Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussion
Conclusion
Materials and Methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(16):3856-3872. doi:10.7150/thno.19981 This issue Cite
Research Paper
Berunda Polypeptides: Multi-Headed Fusion Proteins Promote Subcutaneous Administration of Rapamycin to Breast Cancer In Vivo
Jugal P. Dhandhukia1, Zhe Li1, Santosh Peddi1, Shruti Kakan1, Arjun Mehta2, David Tyrpak1, Jordan Despanie1, J. Andrew MacKay1, 3 ![]()
1. Department of Pharmacology and Pharmaceutical Sciences, University of Southern California, School of Pharmacy, 1985 Zonal Avenue, Los Angeles, CA 90089;
2. Department of Anatomic and Clinical Pathology, Los Angeles County & University of Southern California Medical Center, Los Angeles, CA 90033;
3. Department of Biomedical Engineering, University of Southern California, School of Pharmacy, 1985 Zonal Avenue, Los Angeles, CA 90089.
Received 2017-3-8; Accepted 2017-8-1; Published 2017-8-29
Abstract

Recombinant Elastin-Like Polypeptides (ELPs) serve as attractive scaffolds for nanoformulations because they can be charge-neutral, water soluble, high molecular weight, monodisperse, biodegradable, and decorated with functional proteins. We recently reported that fusion of the FK-506 binding protein 12 (FKBP) to an ELP nanoparticle (FSI) reduces rapamycin (Rapa) toxicity and enables intravenous (IV) therapy in both a xenograft breast cancer model and a murine autoimmune disease model. Rapa has poor solubility, which leads to variable oral bioavailability or drug precipitation following parenteral administration. While IV administration is routine during chemotherapy, cytostatic molecules like Rapa would require repeat administrations in clinical settings. To optimize FKBP/Rapa for subcutaneous (SC) administration, this manuscript expands upon first-generation FSI nanoparticles (Rh ~ 25 nm) and compares them with two second-generation carriers (FA and FAF) that: i) do not self-assemble; ii) retain a hydrodynamic radius (Rh ~ 7 nm) above the renal filtration cutoff; iii) increase tumor accumulation; and iv) have either one (FA) or two (FAF) drug-binding FKBP domains per ELP protein.
Methods: The carriers were compared and evaluated for temperature-concentration phase behavior by UV-Vis spectrophotometry; equilibrium binding and thermodynamics by Isothermal Titration Calorimetry; drug retention and formulation stability by Dialysis and Dynamic Light Scattering; in vitro efficacy using a cell proliferation assay; in vivo efficacy in human MDA-MB-468 orthotopic breast cancer xenografts; downstream target inhibition using western blot; tissue histopathology; and bio-distribution via optical imaging in the orthotopic xenograft mouse model.
Results: Named after the two-headed bird in Hindu mythology, the 'Berunda polypeptide' FAF with molecular weight of 97 kDa and particle size, Rh ~ 7 nm demonstrated polypeptide conformation of a soluble hydrated coiled polymer, retained formulation stability for one month post Rapa loading, eliminated toxicity observed with free Rapa after SC administration, suppressed tumor growth, decreased phosphorylation of a downstream target, and increased tumor accumulation in orthotopic breast tumor xenografts.
Conclusion: This comprehensive manuscript demonstrates the versatility of recombinant protein-polymers to investigate drug carrier architectures. Furthermore, their facilitation of SC administration of poorly soluble drugs, like Rapa, may enable chronic self-administration in patients.
Keywords: elastin-like polypeptides, rapamycin, triple negative breast cancer, drug delivery, bio-distribution, optical imaging.
Introduction
Systemic delivery of potent and hydrophobic drugs remains challenging due to the solubility and toxicity profile associated with such molecules [2-4]. One such drug, sirolimus is the clinical formulation of rapamycin (Rapa), which is indicated as an oral immunosuppressive for renal allograft rejection [5-7] and orphan Lymphangioleiomyomatosis [8, 9]. Despite approval for these treatments, Rapa has low solubility [10] and poor bioavailability [11, 12] accompanied with dose-limiting side effects such as pulmonary- and nephro-toxicity [13, 14], which limits its clinical potential. With the discovery that the mammalian target of rapamycin complex 1 (mTORC1) is inhibited in tumor cells by a complex between Rapa and the FK-506 binding protein 12 (FKBP), Rapa and its structural analogs were developed to treat cancer [15]. However, due to poor oral bioavailability and grade 3-4 adverse events such as skin rashes, anemia, and stomatitis, maintenance of rapalogue therapy remains a challenge [12, 16-18]. Earlier attempts have been made to solubilize and formulate Rapa using organic solvents and emulsions [19-21]; however, there have been side effects reported with the vehicle composition [22, 23]. With the goals of improving the solubility and toxicity profile of Rapa, we recently reported high efficiency drug loading and toxicity-free in vivo efficacy through a recombinant fusion between its native protein receptor, FKBP, and an elastin-like polypeptide (ELP) nanoparticle (FSI) [24, 25].
ELPs are genetically-encoded protein polymers derived from human tropoelastin [26] with the amino acid sequence (Val-Pro-Gly-Xaa-Gly)n where Xaa represents the guest residue and n represents the number of pentameric repeats. ELPs reversibly phase separate above an inverse transition temperature (Tt), which can drive assembly of fused peptides [27, 28]. They make attractive scaffolds for drug delivery applications because they are biodegradable, can be genetically fused with different protein domains, peptides, or therapeutics, and can be purified from prokaryotic systems without chemical synthesis [29]. In our previous report, FKBP was attached to an amphiphilic ELP diblock copolymer (SI) comprised of a hydrophilic ELP at the amino terminus (Xaa = Ser, n = 48 repeats) followed by a hydrophobic ELP (Xaa = Ile, n = 48 repeats) (Table 1). Known as FSI, this fusion protein was purified from E. coli, loaded with Rapa, and evaluated for efficacy using the MDA-MB-468 tumor xenograft model [24]. MDA-MB-468 are triple negative breast cancer cells (TNBCs) lacking estrogen, progesterone and HER2+ receptors [30]. In addition, these cells are devoid of PTEN phosphatase, which results in extensive phosphorylation of AKT kinase and highly active mTORC1 signaling [31, 32]. FSI loaded with Rapa not only improved the solubility of the drug but was also efficacious in suppressing tumor growth when injected intravenously (IV), without the significant systemic toxicity that was observed with free drug treatment [24].
Physicochemical properties of ELP protein polymers with and without FKBP.
| Label | aAmino acid sequence | bMW [kDa] | cMW [kDa] | dPurity [%] | eRh at | fTemperature-concentration phase diagram | ||
|---|---|---|---|---|---|---|---|---|
| 20 °C [nm] | 37 °C [nm] | Slope, m [°C Log(µM)] | Intercept, b [°C] | |||||
| SI | MG(VPGSG)48(VPGIG)48Y | 39.8 | 39.6 | 93.6 | 5.3 ± 1.6 | 22.9 ± 0.5 | 4.7 ± 0.9 | 34.9 ± 1.3 |
| FSI | M-FKBP-G(VPGSG)48(VPGIG)48Y | 51.6 | 51.6 | 94.5 | 6.1 ± 0.1 | 21.3 ± 0.6 | 3.5 ± 0.1 | 29.4 ± 0.2 |
| A192 | MG(VPGAG)192Y | 73.6 | 73.3 | 94.3 | 6.9 ± 0.2 | 6.6 ± 0.0 | 8.4 ± 0.6 | 73.9 ± 0.9 |
| FA | M-FKBP-G(VPGAG)192Y | 85.4 | 85.0 | 98.5 | 8.4 ± 0.1 | 7.8 ± 0.4 | 2.5 ± 6.2 | 61.7 ± 8.9 |
| FAF | M-FKBP-G(VPGAG)192-FKBP | 97.0 | 96.6 | 98.2 | 8.5 ± 0.6 | 7.9 ± 0.2 | 4.3 ± 0.7 | 63.6 ± 0.9 |
aFKBP amino acid sequence:
GVQVETISPGDGRTFPKRGQTCVVHYTGMLEDGKKFDSSRDRNKPFKFMLGKQEVIRGWEEGVAQMSVGQRAKLTISPDYAYGATGHPGIIPPHATLVFDVELLKLE[1]
bExpected MW based on amino acid sequence.
cObserved MW determined by running samples on MALDI-TOF.
dPurity was determined (Equation S1) using SDS-PAGE gel and densitometry analysis of the copper chloride stained gel using ImageJ.
eRh, hydrodynamic radius of 25 µM samples determined by Dynamic Light Scattering (n=3, mean ± SD).
fPhase diagrams for assembly (Figure 2 b, c) were fit with Equation S2. Values represent mean ± 95% CI.
Although FSI-Rapa is efficacious after IV administration, it remained unknown whether the assembly of a nanoparticle was advantageous for subcutaneous (SC) administration. Furthermore, this article explores how the ELP architecture influences the carrier's bio-distribution and toxicity via SC administration. To examine this relationship, this manuscript presents two new second generation carriers designed for enhanced systemic circulation with a comparatively higher molecular weight and a smaller hydrodynamic radius. A new FKBP-ELP carrier was synthesized (FA) by attaching FKBP at the amino terminus of A192 ELP (Xaa = Ala, n = 192 repeats) that remains soluble at physiological temperature. To compare the effect of FKBP valency on drug binding and release, a carrier with two-headed 'Berunda' architecture (FAF) was also synthesized with FKBP domains attached at both the amino and carboxy termini of A192 ELP. With a MW of 73.5 kDa, the backbone ELP A192 was selected due to its favorable biodistribution in orthotopic breast tumors implanted in nude mice when compared to relatively short ELPs or nanoparticle ELPs [33]. The nomenclature, amino acid sequence and physicochemical properties of all the FKBP-ELP carriers evaluated in this manuscript are shown in Table 1. Unlike FSI, FA and FAF are not amphiphilic and remain soluble at physiological temperatures. These carriers were compared for binding thermodynamics, drug retention and stability, in vivo efficacy and bio-distribution in a human breast tumor xenograft model (Figure 1).
Optimization of FKBP-ELP architecture to enhance the stability and efficacy of Rapa delivery. Rapalogues are potent cytostatic molecules with anti-cancer efficacy; however, their poor solubility limits their safety and efficacy by oral and IV delivery. This manuscript describes a new protein-based strategy to deliver Rapa via SC delivery using fusions between the FKBP protein and ELP (Table 1). This side-by-side comparison evaluates soluble ELPs with one (FA) or two (FAF) drug binding domains with a nanoparticle ELP (FSI). While all three carriers can bind Rapa, reduce injection site toxicity, and suppress a human breast cancer xenograft (MDA-MB-468), the Berunda polypeptide named FAF performed best with respect to drug loading, drug retention, formulation stability, tumor efficacy and bio-distribution following SC administration.
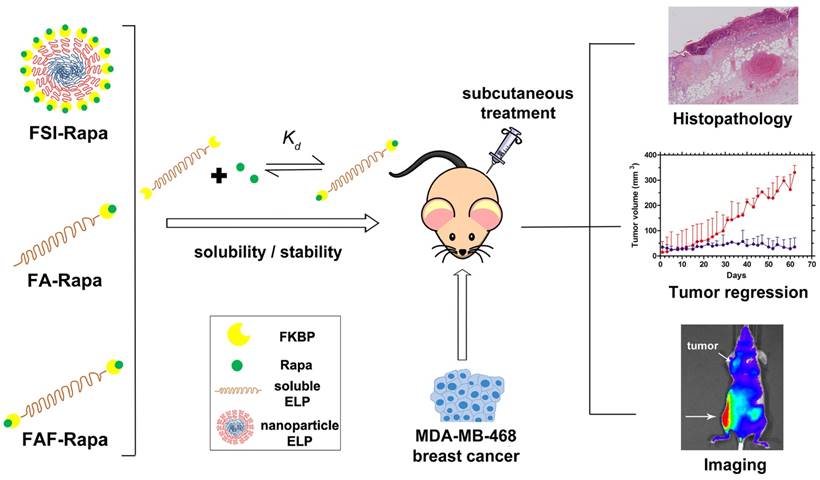
Formulating rapalogues for systemic delivery free of adverse effects remains a challenge [12, 16-18]. To address these challenges, multiple materials have been proposed as advanced drug carriers in cancer models such as poly(lactide-co-glycolide) nanoparticles [34], polyethyleneglycol-block-poly(ɛ-caprolactone) nanoparticles [35], albumin-bound nanoparticles [36] and multi-drug loaded 'triolimus' micelles [37]. Compared to these formulations, the carriers evaluated in this manuscript differ in two fundamental ways. First, while other carriers non-specifically carry the drug in a hydrophobic nanoparticle core or albumin pocket, FKBP-ELP carriers employ Rapa's biological receptor, which has the high specificity/affinity binding necessary to retain the drug for long durations in the body. Secondly, both FKBP and ELP are biodegradable and biocompatible polypeptides that can be produced at high yield and purity through scalable bacterial fermentation. These FKBP-ELP nanoformulations solubilize the drug free of any excipients or organic solvents, thereby eliminating injection site toxicity, which occurs with the free drug formulated using standard emulsions, such as Cremophor-EL. Among the carriers examined herein, FAF performs with the best combination of high drug loading, long drug retention, and particle size stability. While facilitating SC administration of Rapa, FAF further augments tumor accumulation and suppresses tumor growth.
Results and Discussion
Rapa is a highly potent drug with an unfavorable toxicity profile. While its cytostatic activity prevents growth of some tumors, its application in human cancer treatment has been limited by poor bioavailability, rapid clearance, and severe toxicity. Aiming to synthesize a viable and safer Rapa formulation, we recently reported an ELP-based nanoparticle drug carrier (FSI) that suppresses tumor growth in vitro and in vivo following IV administration with reduced side effects [24]. To elucidate the implications of the ELP architecture on the formulation stability and bio-distribution, this manuscript compares a library of related FKBP-ELPs for binding thermodynamics, extended stability, in vivo efficacy and optical imaging following SC administration in a human breast cancer xenograft mouse model.
Physicochemical characterization of FKBP-ELP carriers
All FKBP-ELP fusions were expressed and purified from E. coli. Compared to our previously reported FSI nanoparticles, the second-generation carriers FA and FAF maintain a comparatively smaller particle size at physiological temperature (Table 1). SDS-PAGE was used to determine the purity (Equation S1) of FKBP-ELPs along with their respective ELP backbones (Figure 2a). The precise molecular weights of all the constructs were determined by analyzing samples on a MALDI-TOF mass spectrometer (Table 1). Optical density measurements were used to characterize the temperature-concentration phase diagram of ELPs with and without FKBP (Figure 2b). Fusion of FKBP to ELP minimally influences the ELP phase diagram; furthermore, Tt follows an inverse relationship with logarithm of the ELP concentration. Using the fit parameters (Equation S2, Table 1) for FKBP-ELPs, it is possible to estimate solubility profiles of all FKBP-ELPs at physiological temperatures across concentrations relevant to therapy (1-500 μM). Based on this dataset, it can be extrapolated that FSI assembles into nanoparticles at 37 °C, while FA and FAF remain completely soluble under all concentrations. Very similar ELP phase behavior was also observed after drug loading (Figure 2c).
Physicochemical characterization of FKBP-ELP carriers. (a) Copper-stained SDS-PAGE confirmed the identity and purity of all the constructs with single band corresponding to greater than 90% purity (Table 1). Temperature-concentration phase diagrams of (b) ELPs with and without FKBP and (c) Rapa-loaded FKBP-ELPs measured by determining optical density at 350 nm. ELPs phase separate (A192, FA, FAF) above the transition temperature or assemble nanoparticles (SI, FSI) above the critical micelle temperature (CMT) as shown by the indicated model fits. (d) Hydrodynamic radii (Rh) of ELPs shows nanoparticle assembly only for SI and FSI whereas A192, FA and FAF remain soluble at 37 °C (n = 3, mean ± SD). (e) All Rapa-loaded FKBP-ELPs maintain stable Rh at 37 °C for 24 h (n = 3, mean ± SD).
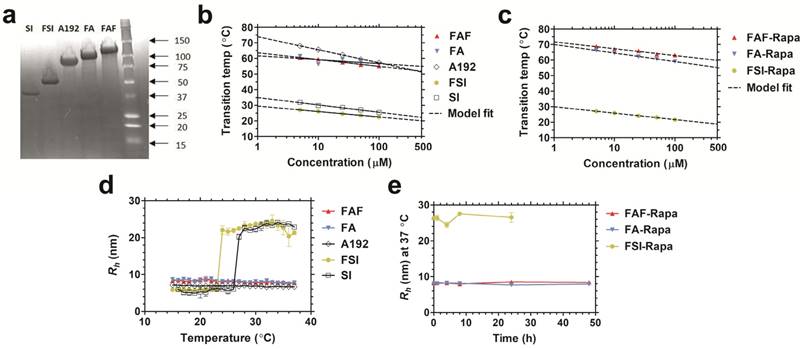
After studying the thermal properties of FKBP-ELPs, the hydrodynamic radii (Rh) of all the constructs were evaluated over a temperature gradient using Dynamic Light Scattering (DLS) (Figure 2d). As shown in the figure, hydrodynamic radii were minimally influenced by fusing FKBP to respective ELP backbones, with SI and FSI assembling nanoparticles above a critical micelle temperature (CMT) of ~25 °C [24]. Unlike SI and FSI, which undergo nanoparticle assembly as previously characterized by DLS and cryo-TEM [24], A192, FA and FAF retain sizes similar to that of linear polymers. To better explore their conformation and solution behavior, these three soluble ELPs were further compared using Size Exclusion Chromatography - Multi Angle Light Scattering (SEC-MALS). The radius of gyration (Rg) and absolute molecular weight were first obtained, and next the Rg/Rh ratio was used to interpret polypeptide conformation (Figure S1). The Rg for A192, FA, and FAF were found to be 11.9 nm ± 0.9 nm, 16.4 nm ± 1.6 nm, and 13.8 nm ± 0.4 nm respectively. Thus, using Rh obtained from DLS (Table 1), their Rg/Rh ratios were 1.72, 1.95 and 1.62 respectively. Rg/Rh ratios ≤ 0.7 are described in literature as spherical micelles [38] whereas higher ratios (1.6-1.8) are exhibited by hydrated polymers (PEG, PNIPA, and PVP) with coil conformations [39]. This data suggests that FA and FAF do not participate in significant peptide-mediated assembly but remain solvated, and mostly retain the random coil conformation for A192. In addition to obtaining the Rg values, the absolute molecular weights of A192, FA, and FAF were also determined as 74.4 kDa, 85.7 kDa, and 96.8 kDa respectively, which are remarkably consistent with values obtained using MALDI-TOF (Table 1). Furthermore, stability of all FKBP-ELPs was evaluated by measuring the particle size after Rapa loading for a period of 48 h at 37 °C (Figure 2e). Over this duration, the hydrodynamic radii remained stable, which demonstrates that drug binding to FKBP does not adversely alter the stability of the FKBP domain and also suggests that the formulation prevents precipitation of the free drug.
FAF binds two Rapa per molecule with a similar affinity to FA and FSI
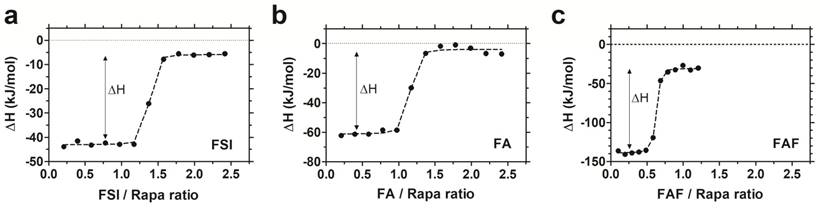
To study the interaction between Rapa and FKBP-ELPs, Isothermal Titration Calorimetry (ITC) was used to evaluate binding affinity and thermodynamics. ITC is a label-free technique to evaluate a wide range of binding energetics including binding stoichiometry, enthalpy (ΔH), entropy (ΔS), Gibbs free energy (ΔG) and affinity (Kd). Due to limitations on Rapa's aqueous solubility, reverse titrations were performed with Rapa in the calorimeter cell, which was titrated against successive injections of FKBP-ELPs. Saturable heat release was observed for all FKBP-ELPs, which suggests specific interaction between Rapa and the FKBP domain (Figure S2). To eliminate the possibility of non-specific interactions between Rapa and the ELP backbone, the experiment was repeated with ELPs lacking the FKBP domain where only heat of dilution was observed (Figure S2). ITC was performed for all FKBP-ELPs, and binding isotherms were obtained as shown in Figure 3. The data was fitted to a 'one set of sites' binding model, which enabled estimation of binding stoichiometry and thermodynamic parameters (Table 2). As observed in Figure 3, the sharp inflection in ΔH on successive injections of FKBP-ELPs occurred just before saturation when the ELP/Rapa ratio was ~1 for FSI and FA. This confirms the stoichiometry of Rapa bound to FSI and FA is ~1, each matching the number of FKBP domains. In contrast, saturation of FAF binding occurred at an ELP/Rapa ratio of ~0.5. This data confirms that both FKBP domains on FAF are functional; furthermore, FAF has twice the drug loading capacity per molecule in comparison to FSI and FA.
FAF has twice the drug loading capacity compared to FSI and FA. Isothermal titration calorimetry was used to quantify the binding stoichiometry and thermodynamic parameters between Rapa (8 µM) in 2.4% v/v DMSO in PBS at 37 °C against increasing titrations of (a) FSI, (b) FA and (c) FAF. An inflection in the ELP/Rapa ratio was ~1 for FSI and FA and ~0.5 for FAF, which is roughly consistent with having 1 (FA, FSI) or 2 (FAF) FKBP domains per ELP. A representative data set is shown from n = 3 per group.
Thermodynamic parameters of FKBP-ELP interaction with Rapa.
| Label | Binding stoichiometry, FKBP-ELP / Rapa | Dissociation constant, Kd (nM) | Enthalpy of binding, ΔH (kJ/mol) | -TΔS (kJ/mol) | Gibbs free energy of binding, ΔG (kJ/mol) |
|---|---|---|---|---|---|
| FSI | 1.33 ± 0.33 | 6.05 ± 0.91 | -38.8 ± 2.1 | 10.0 ± 2.4 | -48.8 ± 0.3 |
| FA | 1.12 ± 0.08 | 7.18 ± 1.42 | -57.6 ± 2.2 | 9.2 ± 1.9 | -48.4 ± 0.5 |
| FAF | 0.58 ± 0.04 | 4.87 ± 1.48 | -103.8 ± 2.9 | 54.4 ± 3.2 | -49.4 ± 0.3 |
All experiments were performed at 37 °C. Binding isotherms were fitted to a 'one set of sites' binding model to generate binding stoichiometry and thermodynamic parameters (n=3, mean ± SD).
Using a displacement assay, FKBP-Rapa interactions have been previously reported to have a dissociation constant of 0.2 nM [40]. ITC of this dataset revealed a slightly higher dissociation constant of ~6 nM for all the FKBP-ELPs (Table 2). The comparatively weaker Kd may be attributed to fusion of ELP to FKBP or the DMSO used to dissolve Rapa in phosphate buffered saline (PBS), both of which could weaken the interaction. Other parameters obtained on fitting the binding isotherms indicated a thermodynamically favorable interaction. Binding enthalpy (ΔH) for FSI and FA were -39 kJ/mol and -58 kJ/mol respectively, indicating exothermic reactions with heat released due to non-covalent association between Rapa and respective FKBP-ELPs. The observed enthalpies of binding are similar to a published dataset that predicts -83 kJ/mol for the binding of purified bovine FKBP12 and Rapa at 37°C at pH 7.0 [41]. A significant change in binding enthalpy of -104 kJ/mol was observed for FAF due to the two-fold increase in binding capacity per ELP. A positive -TΔS of ~10 kJ/mol for both FSI and FA; and 54 kJ/mol was observed for FAF, which revealed an entropic cost associated with Rapa binding to FKBP-ELPs. This could be explained by transitioning of Rapa from a free, unbound state in solution to a more ordered FKBP bound state. However, previous studies have shown expulsion of bound water molecules from the hydrophobic cavity of bovine and human FKBP into bulk water on binding of rapalogues as a favorable entropic contribution [41, 42]. An overall similar negative Gibbs free energy (ΔG), which sums enthalpy and entropy contributions, suggests Rapa binding to FKBP-ELPs remains a thermodynamically favorable interaction [43]. Among all the FKBP-ELPs, FAF showed twice the drug binding stoichiometry and comparatively significant change in binding enthalpy and entropy, which may favor Rapa interactions over that with FSI and FA.
FA and FAF architectures extend stability and drug retention compared to FSI nanoparticles
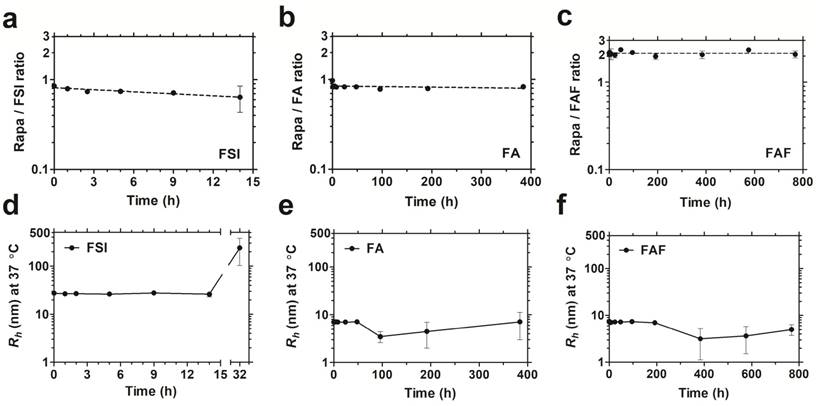
After studying drug binding interactions, Rapa-loaded formulations were evaluated for long-term drug retention and stability by performing dialysis under PBS sink conditions at 37 °C (Figure 4). To control for the loss in FA and FAF after long-duration dialysis against a 20 kDa MWCO membrane, aliquots taken at fixed time intervals were analyzed separately for both Rapa and FKBP-ELP using calibrated RP-HPLC assays, and Rapa/ELP ratios were plotted (Figure 4 a-c). At 0 h post drug loading, FSI and FA, had ~0.9 molecules of Rapa bound per ELP, whereas Berunda FAF had ~2.2 Rapa per ELP, which is consistent with the binding stoichiometry observed by ITC (Figure 3). Surprisingly, this ratio remained approximately unchanged for FAF up to one month. While measuring drug retention, formulations were simultaneously assessed for stability using DLS, and the assay was halted when the population consistent with the initial hydrodynamic radius dropped below 90% by mass (Figure 4 d-f). The FSI-Rapa formulation demonstrated an initial Rh of 26.6 nm ± 1.5 nm, which remained consistent until at least 14 h, after which Rh increased to ~239 nm ± 137 nm. In contrast, FA-Rapa and FAF-Rapa formulations were stable throughout the length of the assay with initial Rh of 6.4 nm ± 1.7 nm and 6.4 nm ± 1.6 nm respectively. Thus, FSI was stable for about one day, FA was stable for about 2 weeks, and FAF was stable for about one month. Together, these data verify that FAF is functionally bi-headed, with two FKBP domains per ELP. Furthermore, both FA and FAF formulations have greater physical stability than observed for the FSI formulation.
The soluble architecture of FA and FAF enhance stability with extended drug retention compared to the FSI nanoparticle. Dialysis was performed under PBS sink conditions at 37 °C to test drug retention and stability of FKBP-ELP formulations. (a) FSI-Rapa and (b) FA-Rapa formulations bound Rapa in ~1:1 ratio compared to (c) FAF-Rapa which bound drug in ~2:1 ratio as quantified by RP-HPLC. Values are presented as mean ± SD (n = 3 per group). DLS was performed at respective time points to study stability of Rapa-loaded FKBP-ELP formulations. (d) FSI-Rapa displayed a stable Rh until 14 h after which its radius began to increase whereas (e) FA-Rapa and (f) FAF-Rapa formulations were stable for 2-4 weeks demonstrating extended stability, strong drug retention, and potential benefits to shelf-life (mean ± SD, n ≥ 3 per time point).
Rapa-loaded FKBP-ELPs are more potent in inhibiting cell proliferation compared to free Rapa in vitro
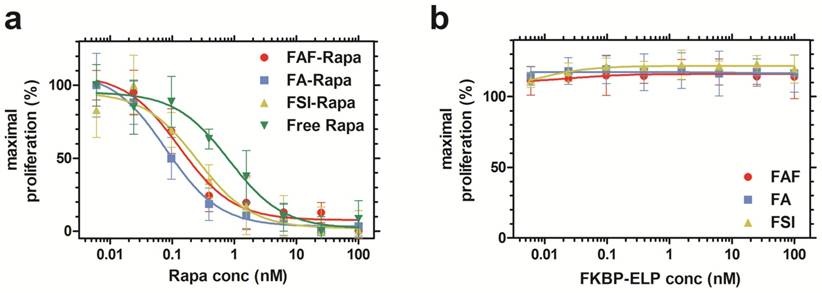
After characterizing the phase behavior, conformation, stability, and drug binding and retention, the in vitro cytostatic efficacy of Rapa with and without FKBP-ELPs was evaluated on MDA-MB-468 cells. All the four groups inhibited cell proliferation in a dose-dependent manner (Figure 5a). There was no significant difference between FAF-Rapa, FA-Rapa and FSI-Rapa, which suggested that ELP architecture does not play a distinguishing role in the cellular accessibility of Rapa in vitro. Instead a variety of architectures of FKBP-ELP carriers (Figure 1) appear to be capable of delivering drug to the cytosol where they inhibit proliferation through intracellular mTORC1. Interestingly, all three Rapa-loaded carriers were significantly more potent than free Rapa. The concentration corresponding to a 50% suppression in proliferation, IC50, for FAF-Rapa, FA-Rapa, FSI-Rapa and free Rapa were found to be 0.14 nM ± 0.05 nM, 0.08 nM ± 0.00 nM, 0.3 nM ± 0.2 nM and 0.84 nM ± 0.10 nM respectively (n = 3, mean ± SD). Tukey's post hoc analysis revealed statistically significant differences between free Rapa and FAF-Rapa, FA-Rapa, and FSI-Rapa (α = 0.05, p = 0.002, 0.0004 and 0.028 respectively). This downward shift in the IC50 with respect to free Rapa suggests that the FKBP-ELPs maintain their interaction with Rapa even in complete medium. Since Rapa binds strongly to albumin (>95%) [44], if the FKBP-ELPs were to rapidly release Rapa during incubation, the liberated drug might be expected to bind the great excess of bovine albumin in the medium and one would thus predict a similar IC50 as that observed for the free drug. Thus, the observation that the IC50 for FKBP-ELPs is significantly less than free Rapa is consistent with long retention (Figure 4) and high affinity interactions with the drug (Table 2). To test any ELP-mediated effect on this assay, the carriers without Rapa were evaluated as controls, which had no influence on cell proliferation (Figure 5b).
Rapa-loaded FKBP-ELPs are more potent in inhibiting MDA-MB-468 proliferation compared to free Rapa. (a) The inhibitory concentration corresponding to 50% suppression of proliferation, IC50, for FAF-Rapa, FA-Rapa, FSI-Rapa and free Rapa were determined by non-linear regression as 0.14 nM ± 0.05 nM, 0.08 nM ± 0.00 nM, 0.3 nM ± 0.2 nM and 0.84 nM ± 0.10 nM respectively (n = 3, mean ± SD). Tukey's post hoc analysis revealed statistically significant differences between free Rapa and FAF-Rapa, FA-Rapa and FSI-Rapa each (α = 0.05, p = 0.002, 0.0004 and 0.028 respectively) (b) Unloaded FKBP-ELPs had no effect on cell viability (n = 3, mean ± SD).
FAF-Rapa demonstrates superior tumor growth suppression in vivo following SC administration
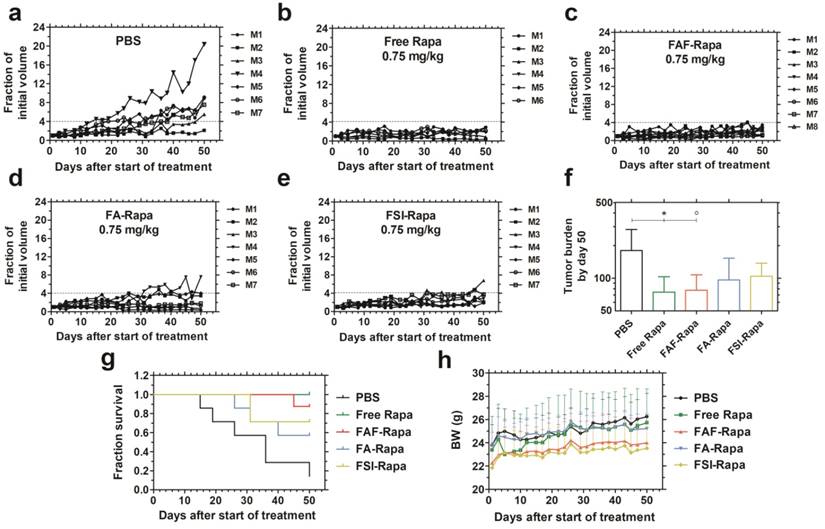
With no difference observed in cell viability between the Rapa-loaded carriers in vitro, the drug's in vivo efficacy was compared using MDA-MB-468 orthotopic breast cancer xenografts in mice. To determine the minimum effective dose, a pilot dose-ranging study was first performed using tail vein administration (Figure S3). A dose escalation regimen of FAF-Rapa (0.025-0.75 mg Rapa/kg body weight) with PBS and an intermediate dose of FSI-Rapa (0.25 mg/kg) [24] was evaluated. Treatment groups responded differentially to the dose regimen. PBS treated mice showed increasing tumor volumes, at rates that varied between subjects (Figure S3 a). Mice treated with FSI-Rapa at 0.25 mg/kg (Figure S3 b) and FAF-Rapa at 0.025-0.25 mg/kg (Figure S3 c-e) showed weak tumor suppression. Only the group treated with the high dose of FAF-Rapa at 0.75 mg/kg (Figure S3 f) showed statistical significance compared to PBS (Tukey's post-hoc analysis, α = 0.05, p = 0.002) (Figure S3 g). With similar efficacy observed between FSI and FAF formulations at 0.25 mg/kg, the dose-ranging study suggested that there is little difference in IV efficacy between the nanoparticle (FSI) and the soluble (FAF) architectures. Having identified that maximum tumor suppression occurs with FAF-Rapa at 0.75 mg/kg, this dose was then selected to compare free Rapa and Rapa-loaded carriers (FSI, FA and FAF) using SC administration (Figure 6).
FAF-Rapa outperforms other carriers in suppressing tumors when injected SC. Athymic nude mice (n = 6-8 per group) with orthotopic 30-180 mm3 MDA-MB-468 tumors were treated three times a week SC with (a) PBS (b) Free Rapa (c) FAF-Rapa (d) FA-Rapa and (e) FSI-Rapa at 0.75 mg/kg dose. (f) Tumor burden (Equation S4) by day 50 (mean ± SD) was statistically significant for the comparisons between PBS and Free Rapa, FAF-Rapa each (Tukey's post-hoc analysis, α = 0.05, *p = 0.03, ○p = 0.03 respectively). (g) Kaplan-Meier analysis shows Free Rapa and FAF-Rapa also had longer survival end points than the PBS treated control (Log-rank post-hoc analysis, α = 0.0125, p = 0.003 and 0.003 respectively). (h) There were no long-term trends in body weight (mean ± SD), except for the free Rapa group which recovered after a mild decrease in the first week of treatment.
The rationale behind exploring the SC administration is to identify opportunities for absorption from the site of injection into the systemic circulation. SC administration could for example be performed by patients at home, whereas IV administration requires more clinical expertise. SC administration is FDA-approved for monoclonal antibodies and other biotherapeutics including insulin [45]. In addition, there is a strong precedent for SC delivery of ELP fusions such as GlymeraTM, a formulation that reached Phase IIB clinical status for Type II diabetes [46, 47]. Bioavailability through the SC route is governed by either direct diffusion into blood capillaries or through the drainage into the lymphatic system, which depends on particle size and surface charge [48]. Based on the differences in hydrodynamic radii and drug loading, the library of FKBP-ELPs (Table 1) was compared to determine the optimal carrier to achieve tumor growth suppression following SC administration.
The comparative tumor regression study consisted of 5 groups: PBS, Free Rapa, FAF-Rapa, FA-Rapa, and FSI-Rapa, all at an equivalent dose of 0.75 mg/kg (n = 6-8 mice/ group). The injections were dosed SC in the right flank above the hind leg. The group treated with PBS exhibited a gradual increase in tumor volumes (Figure 6a). Groups treated with Free Rapa and FAF-Rapa showed strong tumor suppression (Figure 6 b, c) relative to groups treated with FA-Rapa and FSI-Rapa (Figure 6 d, e). FA-Rapa and FSI-Rapa treatments showed no statistical significance compared to PBS (Tukey's post-hoc analysis, α = 0.05, p = 0.095 and 0.38 respectively). Only the groups treated with free Rapa and FAF-Rapa demonstrated statistical significance compared to the PBS group (Tukey's post-hoc analysis, α = 0.05, p = 0.03 and 0.03 respectively) (Figure 6f). In addition to having lower tumor burdens, mice treated with free Rapa and FAF-Rapa had higher survival rates than FA-Rapa and FSI-Rapa groups (Figure 6g). Kaplan-Meier analysis was employed to distinguish the survival rates with a four-fold increase in tumor volume considered as the endpoint. Post-hoc log-rank tests comparing PBS with each of the four treatment groups revealed significant differences with Free Rapa and FAF-Rapa (Log-rank post-hoc analysis, α = 0.0125, p = 0.003 and 0.003 respectively). There was no decrease in body weight across all treatment groups, except for Free Rapa in the first week of treatment (Figure 6h). While it was not possible to distinguish statistical differences in tumor burden or survival directly between FAF and the FA and FSI formulations, the FAF-Rapa formulation exerted the strongest suppression of tumor growth with respect to PBS (Figure 6f). To further elucidate the relationship between SC absorption of FKBP-ELPs and tumor efficacy, the influence of carrier architecture on mTORC1-signaling, injection site toxicity, and bio-distribution was next evaluated in the same tumor xenograft model.
SC treatment with Rapa inhibits a downstream target of the AKT-mTORC1 axis in MDA-MB-468 solid tumors
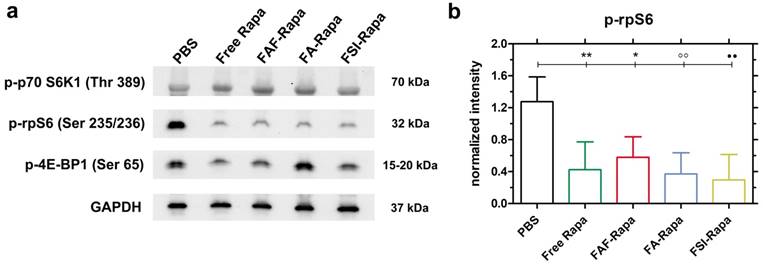
Due to the unexpectedly high stability (Figure 4) and superior SC efficacy of the FAF Berunda architecture (Figure 6), further pharmacological evidence was collected to confirm whether the drug can reach the tumor from the SC site of injection and act on a known molecular target of the mTORC1 pathway. Stimulation of cell surface receptor tyrosine kinases by growth factors is known to activate the PI3K-AKT-mTORC1 pathway in TNBCs, which activates and phosphorylates two major targets: p70 S6 Kinase 1 (p70 S6K1) and 4E-binding protein 1 (4E-BP1) [49]. Phosphorylation of p70 S6K1 further activates and phosphorylates downstream ribosomal S6 protein (rpS6), which plays a role in mRNA translation of proteins including elongation factors and ribosomal proteins. Phosphorylation of the other mTORC1 target 4E-BP1 facilitates cap dependent protein translation required for the G1-to-S phase transition of the cell cycle [32]. Treating MDA-MB-468 cells with Rapa in vitro has been previously shown to inhibit p-p70 S6K1 (Thr 389) and p-4E-BP1 (Ser 65) while strongly suppressing p-rpS6 (Ser 235/236) [50-52]. Based on this pharmacology, at the conclusion of the SC study tumor lysates were probed for p-p70 S6K1, p-4E-BP1 and p-rpS6 (Figure 7). At 24 h after the last SC dose, there was no significant decrease in the tumor level of p-p70 S6K1 and p-4E-BP1 (Figure 7a, Figure S4). In contrast, there was a significant decrease in p-rpS6 levels (Figure 7b) between PBS and free Rapa, FAF-Rapa, FA-Rapa and FSI-Rapa each (Tukey's post-hoc analysis, α = 0.05, p = 0.009, 0.035, 0.005 and 0.003 respectively). Similar to the results observed in the in vitro (Figure 5a) and SC in vivo (Figure 6f) studies, there was no statistical difference between the FKBP-ELP formulations, which suggests all the Rapa-loaded carriers equally suppress p-rpS6 levels.
SC treatment with Rapa inhibits a downstream target of mTORC1. (a) Tumor lysates probed for downstream targets of mTORC1 showed no significant inhibition in p-p70-S6K1 and p-4E-BP1 levels. However, there was a significant decrease in p-rpS6 levels in all treatment groups compared to PBS. A representative western blot is shown from n = 4 per group. (b) 1-way ANOVA on the normalized intensity levels (Equation S5) of p-rpS6 (n = 4, mean ± SD) showed a statistically significant decrease between PBS and Free Rapa, FAF-Rapa, FA-Rapa and FSI-Rapa each (Tukey post-hoc analysis, α = 0.05, **p = 0.009, *p = 0.035, °°p = 0.005, ●●p = 0.003 respectively).
The lack of a detected decrease in p-p70 S6K1 and p-4E-BP1 could be either due to treatment with a transient tumor exposure in vivo following a low dose (0.75 mg/kg) as compared to the constant exposure achieved for in vitro incubation [51]. It is plausible that p-p70 S6K1 and p-4E-BP1 levels recover during the 24 h between the last dose and tumor lysis. Despite this, the significant decrease in p-rpS6 level signifies that Rapa with or without FKBP-ELP can travel from a distal site of SC injection, presumably through systemic circulation, to exert its effect on an orthotopic solid tumor. Since all the treatments worked similarly in dephosphorylating rpS6, we then proceeded to study the relative toxicity and bio-distribution as a function of FKBP-ELP architecture.
Free Rapa treatment is toxic at the site of SC injection
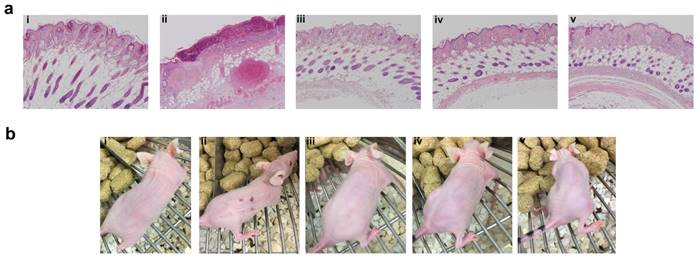
Even though SC administration of free Rapa without any carrier was efficacious in suppressing tumor growth and phosphorylation of rpS6, treatments were not equally tolerated at their injection site (Figure 8). Free Rapa was administered the first week in 100% DMSO, which was not tolerated and produced a small drop in BW. To continue the treatment, starting in the second week, the free Rapa control was instead solubilized using Cremophor-EL as a co-solvent [53], which proved to be tolerated by the mice resulting in a gradual gain of lost BW (Figure 6h). Free Rapa was formulated in DMSO: EtOH: Cremophor-EL: PBS in a 1:1:2:6 % v/v ratio. While the local bruising of free Rapa was less prominent, local edema persisted at each site of injection for one week after every SC injection. In contrast to free Rapa, all the FKBP-ELP formulations showed no external signs of redness or blistering throughout the duration of the study (Figure 8b). At the end of the SC study, all mice were euthanized and organs were evaluated for histopathology by Hematoxylin and Eosin (H & E) staining. Despite 50 days of treatment, there was no abnormal pathophysiology observed in any of the major organs (Figure S5), with the exception of the skin (Figure 8a). The skin section obtained after SC administration of free Rapa group displayed epidermis with focal hyperkeratosis, parakeratosis, and intraepidermal neutrophilic infiltrate with cellular debris. The underlying hair follicles were necrotic and the adipose tissue exhibited hemorrhage and neovascularization. In contrast, the epidermis in all the other treatment groups was intact with keratinizing squamous epithelium. The dermis was observed with connective tissue and sebaceous glands with numerous intact hair follicles. No evidence of inflammation or necrosis was identified with any of the Rapa loaded FKBP-ELP formulations. Though effective in solubilizing free Rapa and inhibiting tumor growth, the Cremophor-EL formulation produced severe toxicity at the site of injection. In contrast, all other sections from mice treated with drug-loaded FKBP-ELPs were devoid of local or systemic toxicity. This strengthens the argument that while free Rapa remains effective as a cytostatic agent, it cannot be administered SC without inducing toxicity; furthermore, suitable drug carriers must both improve its solubility, bioavailability and retain its relative cytostatic efficacy.
FKBP-ELP carrier protects against tissue necrosis during SC administration. (a) After the conclusion of 50 days of SC treatment three times weekly at 0.75 mg/kg Rapa dose, skin sections from the most recent site of injection were excised, paraffin embedded, and evaluated for H & E histopathology. Free Rapa treatment was toxic at site of injection compared to Rapa-loaded FKBP-ELPs. Panels i, ii, iii, iv, and v are from a representative mouse treated with PBS, free Rapa, FAF-Rapa, FA-Rapa and FSI-Rapa respectively (n = 6-8). (b) The relative differences between the SC toxicity of these formulations at the site of injection was also externally evident on day 30 of this study.
Soluble FA and FAF tumor accumulation is greater than FSI nanoparticles following SC administration
To explore the effect of ELP architecture on the SC bio-distribution of FKBP-ELPs, their uptake was tracked in xenograft mice by performing live optical imaging using a near infrared dye, Cyanine5.5 (Cy5.5). FA, FAF and FSI were covalently labelled with Cy5.5 using N-hydroxysuccinimide chemistry at a ratio of ~0.15 Cy5.5/protein (Equation S8) and an equivalent dose of 0.5 mg Cy5.5 / kg body weight was administered SC in the flank above the right hind leg (n = 4 mice/group). The Rh of Cy5.5 labelled FKBP-ELPs was confirmed as stable prior to in vivo administration (Figure S6). In vivo fluorescence was monitored using whole body optical imaging for 48 h from both ventral and dorsal perspectives (Figure 9, Figure S7). For all three formulations, the injection site remained highly fluorescent during the entire duration of the study; which could be due to entrapment of carriers in the lymph nodes. However, FA and FAF moved away from the injection site more rapidly than did FSI nanoparticles (Figure S7). From the ventral perspective, fluorescence accumulation at both the tumor and site of injection were qualitatively visible (Figure 9a). For quantitative comparisons between organs too deep to view using this optical imaging modality, mice were euthanized at 48 h, organs were isolated, and scanned immediately for fluorescence. After correcting for the tissue auto fluorescence observed in control mice without Cy5.5 administration, FA and FAF showed high fluorescence in the kidney, liver, and tumor compared to FSI (Figure 9b). Tukey post-hoc comparisons were performed to test differences between treatment groups within each tissue (Figure 9c). The kidney accumulation of signal was significantly greater for FA and FAF than for FSI (α = 0.05, p = 0.0004, 0.005 respectively). Similarly, the liver accumulation was significantly greater for FA and FAF than for FSI (α = 0.05, p= 0.001, 0.0004 respectively). Matching the qualitative observations (Figure 9a), the tumor accumulation for FA and FAF were also significantly greater than for FSI (α = 0.05, p = 0.0004, 0.004 respectively). Taken together, these observations suggest that soluble FA and FAF yield greater accumulation in the tumor compared to the FSI nanoparticles, which is likely a combination of their more rapid absorption from the SC site of administration, differences in tumor permeability, and also differences in interaction with clearance organs. FA and FAF also exhibited significantly higher tumor localization in live mice at 24 h and 48 h compared to FSI nanoparticles (Figure 9a). To further understand the poor FSI bio-distribution observed with the SC administration, Cy5.5 modified FSI was injected intravenously and compared to its SC counterpart (Figure S8). FSI injected intravenously had significantly higher liver, tumor and spleen accumulation than the SC counterpart (Tukey's post hoc analysis, α = 0.05, p = 0.001, 0.008 and 0.01 respectively) that suggests longer systemic circulation and enhanced bio-distribution are due to higher bio-availability observed with intravenous administrations. There was no difference in kidney accumulation compared to SC injection, which suggests nanoparticles were cleared predominantly by the liver and spleen.
FA and FAF show high accumulation in tumor and clearance organs compared to FSI. (a) MDA-MB-468 tumor implanted nude mice administered SC with Cy5.5 labeled FKBP-ELPs were imaged using IVIS Spectrum. FA and FAF show high tumor fluorescence at 24 h and 48 h compared to FSI. Tumor accumulation and site of injection are shown as indicated by arrows. A representative mouse scan in ventral position at 0, 4, 24 and 48 h is shown from each group of n = 4 mice. (b) Post 48 h whole body scan, mice were immediately euthanized and organs were scanned for fluorescence. A representative mouse is shown from each group of n = 4. (c) The fluorescence intensity in Avg. radiant efficiency (n = 4, mean ± SD) was quantified by drawing ROIs on all the organs. Tukey's multiple post hoc analysis reveals statistical significance between FA and FSI; FAF and FSI in kidneys, liver and tumor respectively (α = 0.05, ***p = 0.0004, **p = 0.005, °°p = 0.001, °°°p = 0.0004, ●●●p = 0.0004, ●●p = 0.004).
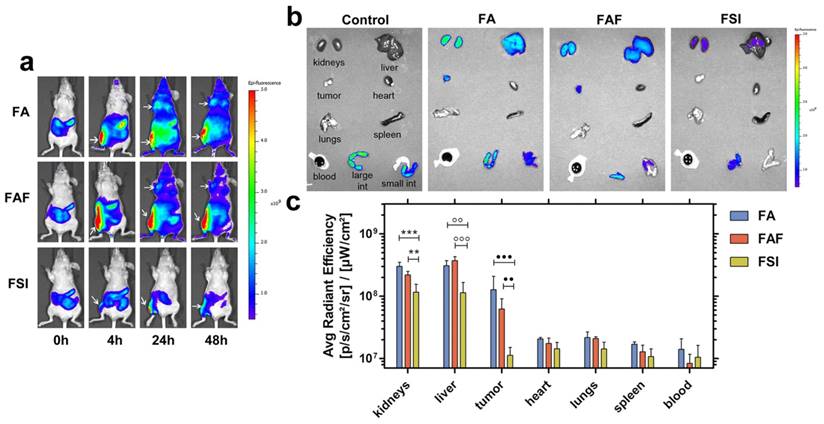
Absorption of biologics from SC injection into systemic circulation occurs either by direct uptake into blood capillaries or through the interstitium into lymphatic vessels [48]. Small peptides and proteins with particle sizes <10 nm and MW ≤16 kDa are more easily taken up by diffusion into blood capillaries whereas high molecular weight protein therapeutics with particle sizes in the range of 10-100 nm are taken up by lymphatic vessels [54, 55]. FA/FAF and FSI are high molecular weight proteins with particle radii ~7 nm and 25 nm respectively (Table 1, Figure 2). Even though all three FKBP-ELPs exhibit favorable characteristics for lymphatic uptake, FA and FAF owing to their smaller particle size exhibit more rapid absorption and thus greater tumor accumulation compared to FSI nanoparticles (Figure 9a). Curiously, these differences in tumor accumulation were not reflected by differences in p-rpS6 levels (Figure 7b). The lack of strong tumor accumulation by FSI with SC administration may suggest its delivery of Rapa by some other mechanism, such as through biodegradation inside macrophages along the lymph absorption pathway [56, 57]. While there is not enough evidence to point to a different mechanism between FA/FAF and FSI efficacy, the observations of higher tumor accumulation for FA/FAF are consistent with our previous report that soluble A192 ELP alone exhibits better tumor penetration and favorable pharmacokinetics compared to an ELP nanoparticle [33]. When Cy5.5 labeled FSI was injected IV, there was enhanced accumulation in liver and tumor compared to FSI injected SC, which is consistent with a reduced bioavailability of FSI during its transport along the lymph absorption pathway. Furthermore, despite the enhanced bio-distribution of FSI observed with IV administration (Figure S8 b, c), the tumor and liver fluorescence was similar to that observed with FA and FAF injected SC (Figure 9 b, c). This observation highlights the advantageous bio-distribution of soluble FA/FAF given SC, which was only matched by FSI when injected IV.
Taken together, these findings suggest that ELPs with soluble conformations (FA and FAF) behave similarly when administered SC and superior to ELPs that assemble nanoparticles (FSI). Increasing the number of ELP repeats beyond 192 pentamers may further extend the plasma half-life of soluble FKBP-ELP formulations due to a favorable increase in MW. In contrast, despite the poor SC bio-distribution of FSI, it retains its ability to suppress phosphorylation of tumor rpS6. All three carriers facilitated toxicity-free administration from an SC site. However, based on the fact that FAF efficacy was highest among the carriers, in addition to its higher drug-loading and long-duration stability, the sum of the data suggests that the Berunda FAF architecture is optimal for SC administration of Rapa. Future studies are required to clarify the differences in bioavailability, clearance, blood half-lives, tumor permeability, FKBP biodegradation, and to explore their fate during lymphatic absorption from an SC site of injection.
Rapa and its analogs are potent small molecule inhibitors of the AKT-mTORC1 pathway, which renders them excellent cytostatic drugs [15, 32]. Currently, there are three FDA approved rapalogues - Sirolimus, Temsirolimus and Everolimus. Sirolimus is formulated only for oral administration via liquid and solid formulations; however, its poor solubility yields low bioavailability with severe side effects [11, 13, 14]. Temsirolimus, an ester prodrug of sirolimus, is formulated as an ethanolic, non-aqueous IV infusion to overcome its poor solubility, which promotes dermatological and hypersensitivity reactions [17, 18]. Formulated as a solid tablet, Everolimus also has a low bioavailability and commonly causes dose-limiting side effects in the clinic [12, 18]. Not unlike chemotherapeutics such as doxorubicin and paclitaxel, the rapalogues are potent, effective drugs that can be further enhanced through better delivery strategies. To explore proteins as potential carriers, we previously reported that a recombinant fusion using FSI to carry Rapa suppresses xenograft tumor growth when administered IV, while reducing evidence of systemic toxicity. While IV administration overcomes low oral bioavailability, the rapalogues would clearly benefit from a parenteral route of self-administration such as SC. This manuscript further advances our approach by exploring a library of protein-polymer architectures to favor systemic drug absorption, tumor localization, and efficacy with SC administration.
Conclusion
Three different architectures for FKBP-ELP fusions were compared for drug loading, binding thermodynamics, stability, in vitro, in vivo efficacy and bio-distribution in an orthotopic model of breast cancer. The soluble two-headed Berunda polypeptide (FAF) had significant advantages over a single-headed (FA) or a nanoparticle formulation (FSI). The Berunda polypeptide improved drug-loading capacity and formulation stability as shown by ITC, dialysis, and light scattering. When all three architectures were compared via SC administration at an equivalent dose, only free Rapa and FAF-Rapa showed statistical significance compared to PBS. Despite these differences, all formulations improved in vitro potency in comparison to free Rapa and all delivered pharmacologically detectable evidence for in vivo suppression of a downstream target of mTORC1. When toxicity was evaluated, free Rapa treatment produced significant necrosis at the site of injection compared to Rapa-loaded FKBP-ELP formulations, which is consistent with other literature demonstrating that free Rapa administration is often associated with dose-limiting toxicity. Molecular imaging confirmed that tumor accumulation of FA and FAF injected SC is significantly higher than for the nanoparticle FSI, which was matched only when FSI was administered IV. These comparisons suggest soluble ELPs (FA, FAF) are preferable to nanoparticles (FSI) for SC administration. While all three carriers maintained drug potency in vitro, the Berunda FAF formulation provides the best combination of increased drug loading, favorable drug binding interactions, enhanced formulation shelf-stability, in vivo tumor accumulation, and growth suppression. This strategy of fusing the FKBP domain to protein-polymers, such as an ELP, effectively opens up a range of interesting delivery options for rapalogues and other potent drugs.
Materials and Methods
ELP gene design and cloning
FA and FSI cloning was performed as previously described [58]. Cloning of FAF was done by fusing an FKBP gene in frame to the 3' end of the gene for FA. To fuse another FKBP gene to the carboxy terminus, a new gene was synthesized on ampicillin-resistant pIDTsmart vector (Integrated DNA technologies, Coralville, IA) with three restriction cut sites: XbaI, BseRI and BamHI. This vector was designed such that the FKBP gene was flanked with cut sites for BseRI and BamHI with XbaI at the 5' end and BamHI at the 3' end of the oligonucleotide. The pIDTsmart vector was double digested with XbaI and BamHI and the FKBP gene was isolated by 1% agarose gel electrophoresis and purification (28-9034-70, GE Healthcare Life Sciences, Marlborough, MA). The FKBP gene was then ligated into a pET25b (+) vector (EMD Millipore, Billerica, MA) double digested with same set of XbaI and BamHI enzymes. In a second cloning step, the modified pET25b (+) vector containing the FKBP gene was double digested with BseRI and BssHII and gel purified. The appropriate fragment containing the FKBP gene was then ligated to the 3' end of the gel purified FA gene, which was isolated from a pET25b (+) vector double digested by AcuI and BssHII. Restriction enzymes were purchased from New England Biolabs® Inc (Ipswich, MA). The in-frame amino acid sequence was confirmed by diagnostic digestion and DNA sequencing.
FKBP-ELP expression and purification
The pET25b (+) vectors encoding genes for ELPs or FKBP-ELPs were transfected into BLR (DE3) E. coli competent cells (69053, EMD Millipore) and plated onto agar with 100 µg/mL ampicillin. A single colony was inoculated in 50 mL autoclaved terrific broth (TB) media (12105, Mo Bio Laboratories, Carlsbad, CA) supplemented with 100 µg/mL carbenicillin and grown overnight at 37 ºC in a shaker incubator. Starter cultures were then amplified in 3-4 L batches supplemented with 100 µg/mL carbenicillin and allowed to grow for 24 h at 37 ºC. Cell lysis was performed as previously described [58] and protein purification was performed using Inverse Transition Cycling [59]. Purified protein in Dulbecco's sterile PBS buffer (PBL01, Caisson labs, Smithfield, UT) was filtered using 200 nm sterile Acrodisc® 25 mm filters (PN 4612, Pall Corporation, Port Washington, NY) and assayed for concentration using Beer Lambert's law:
Protein concentration (M) = [(A280-A350) × dilution factor]/(MEC × l) (Eq. 1)
where M is molar concentration, A280 and A350 are absorbance at 280 and 350 nm respectively, l is the path length (cm) and MEC is the estimated molar extinction coefficient at 280 nm: 1285 M-1cm-1 for plain ELPs; 11585 M-1cm-1 for FA and FSI; and 20190 M-1cm-1 for FAF [60]. Final yields obtained were 50-60 mg/L.
FKBP-ELP physicochemical characterization
The purified fusion proteins were characterized for physicochemical properties using SDS-PAGE, optical density, MALDI-TOF, dynamic light scattering (DLS) and size-exclusion chromatography - multi angle light scattering (SEC-MALS). The detailed procedures are explained in the supplemental methods.
Isothermal titration calorimetry of Rapa interactions with FKBP-ELPs
The drug binding interactions between Rapa (R-5000, LC Laboratories Inc., Woburn, MA) and FKBP-ELPs were studied using ITC (Figure 3) on a MicroCal PEAQ ITC (Malvern Instruments Ltd, Worcestershire, United Kingdom). The reference cell of the calorimeter was filled with water and all binding studies were performed at 37 °C. Reverse titrations were performed with a fixed Rapa concentration in the calorimeter cell and FKBP-ELPs in the titration syringe. The drug and all the FKBP-ELPs were solubilized in the same buffer (2.4% v/v DMSO in PBS) to prevent background heat of release due to differences in buffer composition. Briefly, 300 µl of 8 µM Rapa was carefully loaded into the calorimeter cell and a titration syringe filled with 50 µM FAF or 100 µM FA/ FSI was injected (3 µl) into the calorimeter cell 12 times. The resulting isotherm was fitted to a 'one set of sites' binding model in offset mode using the MicroCal PEAQ ITC analysis software (Malvern Instruments) to estimate affinity (Kd), stoichiometry and thermodynamics (ΔH, ΔS and ΔG).
Drug retention and formulation stability using extended dialysis
Drug loading and formulation preparation for in vivo administration was performed as described in supplemental methods. To prevent non-specific binding of drug into the hydrophobic core of FSI nanoparticles [24] and to achieve a ~1:1 drug binding ratio of Rapa to FSI, drug loading was performed at room temperature for all FKBP-ELPs. 150 µM Rapa loaded FKBP-ELPs were dialyzed in 1:650 PBS sink conditions at 37 ºC using 20 kDa MWCO dialysis cassettes (87735, Thermo Fischer Scientific, Waltham, MA). PBS sink was supplemented with Penicillin-Streptomycin (30-002-CI, Corning®, Corning, NY) and buffer changes were performed 2x on day 1 and every 48 h thereafter. 100 µL aliquots were collected from the cassettes at fixed time intervals and were characterized for Rapa and FKBP-ELP concentrations at 280 nm using RP-HPLC. The assay was halted upon observation of turbidity. The data was fit by non-linear regression using a one-phase exponential decay model with values presented as mean ± SD with 95% CI (n = 3) (Figure 4 a-c). Aliquots withdrawn were filtered using 200 nm sterile Acrodisc® 13 mm filters (PN 4454, Pall Corporation) and evaluated for particle size at ~20-25 µM protein concentration using DLS at 37 °C (n ≥ 3). Rh values for particle populations with greater than 90% by mass were reported as the dominant species (Figure 4 d-f).
Cell proliferation assay
Cell viability was evaluated using the 4-[3-(4-Iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate (WST-1) assay (Figure 5). MDA-MB-468 cells (HTB-132, American Type Tissue Culture Collection, Manassas, VA) were cultured at 37 ºC with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM)/F-12 media (DFL21, Caisson labs) supplemented with 10% v/v heat inactivated fetal bovine serum (FBS, 35-011-CV, Corning®). 3,000 cells in 100 µL media per well were seeded in 96 well flat bottom plates in triplicate. Cells were allowed to adhere for 24 h and media was replaced with fresh media followed by treatment with dilutions of complete media containing free Rapa, Rapa-loaded FKBP-ELPs and/or unloaded FKBP-ELPs. After incubation for 72 h, the WST-1 reagent was incubated with the cells for 3 h according to the manufacturers recommendations (11644807001, Sigma-Aldrich, St. Louis, MO). Tetrazolium conversion to formazan was monitored by absorbance at 440 nm after subtracting for a reference wavelength at 690 nm. The % maximal proliferation was calculated and plotted against Rapa or FKBP-ELP concentration using the following equation:
maximal proliferation (%) = (Atreated-A0)/(Auntreated-A0) x 100 (Eq. 2)
where Atreated is the corrected absorbance in the presence of Rapa and/or unloaded FKBP-ELPs, Auntreated is the corrected absorbance of untreated cells, and A0 is the corrected absorbance of media cultured without cells. The IC50 was determined using non-linear regression in three independent assays (n = 3, mean ± SD). A global one-way ANOVA revealed significant differences among the 4 Rapa-treatment groups (α = 0.05, p = 0.0004). Tukey-Kramer post-hoc analysis was then performed to test significance between the individual groups as reported in the results (Figure 5a).
Tumor regression studies
All animal experiments were conducted as per the guidelines of the American Association of Laboratory Animal Care under approval by the USC Institutional Animal Care and Use Committee. MDA-MB-468 cells were screened for major mouse pathogens and human blood borne pathogens (Charles River) prior to implantation. Procedure for tumor implantation, analysis and IV study is provided in the supplemental methods. Mice were randomized into groups and SC treatment was started with a tumor size of 30-180 mm3. Tumor implanted mice were treated with 150-200 µL injections of PBS, Free Rapa (100% DMSO or DMSO: EtOH: Cremophor-EL: PBS in 1:1:2:6% v/v ratio), FAF-Rapa, FA-Rapa and FSI-Rapa all at a fixed dose of 0.75 mg/kg which amounts to 125 µM FSI or FA and 62.5 µM FAF in final injections (Figure 6). Cremophor-EL was purchased from Sigma-Aldrich (C5135). The study was analyzed by performing 1-way ANOVA on the log10 transformed tumor burden (Equation S4) by the last day of treatment, which showed significant differences among the five 5 groups (α = 0.05, p = 0.019). Tukey-Kramer post-hoc analysis was performed to test significance between the individual groups as reported in the results (Figure 6f). One mouse from the PBS group (M6) was euthanized on day 40 at the humane end point of 1000 mm3 tumor volume; therefore, the tumor growth determined on day 40 for M6 was used to calculate the tumor burden by the last day of treatment. For Kaplan-Meier survival analysis, the time to reach four times the initial volume (fi =4) was determined as the end point. The Log Rank (Mantel-Cox) test performed on the entire study revealed significant difference among the survival curves (α = 0.05, p = 0.002). Individual treatment groups were compared to the PBS treatment group using a Bonferroni correction (α = 0.05/4 comparisons = 0.0125) as reported in the results. Body weights are shown as mean ± SD (Figure 6h).
Protein extraction and tumor western blot analysis
24 h after the last SC in vivo treatment, mice were euthanized and excised tumors from all the groups were flash frozen in liquid nitrogen and stored at -80 °C until processing for western blotting. Tumors weighing more than 50 mg from each group (n = 4) were thawed in tissue protein extraction buffer (78510, Thermo Fischer Scientific) in the presence of protease inhibitor cocktail (P8340, Sigma-Aldrich) and phosphatase inhibitor cocktail II, III (P0044, P5726 respectively, Sigma-Aldrich). Tumor lysate was obtained by performing lysis in TriplePure zirconium prefilled tubes (D1032-30, Benchmark Scientific, Edison, NJ) using BeadblasterTM 24 Microtube Homogenizer (D2400, Benchmark Scientific). Supernatant obtained by cold centrifugation (13000 RPM, 10 min, 4 ºC) of tumor lysate was quantified for total protein concentration using Pierce BCA protein assay kit (23227, Thermo Fischer Scientific). 80 µg total protein was used for quantification of mTORC1 downstream targets using western blot analysis as detailed in supplemental methods. Immunoblotting was performed with primary monoclonal antibodies for p-p70 S6K1, p-4E-BP1, p-rpS6 and GAPDH (Figure 7a). The results were analyzed by performing 1-way ANOVA on the normalized intensity levels of the 3 downstream targets, which showed a significant difference for only p-rpS6 between all the 5 treatment groups (α = 0.05, p = 0.002). Tukey-Kramer post-hoc analysis was then performed to test significance between the individual groups as reported in the results (Figure 7b).
Histopathological tissue examination of Rapa treated mice
After 24 h from the last SC in vivo treatment, mice from all the groups were euthanized and major organs were collected for histopathological examination. Before euthanasia, mice were anaesthetized with 2% v/v isoflurane gas with oxygen for whole body blood perfusion. Heart, lungs, spleen, liver, kidney and skin were excised, and fixed in zinc formalin (5701ZF, Thermo Fischer Scientific) overnight before preserving in 70% ethanol. Appropriate size organ samples were then embedded in paraffin, sectioned in 5 µm thick tissue slices onto glass slides, and stained with hematoxylin and eosin (H & E). The H & E stained tissues were then studied under a microscope for histopathological changes analyzed by an unbiased blinded practicing pathologist (Figure 8a, Figure S5).
In vivo imaging of fluorescently labelled FKBP-ELPs
FA, FAF and FSI were labeled covalently with near infrared dye Cyanine5.5 (Cy5.5) as described in supplemental methods. Tumor-implanted mice were anaesthetized with 2% v/v isoflurane gas with oxygen followed by IV or SC injections of ~0.5 mg/kg Cy5.5 (80 µM) labelled FKBP-ELPs (530 µM) through tail vein or subcutaneous injections in the right flank above the hind leg (n = 4 per group). Whole body dorsal and ventral scans were imaged using the IVIS optical spectrum (Perkin Elmer, Waltham, MA) at 0, 4, 24 and 48 h post injection using 1 sec exposure time and small binning (Figure 9a, Figure S7-S8). Excitation and emission filters for Cy5.5 were chosen to be 640 nm and 700 nm respectively. After 48 h, mice were euthanized and a small volume of blood was immediately withdrawn via cardiac puncture. The carcasses were then dissected and individual organs along with the blood withdrawn earlier were scanned for fluorescence (Figure 9b, Figure S8 b). Images were analyzed using Living Image® software (Perkin Elmer). Regions of Interest (ROI) were drawn on individual organs, and fluorescence from respective ROIs was quantified in Avg. radiant efficiency with units [photons/sec/cm2/sr] / [µW/cm2] and plotted after subtracting respective ROIs from control mice organs without Cy5.5 injections (Figure 9c, Figure S8 c). For the SC study, results were analyzed by performing 1-way ANOVA on the log10 transformed Avg. radiant efficiency (n = 4, mean ± SD), which showed significant differences between all the 3 FKBP-ELPs in kidneys, liver and tumor (α = 0.05, p = 0.0004, 0.0003 and 0.0004 respectively). Tukey-Kramer post-hoc analysis was then performed to test significance between the carriers within each tissue as reported in the results (Figure 9c). For the IV study, results were analyzed by performing paired t-test on the log10 transformed Avg. radiant efficiency (n = 4, mean ± SD), which showed statistically significant differences between FSI_IV and FSI_SC groups in liver, tumor and spleen as shown in the results (Figure S8 c).
Abbreviations
Rapa: Rapamycin; ELP: Elastin-Like Polypeptides; FKBP: FK-506 binding protein 12; SI: S48I48; FSI: FKBP-S48I48; FA: FKBP-A192; FAF: FKBP-A192-FKBP; ITC: inverse transition cycling; Tt: transition temperature; CMT: critical micelle temperature; Rg: radius of gyration; Rh: hydrodynamic radius; MW: molecular weight; IV: intravenous; SC: subcutaneous; TNBC: triple negative breast cancer; mTORC1: mammalian target of rapamycin complex 1; p70 S6K1: p70 S6 kinase 1; rpS6: ribosomal S6 protein; 4E-BP1: 4E-binding protein 1; Cy5.5: Cyanine5.5; ROI: region of interest; DLS: dynamic light scattering; SEC-MALS: size exclusion chromatography - multi angle light scattering; RP-HPLC: reverse phase-high performance liquid chromatography; ITC: isothermal titration calorimetry; H & E: hematoxylin and eosin; IVIS: in vivo imaging system.
Acknowledgements
This work was made possible by University of Southern California (USC), the National Institute of Health R01GM114839 and R01EY026635 to JAM, the USC Whittier Foundation, the USC Ming Hsieh Institute, the USC Stevens Institute, P30 CA014089 to the USC Norris Comprehensive Cancer Center. P30 DK048522 to Liver Histology Core of USC Research Center for Liver Diseases, the USC Molecular Imaging Center, USC Nano Biophysics Core Facility, Translational Research Laboratory at USC School of Pharmacy and Proteomics and Metabolomics Facility, Colorado State University.
Supplementary Material
Supplementary figures and methods.
Competing Interests
J.P.D., S.P., and J.A.M are inventors on a patent describing subcutaneous delivery of small molecules using protein polymer fusions related to this work. All other authors declare no competing financial interests.
References
1. Standaert RF, Galat A, Verdine GL, Schreiber SL. Molecular cloning and overexpression of the human FK506-binding protein FKBP. Nature. 1990;346:671-4
2. Moudi M, Go R, Yien CY, Nazre M. Vinca alkaloids. Int J Prev Med. 2013;4:1231-5
3. Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65:157-70
4. Yared JA, Tkaczuk KH. Update on taxane development: new analogs and new formulations. Drug Des Devel Ther. 2012;6:371-84
5. Kahan BD. Efficacy of sirolimus compared with azathioprine for reduction of acute renal allograft rejection: a randomised multicentre study. The Rapamune US Study Group. Lancet. 2000;356:194-202
6. MacDonald AS, Group RGS. A worldwide, phase III, randomized, controlled, safety and efficacy study of a sirolimus/cyclosporine regimen for prevention of acute rejection in recipients of primary mismatched renal allografts. Transplantation. 2001;71:271-80
7. Dumont FJ, Su Q. Mechanism of action of the immunosuppressant rapamycin. Life Sci. 1996;58:373-95
8. Taveira-DaSilva AM, Hathaway O, Stylianou M, Moss J. Changes in lung function and chylous effusions in patients with lymphangioleiomyomatosis treated with sirolimus. Ann Intern Med. 2011;154:797-805 W-292-3
9. Moir LM. Lymphangioleiomyomatosis: Current understanding and potential treatments. Pharmacol Ther. 2016;158:114-24
10. Simamora P, Alvarez JM, Yalkowsky SH. Solubilization of rapamycin. Int J Pharm. 2001;213:25-9
11. Stenton SB, Partovi N, Ensom MH. Sirolimus: the evidence for clinical pharmacokinetic monitoring. Clin Pharmacokinet. 2005;44:769-86
12. Gabardi S, Baroletti SA. Everolimus: a proliferation signal inhibitor with clinical applications in organ transplantation, oncology, and cardiology. Pharmacotherapy. 2010;30:1044-56
13. Pham PT, Pham PC, Danovitch GM, Ross DJ, Gritsch HA, Kendrick EA. et al. Sirolimus-associated pulmonary toxicity. Transplantation. 2004;77:1215-20
14. Marti HP, Frey FJ. Nephrotoxicity of rapamycin: an emerging problem in clinical medicine. Nephrol Dial Transplant. 2005;20:13-5
15. Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10:868-80
16. de Oliveira MA, Martins EMF, Wang Q, Sonis S, Demetri G, George S. et al. Clinical presentation and management of mTOR inhibitor-associated stomatitis. Oral Oncol. 2011;47:998-1003
17. Gomez-Fernandez C, Garden BC, Wu S, Feldman DR, Lacouture ME. The risk of skin rash and stomatitis with the mammalian target of rapamycin inhibitor temsirolimus: a systematic review of the literature and meta-analysis. Eur J Cancer. 2012;48:340-6
18. Danesi R, Boni JP, Ravaud A. Oral and intravenously administered mTOR inhibitors for metastatic renal cell carcinoma: pharmacokinetic considerations and clinical implications. Cancer Treat Rev. 2013;39:784-92
19. Eshleman JS, Carlson BL, Mladek AC, Kastner BD, Shide KL, Sarkaria JN. Inhibition of the mammalian target of rapamycin sensitizes U87 xenografts to fractionated radiation therapy. Cancer Res. 2002;62:7291-7
20. Hackstein H, Taner T, Logar AJ, Thomson AW. Rapamycin inhibits macropinocytosis and mannose receptor-mediated endocytosis by bone marrow-derived dendritic cells. Blood. 2002;100:1084-7
21. Sun M, Si L, Zhai X, Fan Z, Ma Y, Zhang R. et al. The influence of co-solvents on the stability and bioavailability of rapamycin formulated in self-microemulsifying drug delivery systems. Drug Dev Ind Pharm. 2011;37:986-94
22. Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37:1590-8
23. Hennenfent KL, Govindan R. Novel formulations of taxanes: a review. Old wine in a new bottle? Ann Oncol. 2006;17:735-49
24. Shi P, Aluri S, Lin YA, Shah M, Edman M, Dhandhukia J. et al. Elastin-based protein polymer nanoparticles carrying drug at both corona and core suppress tumor growth in vivo. J Control Release. 2013;171:330-8
25. Shah M, Edman MC, Janga SR, Shi P, Dhandhukia J, Liu S. et al. A rapamycin-binding protein polymer nanoparticle shows potent therapeutic activity in suppressing autoimmune dacryoadenitis in a mouse model of Sjogren's syndrome. J Control Release. 2013;171:269-79
26. Urry DW, Okamoto K, Harris RD, Hendrix CF, Long MM. Synthetic, cross-linked polypentapeptide fo tropoelastin: an anisotropic, fibrillar elastomer. Biochemistry. 1976;15:4083-9
27. Urry DW. Entropic elastic processes in protein mechanisms. I. Elastic structure due to an inverse temperature transition and elasticity due to internal chain dynamics. Journal of protein chemistry. 1988;7:1-34
28. Urry DW. Free energy transduction in polypeptides and proteins based on inverse temperature transitions. Prog Biophys Mol Biol. 1992;57:23-57
29. Despanie J, Dhandhukia JP, Hamm-Alvarez SF, MacKay JA. Elastin-like polypeptides: Therapeutic applications for an emerging class of nanomedicines. J Control Release. 2015
30. Chavez KJ, Garimella SV, Lipkowitz S. Triple negative breast cancer cell lines: one tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010;32:35-48
31. DeGraffenried LA, Fulcher L, Friedrichs WE, Grunwald V, Ray RB, Hidalgo M. Reduced PTEN expression in breast cancer cells confers susceptibility to inhibitors of the PI3 kinase/Akt pathway. Ann Oncol. 2004;15:1510-6
32. Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671-88
33. Janib SM, Liu S, Park R, Pastuszka MK, Shi P, Moses AS. et al. Kinetic quantification of protein polymer nanoparticles using non-invasive imaging. Integr Biol (Camb). 2013;5:183-94
34. Acharya S, Dilnawaz F, Sahoo SK. Targeted epidermal growth factor receptor nanoparticle bioconjugates for breast cancer therapy. Biomaterials. 2009;30:5737-50
35. Doddapaneni BS, Kyryachenko S, Chagani SE, Alany RG, Rao DA, Indra AK. et al. A three-drug nanoscale drug delivery system designed for preferential lymphatic uptake for the treatment of metastatic melanoma. J Control Release. 2015;220:503-14
36. Cirstea D, Hideshima T, Rodig S, Santo L, Pozzi S, Vallet S. et al. Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol Cancer Ther. 2010;9:963-75
37. Hasenstein JR, Shin HC, Kasmerchak K, Buehler D, Kwon GS, Kozak KR. Antitumor activity of Triolimus: a novel multidrug-loaded micelle containing Paclitaxel, Rapamycin, and 17-AAG. Mol Cancer Ther. 2012;11:2233-42
38. Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M. et al. Temperature triggered self-assembly of polypeptides into multivalent spherical micelles. J Am Chem Soc. 2008;130:687-94
39. Matsudo T, Ogawa K, Kokufuta E. Complex formation of protein with different water-soluble synthetic polymers. Biomacromolecules. 2003;4:1794-9
40. Bierer BE, Mattila PS, Standaert RF, Herzenberg LA, Burakoff SJ, Crabtree G. et al. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc Natl Acad Sci U S A. 1990;87:9231-5
41. Connelly PR, Thomson JA. Heat capacity changes and hydrophobic interactions in the binding of FK506 and rapamycin to the FK506 binding protein. Proc Natl Acad Sci U S A. 1992;89:4781-5
42. Wilson KP, Yamashita MM, Sintchak MD, Rotstein SH, Murcko MA, Boger J. et al. Comparative X-ray structures of the major binding protein for the immunosuppressant FK506 (tacrolimus) in unliganded form and in complex with FK506 and rapamycin. Acta crystallographica Section D, Biological crystallography. 1995;51:511-21
43. Bissantz C, Kuhn B, Stahl M. A medicinal chemist's guide to molecular interactions. J Med Chem. 2010;53:5061-84
44. Pfizer. Rapamune product monograph. July. 2014
45. McDonald TA, Zepeda ML, Tomlinson MJ, Bee WH, Ivens IA. Subcutaneous administration of biotherapeutics: current experience in animal models. Curr Opin Mol Ther. 2010;12:461-70
46. Pharmaceuticals P. Phase 2b Multicenter, Randomized, Double-Blind, Placebo- and Active-Controlled, Parallel-Group Study to Assess the PD Response and Safety of Three Dose Levels of Glymera Injection Following 20 Weeks of Weekly SC Dosing in Adults With T2DM. 2012.
47. Amiram M, Luginbuhl KM, Li X, Feinglos MN, Chilkoti A. A depot-forming glucagon-like peptide-1 fusion protein reduces blood glucose for five days with a single injection. J Control Release. 2013;172:144-51
48. Richter WF, Bhansali SG, Morris ME. Mechanistic determinants of biotherapeutics absorption following SC administration. AAPS J. 2012;14:559-70
49. Massihnia D, Galvano A, Fanale D, Perez A, Castiglia M, Incorvaia L. et al. Triple negative breast cancer: shedding light onto the role of pi3k/akt/mtor pathway. Oncotarget. 2016;7:60712-22
50. Mondesire WH, Jian W, Zhang H, Ensor J, Hung MC, Mills GB. et al. Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin Cancer Res. 2004;10:7031-42
51. Noh WC, Mondesire WH, Peng J, Jian W, Zhang H, Dong J. et al. Determinants of rapamycin sensitivity in breast cancer cells. Clin Cancer Res. 2004;10:1013-23
52. Liu T, Yacoub R, Taliaferro-Smith LD, Sun SY, Graham TR, Dolan R. et al. Combinatorial effects of lapatinib and rapamycin in triple-negative breast cancer cells. Mol Cancer Ther. 2011;10:1460-9
53. Chao TC, Chu Z, Tseng LM, Chiou TJ, Hsieh RK, Wang WS. et al. Paclitaxel in a novel formulation containing less Cremophor EL as first-line therapy for advanced breast cancer: a phase II trial. Invest New Drugs. 2005;23:171-7
54. Supersaxo A, Hein WR, Steffen H. Effect of molecular weight on the lymphatic absorption of water-soluble compounds following subcutaneous administration. Pharm Res. 1990;7:167-9
55. Swartz MA. The physiology of the lymphatic system. Adv Drug Deliv Rev. 2001;50:3-20
56. McLennan DN, Porter CJ, Charman SA. Subcutaneous drug delivery and the role of the lymphatics. Drug Discov Today Technol. 2005;2:89-96
57. Porter CJ, Charman SA. Lymphatic transport of proteins after subcutaneous administration. J Pharm Sci. 2000;89:297-310
58. Dhandhukia J, Weitzhandler I, Wang W, MacKay JA. Switchable Elastin-Like Polypeptides that Respond to Chemical Inducers of Dimerization. Biomacromolecules. 2013;14:976-85
59. Hassouneh W, Christensen T, Chilkoti A. Elastin-like polypeptides as a purification tag for recombinant proteins. Curr Protoc Protein Sci. 2010 Chapter 6: Unit 6 11
60. Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411-23
Author contact
![]() Corresponding author: J. Andrew MacKay (jamackayedu)
Corresponding author: J. Andrew MacKay (jamackayedu)