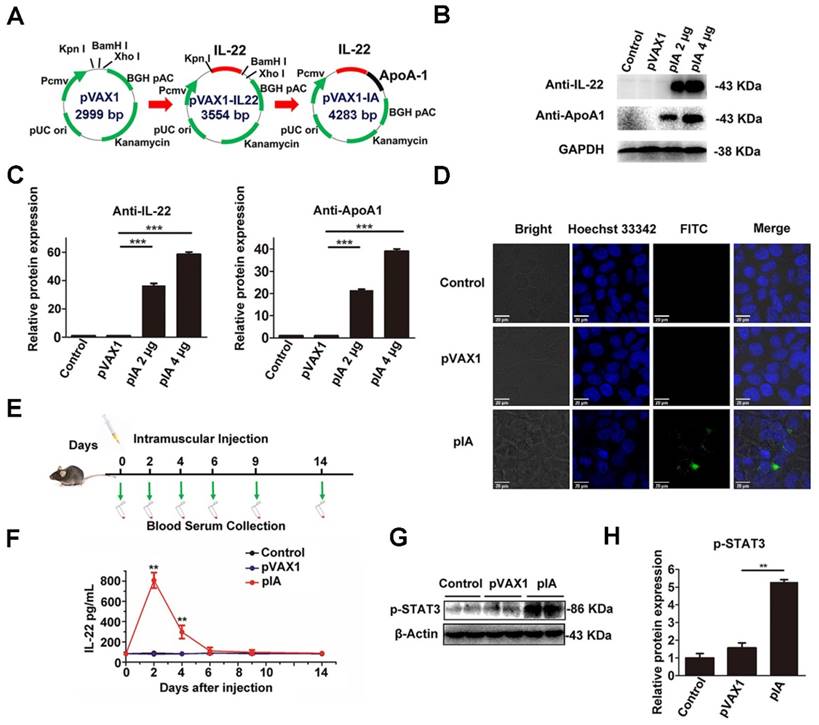
The pIA was transfected into HEK-293T cells by lipofection, and the expression of IL-22 and ApoA-I fusion protein was evaluated by western blot. As anticipated, strong specific bands of IL-22 and ApoA-I fusion protein (43 kD) were detected in lysates of pIA transfected cells but not the pVAX1 vector transfected ones (Figure 1B, C). The localization and expression of the protein expressed by the pIA were determined by immunofluorescence assay. The data revealed a clear signal in the cytoplasm of pIA transfected cells (Figure 1D). In comparison, there was no positive signal in the cells transfected with the pVAX1 vector.
To evaluate the in vivo expression of IL-22 and ApoA-I fusion protein, male C57BL/6 mice were intramuscularly injected with 40 µg of pIA or pVAX1 vector, and the blood samples were collected at the indicated times for ELISA assays (Figure 1E). The IL-22 and ApoA-I fusion protein increased at 2 days and slowly declined, whereas no IL-22 risen was observed in the serum of the mice receiving the PBS and pVAX1 vector (Figure 1F). Furthermore, the phosphorylation of STAT3 was detected in the liver at 3 days after intramuscular injection with a single dose (40 µg) of pIA (Figure 1G, H).
These results indicated that IL-22 and ApoA-I fusion protein in the gene therapy vector could express successfully in vitro and in vivo.
Preparation and characterization of liposome-based pIA complexes
We chose cationic liposomes composed of DOTAP and cholesterol as carriers for IL-22 gene therapy (Figure 2A). Nanoparticle complexes were prepared by using different weight (w/w) ratios of DOTAP/Chol and pIA ranging from 0.5:1 to 22:1. The electrophoretic mobility of liposIA was retarded with the increasing weight of cationic liposome and liposIA even remained at the top of the gel at weight ratios of 12-22:1, suggesting that pIA formed complexes with DOTAP/Chol successfully (Figure 2B, C). As the amounts of carriers increased, the mean diameter of the liposIA complexes decreased gradually, indicating compact condensation between the carrier and pIA (Figure 2D). Moreover, liposIA complexes at the weight ratio of 12:1 showed well dispersed size distribution with a fine polydisperse index value (Figure 2E). The Zeta-potentials of the complexes ranged from -14 mV to 37 mV as the weight ratio increased, indicating that negatively charged pIA was further bound with the primary carriers (Figure 2F).
Preparation and characterization of liposome-based pIA complexes. (A) Composition of liposome-based pIA complexes. (B) Formation analysis of liposome-based pIA complexes at various weight ratios (liposome-pIA) by agarose gel electrophoresis. Lane 1: Marker; Lane 2: naked pIA (N, 5 µg); Lane 3-8: liposIA (each sample contains 5 µg of pIA) with progressively increasing weight of liposome solution. (C) Ethidium bromide exclusion assay of liposIA complexes at various weight ratios (n = 3). (D) Size, (E) Polydispersity index (PDI) and (F) Zeta potential of liposIA. Empty carriers as control (Ctr). (G) Expression of the IA fusion protein in HEK-293T cells mediated by naked pIA and liposIA at various weight ratios (each sample contains 5 µg of pIA), assayed 24 h post transfection (n = 3; mean ± SD; *P < 0.05). (H) Serum stability assay of naked pIA and liposIA. Free pIA and liposIA complexes (5 µg) were separately incubated in 50% fetal bovine serum-containing media at 37°C for the indicated durations and degradation of pIA was investigated by 1.0% agarose gel electrophoresis. (I) Quantification of liposIA and naked pIA stability in the presence of serum as compared to PBS controls (n = 3; mean ± SD; **P < 0.01).
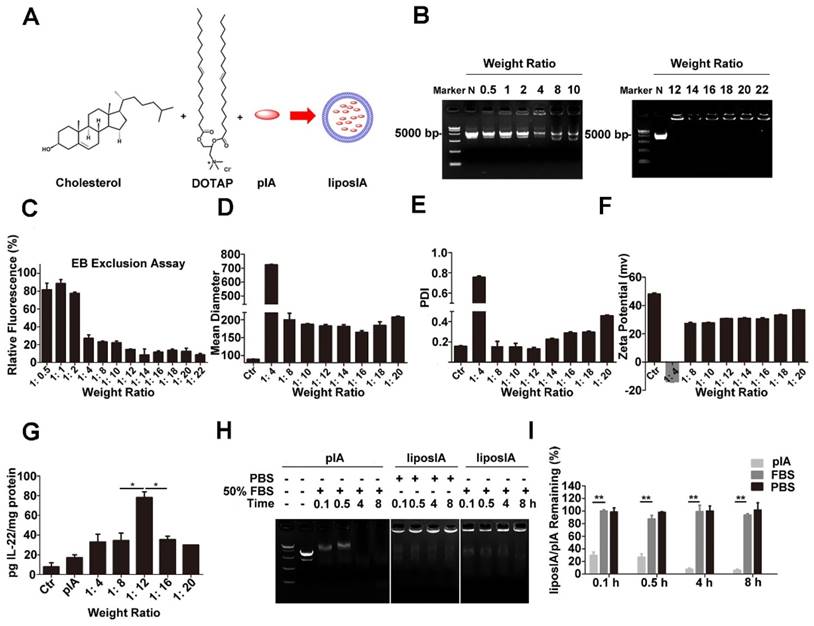
According to the results of particle size, zeta potential and polydisperse index, we selected five different complexes for in vitro protein expression assay. We chose the optimum weight ratio of 12:1, because it could strongly drive IL-22 and ApoA-I fusion protein expression in cell culture (Figure 2G) and remained stable while in fetal bovine serum at 37°C. As shown in Figure 2H and Figure 2I, naked pIA was faint at 0.5 h, and nearly disappeared after 4 h, suggesting that free pIA was prone to be degraded by serum. In contrast, liposIA was well-protected in serum and remained relatively intact even at 8 h. To confirm long-term stability, liposIA was stored for varying times. No statistically significant changes in particle size and polydisperse index was observed for particles stored at 4°C for 1, 2, 4, 5 or 12 days, indicating good stability against aggregation due to its compact structure and optimum charge (Figure S1).
Taken together, our results indicated that the liposIA complexes showed desirable nanoparticle size, fine polydisperse index, highly efficient transfection, and excellent serum and storage stability at the optimum weight ratio of 12:1.
Kinetics, liver targeting and effect on STAT3 activity of liposIA
First, the serum kinetics of liposIA were evaluated. Mice were intramuscularly injected with a single dose of liposome complexes encoding IL-22 and ApoA-I fusion protein (liposIA), IL-22 (liposIL-22), pVAX1 empty vector (liposN), pIL-22 or PBS. The blood samples were collected at the indicated times (Figure 3A). Concentrations of IL-22 in blood serums were measured by ELISA assay. Our results showed that the plasma half-life of IL-22 in mice injected with liposIA was much higher than in that injected with liposIL-22 or pIL-22, indicating that the formulation of IL-22 prolonged its persistence in the circulation (Figure 3B). Next, in order to determine the expression and in vivo distribution of IL-22, heart, liver, lung, spleen, kidney and brain were collected from the sacrificed mice at 1, 2, 3, and 7 days after treatment. The ELISA results showed that IL-22 significantly increased in liposIA-injected mice (Figure 3C, Figure 3D and Figure S2). Furthermore, IL-22 predominantly accumulated in the liver at 2 days post injection (Figure 3D), and slowly declined (Figure S2). Therefore, our results indicated that liposIA had a prolonged serum half-life and could target liver efficiently in vivo.
Kinetics, liver targeting and effect on STAT3 activity of liposIA. (A) Time course of liposN, pIL-22, liposIL-22, liposIA intramuscular injection and blood sample collection. (B) IL-22 serum levels in C57BL/6 mice after a single dose injection with 0.5 mg (contains 38.5 µg plasmid) of liposIA, liposIL-22, pIL-22, liposN or PBS (n = 5; mean ± SD). (C, D) IL-22 concentrations measured by ELISA in selected organs (heart, liver, lungs, spleen, kidney and brain) at 1 or 2 days. (E) Signaling transductions activated by liposIA were analyzed by western blot against phosphorylated and total protein levels of STAT3, actin levels were shown as loading control. (F) Densitometric values of p-STAT3 were quantified and normalized to control (n = 3; mean ± SD; *P < 0.05, **P < 0.01). The values of control were set to 1.
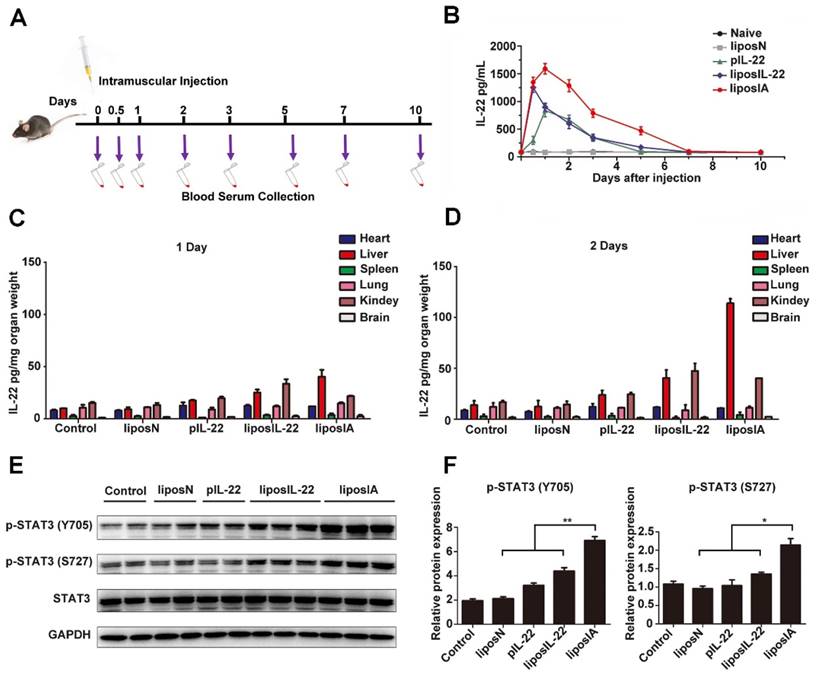
Expression of STAT3 phosphorylation (p-STAT3) was significantly enhanced after injection with a single dose of liposIA (0.5 mg) or liposIL-22 (0.5 mg) at 2 days (Figure 3E). However, liposIA induced a 2-fold stronger expression of STAT3 phosphorylation in liver than liposIL-22, indicating that IL-22 tethered to ApoA-I could deliver IL-22 to the liver and remarkably enhance the efficiency of IL-22 (Figure 3F). Moreover, downstream signaling of STAT3 activation was also checked in other organs such as heart, kidney, spleen, brain, lung, intestine and pancreas (Figure S3 and Figure S4). The activation of STAT3 was negligible in these organs except for a little rise in kidney, which was consistent with the result of IL-22 distribution in vivo (Figure 3D).
To elucidate which cells were the main target of IL-22 and ApoA-I fusion protein in the liver microenvironment, immunohistological results of p-STAT3 in mice liver sections were provided (Figure S5A). In addition, primary hepatocytes and non-parenchymal cells were isolated from C57BL/6 mice after a dose of liposIA at 2 days (Figure S5B, C). The data revealed that hepatocytes, instead of Kupffer cells, endothelial cell or hepatic stellate cells, were the major target cells and were associated with a significant increase in expression of downstream signaling transductions of IL-22 (p-STAT3).
Altogether, our data demonstrated that liposIA could provide IL-22 with prolonged half-life and liver targeting.
Single dose liposIA protected mice from acetaminophen-induced acute liver injury via activating hepatic STAT3/Erk and AKT/mTOR signaling transductions
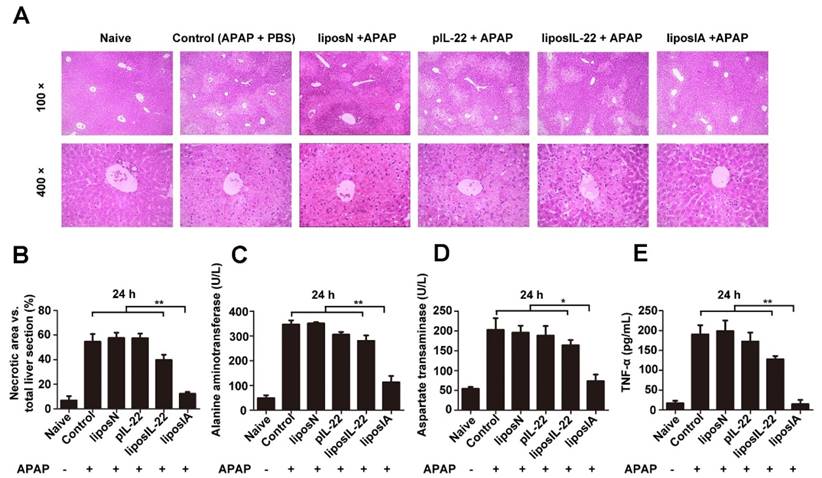
We further evaluate whether liposIA could have a liver-targeted protective potential. C57BL/6 mice were intramuscularly injected with 0.5 mg of liposIA, liposIL-22, pIL-22, liposN or PBS (positive control group) and challenged 48 h later with an injury dose of acetaminophen. After 24 h of APAP intoxication, liver histology showed that APAP injection caused massive necrosis in the livers of liposIL-22, pIL-22, liposN and PBS-treated mice, but only caused spotted necrosis in the livers of liposIA-treated mice (Figure 4A and Figure 4B). As shown in Figure S6, Figure 4C, Figure 4D and Figure 4E, administration of 500 mg/kg of APAP caused significant elevations in serum ALT, AST and TNF-α levels 12 h or 24 h post injection in mice treated with liposIL-22, pIL-22, liposN or PBS. These changes were markedly suppressed in mice treated with liposIA. These results indicated that a single dose of liposIA could attenuate the liver injury caused by APAP.
LiposIA protected mice from acetaminophen-induced acute liver injury. C57BL/6 mice were intramuscularly injected with 0.5 mg (contains 38.5 µg plasmid) of liposIA, liposIL-22, pIL-22, liposN or PBS, followed 48 h later by intraperitoneal injection of APAP (500 mg/kg). Naive mice were the blank control group. Mice were then sacrificed 24 h post APAP injection. (A) Livers were collected for HE staining. (B) Statistical analysis of necrotic areas in HE stained liver sections. (C, D and E) Serum was collected for determination of ALT, AST, and TNF-α levels (n = 5; mean ± SD; **P < 0.01).
To understand the underlying mechanism by which liposIA protects mice from APAP-induced liver injury, we compared activation of liver STAT3 and Erk in liposIA, liposN or PBS-treated groups. We found that the mice treated with liposIA exhibited the upregulated phosphorylation of STAT3 and Erk as well as their downstream protein Bcl-XL and CyclinD-1 in liver when compared to those treated with liposIL-22, pIL-22, liposN or PBS (Figure 5A, Figure 5C and Figure S4).
LiposIA activated hepatic STAT3/Erk and AKT/mTOR signaling transductions. (A) Total liver extracts were prepared and subjected to western blot analysis with various antibodies as indicated. (B) The expression levels of p-mTOR (S2448), p-AKT (S473), p-p70s6K (S371) and p-4EBP1-pT45 were analyzed by western blot. (C) Densitometric values were quantified and normalized to naive group (n = 3; mean ± SD; *P < 0.05, **P < 0.01). The values of naive group were set to 1.
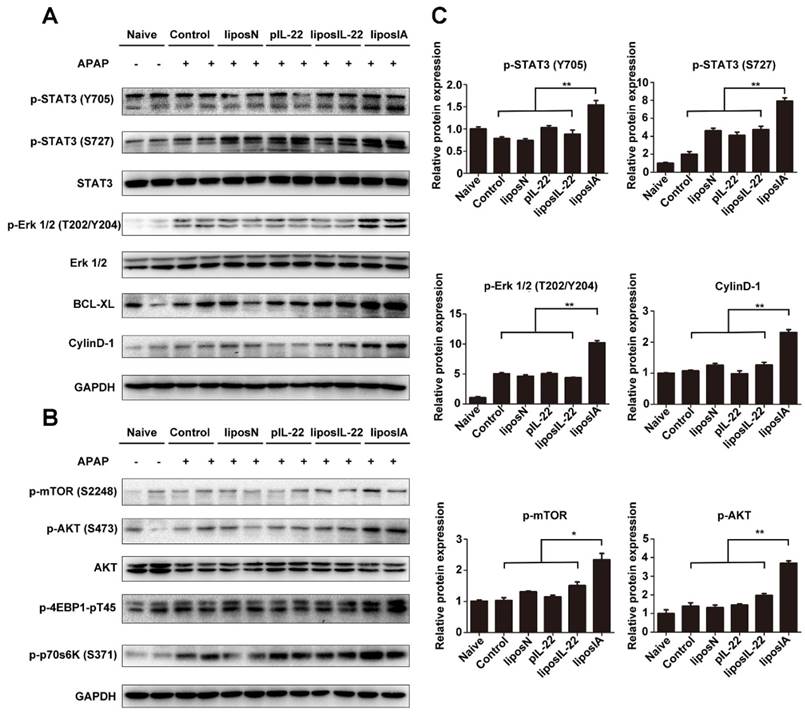
Apart from the STAT3/Erk and its-related signal transductions, IL-22 could also modulate a series of signaling transductions that are related to cell growth and proliferation. The mammalian target of rapamycin (mTOR) is an evolutionarily conserved nutrient-sensing serine/threonine protein kinase that plays a positive regulator of protein synthesis and cell growth. In this work, we investigated whether liposIA affected the status of mTOR signaling transduction. Our results demonstrated that liposIA-treated mice expressed high levels of protein p-mTOR (S2448), as well as its related signaling factors such as p-AKT (S473), p-4EBP1-pT45 and p-p70s6K (S371) when compared with liposIL-22, pIL-22, or liposN-treated mice or naive mice (Figure 5B, Figure S4).
Altogether, our results indicated that a single dose of liposIA could protect mice from acetaminophen-induced acute liver injury with activated hepatic STAT3/Erk and AKT/mTOR signaling transductions.
LiposIA inhibited ROS generation and prevented APAP-induced mitochondrial dysfunction in the liver
APAP overdose triggers liver injury by formation of a reactive metabolite N-acetyl-p-amino-benzoquinone imine (NAPQI), which in turn depletes glutathione (GSH) and causes oxidative stress [24]. We further investigated if ROS concentration or mitochondrial membrane potential changed after APAP and/or liposIA treatment. As detected by ROS dye Mitosox, exposure of C57BL/6 mice to APAP promoted hepatocyte accumulation of intracellular ROS, which was significantly reduced by liposIA (Fig 6A). As an important ROS generator, mitochondrion is known to regulate hepatocyte apoptosis and necrosis [25]. The mitochondrial damage was thus measured by detecting the mitochondrial membrane potential with JC-1 dye. As expected, we found a normal mitochondrial membrane potential (red fluorescence) in the livers of liposIA treated-mice, while mice treated with liposIL-22, pIL-22, liposN or PBS exhibited a decreased mitochondrial membrane potential (Fig 6B). Consistent with these findings, we found that an overdose of APAP led to an obvious elevation in phosphorylation of mitochondrial c-Jun-N-terminal kinase (JNK) and inducible nitric oxide synthase (iNOS), indicating mitochondrial damage by APAP. These changes were alleviated in liposIA-treated mice, suggesting a protective role against APAP-induced mitochondrial damage.
LiposIA inhibited ROS generation and prevented APAP-induced mitochondrial dysfunction in the liver. (A) C57BL/6 mice were intramuscularly injected with 0.5 mg (contains 38.5 µg plasmid) of liposIA, liposIL-22, liposN, pIL-22 or PBS, followed 48 h later by intraperitoneal injection of APAP (500 mg/kg). Naive mice were the blank control group. Mice were then sacrificed 24 h post APAP injection. Livers were collected for special staining. (a) The ROS levels in the liver tissues were visualized by Mitosox staining. (b) Specific mitochondrial dye JC-1 was used to measure the mitochondrial membrane potential within hepatocytes. Red fluorescence represents normal mitochondrial membrane potential whereas green fluorescence indicates low potential. (B) Immunoblot images of iNOS and p-JNK. (C) Densitometric values of p-JNK and iNOS were quantified and normalized to naive group (n = 3; mean ± SD; **P < 0.01). The values of naive group were set to 1.
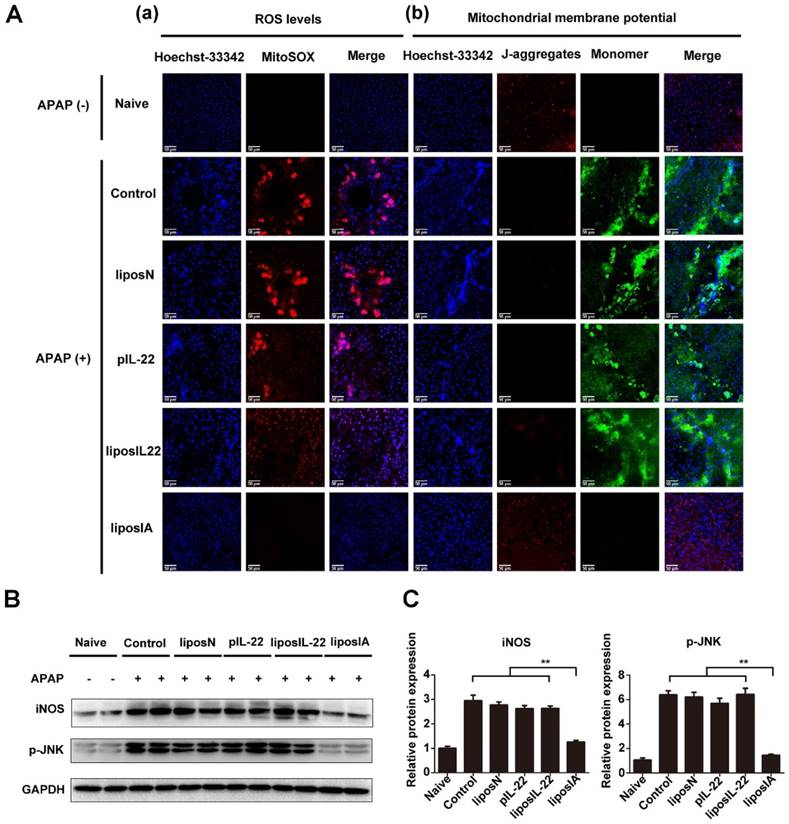
These results indicated that liposIA inhibited ROS generation and prevented mitochondrial dysfunction in acetaminophen-induced acute liver injury.
Liver-targeted therapy with liposIA did not manifest toxicity
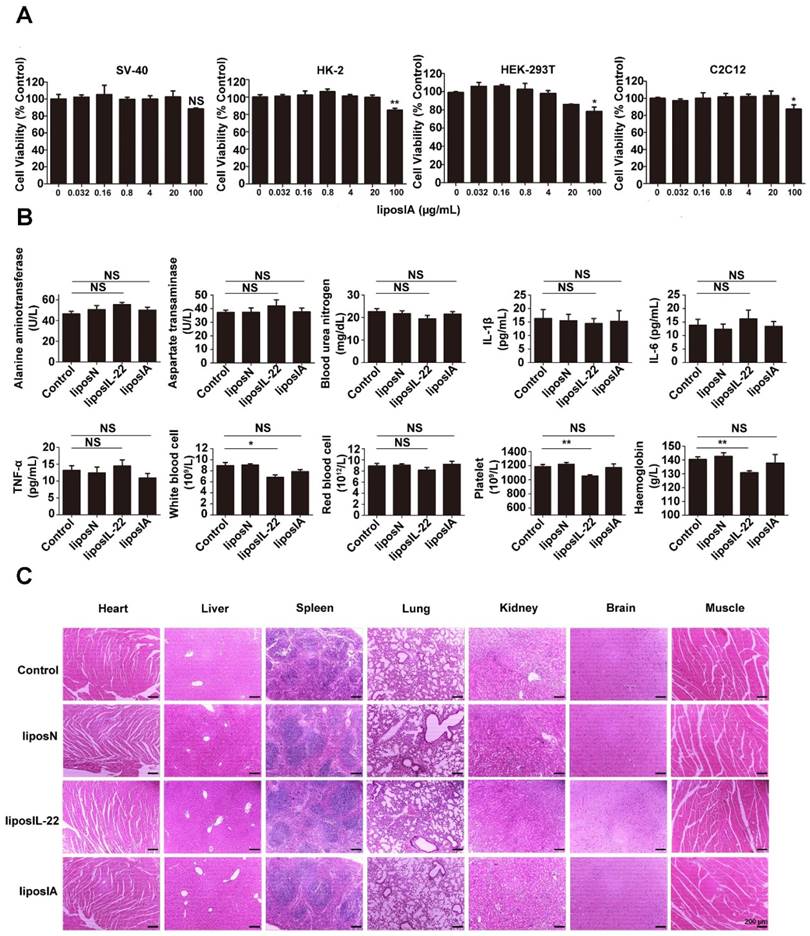
We next evaluated if the intramuscular injection of liposIA would trigger an inflammatory response and systemic toxicity. The MTT assays illustrated that liposIA complexes at various concentrations were almost nontoxic in a broad variety of cell types including SV-40, HK-2, HEK-293T and C2C12 cells (Figure 7A). Furthermore, no abnormalities were evident in hematological markers, including serum ALT, AST, BUN, IL-6, TNFα and IL-1β (Figure 7B). In addition, the complete blood count results showed that red blood cells (RBC), white blood cells (WBC), haemoglobin (HGB), and platelets (PLT) did not differ in mice between the control group and liposIA group. However, the same dose of liposIL-22 induced an obvious decrease in white blood cells, haemoglobin and platelets (Figure 7B). Finally, histological analysis on major organs indicated that liposIA-treated mice showed no significant pathological or histological changes (Figure 7C). These results suggested that liposIA could be used for liver-selective therapy without significant side effects.
Liver-targeted therapy with liposIA did not manifest toxicity. (A) In vitro cytotoxicity of liposIA on SV40, HK-2, HEK-293T and C2C12 cells (n = 3; mean ± SD; *P < 0.05, **P < 0.01). (B) Hematological markers of hepatic or renal toxicity, main cytokines (IL-6, IL-1β and TNF-α) and peripheral blood were assessed 2 days after intramuscular injection of PBS, liposN, liposIL-22, or liposIA (n = 4; mean ± SD; *P < 0.05, **P < 0.01). (C) Representative histopathology for different tissues in C57BL/6 mice 2 days after treatment with 0.5 mg of PBS (control), liposN or liposIA by HE staining.
Discussion
Recently, several lines of evidence showed that IL-22 has a strong protective effect against hepatocellular damage [7-10]. However, potential applications of IL-22 as a liver specific therapeutic strategy must be carefully selected. One particular concern is the pleiotropic effects and inherent potential of IL-22 to contribute to the pathogenesis of psoriasis, rheumatoid arthritis and carcinogenesis [17-20]. In order to overcome the drawbacks of IL-22 in the amelioration of liver injury, we tethered it to ApoA-I in a therapy gene vector for the first time.
Previous studies used viral vectors, hydrodynamic injection and cationic liposomes as the three main methods of gene delivery. However, application of viral vectors has several limitations including limited DNA packaging capacity, immunogenicity, broad tropism, carcinogenesis and complicated vector production, while hydrodynamic injection is not clinically acceptable [26-29]. By contrast, as the properties of cationic liposomes are well understood, liposomes are bright prospects for clinical applications, which have dramatically increased in the past 5 years. In this work, we used a cationic liposome-based therapy approach that expressed an IL-22 and ApoA-I fusion protein by encapsulating a recombinant DNA (liposIA). The biological behaviors of liposIA were assessed and confirmed high transfection efficiency and excellent serum and storage stability, indicating it as a potential gene delivery vector.
Efficient liver-directed therapy requires the development of novel methods with high targeting performance. Moreover, it also depends on targeting objective agents to the right cell types [30]. It has been previously suggested that ApoA-I could accumulate predominantly in the liver due to its interaction with SR-BI [21]. Earlier studies reported that SR-BI was highly expressed in hepatocytes [31-32]. Furthermore, IFNα fused to ApoA-I could prolong its half-life and exhibited hepatic tropism [22]. Consistent with these results, we demonstrated that IL-22 tethered to ApoA-I could benefit the IL-22 with enhanced serum half-life, hepatocyte-targeted distribution and notable efficiency to activate STAT3, indicating that tethered to ApoA-I could improve the efficiency of IL-22.
APAP overdose is currently the leading cause of acute liver failure (ALF). In APAP hepatotoxicity, cell death and subsequent proliferation determines the prognosis of patients, making it urgent to identify suitable protective agents. Numerous in vitro and in vivo researches have indicated that IL‑22 has a strong therapeutic effect in preventing hepatocellular damage. Previous researches showed that hydrodynamic gene delivery of IL-22 could protect mice from concanavalin A, FAS ligand and CCl4-induced hepatic injury [9]. The potential of IL‑22 was also confirmed in mouse models of liver ischemia-reperfusion injury, acute or chronic alcohol-induced liver damage and partial hepatectomy [33-36]. Because of these encouraging results, we evaluated the efficacy of liposIA in acetaminophen-induced acute liver injury. Our data suggested that a single dose of liposIA was able to protect C57BL/6 mice against an injury dose of APAP efficiently, whereas liposIL-22 and pIL-22 could not. It seems possible that these phenomena might be related to the level and the retention time of IL-22 in liver. In addition, although the beneficial role of recombinant murine IL-22 was proved in APAP-induced acute liver injury, the underlying molecular mechanisms are still unclear [10]. In the present study, our data showed that STAT3/Erk signaling transductions were activated, as evidenced by increased p-STAT3 and p-Erk1/2 expression. Consistent with previous reports [37-38], our work also suggested that the mitogenic signaling transduction of Erk was also induced in control and liposN groups in APAP-induced acute liver injury. Meanwhile mTOR signaling transduction was activated in total liver extracts by liposIA as evidenced by increased p-mTOR (S2448), upstream protein p-Akt as well as two substrates p-p70s6K (S371) and p-4EBP1-pT45. Since the STAT3/Erk and Akt/mTOR signaling transductions are significantly associated with cell death and proliferation, we demonstrated for the first time that IL-22 tethered to apolipoprotein A-I alleviated acetaminophen-induced acute liver injury, which may be related to activation of STAT3/Erk and Akt/mTOR signaling transductions.
Generally, the toxicity of APAP is mediated by its conversion to the reactive metabolite NAPQI, which forms adducts on intracellular proteins [39-40]. Subsequently, the adducts could induce nuclear DNA fragmentation, ROS and mitochondrial dysfunction, leading to apoptosis and necrosis of hepatocytes [24-25]. To our best knowledge, there has been no report on the relationship between ROS/mitochondrial dysfunction and the therapeutic effects of IL-22. In consideration of the importance of these factors in the pathogenesis of APAP hepatotoxicity [41-44], it is necessary to elucidate the production of ROS and the mitochondrial membrane potential. In this work, liposIA-treated mice had a low mitochondrial ROS and a normal mitochondrial membrane potential. Besides, a growing number of evidence demonstrates that upregulation of iNOS and p-JNK play a key role in mitochondrial dysfunction and cell death [45-47]. Here, our results indicated a decrease of iNOS and p-JNK in liposIA-treated mice. These results suggested that liposIA inhibited ROS generation and prevented mitochondrial dysfunction in APAP-induced liver injury.
Toxicity of nanoparticle complexes is one of the biggest challenges to their clinical application. Researchers have shown that nanoparticles such as quantum dots and poly-amidoamine dendrimers could accumulated in liver, kidney or lung [48-49]. Meanwhile, they could disrupt platelet functions, initiate blood clot formation, cause acute liver or lung injury and inflammation [50-54]. In this work, a relatively safe administration and a reliable targeting strategy were applied. The data showed that liposIA had a low cell cytotoxicity in a broad variety of cell types in culture. More importantly, it did not initiate inflammatory response or systemic toxicity in vivo. Notably, anemia, moderate oligocythemia and thrombocytopenia were noted after treatment with liposIL-22 in mice while liposIA was associated with minor changes in blood system. These data indicated the relative safety profile of liposIA.
Conclusion
In conclusion, we firstly developed a liver-targeted strategy for IL-22 delivery and for the protection of acetaminophen-induced liver injury. The well-formed liposIA complexes showed desirable size distribution, fine polydisperse index, highly efficient transfection, and excellent serum and storage stability. Moreover, the most important advantage of liposIA was that it could provide IL-22 with prolonged half-life, liver targeting, high efficiency and low toxicity. LiposIA-delivered IL-22 activated STAT3/Erk and Akt/mTOR signaling transductions, inhibited ROS generation and prevented mitochondrial dysfunction, thereby significantly improving the protective efficacy of IL-22 against acetaminophen-induced liver injury in vivo. Thus, the liver-targeted delivery approach of IL-22 introduced here might be regarded as a promising strategy for preventing hepatocellular damage in patients.
Materials and Methods
Plasmids and expression
The GenBank accession no. NM_016971 and no. X07496 were used to synthesize the DNA constructs encoding IL-22 and ApoA-I respectively. All constructs contained a Kozak sequence at the 5' end of the gene. Experiments were performed using pVAX1 vectors (Invitrogen, USA). These DNA sequences were subcloned into the Kpn I, BamH I and Xho I sites of pVAX1 to construct pVAX1-IL22 (pIL-22) and pVAX1-IA (pIA). The plasmid DNA (pDNA) grown in E. coli was isolated using the Endo-Free Plasmid Mega Kit (Axygen, USA).
For in vitro expression studies, transfections were performed using the lipofectamine 2000 (Invitrogen, USA), following the manufacturer's protocols. Briefly, cells were grown to 70-80% confluence in a 35 mm dish and transfected with 2 µg or 4 µg of pDNA. The cells were harvested 1 day after transfection, washed three times with phosphate buffered saline (PBS), and lysed with cell lysis buffer (Beyotime Biotechnology, China). Western blot analysis was used to verify the expression of the IL-22 and ApoA-I fusion protein from 40 µg of harvested cell lysate. For the immunofluorescence assay, HEK-293T cells were grown on coverslips and transfected with 4 µg of pDNA. One day after transfection, the cells were fixed with 4% paraformaldehyde for 15 min and washed three times with PBS. Then the cells were incubated with 0.5% Triton X-100 in PBS for 20 min and washed three times with PBS. Nonspecific binding was then blocked with fetal bovine serum (FBS) at 37°C for 30 min. The slides were then incubated with anti-ApoA-I antibody (Abcam, USA) at 4°C for 12 h. Slides were washed three times with PBST (0.2% Triton X-100 in PBS) and incubated with anti-rabbit IgG-AF488 (Invitrogen, USA) at 37°C for 1 h. After washing, Hoechst 33342 was used to stain the nuclei of all cells. Coverslips were mounted with ProLong Gold antifade reagent (Beyotime Biotechnology, China), and the slides were observed under an inverted confocal microscope. All the procedures were done in the dark. The concentration of DNA was confirmed by NanoDrop measurement (Thermo Scientific, USA).
Cell culture
HEK-293T cells, SV-40 cells, HK-2 cells and C2C12 cells were obtained from Cell Bank of Chinese Academy of Sciences, Shanghai Branch (Shanghai, China). All cells were maintained at 37°C in a humidified atmosphere of 5% CO2 incubator and cultured in Dulbecco's modified eagle's medium (DMEM) (Invitrogen, San Diego, CA, USA) containing 10% heat-inactivated FBS (Invitrogen, San Diego, CA, USA), 100 µg/mL of streptomycin, and 100 U/mL of penicillin (Beyotime Biotechnology, China).
Animals and models of liver injury
Male C57BL/6 mice were obtained from Department of Experimental Animals, School of Pharmacy, Fudan University and used between 6 weeks and 8 weeks of age. All animals were housed in a temperature-controlled, light-cycled facility in accordance with the guidelines evaluated and approved by the ethics committee of Fudan University to ensure the humane animal care and use. The experimental protocols were optimized to avoid pain, discomfort or distress to the mice. The appropriate anesthetics or analgesics were used when necessary. For experimental endpoints or ethical reasons, mice were sacrificed by carbon dioxide asphyxiation followed by cervical dislocation.
The mice were fasted overnight and then were administered with freshly prepared APAP (500 mg/kg body weight, MedChemExpress, USA) intraperitoneally. The serum was collected for determination of alanine aminotransferase (ALT), aspartate transaminase (AST) and tumor necrosis factor (TNF)-α levels at various time points post injection. The mice were then sacrificed, and organs were collected for ELISA and H&E staining.
Nanoparticle formulation and preparation
Thin-film dispersion method was alternatively employed to prepare liposomes. Briefly, 20 mg of DOTAP (Sigma, USA) and 11 mg of cholesterol (Sigma, USA) dissolved in 10 mL of organic solvent (chloroform), and the solvent of lipid solution was evaporated with a rotary evaporator at 25°C until a thin film was formed. The solvent trace was finally evaporated by N2 streaming. The dried lipid film was hydrated in a 5% dextrose solution to give a final concentration of 5 mg/mL (total weight). Then the mixture was sonicated using a probe-type ultrasonicator until a clear suspension was obtained (DOTAP/Chol). The dispersion was sequentially filtered through a 0.22 µm pore-sized microporous membrane. LiposN, liposIL-22 and liposIA complexes were prepared the day before injections. DNA was diluted in 5% dextrose solution, and stock liposomes (DOTAP/Chol) were rapidly added by vortexing for 30 s and then incubated for 0.5 h at room temperature to produce various w/w ratios of nanoparticle complexes. The particle size, zeta potential and polydisperse index of the complexes were measured by Zetasizer Nano (Malven, UK).
Serum stability assay
To evaluate their serum stability, nanoparticles were incubated with PBS supplemented with 50% final concentration of FBS at 37°C. An aliquot of 10 μL was withdrawn at different time points (0.1 h, 0.5 h, 4 h and 8 h) and immediately frozen at -80°C until gel electrophoresis was performed. Samples incubated with PBS, and naked DNA incubated with FBS were used as control groups. The results were analyzed by 1.0% agarose gel electrophoresis and calculated as percent encapsulated DNA obtained in the presence of FBS as compared to controls.
Immunoblotting
Whole-liver protein homogenates were centrifuged at 12000 × g at 4°C for 15 min. Supernatant was collected and BCA Protein Assay was used to determine protein concentration. Equal amounts of protein (30 µg) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a polyvinylidene fluoride membrane. The membrane was blocked with 5% BSA for 2 h. The protein bands were probed with primary antibodies (CST, USA) at 4°C overnight and then incubated with a horseradish peroxidase-conjugated secondary antibody at room temperature for 2 h. The immunoblots were detected using the enhanced chemiluminescence system (Pierce, Rockford, IL, USA) and intensities in the resulting bands were quantified by ChemiDoc software (Bio-Rad, USA).
Cytotoxicity of liposIA on different cells
The cytotoxicity of liposIA on various cell lines was evaluated by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay. Briefly, SV40, HK-2, HEK293T and C2C12 cells were seeded on 96-well plates at 1 × 104 cells per well and cultured for 24 h in a 5% CO2 humidified atmosphere and DMEM with 10% final concentration of FBS at 37°C. Then, the culture medium was refreshed with 100 μL DMEM containing different concentrations of liposIA. After 24 h of incubation, the cells were rinsed with PBS. MTT agent (Sigma, USA) was subsequently added in each well at a concentration of 0.5 mg/mL at 37°C for 4 h. Finally, 100 μL of dimethyl sulfoxide (DMSO) was employed to dissolve formazan by constant shaking, whose absorbance was determined with a microplate reader at 570 nm.
Hematological and serum biochemical analysis
Blood samples were extracted from mice after injection of liposIA at different time points. Hematological parameters were determined by a full automatic blood cell analysis (SYSMEX XT-1000i, Japan). The ALT, AST and blood urea nitrogen (BUN) levels in serum were assayed using a kit from DREW Scientific (NJBI, China). Serum IL-22, TNF-α, IL-6 and IL-1β levels were detected using an ELISA Kit (Multi Sciences, China) following the manufacturer's instructions.
Histological analysis
Heart, liver, spleen, lung, kidney, brain and muscle tissues were collected from mice. To evaluate general morphology, all the excised tissues were fixed in 4% formaldehyde, embedded in paraffin wax, sectioned into 5 μm thick slices, stained with hematoxylin and eosin (HE) and examined by light microscopy for alteration of histological structures.
Assessment of reactive oxygen species (ROS) and mitochondrial membrane potential
ROS assay kit (Beyotime Biotechnology, China) was used to measure intracellular ROS generation in mouse primary hepatocytes. Fluorescent probe Mitosox (10 μM) was added to each sample, and the samples were incubated at 37°C for 20 min. Subsequently, the fluorescence intensity was measured at an excitation and emission wavelength of 485 nm and 530 nm respectively. To measure the mitochondrial membrane potential, 5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) (Beyotime Biotechnology, China), a sensitive fluorescent probe for mitochondrial membrane potential was used. Mouse primary hepatocytes were stained with 10 µg/mL JC-1 at 37°C for 30 min. Then the samples immediately analyzed with a fluorescence microscope (excitation: 488 nm, emission: 535 nm, 595 nm).
Statistical analysis
GraphPad Prism version 5.0 (San Diego, USA) was used for preparation of all graphs and analyses. Data is expressed as mean ± SD, unless otherwise specified. Statistical analysis was performed using the Student's test. *, **, and *** indicated P < 0.05, P < 0.01 and P < 0.001, respectively.
Abbreviation
IL-22: interleukin-22; ApoA-I: apolipoprotein A-I; CCl4: carbon tetrachloride; IL-22BP: interleukin-22's binding protein; HDL: high-density lipoprotein; SR-BI: scavenger receptor class B type I; APAP: acetaminophen; pIL-22: the expression of IL-22 vector; IA: IL-22 and ApoA-I fusion protein; pIA: the expression of IA vector; DOTAP: N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium chloride; Chol: cholesterol; liposIA: therapeutic pIA conjugated with liposome; PDI: polydisperse index value; liposIL-22: therapeutic pIL-22 conjugated with liposome; liposN: pVAX1 empty vector conjugated with liposome; ALT: alanine aminotransferase; AST: aspartate transaminase; TNF-α: and tumor necrosis factor; ROS: reactive oxygen species (ROS); JC-1: 5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolylcarbocyanine iodide; NAPQI: N-acetyl-p-amino-benzoquinone imine; GSH: glutathione; RBC: red blood cells; WBC: white blood cells; HGB: haemoglobin; PLT: platelets.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81573332, 81773620), National Key Basic Research Program of China (2015CB931800, 2013CB932502), Shanghai Science and Technology Funds (14431900200), and Special Research Foundation of State Key Laboratory of Medical Genomics and Collaborative Innovation Center of Systems Biomedicine.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dumoutier L, Van Roost E, Colau D, Renauld JC. Human interleukin-10 related T cell derived inducible factor: molecular cloning and functional characterization as a hepatocyte stimulating factor. Proc Natl Acad Sci USA. 2000;97:10144-10149
2. Xie MH, Aggarwal S, Ho WH, Foster J, Zhang Z, Stinson J. et al. Interleukin (IL)-22, a novel human cytokine that signals through the interferon receptor related proteins CRF2-4 and IL-22R. J Biol Chem. 2000;275:31335-31339
3. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454-467
4. Witte E, Witte K, Warszawska K, Sabat R, Wolk K. Interleukin-22: a cytokine produced by T, NK and NKT cell subsets, with importance in the innate immune defense and tissue protection. Cytokine Growth Factor Rev. 2010;21:365-379
5. Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Biology of interleukin-22. Semin Immunopathol. 2010;32:17-31
6. Zenewicz LA, Flavell RA. Recent advances in IL-22 biology. Int Immunol. 2011;23:159-163
7. Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332-1342
8. Ki SH, Park O, Zheng M, Morales-Ibanez O, Kolls JK, Bataller R. et al. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology. 2010;52:1291-1300
9. Pan H, Hong F, Radaeva S, Gao B. Hydrodynamic gene delivery of interleukin-22 protects the mouse liver from concanavalin A-, carbon tetrachloride- and Fas ligand-induced injury via activation of STAT3. Cell Mol Immunol. 2004;1:43-49
10. Scheiermann P, Bachmann M, Goren I, Zwissler B, Pfeilschifter J, Mühl H. Application of Interleukin-22 Mediates Protection in Experimental Acetaminophen-Induced Acute Liver Injury. Am J Pathol. 2013;182:1108-1113
11. Feng D, Kong X, Weng H, Park O, Wang H, Dooley S. et al. Interleukin-22 promotes proliferation of liver stem/progenitor cells in mice and patients with chronic hepatitis B virus infection. Gastroenterology. 2012;143:188-198
12. Sertorio M, Hou X, Carmo RF, Dessein H, Cabantous S, Abdelwahed M. et al. Interleukin-22 and IL-22 binding protein (IL-22BP) regulate fibrosis and cirrhosis in hepatitis C virus and schistosome infections. Hepatology. 2015;61:1321-1331
13. Weiss B, Wolk K, Grünberg BH, Volk HD, Sterry W, Asadullah K. et al. Cloning of murine IL-22 receptor alpha 2 and comparison with its human counterpart. Genes Immun. 2004;5:330-336
14. Gruenberg BH, Schoenemeyer A, Weiss B, Toschi L, Kunz S, Wolk K. et al. A novel, soluble homologue of the human IL-10 receptor with preferential expression in placenta. Genes Immun. 2001;2:329-334
15. Dumoutier L, Lejeune D, Colau D, Renauld JC. Cloning and characterization of IL-22 binding protein, a natural antagonist of IL-10-related T cell-derived inducible factor/IL-22. J Immunol. 2001;166:7090-7095
16. Xu W, Presnell SR, Parrish-Novak J, Kindsvogel W, Jaspers S, Chen Z. et al. A soluble class II cytokine receptor, IL-22RA2, is a naturally occurring IL-22 antagonist. Proc Natl Acad Sci USA. 2001;98:9511-9516
17. Pan HF, Li XP, Zheng SG, Ye DQ. Emerging role of interleukin-22 in autoimmune diseases. Cytokine Growth Factor Rev. 2013;24:51-57
18. Zhang W, Chen Y, Wei H, Zheng C, Sun R, Zhang J. et al. Antiapoptotic activity of autocrine interleukin-22 and therapeutic effects of interleukin-22-small interfering RNA on human lung cancer xenografts. Clin Cancer Res. 2008;14:6432-6439
19. Park O, Wang H, Weng H, Feigenbaum L, Li H, Yin S. et al. In vivo consequences of liver-specific interleukin-22 expression in mice: implications for human liver disease progression. Hepatology. 2011;54:252-261
20. Jiang R, Tan Z, Deng L, Chen Y, Xia Y, Gao Y. et al. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011;54:900-909
21. Kim SI, Shin D, Choi TH, Lee JC, Cheon GJ, Kim KY. et al. Systemic and specific delivery of small interfering RNAs to the liver mediated by apolipoprotein A-I. Mol Ther. 2007;15:1145-1152
22. Fioravanti J, González I, Medina-Echeverz J, Larrea E, Ardaiz N, González-Aseguinolaza G. et al. Anchoring Interferon Alpha to Apolipoprotein A-I Reduces Hematological Toxicity While Enhancing Immunostimulatory Properties. Hepatology. 2011;53:1864-1873
23. Ding Y, Wang W, Feng M, Wang Y, Zhou J, Ding X. et al. A biomimetic nanovector-mediated targeted cholesterol-conjugated siRNA delivery for tumor gene therapy. Biomaterials. 2012;33:8893-8905
24. McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. et al. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J. Clin. Invest. 2012;122:1574-1583
25. Bhogal RH, Curbishley SM, Weston CJ, Adams DH, Afford SC. Reactive oxygen species mediate human hepatocyte injury during hypoxia/reoxygenation. Liver Transpl. 2010;16:1303-1313
26. Baum C, Kustikova O, Modlich U, Li Z, Fehse B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum Gene Ther. 2006;17:253-263
27. Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nature Rev Genet. 2007;8:573-587
28. Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nature Rev Genet. 2003;4:346-358
29. Bouard D, Alazard-Dany D, Cosset FL. Viral vectors: from virology to transgene expression. Br J Pharmacol. 2009;157:153-165
30. Park JK, Utsumi T, Seo YE, Deng Y, Satoh A, Saltzman WM. et al. Cellular distribution of injected PLGA-nanoparticles in the liver. Nanomedicine. 2016;12:1365-74
31. Fluiter K, van der Westhuijzen DR, van Berkel TJ. In vivo regulation of scavenger receptor BI and the selective uptake of high density lipoprotein cholesteryl esters in rat liver parenchymal and kupffer cells. J Biol Chem. 1998;273:8434-8
32. Malerød L, Juvet K, Gjøen T, Berg T. The expression of scavenger receptor class B, type I (SR-BI) and caveolin-1 in parenchymal and nonparenchymal liver cells. Cell Tissue Res. 2002;307:173-80
33. Ki SH, Park O, Zheng M, Morales-Ibanez O, Kolls JK, Bataller R. et al. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology. 2010;52:1291-1300
34. Xing WW, Zou MJ, Liu S, Xu T, Wang JX, Xu DG. Interleukin-22 protects against acute alcohol-induced hepatotoxicity in mice. Biosci Biotechnol Biochem. 2011;75:1290-1294
35. Chestovich PJ, Uchida Y, Chang W, Ajalat M, Lassman C, Sabat R. et al. Interleukin-22: implications for liver ischemia-reperfusion injury. Transplantation. 2012;93:485-492
36. Ren X, Hu B, Colletti LM. IL-22 is involved in liver regeneration after hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2010;298:G74-G80
37. Liu FC, Lee HC, Liao CC, Li AH, Yu HP. Tropisetron protects against acetaminophen-induced liver injury via suppressing hepatic oxidative stress and modulating the activation of JNK/ERK MAPK pathways. Biomed Res Int. 2016;2016:1952947
38. Furuta K, Yoshida Y, Ogura S, Kurahashi T, Kizu T, Maeda S. et al. Gab1 adaptor protein acts as a gatekeeper to balance hepatocyte death and proliferation during acetaminophen-induced liver injury in mice. Hepatology. 2016;63:1340-1355
39. Cohen SD, Pumford NR, Khairallah EA, Boekelheide K, Pohl LR, Amouzadeh HR. et al. Selective protein covalent binding and target organ toxicity. Toxicol Appl Pharmacol. 1997;143:1-12
40. Jaeschke H, Knight TR, Bajt ML. The role of oxidant stress and reactive nitrogen species in acetaminophen hepatotoxicity. Toxicol Lett. 2003;144:279-288
41. Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89:31-41
42. Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 2008;283:13565-13577
43. Burke AS, MacMillan-Crow LA, Hinson JA. Reactive nitrogen species in acetaminophen-induced mitochondrial damage and toxicity in mouse hepatocytes. Chem Res Toxicol. 2010;23:1286-1292
44. Williams JA, Ni HM, Haynes A, Manley S, Li Y, Jaeschke H. et al. Chronic deletion and acute knockdown of parkin have differential responses to acetaminophen-induced mitophagy and liver injury in mice. J Biol Chem. 2015;290:10934-10946
45. Ding WX, Ni HM, DiFrancesca D, Stolz DB, Yin XM. Bid-dependent generation of oxygen radicals promotes death receptor activation-induced apoptosis in murine hepatocytes. Hepatology. 2004;40:403-413
46. Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307-320
47. Liu Q, Rehman H, Krishnasamy Y, Schnellmann RG, Lemasters JJ, Zhong Z. Improvement of liver injury and survival by JNK2 and iNOS deficiency in liver transplants from cardiac death mice. J Hepatol. 2015;63:68-74
48. Khan MK, Nigavekar SS, Minc LD, Kariapper MS, Nair BM, Lesniak WG. et al. In vivo biodistribution of dendrimers and dendrimer nanocomposites - implications for cancer imaging and therapy. Technol Cancer Res Treat. 2005;4:603-613
49. Tang Y, Han S, Liu H, Chen X, Huang L, Li X. et al. The role of surface chemistry in determining in vivo biodistribution and toxicity of CdSe/ZnS core-shell quantum dots. Biomaterials. 2013;34:8741-8755
50. Roberts JR, Antonini JM, Porter DW, Chapman RS, Scabilloni JF, Young SH. et al. Lung toxicity and biodistribution of Cd/Se-ZnS quantum dots with different surface functional groups after pulmonary exposure in rats. Part Fibre Toxicol. 2013;10:5
51. Li C, Liu H, Sun Y, Wang H, Guo F, Rao S. et al. PAMAM nanoparticles promote acute lung injury by inducing autophagic cell death through the Akt-TSC2-mTOR signaling pathway. J Mol Cell Biol. 2009;1:37-45
52. Fan J, Sun Y, Wang S, Li Y, Zeng X, Cao Z. et al. Inhibition of autophagy overcomes the nanotoxicity elicited by cadmium-based quantum dots. Biomaterials. 2016;78:102-14
53. Li Y, Zhu H, Wang S, Qian X, Fan J, Wang Z. et al. Interplay of oxidative stress and autophagy in PAMAM dendrimers-induced neuronal cell death. Theranostics. 2015;5:1363-77
54. Jones CF, Campbell RA, Brooks AE, Assemi S, Tadjiki S, Thiagarajan G. et al. Cationic PAMAM dendrimers aggressively initiate blood clot formation. ACS nano. 2012;6:9900-10
Author contact
![]() Corresponding author: Dianwen Ju, Tel: +86 21 5198 0037; Fax: +86 21 5198 0036; E-mail: dianwenjuedu.cn
Corresponding author: Dianwen Ju, Tel: +86 21 5198 0037; Fax: +86 21 5198 0036; E-mail: dianwenjuedu.cn