Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(14):3585-3594. doi:10.7150/thno.21471 This issue Cite
Research Paper
Genomic Analysis of Tumor Microenvironment Immune Types across 14 Solid Cancer Types: Immunotherapeutic Implications
Yu-Pei Chen1*, Yu Zhang2*, Jia-Wei Lv1*, Ying-Qin Li1*, Ya-Qin Wang1, Qing-Mei He1, Xiao-Jing Yang1, Ying Sun1, Yan-Ping Mao1, 3, Jing-Ping Yun2, Na Liu1, 4 ![]() , Jun Ma1
, Jun Ma1 ![]()
1. Sun Yat-sen University Cancer Centre, State Key Laboratory of Oncology in South China, Collaborative Innovation Centre for Cancer Medicine, Guangzhou, People's Republic of China;
2. Department of Pathology, Sun Yat-sen University Cancer Center, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, People's Republic of China;
3. Department of Radiation Oncology, University of Michigan, Ann Arbor, MI, United States;
4. Department of Experimental Radiation Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA.
* These authors contributed equally to this work.
Received 2017-6-14; Accepted 2017-7-6; Published 2017-8-22
Abstract

We performed a comprehensive immuno-genomic analysis of tumor microenvironment immune types (TMITs), which is classified into four groups based on PD-L1+CD8A or PD-L1+cytolytic activity (CYT) expression, across a broad spectrum of solid tumors in order to help identify patients who will benefit from anti- PD-1/PD-L1 therapy. The mRNA sequencing data from The Cancer Genome Atlas (TCGA) of 14 solid cancer types representing 6,685 tumor samples was analyzed. TMIT was classified only for those tumor types that both PD-L1 and CD8A/CYT could prefict mutation and/or neoantigen number. The mutational and neoepitope features of the tumor were compared according to the four TMITs. We found that PD-L1/CD8A/CYT subgroups could not distinguish different mutation and neoantigen numbers in certain tumor types such as glioblastoma multiforme, prostate adenocarcinoma, and head and neck and lung squamous cell carcinoma. For the remaining tumor types, compared with TIMT II (low PD-L1 and CD8A/CYT), TIMT I (high PD-L1 and CD8A/CYT) had a significantly higher number of mutations or neoantigens in bladder urothelial carcinoma, breast and cervical cancer, colorectal, stomach and lung adenocarcinoma, and melanoma. In contrast, TMIT I of kidney clear cell, liver hepatocellular, and thyroid carcinoma were negatively correlated with mutation burden or neoantigen numbers. Our findings show that the TMIT stratification proposed could serve as a favorable approach for tailoring optimal immunotherapeutic strategies in certain tumor types. Going forward, it will be important to test the clinical practicability of TMIT based on quantification of immune infiltrates using mRNA-seq to predict clinical response to these and other immunotherapeutic strategies in more different tumors.
Keywords: Tumor microenvironment immune type, Immune checkpoint inhibitors, Biomarker, mRNA sequencing, Mutation burden, Neoantigen.
Introduction
Inhibition of immune checkpoint proteins, primarily CTLA-4 or PD-1/PD-L1 may reduce the ability of the tumor microenvironment to suppress host antitumor immunity [1]. Such immune checkpoint inhibitors have already shown remarkable clinical efficacy in various types of cancers [2-6]. As these immune-targeted therapies increasingly gain widespread clinical usage, it is imperative to identify predictive biomarkers that allow us to tailor immunotherapeutic strategies appropriately. Recent studies suggest that mutation burden, abundant neoantigens, and potential immune gene signatures is associated with a good response to checkpoint inhibitor treatment [7-11]. Still, currently no pretreatment biomarker has been validated to be included in part of the standard-of-care decision making for anti- PD-1/PD-L1 therapy.
PD-L1 expression on tumors is upregulated in response to immune challenge [12-14], which may represent a simple predictive factor for predicting response.[15] However, despite positive expression of PD-L1, not all patients respond well to immune checkpoint treatment [3, 6, 16], suggesting that other microenvironment factors such as tumor-infiltrating lymphocytes (TILs) may also play an important role [11, 17-21]. Considering the clinical relevance of the multifaceted tumor-associated immune response, a comprehensive immuno-genomic analysis of the tumor microenvironment (i.e., PD-L1 expression and TIL infiltration) is critical to deepen our understanding of this phenomenon.
Unfortunately, assessment of TILs in the tumor microenvironment is challenging [22]. Rooney and colleagues devised an RNA-based metric of immune cell cytolytic activity (CYT) by measuring the mRNA expression levels of granzyme A (GZMA) and perforin 1 (PRF1) from The Cancer Genome Atlas (TCGA) solid tumor samples [18]. The study showed that mRNA-seq data constitutes an appropriate model for assessing the tumor microenvironment, as the stromal tissues surrounding the cancer cells would proportionally influence the gene expression profiles of microenvironment in an unbiased manner [18]. As CD8+ cytolytic T cells (CTLs) recruitment by the adaptive immune response plays a crucial role in the antitumor activity of immune checkpoint inhibitors [23], measuring CD8A expression may also reflect immune cell infiltration, while CYT might be influenced by both CD8+ CTL and other immune cells (e.g., natural killer T cells) [18, 23-25].
Recently, it was proposed that classifying tumor microenvironments into four different types (based on the PD-L1 status and presence or absence of TILs) may help us to rationally design ideal combination cancer immunotherapies [14, 22, 25]. For example, PD-L1 positive with TILs are classified as tumor microenvironment immune type (TMIT) I, and would benefit from anti-PD-L1/PD-1 therapies. This proposed stratification highlights the importance of assessing both PD-L1 expression and TIL recruitment. Ock and colleagues performed a pan-cancer analysis to establish a TMIT model based on the PD-L1 and CD8A mRNA expression, and found it predicted response to immune checkpoint inhibitors [25]. The study used the median PD-L1 and CD8A expression levels from all tumor samples as the cut-off values to define the high and low subgroups; however, these cut-offs may not be optimal and the heterogeneity of different tumor types was underestimated, which could cause potential bias. Besides, it has not been investigated which biomarker (CD8A or CYT expression) along with PD-L1 expression may be more suitable for classifying the TMIT in different tumor types.
Therefore, the aims of this study were to assess: (i) the optimal cut-off values for PD-L1/CD8A/CYT expression (i.e., high and low subgroups), (ii) the reasonable classification of TMITs (i.e., based on PD-L1 + CD8A or PD-L1 + CYT), and (iii) the applicability of TMITs to predict response to checkpoint inhibitor treatment (i.e., its correlation with mutation burden and neoantigen number), in each tumor type through using a large-scale mRNA-seq data of TCGA solid tumor samples.
Materials and Methods
Tumor types and datasets
The dataset used comprised mRNA-seq data from The Cancer Genome Atlas (TCGA) tumor samples (data accessed at cBioPortal for Cancer Genomics in December 2016, http://www.cbioportal.org/) [26, 27]. The following solid tumor types were selected: bladder urothelial carcinoma (BLCA, n = 407), breast cancer (BRCA, n = 1,098), cervical cancer (CESC, n = 303), colon and rectal adenocarcinoma (COAD, n = 382), glioblastoma multiforme (GBM, n = 166), head and neck squamous cell carcinoma (HNSC, n = 521), kidney clear cell carcinoma (KIRC, n = 534), liver hepatocellular carcinoma (LIHC, n = 373), lung adenocarcinoma (LUAD, n = 513), lung squamous cell carcinoma (LUSC, n = 501), prostate adenocarcinoma (PRAD, n = 495), skin cutaneous melanoma (SKCM, n = 468), stomach adenocarcinoma (STAD, n = 415), and thyroid carcinoma (THCA, n = 509). Altogether, samples of 14 cancer types (N = 6,685) were included in the analysis (Supplementary Table S1).
All samples were assayed by mRNA-seq, as described by the TCGA Research Network [28]. Gene expression values were represented as RNA-Seq by Expectation Maximization (RSEM) data normalized within each sample to the upper quartile of total reads [29]. The total number of somatic mutations was adopted to assess the mutation burden, as it is convenient and significantly correlated with the number of non-synonymous mutations [25]. As described previously, multiple somatic mutations (including non-synonymous mutations, insertion-deletion mutations, and silent mutations) were counted and summated, and germline mutations without somatic mutations were excluded [25]. The number of neoantigens was referenced in a previous report [18]. COAD was excluded from the current analysis of neoantigens as the neoantigen number was only available for three samples. Clinical information was also obtained from the cBioPortal for Cancer Genomics [26, 27].
Classification of TMIT
CYT was defined as the geometric mean of expression of GZMA and PRF1 in RSEM, as previously reported [18]. The expression of PD-L1, CD8A, and CYT were measured by using log 2-transformed values in RSEM. As per the study by Teng and colleagues [22], we classified TCGA samples of each cancer type into four TMITs by merging the mRNA expression levels of PD-L1 and CD8A, or PD-L1 and CYT as follows: type I, PD-L1 high expression and CD8A/CYT high expression; type II, PD-L1 low expression and CD8A/CYT low expression; type III, PD-L1 high expression and CD8A/CYT low expression; and type IV, PD-L1 low expression and CD8A/CYT high expression.
As mutation and neoantigen numbers correlate with response to checkpoint inhibitor treatment [7-10], we classified the PD-L1/CD8A/CYT expression into high and low subgroups according to these two factors for each cancer type. The Recursive Partitioning and Regression Trees (RPART) was adopted to determine optimal cut-off values of high and low expression of PD-L1/CD8A/CYT based on the log 2-transformed value of the geometric mean of mutation number and neoantigen number. RPART can objectively divide samples at each step into two subgroups for the object variable, which provides maximum discrimination and yields subgroups with relatively homogeneous performance [30].
To assure the practicability of cut-off points, and that each group contained sufficient samples, we set up the algorithm so the cut-off values were determined at intervals of 5% (e.g., 50%, 55%, 60% of samples, and so on), and that each subgroup contained at least 20% of the samples for each tumor type. Due to insufficient neoantigen data for COAD, the log 2-transformed value of the mutation number alone was used as the object variable in RPART. To confirm the superiority of the RPART cut-off value, we also divided PD-L1/CD8A/CYT expression into high and low subgroups using median values, and compared the results. TMIT was classified only for those tumor types with significant differences in mutation and/or neoantigen number in both PD-L1 and CD8A/CYT RPART subgroups.
Statistical analyses
Correlations were evaluated using the Spearman correlation test: the Spearman coefficient was considered to indicate poor correlation if <0.2, moderate if <0.4, relatively strong if <0.6, strong if <0.8, and very strong if >0.8. The significance of the differences between continuous variables (i.e., number of mutations and neoantigens) and categorical variables (i.e., PD-L1/CD8A/CYT subgroups and TMITs) was calculated by the Wilcoxon rank-sum test. The prognostic significance of the four TMITs was estimated using Kaplan-Meier survival curves (log-rank test); the unadjusted Cox proportional hazards model was used to calculate the hazard ratios (HRs) and corresponding 95% confidence intervals (CIs). Statistical significance was set as P-values <0.05 in a two-tailed test. The statistical analyses were performed using SAS version 9.3 (SAS Institute, Cary, NC, USA) and R version 3.2.3 (http://www.r-project.org) using the “rpart” package (version 4.1-10, https://cran.r-project.org/web/packages/rpart/index.html).
Results
Distribution of mutation burden, neoantigens, and PD-L1/CD8A/CYT expression across TCGA tumors
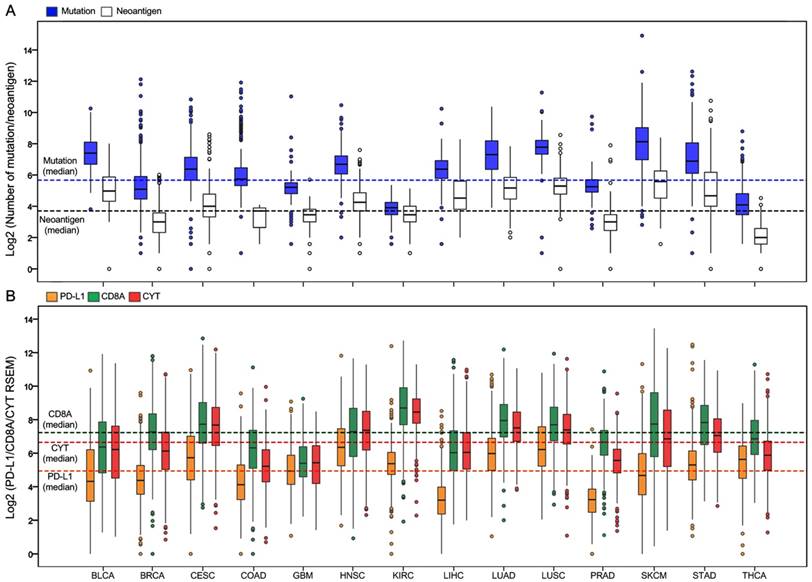
Totally, 6,685 tumor samples from 14 TCGA cancer types were included in the analysis. The sample characteristics are summarized in Supplementary Table S2. Figure 1A presents the log 2-transformed values of the number of mutations and neoantigens according to cancer type. The number of mutations and neoantigens were significantly positively correlated, with a strong or very strong correlation for almost all tumors (R2 > 0.6), except for the LIHC and PRAD (relatively strong), and THCA (moderate; Supplementary Figure S1). Figure 1B shows PD-L1/CD8A/CYT expression according to tumor type.
Distribution of mutation burden, neoantigen number, and PD-L1/CD8A/CYT expression across TCGA cancer types. (A) Boxplot distributions of log 2-transformed values of the number of mutations and neoantigens according to TCGA cancer types. (B) Boxplot distributions of log 2-transformed values of expression of PD-L1 and CD8A, and CYT (cytolytic activity, defined as the geometric mean of GZMA and PRF1 expression in RSEM) according to TCGA cancer types. The dashed lines indicate the median values of all tumor samples.
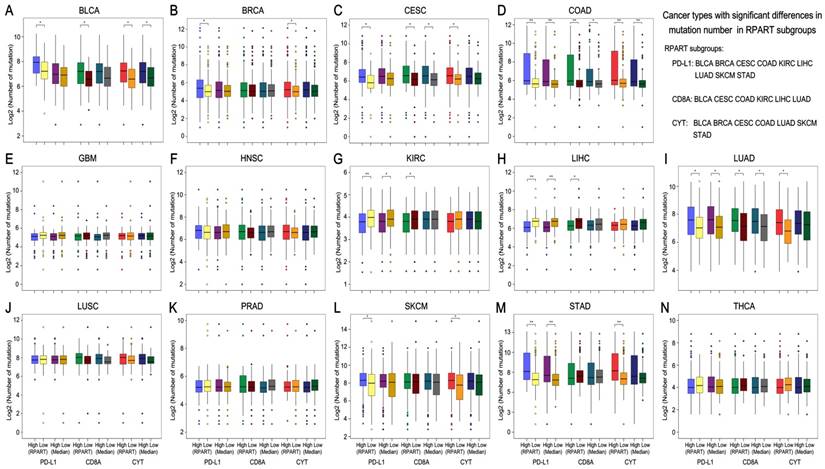
Mutation burden and neoantigen number differ across RPART subgroups of PD-L1/CD8A/CYT in certain tumor types
After the number of mutations and neoantigens were merged as an object variable, the cut-off values of PD-L1, CD8A, and CYT expression for each cancer type were calculated by RPART. These cut-offs varied according to tumor type (Table 1). We then divided the expression of PD-L1, CD8A, and CYT into two subgroups using RPART cut-off or median values, and compared mutation and neoantigen numbers across these subgroups for each tumor type (Figures 2 and 3).
PD-L1/CD8A/CYT expression cut-offs by RPART according to cancer type
| Cancer type | Cut-off by RPART (Log2 RSEM) | |||||
|---|---|---|---|---|---|---|
| PD-L1 | CD8A | CYT | ||||
| Value | Percent of samples (from bottom) | Value | Percent of samples (from bottom) | Value | Percent of samples (from bottom) | |
| BLCA | 6.71 | 80% | 5.79 | 40% | 5.91 | 45% |
| BRCA | 5.07 | 70% | 7.09 | 45% | 5.79 | 40% |
| CESC | 4.10 | 20% | 7.97 | 55% | 7.47 | 45% |
| COAD | 5.30 | 75% | 6.90 | 65% | 6.46 | 80% |
| GBM | 4.76 | 45% | 5.31 | 45% | 5.18 | 45% |
| HNSC | 7.73 | 80% | 5.80 | 25% | 5.83 | 20% |
| KIRC | 5.12 | 40% | 10.28 | 80% | 9.44 | 80% |
| LIHC | 3.20 | 50% | 5.22 | 30% | 7.68 | 80% |
| LUAD | 5.82 | 45% | 8.29 | 60% | 6.48 | 20% |
| LUSC | 6.44 | 55% | 9.02 | 80% | 8.64 | 80% |
| PRAD | 3.86 | 75% | 7.37 | 75% | 6.24 | 75% |
| SKCM | 5.19 | 60% | 8.98 | 65% | 6.05 | 35% |
| STAD | 5.98 | 70% | 7.14 | 35% | 8.06 | 75% |
| THCA | 4.50 | 25% | 5.92 | 25% | 4.76 | 20% |
Abbreviations: BLCA, bladder urothelial carcinoma; BRCA, breast cancer; CESC, cervical cancer; COAD, colon and rectal adenocarcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; IQR, interquartile range; KIRC, kidney clear cell carcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PRAD, prostate adenocarcinoma; RPART, Recursive Partitioning and Regression Trees; RSEM, RNA-Seq by Expectation Maximization; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TCGA, The Cancer Genome Atlas; THCA, thyroid carcinoma.
Mutation burden by subgroups of PD-L1/CD8A/CYT expression across TCGA cancer types. Boxplot distributions of log 2-transformed values of the number of somatic mutations between subgroups of PD-L1/CD8A/CYT expression, as defined by RPART cut-off or median value, according to TCGA cancer types (A-N). For some cancer types, the number of mutations differ significantly in certain RPART subgroups: in PD-L1 subgroups for BLCA, BRCA, CESC, COAD, KIRC, LIHC, LUAD, SKCM, and STAD; in CD8A subgroups for BLCA, CESC, COAD, KIRC, LIHC, and LUAD; and in CYT subgroups for BLCA, BRCA, CESC, COAD, LUAD, SKCM, and STAD. P values are calculated by Wilcoxon rank-sum test (*P < 0.05, **P < 0.001). RPART, Recursive Partitioning and Regression Trees.
Neoantigen number by subgroups of PD-L1/CD8A/CYT expression across TCGA cancer types. Boxplot distributions of log 2-transformed values of the number of neoantigens between subgroups of PD-L1/CD8A/CYT expression, as defined by RPART cutoff or median value, according to TCGA cancer types (A-M). For some cancer types, the number of neoantigens differ significantly in certain RPART subgroups: in PD-L1 subgroups for BLCA, BRCA, CESC, KIRC, LUAD, SKCM, STAD, and THCA; in CD8A subgroups for CESC, KIRC, and LUAD; and in CYT subgroups for BLCA, BRCA, CESC, LUAD, SKCM, STAD, and THCA. P values are calculated by Wilcoxon rank-sum test (*P < 0.05, **P < 0.001). COAD was excluded due to insufficient data. RPART, Recursive Partitioning and Regression Trees.
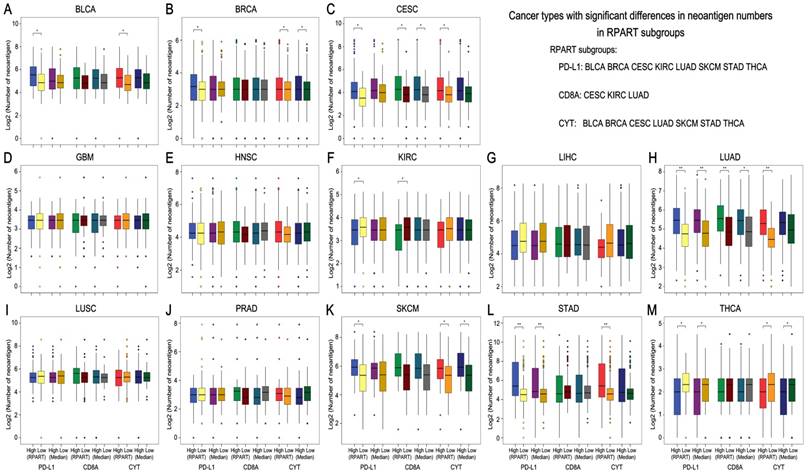
The mutation number differs significantly in both the RPART and Median PD-L1 subgroups for COAD, KIRC, LUAD and STAD; in the CD8A subgroups for CESC, COAD and LUAD; and in the CYT subgroups for BLCA and COAD (Figure 2). Besides, RPART also better distinguished the different mutation number in the PD-L1 subgroups for BLCA, BRCA, CESC, LIHC, and SKCM; in the CD8A subgroups for BLCA, KIRC, and LIHC; and in CYT subgroups for BRCA, CESC, LUAD, SKCM, and STAD (Figure 2). RPART cut-off values perform similarly better for neoantigen numbers as compared to the median values (Figure 3).
In summary, for both mutation and neoantigen number, KIRC differs significantly in the PD-L1 and CD8A subgroups; BLCA, BRCA, SKCM, and STAD differ significantly in the PD-L1 and CYT subgroups; and CESC and LUAD differ significantly in the PD-L1, CD8A, and CYT subgroups. COAD differs significantly in mutation number for the PD-L1, CD8A, and CYT subgroups (no sufficient neoantigen data was available for COAD); LIHC differs significantly in only mutation number for the PD-L1 and CD8A subgroups; and THCA differs significantly in only neoantigen number for the PD-L1 and CYT subgroups. Interestingly, the number of mutations or neoantigens was at a higher level in PD-L1/CD8A/CYT high expression subgroups for these tumors; however, PD-L1 and CD8A/CYT expression were both negatively correlated with mutation or neoantigen numbers for KIRC, LIHC, and THCA (Figures 2G, 2H, and 2N; Figures 3F, 3G, and 3M). Differences in mutation or neoantigen numbers were not observed in the PD-L1/CD8A/CYT subgroups for the remaining tumors (GBM, HNSC, LUSC, and PRAD), which indicates these biomarkers may not serve as good strategy to classify the tumor microenvironment and predict response in these cancer types.
Distribution and clinical significance of TMITs across different tumor types
Based on the abovementioned results, certain tumor samples were divided into four groups of tumor microenvironments according to the RPART cut-off values of PD-L1 and CD8A/CYT expression: PD-L1+CD8A for KIRC and LIHC; PD-L1+CYT for BLCA, BRCA, SKCM, STAD, and THCA; and PD-L1+CD8A/CYT for CESC, COAD, and LUAD. The distributions of TMITs are shown in Supplementary Table S3 and Figures 4A and 4B. In general, the proportion of TIMT I and II were relatively high compared to TIMT III and IV, but it varies across tumor types. The proportions of TMIT I, II, III, and IV in SKCM tumors were 37%, 32%, 3%, and 28%, respectively, which are comparable to those mentioned previously (i.e., 38%, 41%, 1%, and 20%, respectively) [14, 22].
Distribution of TMITs, as well as mutation burden and neoantigen number according to TMIT, across certain cancer types. Proportion of TMITs as defined by PD-L1 and CD8A expression in CESC, COAD, KIRC, LIHC, LUAD (A), and by PD-L1 and CYT expression in BLCA, BRCA, CESC, COAD, LUAD, SKCM, STAD, and THCA (B). Boxplot distributions of log 2-transformed values of the number of mutations/neoantigens according to TMIT, and histogram distributions of TMITs according to mutation/neoantigen status of these cancer types (C-N). P values are calculated by Wilcoxon rank-sum test as compared to TMIT I (*P < 0.05, **P < 0.001). RPART, Recursive Partitioning and Regression Trees; TMIT, tumor microenvironment immune type.
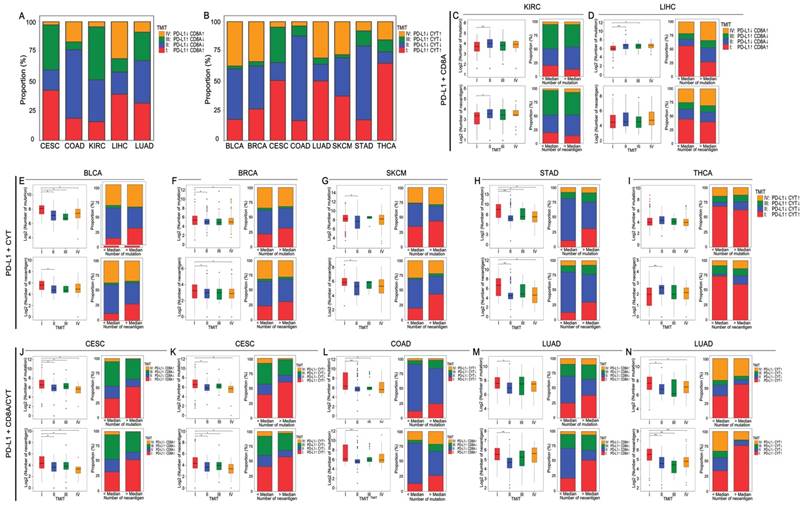
Generally, TMIT I tumors had a significantly higher number of mutations and neoantigens compared with other TMITs, especially TMIT II. Tumor samples with a higher mutation or neoantigen numbers than the median value also tended to have a higher proportion of TMIT I (Figures 4C-N). The insignificant differences in mutation or neoantigen numbers between TIMT I and TIMTs III/IV may be attributed the lack of adequate samples in the subgroups. No significant differences between TMIT groups were observed in mutation number for THCA (Figure 4I), and in neoantigen number for LIHC (Figure 4D), which is in consistent with the abovementioned conclusions. PD-L1+CD8A based TMIT may be optimal for CESC, while PD-L1+CYT based TMIT is better for COAD and LUAD, as it can better distinguish type I from other types in terms of mutation or neoantigen numbers (Figures 4J-N). Besides, as expected, TMIT I of KIRC, LIHC, and THCA were negatively correlated with mutation burden or neoantigen numbers, while TMIT II of these tumor types had higher mutation and neoantigen numbers (Figures 4C, 4D, and 4I).
An overall survival analysis according to TMIT across these TCGA cancer types is shown in Supplementary Figure S2. The prognostic significance of different TIMTs varied in different tumors. Compared with TIMT II, the favorable prognostic effect of TMIT I was the most prominent in SKCM (P < 0.001; Supplementary Figure S2K).
Discussion
Here we present several key aspects for classifying the tumor microenvironment based on PD-L1 and CD8A/CYT mRNA expression determined from RNA-seq data across numerous TCGA solid tumor samples. We present optimal cut-off values for PD-L1/CD8A/CYT expression by using RPART. We found the ability for these cut-off values to distinguish different mutation and neoantigen numbers varies across different cancer types. The PD-L1 and CD8A or CYT cut-offs were then used to classify the tumor microenvironment in the various tumors into four different TIMTs. We found TIMT I was generally associated with a high mutation burden or neoantigen number compared with TMIT II, except in cases of KIRC, LIHC, and THCA. Our results suggest that PD-L1 positivity should be comprehensively interpreted along with other tumor microenvironment factors in different cancer types.
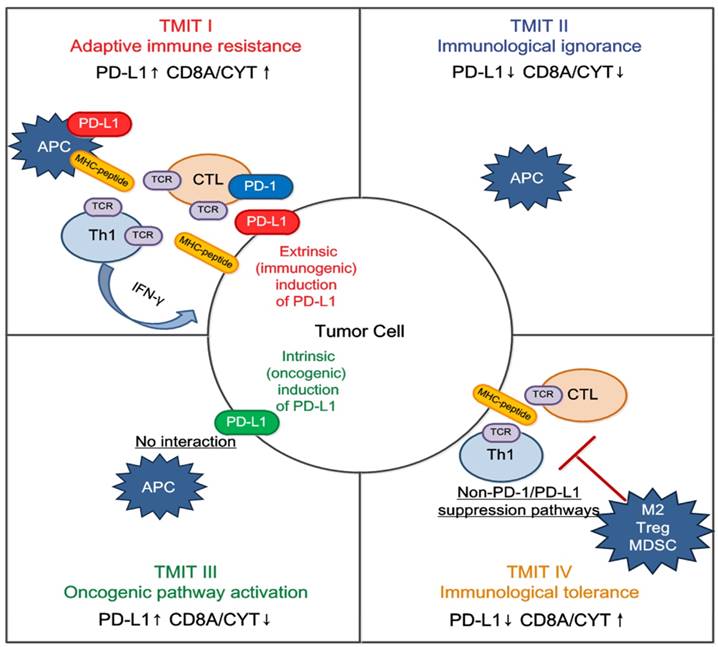
We propose that classification of the tumor microenvironment into four TMITs may be a simple and useful model for tailoring cancer immunotherapeutic modules (Figure 5) [22]. In melanoma, a high proportion of type I (38%) and type II (41%) tumor microenvironments was observed, while types III and IV were observed in only 1% and 20% of tumors respectively [14]. It is suggested that type I tumor immune microenvironments are most likely to benefit from single-agent anti-PD-1/PD-L1 checkpoint blockade, as these tumors have evidence of pre-existing intratumor T cells that are turned off by PD-L1 engagement [22]. Alternatively, TIMT II are predicted to have very poor prognosis considering their lack of detectable immune reaction. Combination therapy designed to attract T cell infiltration into the tumor microenvironment and then stop them being turned off, such as combined anti-CTLA-4 and anti-PD-1/PD-L1, should be considered in this scenario; inducing a type I interferon response would also be an approach [31]. Similarly in TIMT III, in which TILs are absent, it is unlikely that blocking PD-1/PD-L1 will lead to a T cell-mediated cancer response. Therefore, a similar approach to TIMT II patients might be used to try to recruit lymphocytes into tumors. In addition, radiotherapy-mediated immunogenic cell death (to liberate neoantigens) to induce T cell responses in combination with anti PD-1/PD-L1 may be useful in these cases [32]. Finally, for TIMT IV, other suppressive pathways might be dominant and the therapeutic approaches for these pathways are mostly still in the initial stage.
Tailoring cancer immunotherapeutics based on tumor microenvironment immune types (TMITs). The tumor microenvironment has been categorized into four different types based on the expression of PD-L1, and CD8A/CYT: type I (adaptive immune resistance), type II (immunologic ignorance), type III (oncogenic pathway activation), and type IV (immunologic tolerance). The proposed four TMITs are simplistic, but can help us to tailor the most suitable immunotherapeutic strategies. APC, antigen presenting cell; CTL, cytotoxic lymphocyte; IFN, interferon; M2, M2 macrophage; MDSC, myeloid-derived suppressor cell; Th1, T helper 1; Treg, regulatory T cell.
Among the solid cancer types in this study, neither PD-L1, CD8A, nor CYT expression was found to be associated with mutation burden and neoantigen number in GBM, HNSC, LUSC, and PRAD, indicating that these biomarkers may not be able to predict response to anti-PD-L1/PD-1 therapies in these cancer types. Consistent with our results, two recent phase III trials, in which patients with platinum-refractory recurrent HNSC and advanced squamous-cell non-small-cell lung cancer [NSCLC] were treated with the nivolumab (an anti-PD1 monoclonal antibody), found no correlation between response rate and PD-L1 expression level [6, 33]. However, another trial, which evaluated nivolumab versus docetaxel in non-squamous NSCLC (mainly LUAD), showed PD-L1 expression could help predict greater efficacy of nivolumab [34]. This is in agreement with our conclusions, which suggest TMIT may be useful in predicting response in LUAD.
With regards to SKCM, we found the proportion of TMIT I, II, III, and IV were 37%, 32%, 3%, and 28%, respectively, which are comparable to that of a previous report (38%, 41%, 1%, and 20%) [14]. In a phase III study evaluating nivolumab in previously untreated melanoma, the objective response rate was 40% [4], which is similar to the TMIT I proportion of this study (37%). The objective response rate was also higher in patients with PD-L1 positive status than those with negative status (53% vs. 33%). Our analyses also found that TMIT I was associated with higher mutation or neoantigen numbers in BLCA, BRCA, CESC, COAD, and STAD. In published trials assessing anti-PD-1/PD-L1 therapies, the response rates were 26% in metastatic bladder cancer (18% TIMT I for BLCA in this study) [35], 19% in advanced triple-negative breast cancer (27% TIMT I for BRCA) [36], and 23% in advanced gastric cancer (17% TIMT I for STAD) [37]; all these trials observed an increasing probability of response with increasing expression of PD-L1. Meanwhile, in KIRC, LIHC, and THCA, TIMT II had a higher level of mutation burden and neoantigen number. A phase III trial evaluating nivolumab versus everolimus in advanced renal cell carcinoma also observed similar phenomenon: the objective response rate of nivolumab was 25% (35% for TIMT II in this study), and among patients with ≥1% PD-L1 expression detected by immunohistochemistry, the median overall survival was 21.8 months in the nivolumab group versus 27.4 months in those with <1% PD-L1 expression [5]. Similar results were observed when using 5% as the cut-off.
Overall, our results are generally consistent with those observed in clinical trials evaluating checkpoint inhibitor treatment, highlighting that the combination of PD-L1 and CD8A/CYT expression may help better identify subsets of patients who will benefit from anti-PD-1/PD-L1 therapy and avoid any potential toxicities and costs. We eagerly await more studies to validate our conclusions, and to elucidate the underlying mechanisms of these phenomena.
In this study, the RNA-seq data was obtained from the mixed cancer cells and the surrounding tissues. As interpreted by Rooney and colleagues [18], the unintentional contamination of surrounding tissues would actually be advantageous to assessing the infiltration of TILs. Besides, as the presence of TILs or PD-L1 is not a dichotomous variable, there is a need for a quantitative assessment in biopsies to derive the desired predictive information [38]. The assessment of CD8A or CYT expression in a mixture of cancer and stromal cells would be more practical in the clinical setting than the immunohistochemical assessment of TILs, which would be difficult to judge uniformly across different tumor types. Nevertheless, the limitations of this study should be addressed in future. The cut-off values of PD-L1/CD8A/CYT expression require external validation: the optimal classification of TMIT and its ability to predict response to immune checkpoint treatment should be clinically validated. Still, our findings are consistent with previous findings and this study represents an important step toward providing a systematical assessment of tumor microenvironment of most solid cancers.
In summary, the TMIT stratification proposed could serve as a favorable approach for tailoring optimal immunotherapeutic strategies in certain tumor types. Going forward, it will be important to test the clinical practicability of TMIT based on quantification of immune infiltrates using mRNA-seq to predict clinical response to these and other immunotherapeutic strategies in more different tumors.
Abbreviations
BLCA: bladder urothelial carcinoma; BRCA: breast cancer; CESC: cervical cancer; CI: confidence interval; COAD: colon and rectal adenocarcinoma; CYT: cytolytic activity; GBM: glioblastoma multiforme; GZMA: granzyme A; HNSC: head and neck squamous cell carcinoma; HR: hazard ratios; IQR: interquartile range; KIRC: kidney clear cell carcinoma; LIHC: liver hepatocellular carcinoma; LUAD: lung adenocarcinoma; LUSC: lung squamous cell carcinoma; OS: overall survival; PRAD: prostate adenocarcinoma; PRF1: perforin 1; RSEM: RNA-Seq by Expectation Maximization; SKCM: skin cutaneous melanoma; STAD: stomach adenocarcinoma; TCGA: The Cancer Genome Atlas; THCA: thyroid carcinoma; TILs: tumor-infiltrating lymphocytes; TMIT: tumor microenvironment immune types.
Acknowledgements
We would like to thank the staff members of the Cancer Genome Atlas for their involvement in the cBioPortal for Cancer Genomics Program. This work was supported by grants from the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (2014BAI09B10), the National Natural Science Foundation of China (81572658), the Program of Introducing Talents of Discipline to Universities (B14035), the Health & Medical Collaborative Innovation Project of Guangzhou City, China (201400000001), and the Science and Technology Project of Guangzhou City, China (14570006).
Supplementary Material
Additional File 1Figures S1-S2.
Table S1.
Tables S2-S3.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity. 2016;44:955-72
2. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-54
3. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS. et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563-7
4. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L. et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-30
5. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S. et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1803-13
6. Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L. et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375:1856-67
7. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV. et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415-21
8. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124-8
9. Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH. et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24:743-50
10. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69-74
11. Iglesia MD, Parker JS, Hoadley KA, Serody JS, Perou CM, Vincent BG. Genomic Analysis of Immune Cell Infiltrates Across 11 Tumor Types. J Natl Cancer Inst. 2016:108
12. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450-61
13. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT. et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5:200ra116
14. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL. et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37
15. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4
16. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP. et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018-28
17. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH. et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064-74
18. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48-61
19. Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17:e542-e51
20. Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E. et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002;99:16168-73
21. Milne K, Kobel M, Kalloger SE, Barnes RO, Gao D, Gilks CB. et al. Systematic analysis of immune infiltrates in high-grade serous ovarian cancer reveals CD20, FoxP3 and TIA-1 as positive prognostic factors. PloS one. 2009;4:e6412
22. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015;75:2139-45
23. Danilova L, Wang H, Sunshine J, Kaunitz GJ, Cottrell TR, Xu H. et al. Association of PD-1/PD-L axis expression with cytolytic activity, mutational load, and prognosis in melanoma and other solid tumors. Proc Natl Acad Sci U S A. 2016;113:E7769-E77
24. Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol. 2004;172:7335-40
25. Ock CY, Keam B, Kim S, Lee JS, Kim M, Kim TM. et al. Pan-Cancer Immunogenomic Perspective on the Tumor Microenvironment Based on PD-L1 and CD8 T-Cell Infiltration. Clin Cancer Res. 2016;22:2261-70
26. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
27. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
28. Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519-25
29. Palmer C, Diehn M, Alizadeh AA, Brown PO. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genomics. 2006;7:115
30. O'Sullivan B, Huang SH, Su J, Garden AS, Sturgis EM, Dahlstrom K. et al. Development and validation of a staging system for HPV-related oropharyngeal cancer by the International Collaboration on Oropharyngeal cancer Network for Staging (ICON-S): a multicentre cohort study. Lancet Oncol. 2016;17:440-51
31. Bald T, Landsberg J, Lopez-Ramos D, Renn M, Glodde N, Jansen P. et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014;4:674-87
32. Kalbasi A, June CH, Haas N, Vapiwala N. Radiation and immunotherapy: a synergistic combination. J Clin Invest. 2013;123:2756-63
33. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E. et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123-35
34. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE. et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:1627-39
35. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C. et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558-62
36. Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R. et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J Clin Oncol. 2016;34:2460-7
37. Muro K, Chung HC, Shankaran V, Geva R, Catenacci D, Gupta S. et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol. 2016;17:717-26
38. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568-71
Author contact
![]() Corresponding authors: Jun Ma, Sun Yat-sen University Cancer Centre, State Key Laboratory of Oncology in South China, Collaborative Innovation Centre for Cancer Medicine, 651 Dongfeng Road East, Guangzhou 510060, People's Republic of China. Tel.:+86-20-87343469; Fax:+86-20-87343295; E-mail: majun2sysu.edu.cn Na Liu, Sun Yat-sen University Cancer Centre, State Key Laboratory of Oncology in South China, Collaborative Innovation Centre for Cancer Medicine, 651 Dongfeng Road East, Guangzhou 510060, People's Republic of China; Department of Experimental Radiation Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA Tel.:+86-20-87343469; Fax:+86-20-87343295; E-mail: liun1org.cn
Corresponding authors: Jun Ma, Sun Yat-sen University Cancer Centre, State Key Laboratory of Oncology in South China, Collaborative Innovation Centre for Cancer Medicine, 651 Dongfeng Road East, Guangzhou 510060, People's Republic of China. Tel.:+86-20-87343469; Fax:+86-20-87343295; E-mail: majun2sysu.edu.cn Na Liu, Sun Yat-sen University Cancer Centre, State Key Laboratory of Oncology in South China, Collaborative Innovation Centre for Cancer Medicine, 651 Dongfeng Road East, Guangzhou 510060, People's Republic of China; Department of Experimental Radiation Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA Tel.:+86-20-87343469; Fax:+86-20-87343295; E-mail: liun1org.cn