Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(15):3781-3793. doi:10.7150/thno.20892 This issue Cite
Research Paper
Sequential Delivery of Cyclopeptide RA-V and Doxorubicin for Combination Therapy on Resistant Tumor and In Situ Monitoring of Cytochrome c Release
Huachao Chen1*, Yurong Wang1*, Yongrong Yao1, Shenglin Qiao2, Hao Wang2, Ninghua Tan1 ![]()
1. State Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of TCM Evaluation and Translational Research, School of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing 211198, China
2. CAS Key Laboratory for Biological Effects of Nanomaterials and Nanosafety, National Center for Nanoscience and Technology, Beijing 100190, China
*Contributed equally to this work.
Received 2017-5-6; Accepted 2017-7-20; Published 2017-8-23
Abstract

A programmed drug delivery system that can achieve sequential release of multiple therapeutics under different stimulus holds great promise to enhance the treatment efficacy and overcome multi-drug resistance (MDR) in tumor. Herein, multi-organelle-targeted and pH/ cytochrome c (Cyt c) dual-responsive nanoparticles were designed for combination therapy on resistant tumor. In this system (designated DGLipo NPs), doxorubicin (Dox) was intercalated into the DNA duplex containing a Cyt c aptamer, which subsequently loaded in the dendrigraftpoly-L-lysines (DGL) cores of DGLipo NPs, while cyclopeptide RA-V was doped into the pH-sensitive liposomal shells. After dual modification with c(RGDfK) and mitochondria-penetrating peptide (MPP), DGLipo NPs could successively deliver the two drugs into lysosome and mitochondria of cancer cells, and achieve sequential drug release in virtue of the unique characteristic of these two organelles. The organelle-specific and spatiotemporally controlled release of Dox and RA-V led to enhanced therapeutic outcomes in MDR tumor. More significantly, the DGLipo NPs were successfully applied to monitor Cyt c release during mitochondria-mediated apoptotic process. This work represents a versatile strategy for precise combination therapy against resistant tumor with spatiotemporal control, and provides a potential tool for Cyt c-related apoptotic studies.
Keywords: cancer, sequential drug delivery, combination therapy, cytochrome c, cyclopeptide RA-V, doxorubicin
Introduction
Combination therapy shows great prospect for highly efficient treatment of cancer, due to its superiority in achieving synergistic effects, overcoming multi-drug resistance (MDR) and decreasing non-specific toxicity [1]. A multiple-medication therapy would have inherent pharmacological and pharmacokinetic advantages over monotherapy, because various pathogenic factors are involved in cancer that hitting a single target is not sufficient to treat it [2]. However, traditional systemic combination therapies, which simply mix different drugs into the same formulation, usually lead to poorly controlled dosing and inconsistent in vivo biodistribution of drugs [3]. To overcome the limits of combination cancer therapy, efforts have been made to develop smart nano-platforms for ratiometric delivery and synchronized release of multiple drugs with precise spatiotemporally control [4]. In particular, the design of drug-delivery nanocarriers that enable time or stimulus-programmed sequential release of multiple therapeutic agents to their distinct targets is of much importance for nano-based combination therapy [5]. In virtue of various stimuli-responsive motifs and subcellular organelle-targeting strategy, programmable sequential delivery nano-systems can successfully combine different antitumor mechanisms of multiple drugs, avoid MDR and achieve precise and flexible control of intracellular drug release [6].
Programmable sequential delivery nano-systems were mainly used for co-delivery of traditional chemotherapeutic drugs, which have major limitations such as toxicity and chemotherapy resistance in the treatment of patients [7]. For example, doxorubicin (Dox), a widely used chemotherapy agent in clinical practice, is severely limited by the development of MDR of cancer cells [8]. Therefore, the search for new chemotherapeutics, which are capable of improving the efficacy of Dox in the MDR cancer cells are thus desired. Plant-derived natural products are rich sources of anti-tumor compounds, such as taxol, camptothecin, and so on [9]. Recently, RA-V (deoxybouvardin), a unique natural cyclopeptide which has been isolated from the medical plant Rubia yunnanensis, has been reported to exhibited potent anti-tumor activities in a variety of cancer cell lines [10]. In particular, RA-V could induce cell apoptosis via mitochondrial pathway and then promote the loss of mitochondrial membrane potential (MMP) and the release of cytochrome c (Cyt c) [11]. The apoptosis resistance is normally caused by the higher MMPs in MDR cells compared with drug sensitive cells [12], which could be attributed to the differences in the structure of fatty acid composition, resistance-related membrane protein expression, ion conductivity, or metabolic regulation [13]. Thus, RA-V is a promising anticancer agent for constructing programmable sequential delivery system together with Dox to overcome MDR by altering the MMP.
In addition, Cyt c release is also involved in the RA-V-induced mitochondrial apoptotic pathway [14]. Cyt c, a major mediator in cell apoptosis, plays essential role in apoptotic signaling [15]. It is normally bound to the inner mitochondrial membrane, and released from the intermembrane space of mitochondria to cytosol when the mitochondrial pathway of apoptosis is initiated [16]. Since Cyt c release is highly specific events in apoptotic signaling, real-time imaging of Cyt c translocation in situ could be very useful for monitoring the progression to apoptosis, investigating the apoptotic signaling, exploring the exact anti-tumor mechanism of RA-V and developing drugs to modulate apoptotic pathways in living cells [17]. However, there are limited methods to specifically and dynamically detect Cyt c translocation in apoptotic cells. Moreover, smart co-delivery system that can realize simultaneous monitoring Cyt c release and inducing tumor cell apoptosis via mitochondria pathway has not been reported.
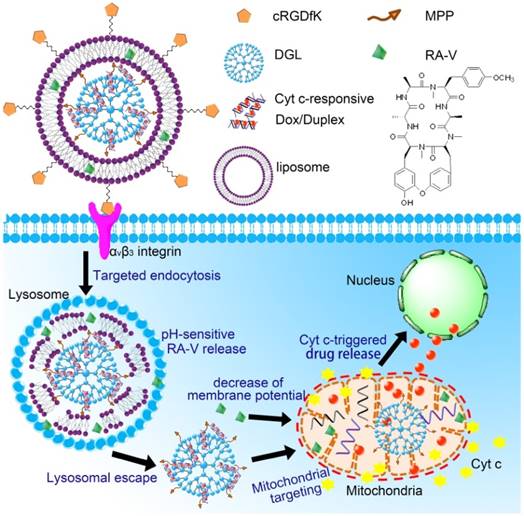
We herein developed a pH/Cyt c dual-responsive drug delivery system consisting of a liposomal shell and a dendritic poly(L-lysines) (DGL) core for site-specific sequential release of RA-V and Dox at the subcellular level, and for real-time monitoring of Cyt c release in apoptosis process (Scheme 1). In this system, two separate depots were incorporated into a nanoparticle to achieve sequential release of two cargos under different endogenous stimulus. Firstly, Dox was intercalated into the DNA duplex hybridized by the Cyt c aptamer [18] and its complementary single stranded DNA via noncovalent interaction [19]. The Dox/Duplex complex was condensed by DGL to form self-assembling nanoparticles (Dox/DGL). To achieve mitochondria targeted properties, a mitochondria-penetrating peptide (MPP) as the targeting ligand [20] was linked to Dox/DGL. Secondly, the MPP-modifide Dox/DGL (Dox/MPP-DGL) was encapsulated into the aqueous core and RA-V was doped into the shell of pH-sensitive liposomes. To further improve the cancer selectivity, a cyclic pentapeptide c(RGDfK) [21] was conjugated on the surface of liposomes. The obtained DGL-liposome nanoparticles (DGLipo NPs) could selectively entered αvβ3 integrin-rich cells via receptor-mediated endocytosis into the lysosome, in which the acidic microenvironment degrade the liposomes to release RA-V and Dox/MPP-DGL. In particular, DGL possess a unique capability that functions as a proton sponge to cause osmotic swelling and rupture of the lysosome membrane [22]. So Dox/MPP-DGL can successfully escape from lysosome and subsequently deliver Dox/Duplex into mitochondria in virtue of MPP. As a result, the high levels of Cyt c in mitochondria induce dissociation of the DNA duplex, thereby causes rapid release. As the fluorescence and cytotoxicity of Dox was quenched when inserted into the duplex, the Dox release could kill cancer cells in coordination with RA-V, as well as monitoring the translocation of Cyt c from mitochondria into the cytoplasm during RA-V-induced cell apoptosis.
Schematic illustration of DGLipo NPs for site-specific sequential release of RA-V and Dox, and real-time monitoring of Cyt c release in apoptosis process.
Materials and methods
Materials
1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) was purchased from Avanti Polar Lipids (USA). Cholesteryl hemisuccinate (CHEMS) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were obtained from Sigma-Aldrich (St. Louis, MO, USA). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[cRGD(poly ethylene glycol)-2000] (DSPE-mPEG2000-cRGD) was purchased from Nanocs (USA). Mitochondria-penetrating peptide (MPP) (MLRAALSTARRGPRLSRLL) with a cysteine was obtained from Sangon Biotech Ltd. Co (Shanghai, China). Dendrigraft poly-L-lysines (DGL) generation 3 with 123 lysine groups was purchased from Colcom, France. α-Malemidyl-ω-N-hydroxysuccinimidyl polyethylene glycol (NHS-PEG-MAL, MW 500) was obtained from Jenkem Technology (Beijing, China). RA-V was isolated from Rubia yunnanensis as described earlier [10]. Cyt c aptamer (CCG TGT CTG GGG CCG ACC GGC GCA TTG GGT ACG TTG TTG CTT TTT TTT), single-stranded cDNA of Cyt c aptamer, and the control DNA sequence that has not Cyt c-responsiveness (ATG ATG CAT CAT CTC TGA AGT AGC GCC GCC GTA TAC TCA) were purchased from Sangon Biotech Ltd. Co (Shanghai, China). Doxorubicin (Dox) hydrochloride and Annexin V-FITC/PI cell apoptosis kit were purchased from KeyGen Biotech. Co. Ltd. (Nanjing, China). Hoechst 33342, LysoTracker Green, MitoTracker Green, GolgiTracker Green were all obtained from Invitrogen (Carlsbad, CA, USA). Ultra-pure water was prepared using a Millipore Simplicity System (Millipore, Bedford, USA).
Synthesis and characterization of Dox/MPP-DGL
DGL was reacted with NHS-PEG-MAL (MW 500) at a ratio of 1:10 (mol/mol) in phosphate buffer (pH 8.0) for 2 h at room temperature. The primary amino groups on the surface of DGL were specifically reacted with the terminal NHS groups of the PEG derivative. The resulting conjugate, DGL-PEG, was purified by Amicon ultra centrifugal filter units, and dissolved in phosphate buffer (pH 7.0). Then DGL-PEG was reacted with MPP in PBS (pH 7.0) for 24 h at room temperature. The MAL groups of DGL-PEG were specifically reacted with the thiol groups of the peptide, yielding the DGL-PEG-MPP vectors.
The Dox/Duplex complex was prepared by incubating Dox with a hybridized duplex of Cyt c aptamer and its cDNA at a molar ratio of 1:1 for 15 min. DGL-PEG-MPP was diluted to 600 mg mL-1 in phosphate buffer (pH 7.4). Dox/Duplex complex solution (100 mg mL-1, 50 mM sodium sulfate solution) was added at a mass ratio of 6:1 (DGL to DNA) and immediately vortexed for 30 s at room temperature. The external amino groups of DGL could encapsulate DNA through electric interactions. Thus, the Dox/Duplex was compacted and encapsulated by the dendrimers.
The Cyt c aptamer in Dox/MPP-DGL was replaced by the control DNA sequence that has not Cyt c-responsiveness for preparing Dox/MPP-cDGL. Dox/MPP-cDGL was obtained by the same method described above, except that duplex hybridized by the control DNA sequence that has not Cyt c-responsiveness and its cDNA at a molar ratio of 1:1 were added to achieve Dox/Duplex complex.
The transmission electron microscopy (TEM, JEM-2100) measurement of Dox/MPP-DGL was prepared by dropping the solution onto a carbon-coated copper grid following negative staining with 2.0% (w/v) phosphotungstic acid. The particle size and size distribution of Dox/MPP-DGL were measured by dynamic light scattering (DLS) (a Mastersizer 2000 particle size analyzer) with a fixed scattering angle of 90°. Zeta potential measurement was performed at 25 °C on a Malvern Zeta Sizer-Nano Z instrument.
Fabrication and characterization of DGLipo NPs
Liposomes were prepared by the thin film hydration method [23]. Briefly, DOPE, CHEMS, DSPE-mPEG2000-cRGD and RA-V with a molar ratio of 50:50:0.5:0.5 were first dissolved in 3 mL of chloroform in a 100 mL round-bottom flask and then dried by a rotary evaporator under vacuum at 40 °C to obtain a thin lipid film. The lipid film was hydrated with 10 mL 0.2 mg mL-1 Dox/MPP-DGL suspension by rotating the round-bottom flask at 180 rpm at 60 °C. After the lipid film was completely hydrated with brief sonication, and the liposomes were extruded through a 100 nm membrane. The product was purified by dialysis membrane (Mn = 100 kDa, Millpore) for 24 h and rinsing with distilled water to remove free Dox/MPP-DGL. Similar procedures were used to prepare DGLipo NPs (without RGD).
Encapsulation Efficiency (EE) was determined using the previously reported method [24]. After destruction of the DGLipo NPs with Triton X-100, 0.5 mL of 1 mg mL-1 DGLipo NPs dispersion was filtated through a centrifugal filter (Mn = 30 kDa, Millpore) at 4000 rpm. The amount of RA-V was determined at 220 nm by a UV detector using the corresponding standard calibration curve. EE was calculated using the following equation:
EE (%)= (the amount of loaded RA-V)/(the amount of added RA-V )×100%
Loading capacity (LC) is defined as the percent ratio of RA-V in DGLipo NPs.
The particle size distribution of DGLipo NPs was determined by dynamic light scattering (DLS) using a Mastersizer 2000 particle size analyzer with a fixed scattering angle of 90°. Zeta potential measurement was performed at 25 °C on a Malvern Zeta Sizer-Nano Z instrument.
Stability study of Dox/Duplex and RA-V in lysosome
As the microenvironment of lysosome is acidic (pH 5-6), the stability of the Dox/Duplex and RA-V were investigated at pH 5.0 and pH 6.0. The fluorescence of Dox was quenched when intercalated into double-stranded DNA, and has not increased over 24 h incubation at pH 6.0 and pH 5.0, suggesting the stability of Dox/Duplex in acid condition. The stability of RA-V was also investigated by HPLC, which showed that no degradation was observed over 24 h incubation at pH 6.0 and pH 5.0.
In vitro payload release profiles
The in vitro payload release profiles were investigate by equilibrium dialysis method [25]. The profiles for in vitro release of Dox were established by dialysis of the Dox/MPP-DGL suspensions in HEPES buffer solution. Briefly, 3 mL Dox/MPP-DGL suspensions (50 μg mL-1) were dialyzed against 15 mL HEPES buffer (molecular-weight cut off: 12000) and gently shaken in a thermostatic rotary shaker at 100 rpm and 37 °C. Samples were removed at different intervals, and an equal amount of the same medium was added to maintain a constant volume. The amount of Dox released from Dox/MPP-DGL was analyzed by using a fluorescence spectrometer. The percentage of Dox released was calculated using: % release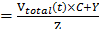 (Vtotal (t) is the remaining volume in the container at time t; C is the concentration of Dox determined from fluorescence spectrometer, Y is the amount of Dox that has already been collected, and Z is the total amount of Dox at t = 0 present in the dialysis bag).
(Vtotal (t) is the remaining volume in the container at time t; C is the concentration of Dox determined from fluorescence spectrometer, Y is the amount of Dox that has already been collected, and Z is the total amount of Dox at t = 0 present in the dialysis bag).
Cell culture and confocal imaging
Human breast cancer cell line (MCF-7 cells), Human cervical carcinoma cell line (HeLa cells) and drug-insensitive HeLa/MDR cells were cultured in glass bottom dishes. Cells were seeded at a density of 1×106 cells mL-1 in Dulbucco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, NaHCO3 (2 g L-1) and 100 units/mL of penicillin. Cells were maintained at 37 °C in a humidified 5% CO2/95% air atmosphere.
For intracellular fluorescence imaging of cells, the incubation media was removed and the cells were rinsed three times with PBS. Then the cells were incubated in the working solution at room temperature. After removing the solution, we washed the dish three times with PBS. The confocal images of the cells were obtained using a ZEISS Laser Scanning Microscope (Zeiss LSM 710) with a 63×oil-immersion objective.
Immunostaining assay
HeLa cells were incubated with 50 μg mL-1 DGLipo NPs for 2 h, and further incubated with fresh culture medium for additional 1 h, 1.5 h and 2 h, respectively. After fixed in 3.7% formaldehyde/PBS solution, the cells were treated in 0.1% Triton X-100/PBS. Primary Cyt c antibody were diluted in 1% bovine serum albumin (BSA)/PBS and incubated with the cells at 4 °C for 12 h. The FITC-labeled secondary Cyt c antibody were diluted in 1% BSA/PBS and incubated at 25 °C for 2 h. The confocal images of the cells were obtained using a ZEISS Laser Scanning Microscope (Zeiss LSM 710) with a 63×oil-immersion objective.
In vitro cytotoxicity assay
The cytotoxicity of empty DGLipo NPs, free RA-V, free Dox, and DGLipo NPs was tested by the MTT [3-(4,5-divmethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay [26]. Briefly, HeLa cells and HeLa/MDR cells were inoculated in 96-well plates, respectively, and incubated in DMEM over night. Empty DGLipo NPs, free RA-V, free Dox, and DGLipo NPs with specific concentrations were added to the culture medium respectively. The cells were incubated for an additional 48 h and MTT (20 mL, 5 mg mL-1 in PBS) was then added to each well. After 4-h incubation, the medium containing unreacted MTT was removed carefully, and 150 μL of DMSO was added to each well to dissolve the formazan crystals. After 1 h the absorbance (Abs.) was measured at 490 nm in a TRITURUS microplate reader. The cell viability was then determined by the following equation: Cell viability (%) = (mean of Abs. value of treatment group/mean Abs. value of control) × 100%. Calculation of the half lethal dose (IC50) values was done according to Huber and Koella [27].
In vivo imaging study
The HeLa cells (5 ×106 cells) were transplanted subcutaneously into the 8-week female BALB/c nude mice by using a stereotactic fixation device with mouse adaptor. DGLipo NPs, DGLipo NPs (no RGD), Dox/MPP-DGL and Dox/Duplex were injected through the tail vein of tumor-bearing nude mice at Dox dosage of 1 mg/kg. At 2, 6, and 24 h, the mice were anesthetized and placed in the chamber of an in vivo imaging system (CRi, MA, USA). After the 24 h scanning, the mice were euthanized and the tumor, heart, liver, spleen, lung and kidney were excised for ex vivo imaging.
Targeted treatment on subcutaneous tumor model and evaluation of therapeutic effect
In vivo targeted treatment was performed using HeLa tumor-bearing mice. The mice were randomly divided into four groups and treated i.v. with saline, free Dox, free RA-V, and DGLipo NPs at Dox dosage of 1 mg kg-1, respectively. Each group contains six mice. From Day 0, the mice were intravenously injected with every three days for 15 days, and tumors were measured by a caliper every three days. The therapeutic effects were evaluated by monitoring the changes of relative tumor volumes and H&E apoptosis staining.
Results and discussion
Characterization of Dox/MPP-DGL and DGLipo NPs
To prepare Dox/MPP-DGL, the Dox/Duplex complex was condensed by MPP-modified DGL to form nanoparticles. The loading capacity and the encapsulation efficiency of Dox in Dox/ MPP-DGL were determined with fluorescence spectroscopy to be about 6% and 68%, respectively. The morphology of Dox/MPP-DGL was analyzed by transmission electron microscopy (TEM). TEM observations demonstrated that Dox/MPP-DGL were well dispersed with spherical shape (Figure S1A). The dynamic light scattering (DLS) showed that Dox/MPP-DGL had a unimodal distribution whose average hydrodynamic sizes was approximately 32 nm (Figure S1B). As the diameter of mitochondria is about 0.5-10 μm [28], the small size of Dox/ MPP-DGL was suitable for mitochondria-targeted drug delivery. The zeta potentials of Dox/ MPP-DGL before and after MPP-modification were 3.5 ± 0.1 mV and 6 ± 0.3 mV, indicating the presence of positively charged MPP on the surface.
The obtained Dox/MPP-DGL was encapsulated into the aqueous core and RA-V was embedded into the lipid bilayers of pH-sensitive liposomes for preparing DGLipo NPs. The loading capacity and the encapsulation efficiency of RA-V in DGLipo NPs were determined with HPLC to be about 3% and 65%, respectively. The TEM image of the resulting DGLipo NPs showed well dispersed spherical shape (Figure S1C) and degraded in acidic environment (Figure S2). The size of DGLipo NPs was relatively uniform, and the average hydrodynamic diameter was measured to be 110 nm by DLS (Figure S1D). Moreover, the size of DGLipo NPs did not change for at least 6 days after incubation with two commonly used biological media (Figure S3), suggesting the stability of DGLipo NPs in physiological environments. The zeta potentials of DGLipo NPs (without RGD) and DGLipo NPs were measured to be 20 ± 0.8 mV and 29 ± 0.6 mV, respectively. The increase of zeta potential was due to the positive charge from arginine in the peptide, confirming the presence of c(RGDfK) at the surface. The positive charge may make it easier to bind with cell membrane, which could facilitate the cellular internalization of DGLipo NPs via endocytosis [29].
Fluorescence response of Dox/Duplex
The Dox/Duplex complex was formulated by insertion Dox into the DNA duplex which contained a Cyt c-specific aptamer and its complementary DNA. The fluorescence of Dox was quenched when intercalated into double-stranded DNA owing to fluorescence resonance energy transfer mechanism, and Cyt c could induce the DNA duplex to dissociate, leading to fluorescence increase along with the course of Dox release. To investigate the responsiveness of Dox/Duplex to Cyt c, the fluorescence spectral variation of Dox/Duplex treated with different concentrations of Cyt c was examined. As can be seen from Figure S4A, the fluorescence of Dox/Duplex enhanced gradually upon adding increasing amount of Cyt c, suggesting the release of Dox triggered by Cyt c. The fluorescence response range was from 0 μM to 10 μM, covering the literature values (1-10 μM) for cytosolic Cyt c in apoptotic cells [30], which suggest the potential application of Dox/Duplex in the cytochrome c release monitoring in live cells.
Taking the complexity of the intracellular environment into consideration, the specificity of Dox/Duplex toward Cyt c was evaluated. The responsiveness of Dox/Duplex to various potential interfering species (100 μM) possibly coexisting with Cyt c such as adenosine triphosphate (ATP) , vascular endothelial growth factor (VEGF), ferritin, cysteine, human serum albumin (HSA), isoascorbic acid (AA), platelet-derived growth factor B-chain (PDGF), insulin, and immunoglobulin G (IgG) was detected. As shown in Figure S4B, the fluorescence intensity of Dox/Duplex increased significantly in the presence of Cyt c, whereas no obvious changes of fluorescence intensity were observed upon reaction with these potential interfering species. The experimental results showed that Dox/Duplex is highly selective for Cyt c over the other biologically related species, which should be ascribed to the specific binding of the Cyt c and its aptamer.
In vitro release of Dox from Dox/MPP-DGL and RA-V from DGLipo NPs
In this study, RA-V and Dox were incorporated into a pH/Cyt c dual-responsive delivery system (DGLipo NPs) for programmed sequential drug release. The DGLipo NPs were consisting of Cyt c-responsive Dox/MPP-DGL in aqueous core and RA-V in the pH-sensitive liposomal shell. Firstly, the in vitro release kinetics of Dox from Dox/MPP-DGL was investigated by fluorescence assay (Figure 1A). Cumulative release profiles exhibit that Dox was released from Dox/MPP-DGL fastly in the presence of 10 μM Cyt c, reaching a plateau after 8-h incubation. Approximately 70% of Dox originally loaded in Dox/MPP-DGL were released after incubation with 10 μM Cyt c for 24 h. In contrast, the release rate of Dox was much lower without Cyt c-incubation and only about 10% of Dox originally loaded in Dox/MPP-DGL were released within 24 h, suggesting that the drug release possessed Cyt c specificity. To further validate that the Cyt c-triggered drug release was due to the specific binding of Cyt c and its aptamer, the Cyt c aptamer in Dox/MPP-DGL was replaced by the control DNA sequence that has not Cyt c-responsiveness for preparing Dox/MPP-cDGL as a reference. The release profile of Dox/MPP-cDGL was also determined by the same method. As shown in Figure 1A, a much lower releasing of Dox from Dox/MPP-cDGL compared to Dox/MPP-DGL after incubation with 10 μM Cyt c for 24 h, signifying the Cyt c aptamer played a key role in Cyt c-triggered drug release.
(A) In vitro release profiles of Dox from Dox/MPP-DGL or Dox/MPP-cDGL incubated with and without 10 μM Cyt c. (B) In vitro release profiles of RA-V from DGLipo NPs at pH 7.4, 6.0 and 5.0. Data are means ± SD (n = 3), ***P < 0.001 compared to other groups using a one-way ANOVA.
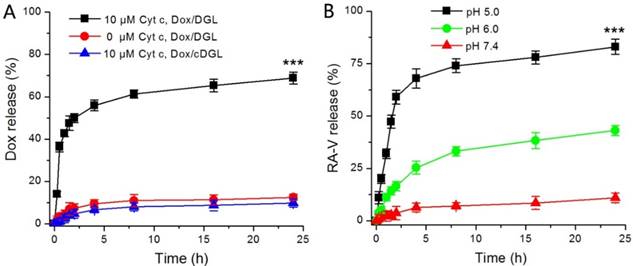
Next, the pH-mediated release of RA-V from DGLipo NPs was investigated under different conditions (Figure 1B). At pH 7.4, DGLipo NPs was stable and only small amounts of RA-V was released over 24 h incubation. However, the release of RA-V was significantly accelerated at pH 6.0, reaching a plateau after about 8-h incubation. Approximately 38% of RA-V released in 24 h under this condition, due to pH-induced degradation of DOPE in DGLipo NPs. Notably, the fastest and most complete drug release was observed at pH 5.0. The accumulatively released RA-V could reached about 85% after incubation for 24 h. As the microenvironment of lysosomes is acidic (pH 5-6), DGLipo NPs could be an effective pH-controlled nanocarrier for targeted drug release in lysosomes of tumor cells. Above all, DGLipo NP achieved precisely trigger smart release in response to pH/Cyt c-dual stimuli, thereby causing an enhanced subcellular combination therapy efficiency.
Cellular selectivity
In order to visualize and localize DGLipo NPs in living cells, Cy5.5 NHS ester, a lipophilic dye, was doped into the liposomal shell as a fluorescence tracker. To investigate the selectivity of DGLipo NPs toward αvβ3 integrin-rich tumor cells, the cellular uptake of DGLipo NPs were monitored by confocal fluorescence imaging. After incubating with HeLa cells for 1 h, some fluorescent spots were observed within the cells. The fluorescence intensity increased with incubation time, and reached a maximum after 2 h, indicating DGLipo NPs were efficiently uptaken by HeLa cells. In contrast, MCF-7 cells, in which the expression level of αvβ3 integrin is much lower than HeLa cells [31], showed negligible fluorescent signal as the incubation time prolonged to 2 h under the same condition (Figure 2). The results demonstrated the specific recognization capability of DGLipo NPs for αvβ3 integrin-overexpressed cancer cells, which was further confirmed by flow cytometry assay.
Real-time confocal fluorescence imaging of HeLa cells or MCF-7 cells with different treatments for 1-2 h. Scale bars: 20 μm.
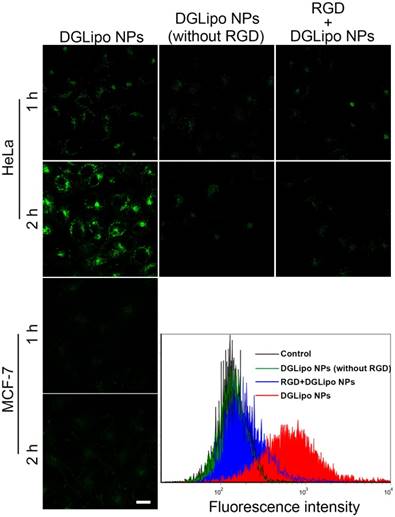
To further confirm that the selectivity of DGLipo NPs was attributed to the specific recognization of c(RGDfK) to αvβ3 integrin on cancer cells, the internalization of DGLipo NPs (without RGD) into HeLa cells was investigated as a reference. As shown in Figure 2, the intracellular fluorescence was much weaker compared with that of DGLipo NPs treated cells, which indicated that c(RGDfK) played a key role in tumor targeting. As an additional control, HeLa cells were treated with excessive free c(RGDfK) before the DGLipo NPs incubation. Negligible intracellular fluorescence was observed after 2-h incubation, showing that the internalization of DGLipo NPs was obviously suppressed. The results verified that the targeting specificity of DGLipo NPs to HeLa cells was due to the specific binding of c(RGDfK) to the αvβ3 integrin on the surface of cells.
Live cell imaging of drug release from DGLipo NPs
The intracellular delivery of DGLipo NPs was real-time monitored by confocal fluorescence imaging (Figure S5). HeLa cells incubated with 50 μg mL-1 DGLipo NPs for 2h showed remarkable fluorescence of Cy 5.5 and co-localized well with LysoTracker Blue, indicating that DGLipo NPs were efficiently uptaken by HeLa cells via endocytosis into the lysosome. After washed the excess DGLipo NPs and subsequently incubated with fresh culture medium for an additional 1 h, no obvious change in fluorescence intensity of Cy 5.5 was observed, whereas the fluorescence of Dox became much stronger in discrete subcellular locations. To further determine the release site of Dox from DGLipo NPs, co-staining experiments with commercially available organelle probe has been carried out. As seen from the overlay images, Dox was released mainly into mitochondria, whereas negligible portion of Dox was distributed in lysosome, endoplasmic reticulum or nucleus (Figure S6). The result suggested that the liposome shells of DGLipo NPs were degraded in lysosome, thereby released Dox/MPP-DGL into mitochondria in virtue of the target ability of MPP. Subsequently, Dox/MPP-DGL dissociated in the high concentrations of Cyt c in mitochondria and released intercalating Dox promptly, achieving the activation of fluorescence and cytotoxicity. After an additional 2 h of incubation, the mitochondrial fluorescence gradually decreased, while the cytoplasm fluorescence became stronger (Figure S5), suggesting that the Dox transported to the cytosol along with the release of Cyt c in apoptosis process. As the incubation time extended to 4 h, the released Dox gradually accumulated in the nuclei due to the nucleus-targeting (Figure S7). Overall, the real-time confocal images revealed that DGLipo NPs could realize precise and flexible control of intracellular drug delivery into different sites of tumor cells, which holds great potential to generate ideal combination therapy efficiency.
Real-time monitoring of Cyt c release during mitochondria-mediated apoptosis
Recent studies showed that RA-V can efficiently induce apoptosis of cancer cell through mitochondrial pathway, leading to the loss of mitochondrial membrane potential and the release of Cyt c [32]. In this study, RA-V was chosen as a mitochondria-mediated apoptosis inducer combining with Dox to improve the antitumor activity and perform time-dependent imaging of Cyt c release. To investigate the ability of DGLipo NPs to promote mitochondria-mediated apoptosis in cancer cells, an early apoptosis probe 5,5',6,6'-tetrachloro-1,1',3,3'- tetraethylbenzimidazolylcarbocyanine-iodide (JC-1) [33], which can indicate the changes of mitochondrial transmembrane potential was adopted. After treatment with DGLipo NPs for 4 h, the mitochondrial membrane potential of cells significantly decreased, suggesting that DGLipo NPs effectively induced apoptosis via mitochondrial pathway. To further validate the apoptosis pathway was specifically induced by RA-V, cells treated with DGLipo NPs without RA-V have also been investigated by the JC-1 assay. In contrast, there was no significant effect for DGLipo NPs without RA-V on HeLa cells after 4-h incubation (Figure S8), indicating that RA-V was involved in altering the mitochondrial membrane potential to kill tumor cells.
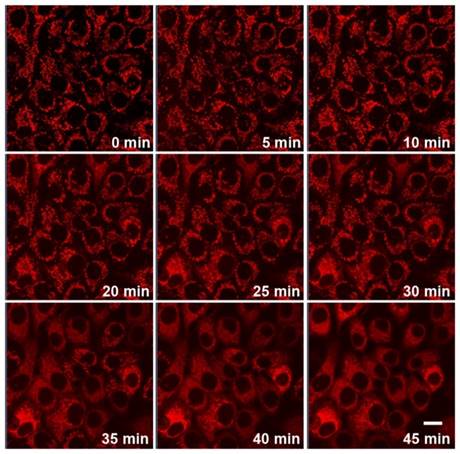
Since Cyt c release is highly specific events in apoptotic signaling, real-time imaging of Cyt c translocation provides an efficient protocol for therapeutic monitoring and visualization of mitochondrial cell-death pathways. The unique Cyt c-activatable fluorescence of DGLipo NPs made it suitable to monitor the Cyt c release dynamically. To verify this capability, the time-dependent fluorescence responses of DGLipo NPs in HeLa cells were tracked in real-time by confocal fluorescence imaging. When HeLa cells incubated with 50 μg mL-1 DGLipo NPs for 2 h, and further incubated with fresh culture medium for additional 1 h, cells displayed intense fluorescence in mitochondria (Figure S6), showing rapid release of Dox from DGLipo NPs triggered by Cyt c. With the time increasing, the mitochondrial fluorescence gradually decreased, and the cytoplasm fluorescence became brighter, suggesting that the RA-V in DGLipo NPs induced Cyt c release from mitochondria to the cytosol during the apoptosis process (Figure 3 and Supplementary Video), which was further proved by immunostaining (Figure S9) and co-localization real-time imaging of DGLipo NPs and MitoTracker (Figure S10). To confirm the potential application of DGLipo NPs in the Cyt c release monitoring in live cells, the nanomaterial prepared with Dox/MPP-cDLG (cDGLipo NPs) were employed as a negative control. Negligible intracellular fluorescence was observed after 2-h incubation with cDGLipo NPs (Figure S11), which suggest that the capacity of DGLipo NPs for the Cyt c-release monitoring was attributed to the specific interaction between Cyt c and its aptamer. These results indicated that DGLipo NPs could realize real-time imaging of Cyt c translocation in situ, providing a convenient method for therapeutic monitoring and investigation of mitochondrial cell-death pathways.
Real-time fluorescence imaging of Cyt c release in HeLa cells incubated with 50 μg mL-1 DGLipo NPs for 2 h, and further incubated with fresh culture medium for additional 1 h. Scale bars: 20 μm.
Overcoming multi-drug resistance in cancer cells by DGLipo NPs
MDR, the principal mechanism by which many cancers develop resistance to chemotherapeutic drugs, poses a significant obstacle to many forms of chemotherapy. As one of the most widely used chemotherapeutics, Dox is severely limited by the development of MDR of cancer cells. It was recently reported that programmable sequential delivery nano-systems, which can accommodate multiple drugs and minimize adverse side effects, may be a new avenue to overcome tumor MDR [34]. Thus, RA-V was incorporated into DGLipo NPs with Dox to construct a synergistic release system for highly efficient chemotherapy in MDR cancer cells.
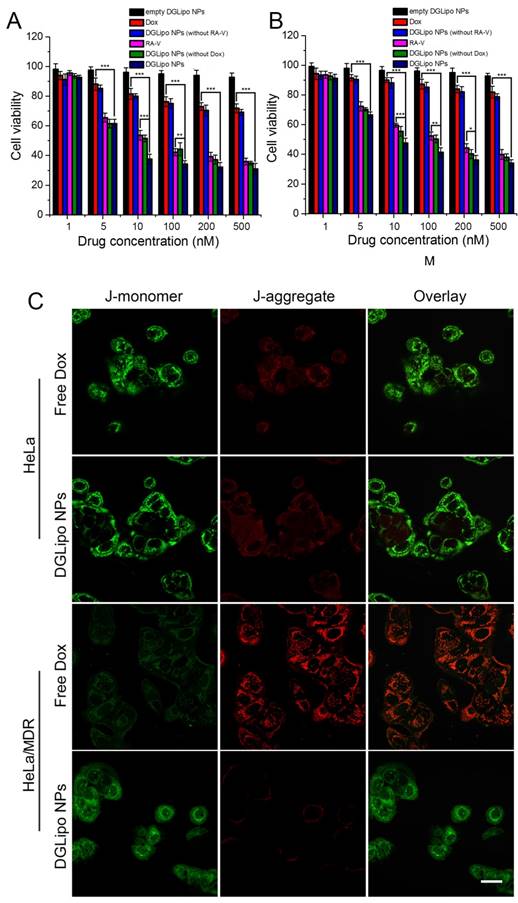
The in vitro antitumor activity of DGLipo NPs was firstly assessed by the MTT assay. As shown in Figure 4A, the in vitro cytotoxicity of DGLipo NPs in HeLa cells was relatively higher than that of free Dox, free RA-V, DGLipo NPs (without Dox) and DGLipo NPs (without RA-V). Since empty DGLipo NPs exhibited negligible cytotoxicity, it can be concluded that combining RA-V and Dox into a multi-organelle targeted co-delivery system with precise spatiotemporal control can improve the antitumor activity of Dox and RA-V. To investigate the ability of DGLipo NPs to overcome MDR, Dox-resistant HeLa/MDR cells were also adopted in the MTT assay. For drug-insensitive HeLa/MDR cells, free Dox showed negligible cytotoxicity after 24 h (Figure S12), whereas prominent apoptosis was observed after treatment with DGLipo NPs under the same condition (Figure 4B). The results were further confirmed by the JC-1 assay (Figure 4C), suggesting that DGLipo NPs could successfully resolve MDR problem by exploring the unique characteristics of different organelle as triggers to realize synergistic drug release in cancer cells.
MTT assay of (A) HeLa cells in the presence of different concentrations of empty DGLipo NPs (IC50>500 nM), free RA-V (IC50=37 nM), DGLipo NPs (without Dox) (IC50=30 nM), free Dox (IC50=1800 nM), DGLipo NPs (without RA-V) (IC50>500 nM) and DGLipo NPs (IC50=8 nM). (B) HeLa/MDR cells in the presence of different concentrations of empty DGLipo NPs (IC50>500 nM), free RA-V (IC50=130 nM), DGLipo NPs (without Dox) (IC50=87 nM), free Dox (IC50=10000 nM), DGLipo NPs (without RA-V) (IC50>500 nM) and DGLipo NPs (IC50=10 nM). Data are means ± SD (n = 3), *P < 0.05, **P < 0.01, ***P < 0.001. (C) Confocal fluorescence images of apoptosis by the JC-1 assay in drug-sensitive HeLa and drug resistance HeLa/MDR cells treated with free Dox or DGLipo NPs for 24 h. Scale bars: 40 μm.
In vivo targeted imaging and therapy on subcutaneous tumor-bearing mice
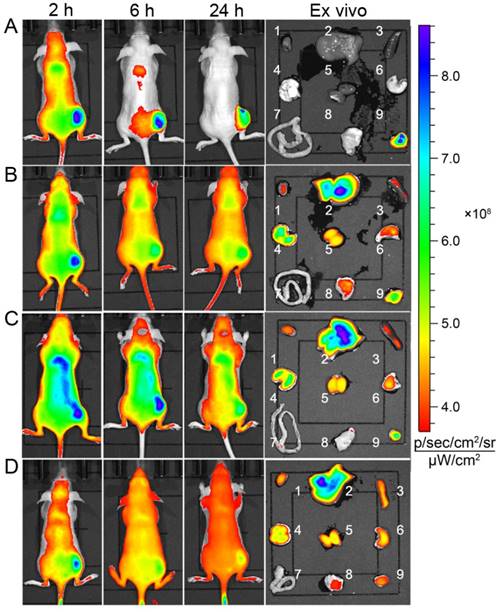
The in vivo targeting capability of DGLipo NPs to tumor was examined in HeLa tumor-bearing BALB/c nude mice. After intravenous (i.v.) injection of DGLipo NPs into the subcutaneous HeLa tumor-bearing mice, the tumor tissue could be distinguished from the surrounding normal tissues at 6 h post injection. As time increased, the fluorescence in tumor region became stronger and reached a maximum at 24 h post injection. The ex vivo fluorescence images showed that the tumor tissue displayed bright fluorescence, while other organs exhibited negligible fluorescence (Figure 5A), demonstrating the tumor-targeted imaging capability of DGLipo NPs in vivo. In contrast, the fluorescence signal at tumor site was still very weak within 24 h after intravenous injection with DGLipo NPs (without RGD) (Figure 5B), proving the essential role of the c(RGDfK) in cancer targeting. Moreover, in vivo fluorescence imaging of HeLa tumor-bearing mice treated with Dox/ MPP-DGL and Dox/Duplex was also carried out. Similar to DGLipo NPs (no RGD) treated mice, no distinct fluorescence signal was observed in tumor site at 24 h post injection (Figure 5C and Figure 5D). These results indicate that DGLipo NPs exhibited high specificity to tumor site in vivo take advantage of the presence of c(RGDfK) at their surface and αvβ3 integrin overexpressed on HeLa cells.
Time-dependent in vivo and ex vivo fluorescence images (1: heart; 2: liver; 3: spleen; 4: lung; 5: kidney; 6: stomach; 7: intestines; 8: muscle; 9: tumor) of subcutaneous HeLa tumor-bearing mice after i.v. injection of (A) DGLipo NPs, (B) DGLipo NPs (without RGD), (C) Dox/MPP-DGL or (D) Dox/Duplex. The order of red, orange, yellow, green, and blue corresponds to the successive increase in fluorescence intensity. The fluorescence images were acquired using IVIS Spectrum instrument equipped with 675/30 nm excitation and 720/20 nm emission filters. The color bar indicates the fluorescence radiant efficiency, multiplied by 108.
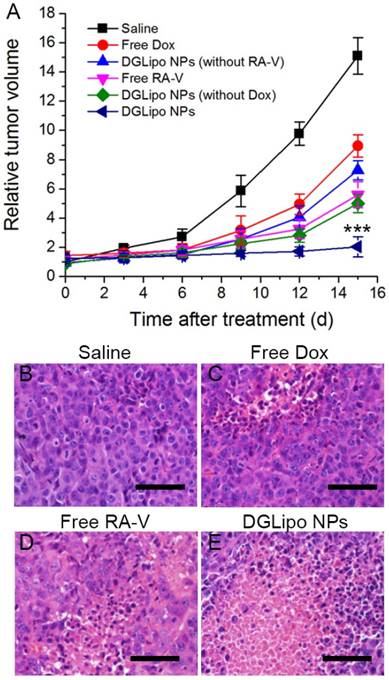
Based on the excellent therapeutic efficacy in vitro, the pH/Cyt c dual-responsive nano-carrier was expected to have great potential as a novel sequential drug-release system for highly efficient cancer therapy in vivo. The in vivo targeted antitumor efficiency of DGLipo NPs was assessed in HeLa tumor-bearing mice by monitoring the tumor volumes over a period of 15 days. From day 0, the mice were intravenously injected with DGLipo NPs every three days, and the tumor volume was measured after treatment. The tumor growth almost completely inhibited after the administration of DGLipo NPs, whereas the tumor growth inhibitions were much lower in the mice treated with the saline, free Dox or free RA-V (Figure 6A). And the tumor volume was decreased in vivo when improve the DGLipo NPs dose (Figure S13). Moreover, the in vivo therapeutic efficacy of DGLipo NPs (without RA-V) and DGLipo NPs (without Dox) were also investigated, showing lower antitumor effect than DGLipo NPs. Thus, it can be concluded the benefits of DGLipo NPs for combination therapy. To further confirm the high antitumor efficacy of DGLipo NPs in vivo, tumor cell death was assessed by hematoxylin and eosin (H&E) staining on tissue sections after different treatment. As shown in Figure 6B-D, H&E staining images exhibited no obvious cell death in the tumor tissues from saline-treated mice, and there were only a small portion of apoptosis cells present in the tumors treated with free Dox or free RA-V. In contrast, a large area of necrosis was observed in histological sections from the tumors of DGLipo NPs-treated mice (Figure 6E), indicating the efficient therapeutic efficacy in vivo of DGLipo NPs. Moreover, the antitumor efficacy of DGLipo NPs was also assessed in HeLa/MDR tumor-bearing mice. The results showed that DGLipo NPs could significantly inhibit the growth of tumor in HeLa/MDR tumor-bearing mice (Figure S14), indicating the capability of Dox/Mito-DGL for overcoming MDR in cancer cells.
(A) Change of relative tumor volume (V/V0) upon different treatments. Data are means ± SD (n = 6), ***P < 0.001 compared to other groups using a one-way ANOVA. Tumors at 24 h post-treatment and corresponding H&E staining of tumor slides: (B) saline injection; (C) free Dox injection; (D) free RA-V injection; (E) DGLipo NPs injection. Scale bars: 100 μm.
Conclusion
In summary, we have successfully constructed a multi-organelle-targeting sequential drug delivery system (DGLipo NPs) for highly efficient combination therapy on MDR tumor and real-time monitoring of Cyt c release in apoptosis process. The modification with c(RGDfK) on the liposomal shell and MPP on the DGL core render DGLipo NPs a high selectivity for site-specific drug delivery at the subcellular level. And the pH/Cyt c dual-responsive feature led to precisely trigger synergistic release of RA-V and Dox, and caused an enhanced subcellular combination therapy efficiency, which significantly overcome the MDR problem in tumor. More importantly, this is the first time that a smart theranostic system can simultaneously realize dynamic visualization of the Cyt c release in situ and inducing tumor cell apoptosis via mitochondria pathway. Overall, this work provides new strategy for precise combination therapy against resistant tumor and contributes to the acquirement of deep insight into the mechanisms of Cyt c-related apoptotic signaling pathway.
Supplementary Material
Additional File 1Supplementary Figures.
Video S1.
Acknowledgements
We thank the National Natural Science Foundation of China (21602255 and 31470428), Natural Science Foundation of Jiangsu Province (BK20160745 and BK20160766), the Program for Jiangsu Province Innovative Research Team, the Foundation of Chinese Academy of Sciences (XDA09030301-4) and the Foundation of High-level Talent Introduction of China Pharmaceutical University for financial support.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jiang T, Mo R, Bellotti A. et al. Gel-liposome-mediated co-delivery of anticancer membrane-associated proteins and small-molecule drugs for enhanced therapeutic efficacy. Adv Funct Mater. 2014;24:2295-2304
2. Jiang T, Sun W, Zhu Q. et al. Furin-mediated sequential delivery of anticancer cytokine and small-molecule drug shuttled by graphene. Adv Mater. 2015;27:1021-1028
3. Cheng R, Meng F, Deng C. et al. Dual and multi-stimuli-responsive polymeric nanoparticles for programmed site-specific drug delivery. Biomaterials. 2013;34:3647-3657
4. An X, Zhu A, Luo H. et al. Rational design of multi-stimuli-responsive nanoparticles for precise cancer therapy. ACS Nano. 2016;10:5947-5958
5. (a) Kim SH, Shum HC, Kim JW. et al. Multiple polymersomes for programmed release of multiple components. J Am Chem Soc. 2011; 133: 15165-15171. (b) Lu Y, Aimetti AA, Langer R, et al. Bioresponsive materials. Nat Rev Mater. 2016; 1: 16075-16092. (c) Wang S, Huang P, Chen X. Hierarchical targeting strategy for enhanced tumor tissue accumulation/retention and cellular internalization. Adv Mater. 2016; 28:7340-7364. (d) Pacardo DB, Ligler FS, Gu Z. Programmable nanomedicine: synergistic and sequential drug delivery systems. Nanoscale. 2015;7:3381-3391
6. Díez P, Sánchez A, de la Torre C. et al. Neoglycoenzyme-gated mesoporous silica nanoparticles: toward the design of nanodevices for pulsatile programmed dequential delivery. ACS Appl Mater Interfaces. 2016;8:7657-7665
7. Wang H, Zhao Y, Wu Y. et al. Enhanced anti-tumor efficacy by co-delivery of doxorubicin and paclitaxel with amphiphilic methoxy PEG-PLGA copolymer nanoparticles. Biomaterials. 2011;32:8281-8290
8. Yang D, Wang T, Su Z. et al. Reversing cancer multidrug resistance in xenograft models via orchestrating multiple actions of functional mesoporous silica nanoparticles. ACS Appl Mater Interfaces. 2016;8:22431-22441
9. Tan NH, Zhou J. Plant cyclopeptides. Chem Rev. 2006;106:840-895
10. Fan JT, Su J, Peng YM. et al. Rubiyunnanins C-H, cytotoxic cyclic hexapeptides from Rubia yunnanensis inhibiting nitric oxide production and NF-kB activation. Bioorg Med Chem. 2010;18:8226-8234
11. Fang XY, Chen W, Fan JT. et al. Plant cyclopeptide RA-V kills human breast cancer cells by inducing mitochondria-mediated apoptosis through blocking PDK1-AKT interaction. Toxicol Appl Pharmacol. 2013;267:95-103
12. Xue X, You S, Zhang Q. et al. Mitaplatin increases sensitivity of tumor cells to cisplatin by inducing mitochondrial dysfunction. Mol Pharm. 2012;9:634-644
13. Liang XJ, Taylor B, Cardarelli C. et al. Dfferent roles for K+ channels in cisplatin-resistant cell lines argue against a critical role for these channels in cisplatin resistance. Anticancer Res. 2005;25:4113-4122
14. Qiao ZY, Zhang D, Hou CY. et al. A pH-responsive natural cyclopeptide RA-V drug formulation for improved breast cancer therapy. J Mater Chem B. 2015;3:4514-4523
15. Chen TT, Tian X, Liu CL. et al. Fluorescence activation imaging of cytochrome c released from mitochondria using aptameric nanosensor. J Am Chem Soc. 2015;137:982-989
16. Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626-629
17. Yue GG, Fan JT, Lee JK. et al. Cyclopeptide RA-V inhibits angiogenesis by down-regulating ERK1/2 phosphorylation in HUVEC and HMEC-1 dndothelial cells. Br J Pharmacol. 2011;164:1883-1898
18. Ju E, Li Z, Liu Z. et al. Near-infrared light-triggered drug-delivery vehicle for mitochondria-targeted chemo-photothermal therapy. ACS Appl Mater Interfaces. 2014;6:4364-4370
19. Wang AH, Gao YG, Liaw YC. et al. Formaldehyde cross-links daunorubicin and DNA dfficiently: HPLC and X-ray diffraction studies. Biochemistry. 1991;30:3812-3815
20. Rin Jean S, Tulumello DV, Wisnovsky SP. et al. Molecular vehicles for mitochondrial chemical biology and drug delivery. ACS Chem Biol. 2014;9:323-333
21. Graf N, Bielenberg DR, Kolishetti N. et al. Lippard SJ. αVβ3 integrin-targeted PLGA-PEG nanoparticles for enhanced anti-tumor efficacy of a Pt(IV) prodrug. ACS Nano. 2012;6:4530-4539
22. Hofman J, Buncek M, Haluza R. et al. In vitro transfection mediated by dendrigraft poly(L-lysines): the effect of structure and molecule size. Macromol Biosci. 2013;13:167-176
23. Calle D, Negri V, Ballesteros P. et al. Magnetoliposomes loaded with poly-unsaturated fatty acids as novel theranostic anti-inflammatory formulations. Theranostics. 2015;5:489-503
24. Chen H, Tian J, Liu D. et al. Dual aptamer modified dendrigraft poly-L-lysines nanoparticle for overcoming multi-drug resistance through mitochondrial targeting. J Mater Chem B. 2017;5:972-979
25. Ke CJ, Su TY, Chen HL. et al. Smart multifunctional hollow microspheres for the quick release of drugs in intracellular lysosomal compartments. Angew Chem Int Ed Engl. 2011;50:8086-8089
26. Tolosa L, Donato MT, Gómez-Lechón MJ. General cytotoxicity assessment by means of the MTT assay. Methods Mol Biol. 2015;1250:333-348
27. Huber W, Koella JC. A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop. 1993;55:257-261
28. Posakony JW, England JM, Attardi G. Mitochondrial growth and division during the cell cycle in HeLa cells. J Cell Biol. 1977;74:468-491
29. Hofman J, Buncek M, Haluza R. et al. In vitro transfection mediated by dendrigraft poly(L-lysines): the effect of structure and molecule size. Macromol Biosci. 2013;13:167-176
30. Waterhouse NJ, Goldstein JC, von Ahsen O. et al. Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol. 2001;153:319-328
31. Yuan Y, Kwok RT, Tang BZ. et al. Targeted theranostic platinum (IV) prodrug with a builtin aggregation-induced emission light-up apoptosis sensor for noninvasive early evaluation of its therapeutic responses in situ. J Am Chem Soc. 2014;136:2546-2554
32. Leung HW, Wang Z, Yue GG. et al. Cyclopeptide RA-V inhibits cell adhesion and invasion in both estrogen receptor positive and negative breast cancer cells via PI3K/AKT and NF-κB signaling pathways. Biochim Biophys Acta. 2015;1853:1827-1840
33. Dhar S, Lippard SJ. Mitaplatin, A potent fusion of cisplatin and the orphan drug dichloroacetate. Proc Natl Acad Sci U S A. 2009;106:22199-22204
34. Han N, Zhao Q, Wan L. et al. Hybrid lipid-capped mesoporous dilica for stimuli-responsive drug release and overcoming multidrug resistance. ACS Appl Mater Interfaces. 2015;7:3342-3351
Author contact
![]() Corresponding author: Ninghua Tan, Email: nhtanedu.cn
Corresponding author: Ninghua Tan, Email: nhtanedu.cn