Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Conclusion
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(13):3306-3318. doi:10.7150/thno.20242 This issue Cite
Research Paper
A Nanosystem of Amphiphilic Oligopeptide-Drug Conjugate Actualizing Both αvβ3 Targeting and Reduction-Triggered Release for Maytansinoid
Yanqin Liang1, 2*, Suxin Li2*, Xiaoyou Wang2, Bo He2, Bing He2, Wenbing Dai2, Hua Zhang2, Xueqing Wang2, Yiguang Wang1, 2, Demin Zhou1, Qiang Zhang1, 2 ![]()
1. State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China;
2. Beijing Key Laboratory of Molecular Pharmaceutics, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China.
* Both authors contributed equally to this manuscript.
Received 2017-3-24; Accepted 2017-6-16; Published 2017-7-23
Abstract

To design a prodrug-based self-assembling nanosystem with both ligand targeting and stimuli-responsive features, and elucidate the superiority of each targeting strategy and the synergistic effect between them, we synthesized four small molecule amphiphilic peptide-drug conjugates (APDCs) using maytansinoid (DM1) as a cytotoxic agent, cRGDfK as a homing peptide, and disulfide (SS) or thioether (SMCC) as linker. Owing to their amphiphilicity, the APDCs could self-assemble into nanoparticles (APDC@NPs) which were evaluated in vitro in three different cell lines and in vivo in tumor-bearing C57BL/6 mice. The RSSD@NPs showed the strongest interaction with αvβ3 integrin, highest cell uptake and intracellular free drug level, and best antitumor efficacy in vitro and in vivo, while it shared the same goodness with other test nanosystems in terms of high drug loading, EPR effect and free of potentially toxic polymers. Especially, the in vivo efficacy of RSSD@NPs was 2 fold of free DM1 which is too cytotoxic to be a drug, while the active targeted APDC@NPs demonstrated acceptable system, tissue and blood compatibility. In αvβ3-positive cells or tumors, the RGD targeting contributed much more than disulfide in anticancer effect. The maximum synergism of the two strategies reached to 22 fold in vitro and 3 fold in vivo. Generally, the active targeting, prodrug and nanosystem could significantly decrease the toxicity of free DM1 and improve its therapy outcome via combining active targeting, prodrug and nanopreparation, especially the dual targeting strategies and their synergism.
Keywords: Amphiphilic peptide-drug conjugate, αvβ3-targeted nanoparticles, reduction-triggered drug release, maytansinoid DM1, endocytosis, antitumor therapy.
Introduction
Cancer is the leading cause of death around the world which severely threatens human health [1]. Chemotherapy is one of major choices since most tumors are defined as the chronic diseases by WHO. However, conventional chemotherapy faces pressing challenges in terms of poor efficacy and serious systemic toxicity possibly due to the nonspecific biodistribution of most chemodrugs [2]. Therefore, the development of targeted drug delivery systems (TDDS) which can site-specifically deliver antitumor drugs to tumor sites is essential for cancer therapy [3]. Nanoparticulate-based drug delivery systems (nano-DDS) have experienced tremendous development and are expected to bring radical solutions to the limitations of conventional chemotherapy [4], because they improve the tumor accumulation and safety of chemodrugs [5]. Nanomedicines [6-8] including liposomes [9], water-soluble polymers [10], vesicles [11], dendrimers [12], polymer nanoparticles or micelles [13, 14], inorganic nanoparticles [15], and some hybrid organic-inorganic nanosystems [16], have been studied extensively for this purpose. However, the general outcome of antitumor nanomedicines after past few decades seems far from satisfactory, and one of the reasons seems the inferior delivery efficiency [17]. Also, the toxicities potentially arising from the synthetic materials limit their applications [18]. Besides, nanocarriers usually provide low drug loading since themselves constitute a high proportion of nanoparticles.
For one hand, it is well-addressed that the targeting strategies such ligand modification and stimuli-responsive delivery can improve the delivery efficiency of antitumor drugs [19, 20], and some of targeted delivery systems have already moved into clinical studies [21]. On the other hand, some prodrug-based self-assembling nanosystems, without use of polymer carriers, may achieve higher drug loading and possess higher safety [22], while are more favorable in terms of EPR effect [23]. It seems better to combine these two strategies [24-26]. Yan et al. linked a hydrophilic anticancer drug to a hydrophobic one via a hydrolyzable ester linkage [18]. He et al. developed a conjugate of paclitaxel and oleic acid through a thioether- or dithioether- linker [23]. In these reports, the prodrugs could self-assembled into nanoparticles in water, and demonstrated improvement in drug loading, tumor accumulation and antitumor effect. Cui et al. conjugated a hydrophobic anticancer drug to a β-sheet-forming peptide through a reducible disulfylbutyrate linker, and such a prodrug with high drug loading exhibited greater cytotoxicity [27]. But, there was currently very limited report on the prodrug-based self-assembling nanosystems using both ligand targeting and stimuli-response strategy. In general, a self-assembled nanosystem based on homing peptide/stimuli-responsive linker/ antitumor drug might be provided with superiorities of active targeting, prodrugs and nano-DDS.
More significantly, there are some very critical and interesting issues related which are not clarified so far. For instance, the ligand modification and stimuli-responsive delivery are extensively investigated [28, 29], respectively, in both field of nanomedicines and prodrugs. However, it is still hard to answer now which strategy is more favorable, especially in the circumstance of prodrug-based nanosystem. Additionally, it is still un-known currently if there exist any synergistic effects between these two approaches when used together in such a prodrug-based nanosystem. Obviously, elucidating these questions is very crucial for the future design and development of related delivery systems.
Here, we design and construct a series of novel peptide-linker-drug conjugates. The maytansinoid DM1 (DM1), a microtubule inhibitor similar to vinca alkaloids [30, 31] was selected as the powerful cytotoxic agent, in addition, the toxicity of free DM1 was significant, as evidenced by the body weight loss, hepatic cytotoxicity, decrease of WBC count and organic damages [32]. Two types of linkers, the cleavable disulfide (SS) and the uncleavable thioether (SMCC), were utilized for comparison. While, the cRGDfK sequence was chosen as a homing peptide and the cRPQfK sequence was used as a garbled control [33]. By the way, these two peptides are very similar in terms of amino acid number, molecular weight, solubility, cyclic structure and so on, and details as shown in the Supporting Information (Materials and Figure S1-S4). Owing to the hydrophilic peptides and hydrophobic DM1 (Scheme 1A), the synthesized small molecule APDCs could self-assemble into nanoparticles (recorded as APDC@NPs) via a nanoprecipitation process [34], resulting in high drug loading since DM1 itself is the main component of the nanoparticles. It is hypothesized that the designed APDC@NPs can deliver themselves into tumor tissues by passive accumulation [35], internalize into tumor cells through the receptor-mediated endocytosis pathway (Scheme 1B) [36], and reduction-triggered release DM1 in the highly reductive endosome or cytosol via the cleavage of disulfide, while keeping stable in plasma [37].
(A) Chemical structure of small molecule amphiphilic peptide-drug conjugates (APDCs): cRGD-SMCC-DM1 (RCCD), cRGD-SS-DM1 (RSSD), cRPQ-SMCC-DM1 (QCCD), and cRPQ-SS-DM1 (QSSD). (B) Schematic illustrations of the self-assembly of APDC nanoparticles (APDC@NPs), the accumulation of APDC@NPs at the tumor site by the EPR effect, their uptake by tumor cells or tumor angiogenesis endothelial cells by αvβ3 receptor-mediated endocytosis, and the triggered intracellular drug release from APDC@NPs.
For the proof-of-concept, four types of APDCs@NPs, including cRGD-SMCC-DM1 (RCCD), cRGD-SS-DM1 (RSSD), cRPQ-SMCC-DM1 (QCCD) and cRPQ-SS-DM1 (QSSD), were chemically synthetized (Figures S1-S4), characterized (Figures S5-S9). Then, they were evaluated in vitro in three different cell lines including αvβ3-positive melanoma B16 cells [38] and human umbilical vein endothelial cells (HUVEC) [39], and αvβ3- negative MCF-7 cells, and in vivo in the B16 tumor-bearing C57BL/6 mice. Especially, the superiority of each targeting strategy as well as the synergistic effect between them was carefully investigated via the parallel comparisons of the four nanosystems throughout of the whole study.
Materials and Methods
Synthesis of APDCs
In our study, we synthesized four types of small molecule APDCs. The uncleavable thioether-linked APDCs (cRGD-SMCC-DM1, cRPQ-SMCC-DM1) were synthesized by a nucleophilic substitution reaction between the NHS amino group of DM1 and the N-terminal group of cyclic peptide, and the cleavable disulfide-linked APDCs (cRGD-SS-DM1, cRPQ-SS-DM1) were obtained with a three-step method. Details of all APDCs preparations, characterizations, and experimental methods were shown in the Supporting Information.
Fabrication of APDC@NPs
All APDC@NPs were prepared according to the nanoprecipitation method [4]. Briefly, the APDC was dissolved in DMSO and stirred uniformly before use. In the process of stirring, the DMSO solution of APDC was added dropwise into distilled water, and then the sample was sonicated using a supersonic cell disruptor (JY92-2D, China) for 10 min to obtain APDC@NPs. The dimensions and zeta potential of various APDC@NPs were measured in triplicate by a dynamic light scattering (DLS) analysis using a Malvern Zetasizer Nano ZS (Malvern, Malvern, United Kingdom) at 25 oC. The shape and surface morphology of various APDC@NPs were observed through transmission electron microscope (TEM, JEOL, JEM-1400, Japan). The aqueous solution storage and plasma solution stability of various APDC@NPs were measured using DLS.
In vitro reduction-triggered drug release from APDC@NPs
The reduction-triggered drug-release behavior of DM1 from active targeting APDC@NPs was determined in the same way as that of leakage test. Briefly, these APDC@NPs were mixed with certain release medium and then transferred to a dialysis bag (MWCO=1000 Da). Then, dialysis bags of each formulation (0.8 mL) were immersed in PBS (containing 0.5% SDS, with or without 50 mM DTT, 16 mL) at 37 °C with gentle shaking (100 rpm). Aliquots of release medium were withdrawn and replenished with equal volume of PBS at predetermined time points (1, 2, 4, 6, 8, 12, 24, 48 h). The amount of released DM1 was measured using the HPLC method and the release percentage was calculated according to the total content. A high performance liquid chromatography (HPLC, Shimadzu, LC-10AT system, Japan) was used to determine the drug contents in different samples. Briefly, the samples were conducted using an ODS column (Phenomenex® C18, 5 μm, 250 × 4.6 mm) and an UV detector set at 232 nm by a mixture of water and acetonitrile with a flow rate at 1.0 mL/min, first equilibrated at 45% acetonitrile, followed by a gradient from 45% acetonitrile to 90% acetonitrile over 28 min, and back to 45% acetonitrile in 2 min eventually. The column was allowed to reequilibrate for another 10 min prior to the next injection and the injection volume was 50 μL.
Cellular uptake studies
The cellular uptake behaviors were studied in three different cell lines including B16, MCF-7 and HUVEC cells using a HPLC analysis. Each experiment was carried out in triplicate. In brief, the tested cells were plated in different 6-well plates at 5.0 × 105 cells per well in 2 mL of complete RPMI-1640 or Ham's F12K-Medium and cultured for 24 h, and then treated with different APDC formulations (100 nM) at 37 °C. After 3 h of incubation, the cells were rinsed with cold PBS thrice, lysised by DMSO, ultrasonicated and centrifugated to extract intracellular drugs successively. The supernate was collected and injected into the HPLC system to determine the content of the free DM1 or APDC@NPs in each sample according to the distinct peaks.
Exploring endocytosis pathways using inhibitors
To explore the endocytosis pathway of various APDC@NPs, the integrin αvβ3 receptor-overexpressing B16 cells were chosen for study. Briefly, B16 cells were seeded in glass bottom dishes and pre-incubated with different cellular uptake inhibitors including dynasore (240 μM), Cytochalasin D (0.5 μM), or cRGD (20 μM) for 1 h at 37 °C. The cells were then treated with different Cy3.5-loaded APDC formulations (100 nM) in the presence of inhibitors. After 3 h of incubation, the cells were rinsed with cold PBS thrice, fixed by paraformaldehyde, and nuclei-stained by Hoechst 33258 successively. The cells were visualized via a Leica TCS SP8 laser scanning confocal microscope (Heidelberg). Cy3.5 and Hoechst 33258 were excited at 591 nm and 350 nm, and their emissions were collected at 610 nm and 460 nm respectively.
In vitro cytotoxicity studies
The in vitro cytotoxicity of various APDC@NPs was evaluated by sulforhodamine B (SRB) colorimetric assay of B16, MCF-7 and HUVEC cells. Briefly, B16, MCF-7 or HUVEC cells were seeded in a 96-well plate at a density of 5000 cells/well, and then pre-treated with or without GSH-OEt (10 mM) for 2 h to change the intracellular level of GSH. Next, the medium containing GSH-OEt was removed and serial concentrations (calculated by free DM1) of different APDC formulations were added into the wells. After further incubation for 48 h at 37 °C, the cells were fixed with 10% cold trichloroacetic acid at 4 °C for 1 h, stained with 0.4% SRB, and washed with 1% acetic acid and air drying successively. Subsequently, the fixed cells were stained with 0.4% SRB for 30 min, followed by washing with 1% acetic acid and air drying successively. Finally, the cellular bound SRB was dissolved by 10 mM Tris base, and the absorbance at 540 nm was determined using a 96-well plate reader (Thermo Scientific, Multiskan FC, USA). All data were calculated as the percentages of viable cells relative to the survival of control group (cells treated with medium) and presented as the mean ± SD (n = 3).
Cell cycle assay
B16 cells and MCF-7 cells were seeded in 12-well plates and treated with different APDC formulations (50 nM, calculated by free DM1) diluted with RPMI-1640 medium, while HUVEC cells with Ham's F12K-Medium for 12 h at 37 °C. The cells incubated with fresh medium were used as the control. Subsequently, the cells were rinsed with cold PBS thrice and detached by trypsinization. Cells were then collected after a gentle centrifugation at 1500 rpm for 5 min and fixed in 70% ethanol over night at 4 °C. The suspended cells were centrifuged at 1500 rpm for 5 min to remove residual ethanol and suspended in 0.3 mL of PBS containing DNase-free RNase A along with 0.1% of Triton X-100 at 37 °C for 30 min. Cell cycle in each sample was finally analysed by FCM (FACS Calibur, BD) with 10,000 events collected.
In vivo antitumor efficiency and safety studies
The B16 tumor model was established by subcutaneous inoculation of 1×106 B16 cells in the right flanks of C57BL/6 mice. The mice were randomly divided into six groups (six mice each) when the tumor volume reached approximately 80 mm3. 0.2 mL of various APDC formulations (400 μg/kg for the content of DM1) suspended in saline were injected via the tail vein every other day for a total of five times according to their pre-determined groups, and the mice with saline-administration were chosen as the control. Tumor volumes and body weights of the animals were recorded every other day calculated using the following formula: V = 1/2 × (major axis) × (minor axis)2. The mice were further observed for another week after the last administration. On the 25th day after implantation, 500 μL of blood sample was also collected without heparinization and then centrifuged at 4000 rpm for 5 min to get serum. The activities of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured as typical biochemical markers to evaluate hepatic function. 20 μL of the peripheral blood of each mouse was collected to assess the following: white blood cells (WBC), red blood cells (RBC), platelets (PLT) via autoanalyzer (MEK-6318K, Nihon Kohden). Subsequently, mice were sacrificed by cervical dislocation, the tumors and major organs including heart, liver, spleen, kidney, and lung were excised. The tumors were accurately weighed to evaluate in vivo anti-tumor efficacy. For the histopathological analysis, the excised organs were fixed in 10% neutral formalin, embedded in paraffin, and then the 5 μm sections were prepared. Finally, paraffin sections were stained with H&E to make the histopathological analysis via optical microscope observation. In addition, the TUNEL assay and the immunohistochemical analysis were further adopted to assess the antitumor efficacy and mechanism of various APDC@NPs, and the details of experimental methods are in the Supporting Information.
Results and Discussion
Fabrication and characterization of APDC@NPs
Four types of small molecule APDCs, cRGD-SMCC-DM1 (RCCD), cRPQ-SMCC-DM1 (QCCD), cRGD-SS-DM1 (RSSD) and cRPQ-SS-DM1 (QSSD) were synthesized (Figures S1-S4), and the chemical structures of APDCs were confirmed by 1H nuclear magnetic resonance spectroscopy (1H NMR), Fourier transform infrared spectroscopy (FTIR), Electrospray ionization mass spectrometry (ESI-MS), Micro-Raman imaging spectrometer (Raman) and Ultraviolet-visible spectrophotometer (UV-vis) analysis as shown in the Supporting Information Figures S5-S9. All experiments demonstrated the successful synthesis of four types of designed APDCs.
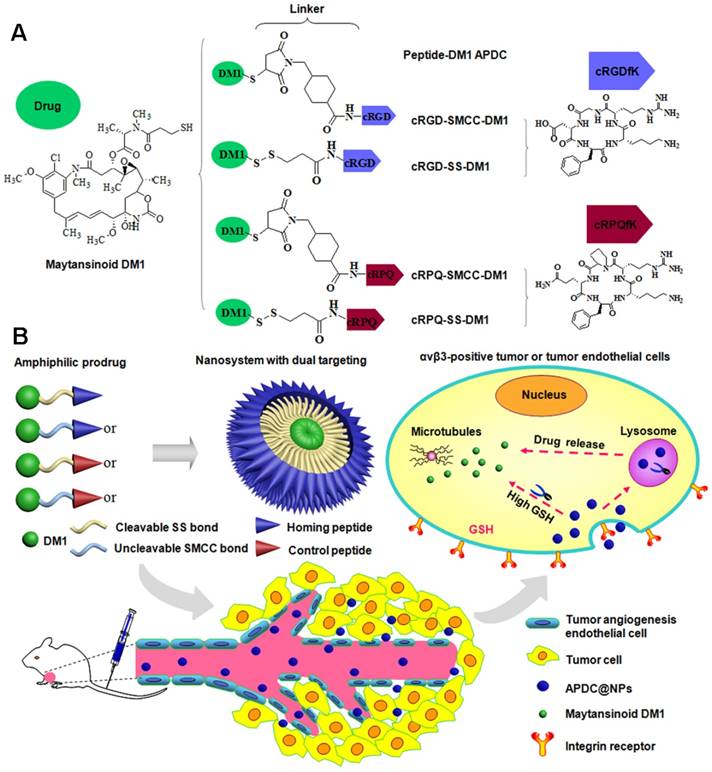
The inherent amphiphilicity of the peptide-DM1 conjugates rendered themselves self-assembling into nanoparticles in water at very gentle condition. The particle size, PDI, zeta potential, and micromorphology of various APDC@NPs were examined by Zetasizer and TEM. As seen in Figure 1A, the obtained solutions of APDC@NPs were stable and almost colorless, and the average hydrodynamic diameter of RCCD, RSSD, QCCD and QSSD nanoparticles were 138.7nm, 82.6 nm, 104.6 nm and 106.8 nm, respectively, indicating the formation of nanoparticles. The size distribution was narrow and in unimodal model. The TEM images in Figures 1B-E showed that the morphology of the APDC@NPs were similar spherical. Consistent with DLS study, the particle size of all APDC@NPs in TEM images were less than 150 nm, which might be favorable for the EPR effect based tumor accumulation [40]. The surface charges of APDC@NPs were all positive (Figure S10), which might facilitate the interaction between APDC@NPs and cell membrane [41]. According to the change of particle size, all the nano-DDS represented excellent serum stability for more than 48 h (Figure S11), which might be fatal for them to keep constant before arriving to tumor sites. In addition, the diameter, PDI and zeta potential of the four APDC@NPs were relatively stable, especially for RSSD@NPs after storage at 4°C for 16 days (Figures S12-S14). The in vitro reduction-triggered drug-release of APDC@NPs were evaluated by dialysis in PBS (pH 7.4) with 0.5% sodium dodecyl sulfate containing (or not) 50 mM reducing agent at 37 °C. A HPLC method was established, which could efficiently detect free DM1 and APDCs (Figure S15). As shown in Figure 1F, the release of DM1 from RSSD@NPs obviously accelerated once the reducing agent DTT was added, from 27.0% to 78.6% within 48 hours. There was about 20% of drug release within the same period of time even without reducer, likely because disulfide bond is unstable [42]. In contrast, the drug release from RCCD@NPs was independent from the reducing agent, as the percentages of cumulative release remained below 4.0% in the condition of reduction or not. Generally, here we demonstrated the reduction-responsive drug release mechanism based on the cleavage of the disulfide bonds for the designed RSSD@NPs but not thioether linker for the prepared RCCD@NPs (Figure 1G).
(A) The diameter distributions, average hydrodynamic particle sizes (DRCCD=138.7 nm, DRSSD=82.2 nm, DQCCD=104.6 nm, DQSSD=106.8 nm), and polydispersity indexes (PDIRCCD=0.142, PDIRSSD=0.205, PDIQCCD=0.206, PDIQSSD=0.247) of the APDC@NPs by DLS analyses. Inset: The digital photographs of APDC@NPs' solution. (B-E) The morphology of RCCD@NPs, RSSD@NPs, QCCD@NPs and QSSD@NPs by TEM images. Scale bars=100nm. (F) Reduction-triggered drug release from the active targeting RCCD@NPs and RSSD@NPs with or without the presence of reducing agent DTT at 37 °C in PBS (pH 7.4, n=3). (G) Reduction-based drug release mechanisms of thioether-linked and disulfide-linked APDC@NPs.
To confirm the specific interaction of APDC@NPs with cells, we utilized the surface plasmon resonance (SPR) to detect the molecular affinity in real-time [43]. Briefly, integrin αvβ3 were immobilized on the sensor surface via covalent binding. With the subsequent flow of phosphate buffer containing various APDC@NPs, the response (RU) signals were recorded and are shown in Figure S16. Compared with the passive QCCD@NPs and QSSD@NPs, the active RCCD@NPs and RSSD@NPs triggered higher responses, indicating stronger binding between active APDC@NPs and integrin αvβ3 highly expressed tumor cells.
Endocytosis patterns, pathway and mechanism of APDC@NPs
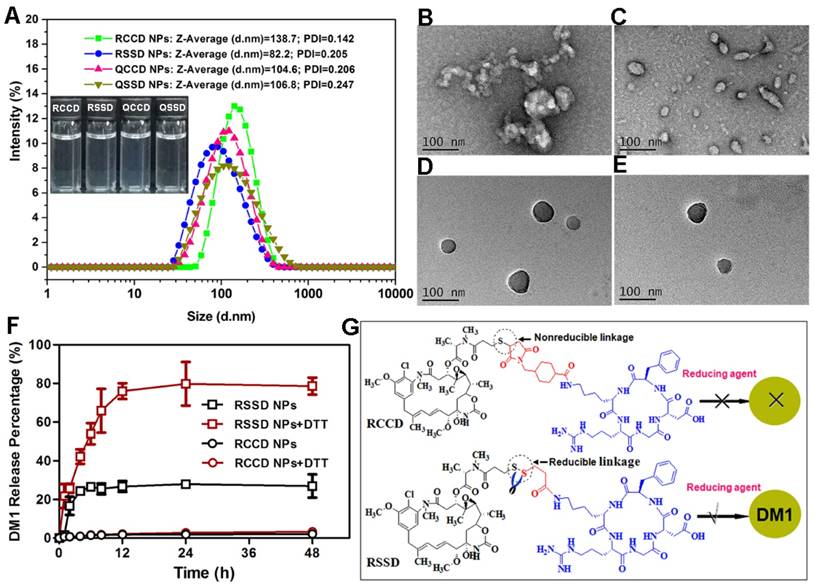
Cellular endocytosis was then conducted in B16 cells, MCF-7 cells and HUVEC quantitatively through a previously established HPLC method which could distinguish released free drug from intact APDC@NPs during the cellular uptake process(Figure S17), and the results are shown in Figures 2A-C and Figures S18-S20. Firstly, it was clear that the cellular uptake of nanoparticles was more in RGD-modified groups than that in RPQ-modified groups in αvβ3-positive cell lines (B16 cells and HUVEC), while there was no such difference in αvβ3-negative cell line (MCF-7 cells). In fact, RCCD@NPs and RSSD@NPs respectively exhibited an 86.6% and 62.4% higher intracellular content than the QCCD@NPs and QSSD@NPs in B16 cells. Secondly, the intracellular level of free DM1 was higher in disulfide containing groups than that in thioether containing groups, independent of the cell types or the expression of αvβ3. The percentage of free DM1 in RSSD@NPs was about two folds of that in RCCD@NPs (62.5% and 35.0%, respectively) inside B16 cells. Finally, it was noticed that the non-specific uptake of nanoparticles by MCF-7 cells was significant, independent of RGD modification. In addition, we analyzed that cellular uptake might be attributed to the high drug loading capacity of RSSD@NPs, in accordance with calculation, drug in a single particle of RCCD@NPs, RSSD@NPs, QCCD@NPs and QSSD@NPs content accounted for 47.3%, 51.7%, 45.7% and 49.8%, respectively. That is to say, when cells took up equal amounts of particles, RSSD@NPs could deliver more drugs into cells than other APDC@NPs in a single particle. In general, the endocytosis level of APDC@NPs varied with peptide ligands and cell lines, and they experienced disparate fates due to different linkers after internalization into cells. In αvβ3-positive cell lines (Figure S19), it was found that cRGDfK contributed greater than disulfide linkage in terms of the intracellular level of free DM1, and the combination of RGD and disulfide led to the highest intracellular level of free DM1.
Intracellular concentrations of DM1 determined by HPLC in (A) B16 cells, (B) MCF-7 cells, and (C) HUVEC after treatments with different APDC@NPs at a drug dose of 100 nM for 3 h (n=3), *p < 0.05, p = ns versus the uptake of passive cRPQfK group. (D) Confocal images of B16 cells treated by various APDC@NPs for 3 h after pre-incubation with different inhibitors at 37 °C. Red represents the Cy3.5-loaded APDC@NPs and blue represents the nucleus stained by Hoechst 33258. Scale bars are 50 μm.
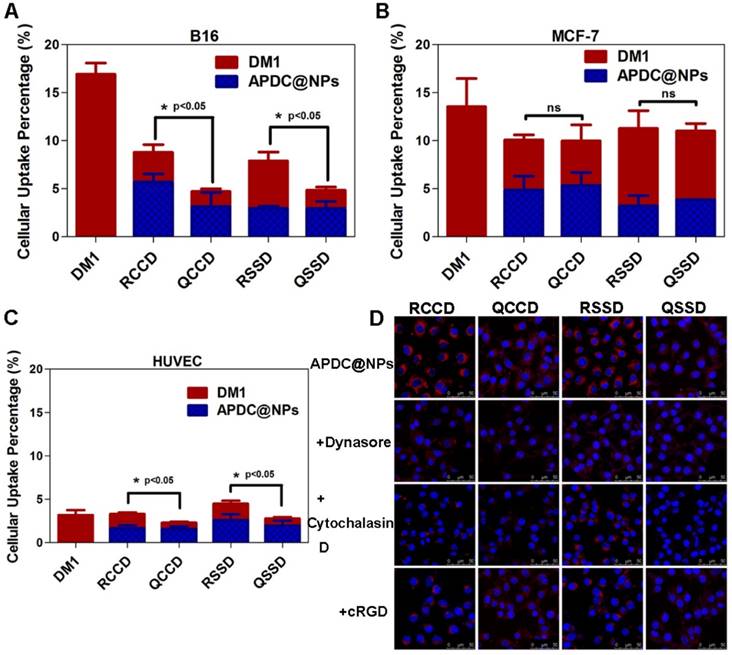
To further investigate the endocytosis pathway of various APDC@NPs, Cy3.5, a near-infrared fluorescence (NIRF) dye, was loaded in APDC@NPs. The characteristics of the Cy3.5-loaded APDC@NPs are given in Figure S21, and DLS observation showed that the size distribution of APDC@NPs was almost unchanged after encapsulation with Cy3.5. Then two of the most classical endocytosis inhibitors, dynasore (the inhibitor of dynamin) [44] and cytochalasin D (the inhibitor of actin depolymerizing) [45], were tested in the endocytosis study, and both of them were found to decrease the intracellular fluorescence of Cy3.5-loaded APDC@NPs (Figure 2D). This revealed a dynamin and actin depolymerizing involved endocytosis pathway. Moreover, the intracellular signals of two active APDC@NPs (RCCD@NPs and RSSD@NPs) also weakened after the pre-treatment with free cRGDfK (Figure 2D), clearly demonstrating a mechanism of receptor-mediated endocytosis.
Impact of APDC@NPs on cell viability and cell cycle
At first, the expression of integrin αvβ3 was confirmed in the three test cell lines by immunofluorescence method (Figure S22). Then, the impact of APDC@NPs on cell viability was investigated by SRB assay using free DM1 as the control. As seen in Figures 3A-C, Figures S23-S25 and Table S1, the IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern. However, there was no significant difference in the IC50 values between passive and active APDC@NPs on MCF-7 cells. Namely, the IC50 of RGD containing APDC@NPs was less than that of RPQ linking APDC@NPs in αvβ3-positive cell lines, and no such difference was found in αvβ3-negative cells. Importantly, from QCCD@NPs to QSSD@NPs, the cytotoxicity in B16 cells increased 0.6 fold, while from QCCD@NPs to RCCD@NPs, the cytotoxicity enhanced 2.8 fold, suggesting the higher impact of RGD modification than sulfide bond. Furthermore, from QCCD@NPs to RSSD@NPs, the in vitro antitumor efficacy in the same cell line ascended 17.2 fold, which clearly demonstrated a synergetic effect between the RGD modification and the reduction-triggered drug release.
Cytotoxicity of free DM1 and various APDC@NPs against (A) B16 cells, (B) MCF-7 cells, and (C) HUVEC cells after 48 h incubation by SRB assay (n=3). The impact of various APDC@NPs with or without pre-treatment of reducing agent GSH-OEt on the cell viability of (D, E) B16 cells, (F, G) MCF-7 cells, and (H, I) HUVEC cells after 48 h incubation by SRB method. Each bar denotes mean ± SD (n=3). (J) The cell cycle of B16 cells, MCF-7 cells, and HUVEC cells after incubation with culture medium, free DM1, RCCD@NPs, RSSD@NPs, QCCD@NPs and QSSD@NPs, respectively, at a dose of 50 nM (calculated by free DM1) for 12 h.
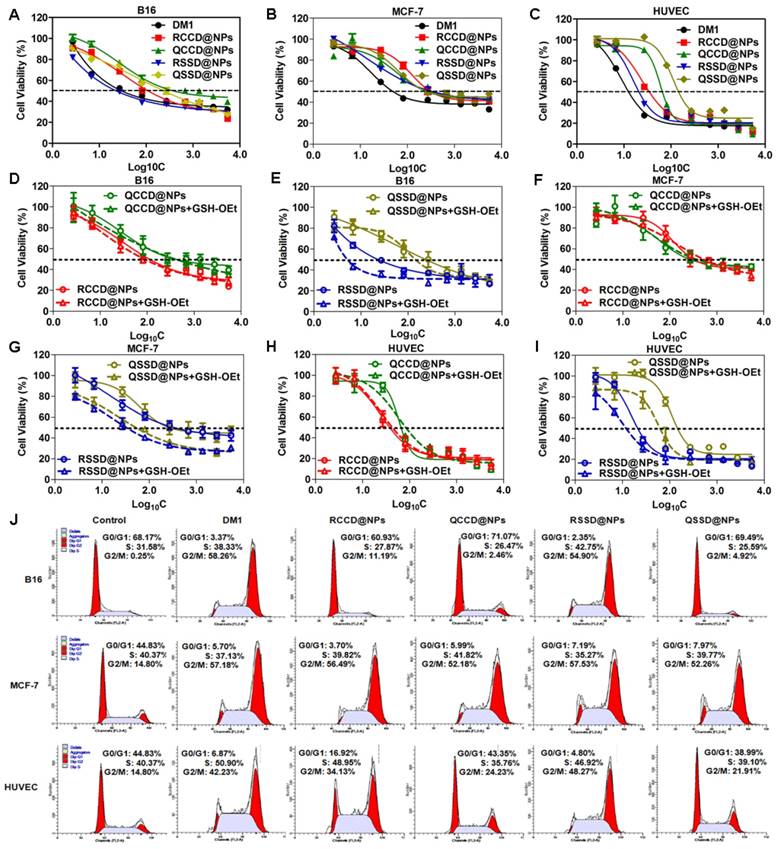
We also studied the environment-sensitivity of various APDC@NPs by comparing their cytotoxicity with or without the reducing agent GSH-OEt which could elevate the intracellular level of GSH and lead to a higher reductive condition. The viability of B16 cells in the groups of RSSD@NPs and QSSD@NPs decreased significantly after the pretreatment of GSH-OEt. In contrast, the cytotoxicity of RCCD@NPs and QCCD@NPs kept unchanged despite the reductive intracellular environment, indicating the inert nature of the thioether linkage. With or without GSH-OEt, the cytotoxicity of APDC@NPs on HUVEC cells and MCF-7 cells was similar with that on B16 cells (Figures 3D-I, Figures S23-S25 and Table S1). In brief, the cytotoxicity of APDC@NPs enhanced significantly in disulfide containing systems than that in thioether containing groups, independent of the cell types and peptide modifications, which was consistent with the level of intracellular free DM1.
To clarify the antitumor mechanism of various APDC@NPs, the cell cycles were analyzed by measuring DNA content via flow cytometry (Figure 3J). Ng et al. reported that increased G2-M phase arrest indicated cell division inhibition and cell growth restraint [46]. Here, when three cell lines were treated with free DM1, RCCD@NPs, RSSD@NPs, QCCD@NPs, and QSSD@NPs for 12 h, the percentage of G2-M phase on B16 cells was 58.26%, 11.19%, 54.90%, 2.46% and 4.92%, the values on MCF-7 cells was 57.18%, 56.49%, 57.53%, 52.18% and 52.26%, and those on HUVEC cells was 42.23%, 34.13%, 48.27%, 24.23% and 21.91%, respectively. It was found in αvβ3-positive cell lines that the percentage of G2-M phase in RSSD@NPs and RCCD@NPs was higher than that in QSSD@NPs and QCCD@NPs, respectively, while the highest value was found in RSSD@NPs. Generally, such tendency was more striking in B16 cells than in HUVEC cells, and in both cell lines, the percentage of G2-M phase in RSSD@NPs was comparable with free DM1. As shown in Figure S26-S28, the percentage of G2-M phase on B16 cells did not increase from QCCD@NPs to QSSD@NPs, but enhanced 3.6 fold from QCCD@NPs to RCCD@NPs, while the value elevated 22.3 fold from QCCD@NPs to RSSD@NPs, which revealed the facts again: bigger contribution of RGD targeting than disulfide cleavable linkage, as well as the synergy between these two functionlizations. On the other hand, there was no significant difference in cell cycles among all nanoparticle systems in αvβ3-negative cells. These results were consistent with the in vitro cytotoxicity above.
In vivo antitumor efficacy of APDC@NPs
A B16 tumor-bearing C57BL/6 mouse model was generated to evaluate the antitumor efficacy of various APDC@NPs in vivo [43]. Tumor volume, weight and size were all recorded accordingly. It was demonstrated here, RCCD@NPs and RSSD@NPs exhibited significantly better tumor-growth inhibition compared with that of the free DM1, QCCD@NPs or QSSD@NPs (Figure 4A). RSSD@NPs showed the most suppression on B16 tumor up to 25 days, and resulted in a tumor volume 4 times smaller than the saline group at the end of the experiment. The final tumor volumes in various nano-DDS groups ranked from the greatest to the least: QCCD@NPs, QSSD@NPs>DM1>RCCD@NPs>RSSD@NPs. Almost the same trends were found in terms of tumor weight and tumor size (Figures 4B, C). As seen in Figures 4A and 4B, the in vivo efficacy of RSSD@NPs is 2 fold of free DM1 which is very powerful and actually too cytotoxic to be a drug. By the comparison between RSSD@NPs and QCCD@NPs in Figures 4A and 4B, it could be seen that the synergism between the two functionlizations reached to about 3 fold in vivo. Again, the effect of targeting peptide was superior to that of cleavable linkage, as there was no significant difference among QSSD@NPs, QCCD@NPs and saline, while the RSSD@NPs and RCCD@NPs presented very significant cancer inhibition (Figure 4B).
(A) Tumor growth curves of B16 tumor-bearing C57BL/6 mice after intravenous administrations of saline, free DM1, and various APDC@NPs (n=6). All formulations were given every other day for a total of five times via tail vein at the dosage of 400 μg/kg (calculated by free DM1). Black arrows indicate the time for injection after tumor cell inoculation. **p < 0.01 versus the saline control, &&p < 0.01 versus the free DM1 group and ##p < 0.01 versus the passive cRPQfK group. (B) The weight of the excised tumor masses from different treatment groups. Each bar represents mean ± SD (n = 6). *p < 0.05 and **p < 0.01 versus the saline control. (C) The tumor photoprints of different treatment groups after tumors were excised at the end of the test (n=6). Scale bar = 2 cm. (D) Confocal images of TUNEL assay for the detection of apoptotic cells in tumor tissue sections from different treatment groups. DNA strand breaks were labeled with FITC (green), and nuclei were stained with Hoechst 33258 (blue). Apoptotic cells exhibited the co-localization of these two labels.
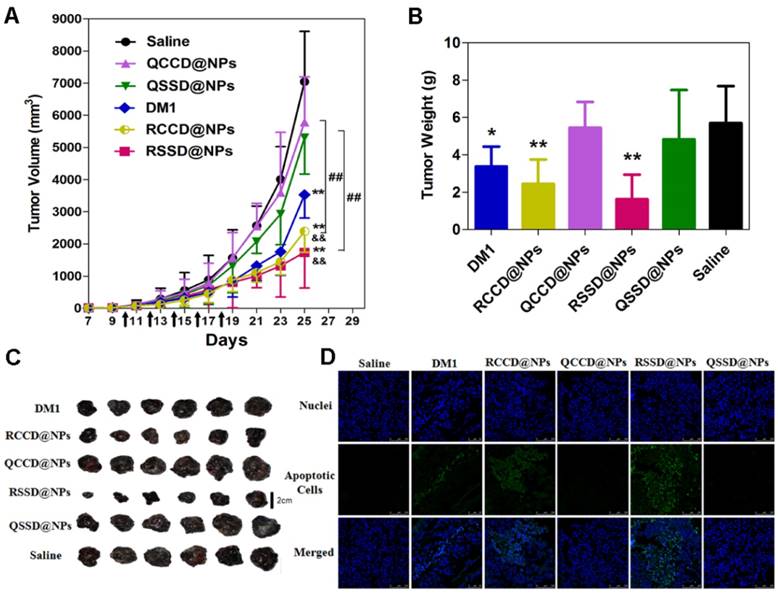
The TUNEL assay was also conducted to count the apoptotic cells in tumor slices. The study provided an order of apoptotic cell numbers opposite to tumor volume (Figure 4D), but concluded the same sequence for the antitumor ability of various nanosystems. From the above-mentioned observations, it was clear that the active nanomedicnes (RCCD@NPs, RSSD@NPs) were more favorable in tumor inhibition than the passive QCCD@NPs and QSSD@NPs). The contribution of RGD-mediated targeting was more than that of sulfide-triggered drug release. The in vivo efficacy of various APDC@NPs against B16 tumor was in accordance with their intracellular free DM1 levels and the in vitro cytotoxicity in B16 cells (Figure 3A and Figure S23). The significant tumor inhibition of RSSD@NPs might be explained by the synergy of enhanced tumor accumulation based on EPR effect of nanosystem, elevated endocytosis via RGD-mediated binding, and reduction-triggered DM1 release from sulfide conjugated prodrug.
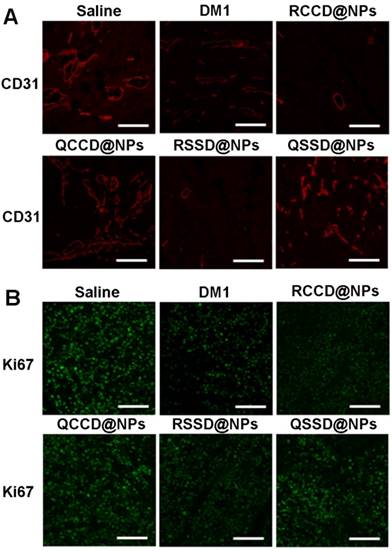
The immunohistochemical analysis was further adopted to assess the antitumor efficacy and mechanism of various APDC@NPs. It was clearly indicated that the active RCCD@NPs and RSSD@NPs exhibited obvious antiangiogenesis effect, as only a few scattered CD31-labelled neovasculature could be observed in tumor sections, while the passive QCCD@NPs and QSSD@NPs showed high fluorescent intensity comparable with the saline control (Figure 5A and Figure S29). Meanwhile, the anti-proliferation effect was also analyzed by under CLSM employing Ki67 as a biomarker [47], and an obvious inhibition on cell proliferation could be detected by two types of active RCCD@NPs and RSSD@NPs, as indicated by much lower density of proliferating cells compared with other groups (Figure 5B and Figure S30).
(A) Fluorescent images of immunostaining with anti-CD31 for detecting tumor angiogenesis in tumor tissue after treatment of B16 tumor-bearing C57BL/6 mice with various APDC@NPs. Red represents the expression of CD31 on a tumor angiogenesis (scale bar = 100 μm). (B) Fluorescent images of proliferating cells in tumor sections from different treatment groups. Tumor sections were immunostained with anti-Ki-67 nuclear antigen for cell proliferation. Green represents the expression of Ki67 on proliferating cells (scale bar = 100 μm).
Safety examination of APDC@NPs
The toxicity of various APDC@NPs was assessed by analyzing their effect on body weight, liver function, leucopenia and myelosuppression, and the histological change of major organs [48]. Firstly, no significant loss of body weight during the animal test was observed in all APDC nanosystems, suggesting their low systemic toxicity, and only the free DM1 caused significant body weight loss compared with the saline (Figure 6A). Second, in the test of liver function with circulating alanine aminotransferase (ALT) and aspartate aminotransferase (AST) as the biomarkers, most of nanoparticles including RSSD@NPs showed limited influence, except that free DM1 and QCCD@NPs exhibited obvious hepatic cytolysis (Figures 6B,C). Leucopenia and myelosuppression are known as two serious safety issues during DM1 chemotherapy. Again, the counts of white blood cells (WBC) were found normal for most APDC@NPs, while free DM1 decreased the count and QCCD@NPs enhanced it significantly compared with the normal value. Additionally, all treatments did not affect the RBC and PLC counts significantly (Figures 6D-F). Last, the H&E staining of major organs, including the heart, liver, spleen, lung, and kidney, demonstrated that the passive QCCD@NPs and QSSD@NPs as well as free DM1 caused severer organic damages as characterized by the presence of hypertrophy or adherent lobes in the liver, minimal steatosis in the spleen, and alveolar collapse and pulmonary bulla in the lung sections, while only mild and negligible changes were observed in active APDC@NPs (Figure 6G).
(A) Body weight changes of B16 tumor-bearing C57BL/6 mice after different treatments during anti-tumor efficacy study (n=6). *p < 0.05 versus the saline control. Black arrows indicate the time for injection after tumor cell inoculation. (B, C) ALT and AST levels in B16 tumor-bearing C57BL/6 mice treated by various APDC@NPs (n=6). *p < 0.05 and **p < 0.01 versus the saline control. (D-F) The counts of blood cells (WBC), red blood cells (WBC) and platelet (PLT) in different treatment groups at the end of test. Each bar represents mean ± SD (n = 6). *p < 0.05 and **p < 0.01 versus the saline control. (G) Histological analysis of major tissues after treatment with various APDC@NPs (200×). Arrows point out the pathologic regions.
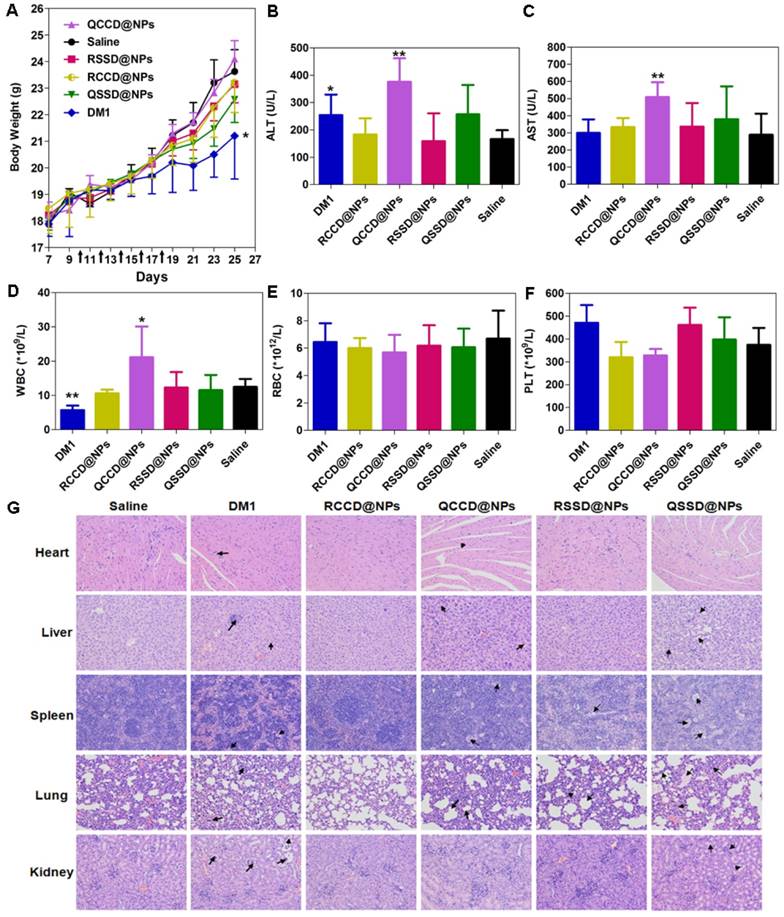
It is well established that free DM1 could not be used directly despite its powerful activity, mainly due to its severe toxicity [49]. The accepted system, tissue and blood compatibility of active APDC nanosystems might be resulted from their enhanced accumulation in tumor and reduced distribution in normal tissues. However, the safety of QCCD@NPs and QSSD@NPs seem need further concerns.
Conclusion
In this work, the successful synthesis of four designed small molecule amphiphilic peptide-drug conjugates (APDCs) through the linkage of a hydrophobic drug and a hydrophilic homing peptide via a microenvironment-responsive bond, and the formation of their nanoparticles were confirmed by various spectroscopies, particle size or morphology. All APDC@NPs exhibited good serum stability and storage stability, while the active ones showed reduction-triggered drug release and higher SPR responses to αvβ3 integrin. Additionally, the active APDC@NPs showed stronger tumor inhibition than the passive ones, in terms of induction of tumor cell apoptosis, anti-angiogenesis, anti-proliferation and tumor suppression.
Based on all findings above, it was concluded that the self-assembling nanosystem based on amphiphilic peptide-linker-drug conjugate did significantly decrease the toxicity of free DM1 and greatly improve its therapy outcome, largely due to the dual targeting strategies and their synergism. In this way, we somehow demonstrated the significance of combining active targeting, prodrug and nanomedicine, providing an insight for the design and development of active targeting nanopreparations in the further.
Acknowledgements
The work was supported by the National Natural Science Foundation of China (81130059, 81503000), the National Basic Research Program of China (2015CB932100), the Innovation Team of the Ministry of Education (BMU20110263), and the China Postdoctoral Science Foundation funded project (2016M591028).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Global Burden of Disease Cancer Collaboration, Fitzmaurice C, Dicker D. et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015;1(4):505-527
2. Ashley CE, Carnes EC, Phillips GK. et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat Mater. 2011;10(5):389-397
3. Choi KY, Yoon HY, Kim JH. et al. Smart nanocarrier based on PEGylated hyaluronic acid for cancer therapy. ACS Nano. 2011;5(11):8591-8599
4. Wang Y, Liu D, Zheng Q. et al. Disulfide bond bridge insertion turns hydrophobic anticancer prodrugs into self-assembled nanomedicines. Nano Lett. 2014;14(10):5577-5583
5. Mikhaylov G, Mikac U, Magaeva AA. et al. Ferri-liposomes as an MRI-visible drug-delivery system for targeting tumours and their microenvironment. Nat Nanotech. 2011;6(9):594-602
6. Saenz del Burgo L, Pedraz JL, Orive G. Advanced nanovehicles for cancer management. Drug Discov Today. 2014;19(10):1659-1670
7. Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771-782
8. Zhang F, Zhang S, Pollack SF. et al. Improving paclitaxel delivery: in vitro and in vivo characterization of PEGylated polyphosphoester-based nanocarriers. J Am Chem Soc. 2015;137(5):2056-2066
9. Spring BQ, Bryan Sears R, Zheng LZ. et al. A photoactivable multi-inhibitor nanoliposome for tumour control and simultaneous inhibition of treatment escape pathways. Nat Nanotechnol. 2016;11(4):378-387
10. Kiick KL. Materials Science. Polymer therapeutics. Science. 2007;317(5842):1182-1183
11. Holme MN, Fedotenko IA, Abegg D. et al. Shear-stress sensitive lenticular vesicles for targeted drug delivery. Nat Nanotechnol. 2012;7(8):536-543
12. Lee CC, MacKay JA, Fréchet JM. et al. Designing dendrimers for biological applications. Nat Biotechnol. 2005;23(12):1517-1526
13. Akcora P, Liu H, Kumar SK. et al. Anisotropic self-assembly of spherical polymer-grafted nanoparticles. Nat Mater. 2009;8(4):354-359
14. Hernández-Cruz O, Avila-Gutierrez L, Zolotukhin MG. et al. Spontaneous, solvent-free, polymer-templated, solid-solid transformation of thin metal films into nanoparticles. Nano Lett. 2016;16(9):5420-5425
15. Han W, Byun M, Li B. et al. A simple route to hierarchically assembled micelles and inorganic nanoparticles. Angew Chem Int Ed Engl. 2012;51(50):12588-12592
16. Horcajada P, Chalati T, Serre C. et al. Porous metal-organic-framework nanoscale carriers as a potential platform for drug delivery and imaging. Nat Mater. 2010;9(2):172-178
17. McNeil SE. Evaluation of nanomedicines: stick to the basics. Nat Rev Mater. 2016;1:16073
18. Huang P, Wang D, Su Y. et al. Combination of small molecule prodrug and nanodrug delivery: amphiphilic drug-drug conjugate for cancer therapy. J Am Chem Soc. 2014;136(33):11748-11756
19. Wang X, Li S, Shi Y. et al. The development of site-specific drug delivery nanocarriers based on receptor mediation. J Control Release. 2014;193:139-153
20. Wang Y, Cheetham AG, Angacian G. et al. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv Drug Deliv Rev. 2017;110-111:112-126
21. van der Meel R, Vehmeijer LJ, Kok RJ. et al. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status. Adv Drug Deliv Rev. 2013;65(10):1284-1298
22. Su H, Koo JM, Cui H. One-component nanomedicine. J Control Release. 2015;219:383-395
23. Luo C, Sun J, Liu D. et al. Self-assembled redox dual-responsive prodrug-nanosystem formed by single thioether-bridged paclitaxel-fatty acid conjugate for cancer chemotherapy. Nano Lett. 2016;16(9):5401-5408
24. Rautio J, Kumpulainen H, Heimbach T. et al. Prodrugs: design and clinical applications. Nat Rev Drug Discov. 2008;7(3):255-270
25. Lee JH, Sahu A, Choi WI. et al. ZOT-derived peptide and chitosan functionalized nanocarrier for oral delivery of protein drug. Biomaterials. 2016;103:160-169
26. Nakagawa O, Ming X, Huang L. et al. Targeted intracellular delivery of antisense oligonucleotides via conjugation with small-molecule ligands. J Am Chem Soc. 2010;132(26):8848-8849
27. Cheetham AG, Zhang P, Lin YA. et al. Supramolecular nanostructures formed by anticancer drug assembly. J Am Chem Soc. 2013;135(8):2907-2910
28. Dissanayake S, Denny WA, Gamage S. et al. Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. J Control Release. 2017;250:62-76
29. Du J, Lane LA, Nie S. Stimuli-responsive nanoparticles for targeting the tumor microenvironment. J Control Release. 2015;219:205-214
30. Remillard S, Rebhun LI, Howie GA. et al. Antimitotic activity of the potent tumor inhibitor maytansine. Science. 1975;189(4207):1002-1005
31. Chari RV, Martell BA, Gross JL. et al. Immunoconjugates containing novel maytansinoids: promising anticancer drugs. Cancer Res. 1992;52(1):127-131
32. Perrino E, Steiner M, Krall N. et al. Curative properties of noninternalizing antibody-drug conjugates based on maytansinoids. Cancer Res. 2014;74(9):2569-2578
33. Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol. 1996;12:697-715
34. Song Q, Wang X, Wang Y. et al. Reduction responsive self-assembled nanoparticles based on disulfide-linked drug-drug conjugate with high drug loading and antitumor efficacy. Mol Pharm. 2016;13(1):190-201
35. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941-951
36. Jiang W, Kim BY, Rutka JT. et al. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3(3):145-150
37. Chuan X, Song Q, Lin J. et al. Novel free-paclitaxel-loaded redox-responsive nanoparticles based on a disulfide-linked poly(ethylene glycol)-drug conjugate for intracellular drug delivery: Synthesis, characterization, and antitumor activity in vitro and in vivo. Mol Pharm. 2014;11(10):3656-3670
38. McQuade P, KnIght LC. Radiopharmaceuticals for targeting the angiogenesis marker alpha (v) beta (3). Q J Nucl Med. 2003;47(3):209-220
39. Chen X, Sievers E, Hou Y. et al. Integrin alpha v beta 3-targeted imaging of lung cancer. Neoplasia. 2005;7(3):271-279
40. Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136-151
41. Liu L, Xu K, Wang H. et al. Self-assembled cationic peptide nanoparticles as an efficient antimicrobial agent. Nat Nanotechnol. 2009;4(7):457-463
42. Krall N, Pretto F, Decurtins W. et al. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl. 2014;53(16):4231-4235
43. Guo Z, He B, Jin H. et al. Targeting efficiency of RGD-modified nanocarriers with different ligand intervals in response to integrin αvβ3 clustering. Biomaterials. 2014;35(23):6106-6117
44. Kleyman TR, Cragoe EJ Jr. Amiloride and its analogs as tools in the study of ion transport. J Membr Biol. 1988;105(1):1-21
45. Goddette DW, Frieden C. Actin polymerization. The mechanism of action of cytochalasin D. J Biol Chem. 1986;261(34):15974-15980
46. Ng SSW, Tsao MS, Chow S. et al. Inhibition of phosphatidylinositide 3-kinase enhances gemcitabine-induced apoptosis in human pancreatic cancer cells. Cancer Res. 2000;60(19):5451-5455
47. Liang L, Lin SW, Dai W. et al. Novel cathepsin B-sensitive paclitaxel conjugate: Higher water solubility, better efficacy and lower toxicity. J Control Release. 2012;160(3):618-629
48. Li S, Wang K, Shi Y. et al. Novel biological functions of ZIF-NP as a delivery vehicle: High pulmonary accumulation, favorable biocompatibility, and improved therapeutic outcome. Adv Funct Mater. 2016;26(16):2715-2727
49. Lewis Phillips GD, Li G, Dugger DL. et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280-9290
Author contact
![]() Corresponding author: Qiang Zhang, Telephone: +86 10 82802791, Fax: +86 10 82802791, E-mail address: zqdodoedu.cn
Corresponding author: Qiang Zhang, Telephone: +86 10 82802791, Fax: +86 10 82802791, E-mail address: zqdodoedu.cn