Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Material and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(13):3293-3305. doi:10.7150/thno.19988 This issue Cite
Research Paper
EGLN1/c-Myc Induced Lymphoid-Specific Helicase Inhibits Ferroptosis through Lipid Metabolic Gene Expression Changes
Yiqun Jiang1, 2, 3, Chao Mao1, 2, Rui Yang1, 2, Bin Yan1, 2, 4, Ying Shi1, 2, Xiaoli Liu1, 2, Weiwei Lai1, 2, Yating Liu1, 2, Xiang Wang3, Desheng Xiao5, Hu Zhou6, Yan Cheng7, Fenglei Yu3, Ya Cao1, 2, Shuang Liu4, Qin Yan8, Yongguang Tao1, 2, 3, 4 ![]()
1. Key Laboratory of Carcinogenesis and Cancer Invasion, Ministry of Education, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, Hunan, 410008 China;
2. Cancer Research Institute, Central South University, 110 Xiangya Road, Changsha, Hunan, 410078 China;
3. Department of Thoracic Surgery, Second Xiangya Hospital, Central South University, Changsha, China;
4. Institute of Medical Sciences, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, Hunan, 410008 China;
5. Department of Pathology, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, Hunan, 410008 China;
6. Shanghai Institute of Material Medica, Chinese Academy of Sciences (CAS), 555 Zu Chongzhi Road, Zhangjiang Hi-Tech Park, Shanghai, 201203, China;
7. Department of Pharmacology, School of Pharmaceutical Sciences, Central South University, Changsha, Hunan 410078 China;
8. Department of Pathology, Yale School of Medicine, New Haven, CT, 06520 U.S.A.
Received 2017-3-8; Accepted 2017-6-15; Published 2017-7-23
Abstract

Ferroptosis is a newly discovered form of non-apoptotic cell death in multiple human diseases. However, the epigenetic mechanisms underlying ferroptosis remain poorly defined. First, we demonstrated that lymphoid-specific helicase (LSH), which is a DNA methylation modifier, interacted with WDR76 to inhibit ferroptosis by activating lipid metabolism-associated genes, including GLUT1, and ferroptosis related genes SCD1 and FADS2, in turn, involved in the Warburg effect. WDR76 targeted these genes expression in dependent manner of LSH and chromatin modification in DNA methylation and histone modification. These effects were dependent on iron and lipid reactive oxygen species. We further demonstrated that EGLN1 and c-Myc directly activated the expression of LSH by inhibiting HIF-1α. Finally, we demonstrated that LSH functioned as an oncogene in lung cancer in vitro and in vivo. Therefore, our study elucidates the molecular basis of the c-Myc/EGLN1-mediated induction of LSH expression that inhibits ferroptosis, which can be exploited for the development of therapeutic strategies targeting ferroptosis for the treatment of cancer.
Keywords: Ferroptosis, LSH, WDR76, EGLN1, PHD2, c-Myc, HIF-1α, SCD1, FADS2, Iron, Lipid reactive oxygen species, Metabolism, Lung cancer.
Introduction
Reprogramming of cellular metabolism, including the regulation of apoptotic and necrotic cell death, is necessary for tumorigenesis [1-3]. Ferroptosis, which is a newly discovered mode of non-apoptotic cell death, involves a metabolic dysfunction that results in the intracellular metabolic process glutaminolysis and the production of iron-dependent reactive oxygen species (ROS), the iron carrier protein transferrin, and other related regulators, such as glutathione peroxidase 4 (GPX4) and p53 [4-7]. Iron and iron derivatives are essential for the functioning of ROS-producing enzymes, suggesting that iron might function as a trigger or mediator of cell death signaling, particularly in ferroptosis [4, 8], further indicating that iron-containing enzymes are essential for ferroptosis. The iron-, oxygen-, and 2-oxoglutarate-dependent dioxygenase family includes three members of prolyl hydroxylase domain (PHD) enzymes (PHD 1, 2 and-3), which are also known as EGLN-2, -1 and -3 respectively. EGLN2 is essential for cell death as a candidate driver of iron chelation-mediated inhibition of cell death [9]. Investigations are required to determine whether and how the iron-dependent hydroxylase activity of EGLNs contributes to ferroptosis.
The EGLNs catalyze hypoxia inducible transcriptional factor (HIF) prolyl hydroxylation, leading to the degradation of the oxygen sensors HIF‐1α and HIF‐2α that are necessary for the function of the HIFs, whereas HIF-1 regulates the oxygen-dependent metabolism of glucose and glutamine [10, 11]. HIF-1 regulates oxygen-dependent glucose, glutamine metabolism and EGLN activity [12-14]. Interestingly, lactate, a common product of anaerobic metabolism, promotes a HIF-dependent hypoxic response, which subsequently triggers signals for cell growth and angiogenesis [15]. Mitochondrial ROS inhibit ELGNs to induce HIF [12]. The relationship between ferroptosis-associated lipid ROS production and EGLN activity remains unclear. The function of HIF in tumors is well characterized, but our understanding of the function of EGLNs in tumors is limited.
Chromatin-modifying enzymes utilize key components of core metabolic pathways as co-factors and substrates for signal integration and nuclear adaption to environmental changes [16-18]; however, the mechanisms by which the signal transduction pathways directly communicate with chromatin to change the epigenetic landscape are poorly understood. Lymphoid-specific helicase (LSH), which is a protein that belongs to the SNF2 family of chromatin-remodeling ATPases, is critical for the normal development of plants and mammals because it establishes correct DNA methylation levels and patterns [19-22]. LSH maintains genome stability in mammalian somatic cells [23, 24]; however, the role of LSH in ferroptosis remains unknown.
Lung cancer is a leading cause of death worldwide and is the first leading cause of death in China, resulting in more than 2.8 million deaths in 2015, more than 40% of which involved lung adenocarcinomas [25]. Furthermore, lung cancer can be divided into small cell lung cancer and non-small cell lung cancer (NSCLC), including adenocarcinoma (ADC) and squamous cell carcinoma (SCC), which account for 80% to 85% of all lung cancer cases.[26]. Epigenetic modifications, including chromatin modifications, play important roles in the development and progression of NSCLC [26, 27]. Although cancer epigenetic discoveries over the past decade have revealed numerous epigenetic modifiers that are involved in the progression of various cancers [28], the role of chromatin remodeling in lung cancer remains unclear.
In this study, we investigated the potential impact of LSH on ferroptosis and lung cancer. We found that LSH is an oncogene and regulates ferroptosis in lung cancer. Mechanistically, LSH epigenetically increases the expression level of metabolic genes that decrease ferroptosis. Moreover, EGLN1 is essential for the induction of the expression of LSH by counteracting the recruitment of HIF-1α rather than that of c-Myc.
Material and Methods
Cell culture, antibodies, chemicals, plasmids and siRNAs
The normal lung cell lines MRC-5 (ATCC: CCL-171™) and HBE (ATCC: CRL-2741™) were purchased from ATCC. The lung cancer cell lines A549 (ATCC: CCL-185™), H358 (ATCC: CRL-5807™), and H522 (ATCC: CRL-5810™) were obtained from ATCC. The lung cancer cell lines PC9, 95C and 95D were obtained from the Cancer Research Institute of Central South University. The A549 cells were maintained in DMEM/F12 1:1(Hyclone); 293T cells were maintained in DMEM (Gibco); other cells were maintained in RPMI 1640 (Gibco). All media were supplemented with 10% (V/V) FBS. All cell lines were maintained at 37°C with 5% CO2. The cell lines were negative for mycoplasma contamination. All cell lines were passaged less than 10 times after their initial revival from frozen stocks. All cell lines were authenticated by short tandem repeat profiling prior to use.
The primary antibodies for LSH and WDR76 were purchased from Santa Cruz, the primary antibodies for EGLN1, EGLN3 and HIF1α were obtained from Novus (Littleton, CO), the primary antibodies for SCD1 and FADS2 were purchased from Origen (Rockville, MD); the primary antibody for β-acting was purchased from Sigma-Aldrich (St. Louis, MO). The chemicals (erastin, ferrostatin-1, DMOG and BAY 85-3934) were purchased from Selleck (Houston, TX).
The LSH lentiviral construct was generated by inserting the LSH cDNA into plvx-EF1a-puro vector (Clontech, Mountain View, CA). FLAG-EglN1-pLenti6 was a gift from William Kaelin (Addgene plasmid # 36949). The Lentiviral shRNA clones targeting human LSH, FADS2, SCD1, and EGLN1 and the non-targeting control construct were purchased from Genechem (www.genechem.com.cn) (Shanghai, China).
Western blot analysis and Co- Immunoprecipitation (Co-IP) assay
Details regarding the Western blot analysis and Co- Immunoprecipitation (Co-IP) assay have been previously described [29], and the detailed procedure is listed into the supplementary Materials and Methods.
Cell proliferation assays, migration and invasion assays and plate-colony formation assays, quantitative real-time PCR and RNA sequencing
The details of these procedures have been previously described [29, 30], and the detailed procedure of the RNA sequencing is listed in the supplementary Materials and Methods. The primer sequences used are listed in the supplementary Table S1. The mean ± SD of three independent experiments is shown.
Immunohistochemical (IHC) analysis of lung cancer biopsies, and immunofluorescence assay
The IHC analyses were generally performed as previously described [29, 30]. The lung cancer biopsies, which were validated by a pathologist (Dr. Desheng Xiao of Xiangya Hospital), were obtained from the Department of Pathology at Xiangya Hospital. The lung cancer tissue array was purchased from Pantomics (Richmond, CA, USA).
Measurement of total ROS, Lipid ROS, and intracellular iron
The details of the procedures have been previously described [4, 8]. The detailed procedure is listed in the supplementary Materials and Methods.
Chromatin immunoprecipitation (ChIP) assay and hydroxymethylated DNA Immunoprecipitation (hMeDIP) assay
The ChIP and hMeDIP assays were performed as previously described [29, 30]. The ChIP DNA was analyzed by qPCR using SYBR Green (Bio-Rad) on an ABI-7500 (Applied Biosystems) using the primers listed in supplementary Table S2. The primers were used to amplify in the promoter regions of selected genes. The antibodies were used as indicated.
Glucose uptake and lactate production measurement
Cells (5×105) were seeded into 6-well plates and after incubated for 4 h. The medium was discarded, and the cells were incubated in fresh medium for 8 h. The glucose and lactate levels were measured (Automatic Biochemical Analyzer, 7170A; HITACHI) at the Clinical Biochemical Laboratory of Xiangya Hospital (Changsha, China).
Nude mice and study approval
The xenograft tumor formation was generally performed as previously described [30]. The SCID Mice were purchased from the Hunan SJA Laboratory Animal Co., Ltd. (Changsha, China). The procedural details are listed in the supplementary Materials and Methods.
Statistical analyses
All experiments, except for those that involved the nude mice, were repeated at least three times. The results are shown as mean ± SD or SEM. All statistical analyses were performed using the Prism 6.0 GraphPad software. A p value of less than 0.05 was considered to be statistically significant.
Results
LSH activates metabolic gene expression at the transcriptional level
We first assessed the expression of LSH in a panel of lung cells using a Western blot analysis (Figure 1A) and found an increased expression of LSH in the lung cancer cell lines compared to that in the normal lung cells in which LSH was expressed at a low level; then, we selected three lung cancer cell lines, i.e., A549, H358 and PC9, for the subsequent studies. To gain insights into the LSH function, we used RNA-sequencing to analyze gene expression changes in the cells by comparing the stable expression of LSH to PC9 lung cells transfected with a control vector (Supplementary Figure S1A). We obtained an average of 8.3 million unique mapped reads and 16995 unique transcripts per condition from two independent biological replicates (fragments per kilobase of exon per million reads (FPKM) >1 in both replicates). After processing the data and averaging the replicates, compared to the control cells, we identified 855 mRNAs that were increased by two-fold (upregulated) and 858 mRNAs that were decreased by two-fold (down-regulated) in the LSH expression group (Figure 1B). A volcano plot further showed 305 upregulated and 423 down-regulated genes (Supplementary Figure S1B). Moreover, the gene ontology analysis identified a significant enrichment in pathways related to metabolic processes (Figure 1C).
LSH affects metabolic gene expressions at the transcriptional level. (A) A Western blot analysis was used to detect the expression of LSH in a panel of lung cancer cells. (B) LSH induced the down-regulation of 858 genes and the upregulation of 855 genes in the H358 cells. (C) The genes that were positively correlated with LSH in the H358 cells were significantly involved in the regulation of cellular processes and metabolic processes according to the gene ontology analysis using DAVID. (D-G) RT-qPCR analysis of GLUT family associated genes (D, E) and FADS family associated genes (F, G) in LSH-overexpressing H358 cells (D, F) and LSH depletion in A549 cells (E, G). (H-K) Glucose (H, J) and lactate (I, K) levels were determined in the culture medium in H358 and PC9 with LSH overexpression (H, I), and in A549 cells with LSH depletion (J, K). * P<0.05, ** P<0.01, *** P<0.001.
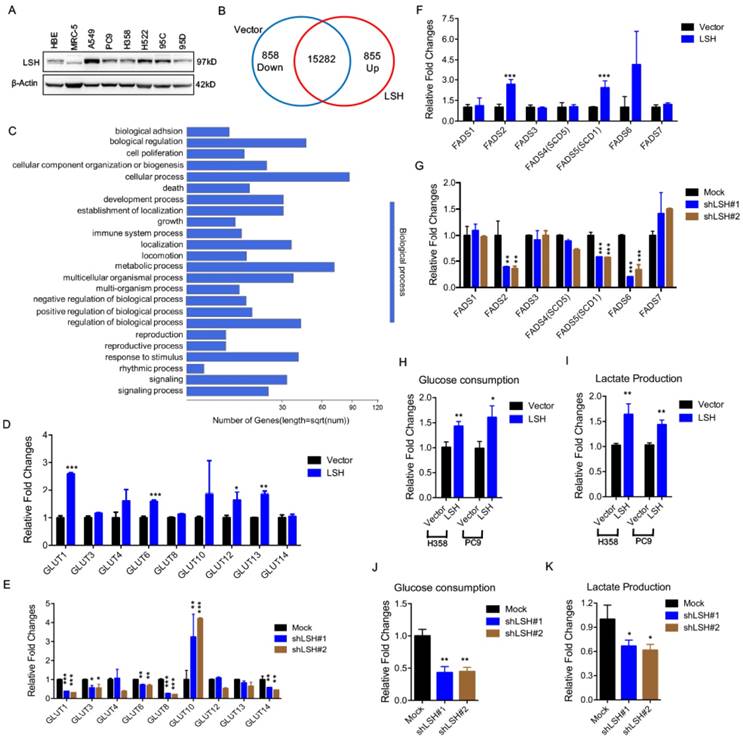
Because LSH is potentially linked to metabolic genes, we selected two gene subtypes and assessed their expression. The first group comprised glucose transporters (GLUTs), which were important in glucose transport, and the other group comprised fatty acid desaturase (FADSs), which were dependent on NAPDH [31]. To substantiate our findings, we stably overexpressed LSH in the H358 lung cancer cell lines (H358-LSH) (Supplementary Figure S1C). Consistent with the data from PC9 cells, we found that overexpression of LSH significantly increased GLUT1, GLUT6, GLUT12 and GLUT13 expression (Figure 1D). To validate the role of LSH in the metabolic gene expressions, we stably knocked down LSH in A549 cancer cells. The knockdown approach successfully reduced the LSH protein to less than 10% (Supplementary Figure S1D), and the knockdown of LSH decreased the GLUT1, GLUT3, GLUT6, GLUT8 and GLUT14 expression levels (Figure 1E). Finally, we found that the LSH overexpression significantly increased the expression levels of FADS2 (fatty acid desaturase 2) and FADS5 (also known as sterol-CoA desaturase 1, SCD1). Conversely, we found that the depletion of LSH decreased the expression of FADS2, SCD1 and FADS6 (Figure 1F-G). Furthermore, we confirmed that LSH promoted glucose consumption and lactate production that was associated with the Warburg effect (Figure 1H-I), whereas depletion of LSH decreased glucose consumption and lactate production (Figure 1J-K). These results are consistent with a function role for LSH in these processes and the observed phenotype. Taken together, LSH affects the metabolic gene expression as well as the Warburg effect.
WDR76 increases metabolic gene expression through epigenetic regulation in dependent of LSH
WDR76 is a nuclear WD40 protein, whose function is poorly characterized in mammals. However, WDR40 always functions an active marker for histone modification (i.e. H3K4Me3). Interestingly, our immunofluorescence analysis revealed that WDR76 co-localized with LSH in A549 and H522 cells in the nuclei (Figure 2A). These observations suggest that WDR76 may directly interact with LSH in human cells. Then, we confirmed the interaction of endogenous LSH and WDR76 by Co-IP assay with anti-LSH antibodies in A549 and H522 cells (Figure 2B). We further confirmed the interaction of endogenous LSH and WDR76 by co-immunoprecipitation assay with anti-LSH and anti-WDR76 antibodies in A549 and H522 cells (Figure 2C).
LSH recruits WDR76 to directly regulate metabolic genes. (A) A549 and H358 cells were assessed to determine the co-localization of LSH and WDR76. (B, C) Co-immunoprecipitation of LSH and WDR76 in A549 cell lysates with anti-LSH antibody (B), and with anti-WDR76 antibody (C) in A549 and H522 cells. (D) ChIP analysis with anti-WDR76 indicated that WDR76 recruitment to the promoters of GLUT1, SCD1 and FADS2 antibodies. (E) ChIP analysis with anti-Flag M2 agarose revealed LSH recruitment to the promoters of GLUT1, SCD1 and FADS2. (F) Re-ChIP assays of Flag-LSH and WDR76 showed WDR76 binding to the promoters of GLUT1, SCD1 and FADS2. (G) ChIP analysis with anti-WDR76 indicated that WDR76 enrichment decreased in the indicated promoters after depletion of LSH. * P<0.05, ** P<0.01, *** P<0.001.
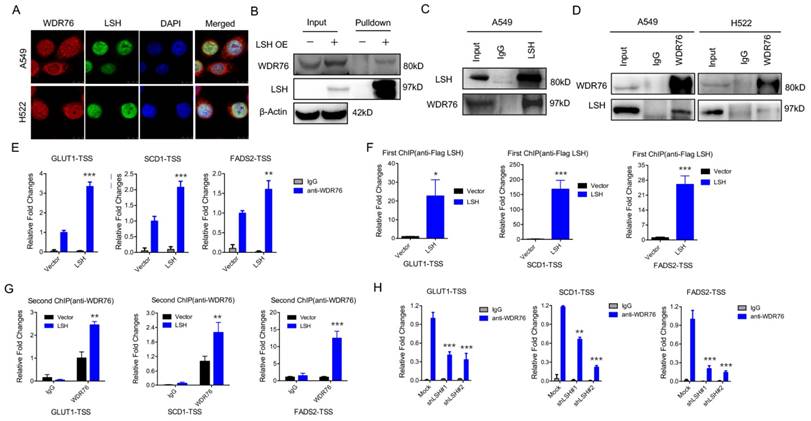
Because LSH is localized in the nucleus and acts as a chromatin modifier dependently and independently of DNA methylation [32, 33], we performed chromatin immunoprecipitation (ChIP) assays to determine whether LSH could directly bind to the promoters of GLUT1, SCD1 and FADS2 (Supplementary Figure S2A). We found that WDR76 was directly associated with the indicated transcription start sites (TSSs) whereas overexpression of LSH promoted the enrichment of WDR76 to the indicated promoters (Figure 2D). Similarly, LSH was also directly enriched to the indicated TSSs (Figure 2E). Furthermore, using Re-ChIP assay, we further found that WDR76 was recruited to the promoters of GLUT1, SCD1 and FADS2 and that LSH promoted the enrichment of WDR76 at the TSSs of GLUT1, SCD1 and FADS2 (Figure 2F). Finally, the enrichment of WDR76 to the indicated genes decreased after depletion of LSH in A549 cells (Figure 2G), indicating that WDR76 binds to metabolic gene promoter in dependent of LSH.
Our histone modification analysis in the presence of LSH showed that LSH slightly changed global levels on different histone modifications, including H3K4Me3, H3K27Me3 and H3K9Me3 (Supplementary Figure S2B-D). Moreover, the active chromatin marker, H3K4Me3, was significantly higher at the GLUT1, SCD1 and FADS2 promoters in the presence of LSH (Supplementary Figure S3A), whereas the repressive chromatin marker, H3K27Me3, decreased at the GLUT1, SCD1 and FADS2 promoters (Supplementary Figure S3B) upon LSH1 overexpression. LSH is a reader of 5-(hydroxyl) methylcytosine (5-hmC) in mice [34]. Furthermore, hMeDIP showed that LSH increased the 5-hmC levels at the GLUT1, SCD1 and FADS2 promoters in H358 cells (Supplementary Figure S3C) and PC9 cells (Supplementary Figure S3D). Finally, a Kaplan-Meier plotter that was performed on a cohort of lung cancers showed that lower WDR76 expression was associated with overall survival in all lung cancers (Supplementary Figure S3E). Similar findings were observed for GLUT1, SCD1 and FADS2, where lower expression was associated with overall survival in all lung cancers (Supplementary Figure S3E).
LSH inhibits ferroptosis by decreasing the intracellular levels of iron and lipid ROS
The LSH effects on the metabolic genes led us to characterize the role of LSH in the novel ferroptotic mode of non-apoptotic cell death, which is associated with metabolic dysfunction, by assessing erastin-induced growth inhibition, in the presence and absence of ferrostatin, an inhibitor of erastin. First, we demonstrated that erastin-induced cell death was dose-dependent (Supplementary Figure S4A), and independent of apoptosis (Supplementary Figure S5B); then we observed that LSH overexpression decreased the erastin-induced growth inhibition of H358 cells (Figure 3A) and PC9 cells (Figure 3B). The depletion of LSH increased the erastin-induced growth inhibition in the A549 cells (Figure 3C), suggesting that LSH might inhibit erastin-induced cancer cell death. The intracellular concentrations of iron and lipid ROS are both surrogate markers for ferroptosis [4, 8]. We found that LSH decreased the intracellular concentrations of iron in H358 and PC9 cells (Figure 3D-E), but LSH depletion increased the intracellular concentrations of iron in the A549 cells (Figure 3F-G).
LSH inhibits ferroptosis. (A,B) Response of H358 (A) and PC9 (B) cells with or without LSH overexpression to erastin (5μM) ± Ferrostatin(1μM). (C) Response of A549 cells to erastin (5μM) ± Ferrostatin(1μM) after LSH depletion. Viability was assessed by Alamar blue and represents the mean ± SD from three independent biological replicate experiments. (D-G) The total iron levels (D, F) and ferrous iron level (E, G) were analyzed in H358 cells and PC9 cells with or without LSH overexpression (D, E) and A549 cells with or without LSH depletion (F, G). (H-J) Lipid ROS was measured by C11-BODIPY staining coupled to flow cytometry in erastin-treated H358 (H) and PC9 (I) cells, and A549 cells after the LSH depletion. * P<0.05, ** P<0.01, *** P<0.001.
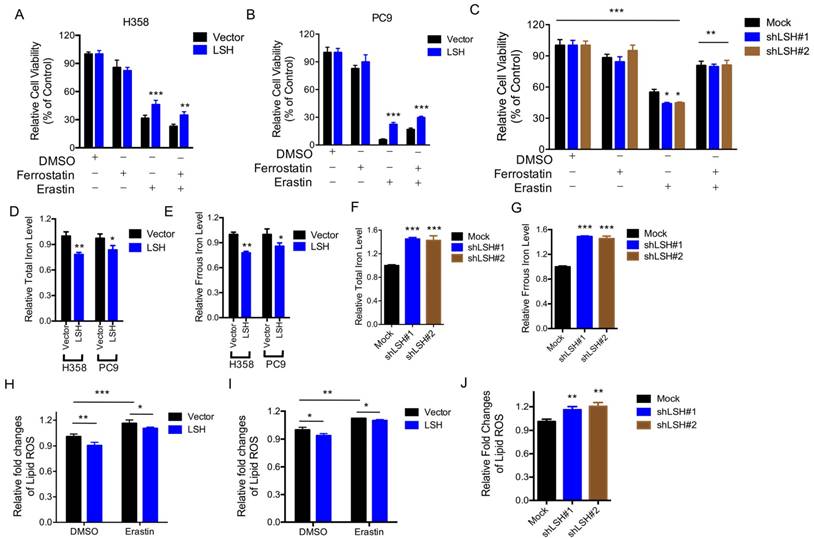
Furthermore, we found that lipid ROS levels increased after the erastin treatment, and LSH decreased the erastin-induced lipid ROS in the H358 cells (Figure 3H) and PC9 cells (Figure 3I). The depletion of LSH increased lipid ROS levels in the A549 cells (Figure 3J). Moreover, we found that total ROS were decreased in the presence of LSH in H358 cells (Supplementary Figure S5A-B) and PC9 cells (Supplementary Figure S5C-D). Thus, these findings indicate that the LSH-mediated inhibition of erastin-induced ferroptosis in cancer cells is dependent on iron and lipid ROS.
Retinoic acid promotes ferroptosis through the attenuation of LSH expression
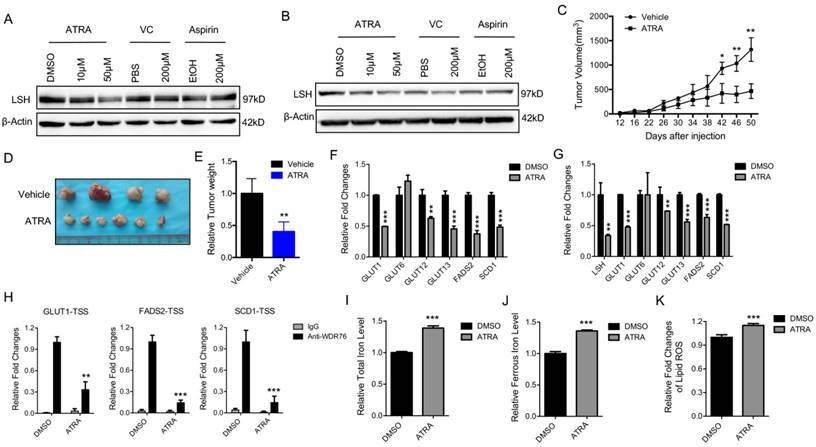
Retinoic acid (ATRA), a metabolite of Vitamin A, has a critical function in initiating the lineage differentiation of embryonic stem cells (ESCs), where LSH plays a critical role in the maintenance of the stem properties [35, 36]. After treating the A549 and H522 cells with various chemicals for 72 h, we found that ATRA, but not vitamin C or aspirin, decreased the expression of LSH (Figure 4A-B). Both vitamin E and erastin did not change LSH expression (Supplementary Figure S6A-B). Furthermore, we found that ATRA reduced A549 tumor volumes (Figure 4C), tumor formation (Figure 4D) and tumor weights (Figure 4E) at the indicated times in the A549 cells. To address the potential roles of ATRA in the xenograft tumors, we assessed the expression levels of metabolism-associated genes, including SCD1 and FADS2. We found that most of these genes decreased after ATRA treatment (Figure 4F). Similar observations were made in the cultured A549 cells after ATRA treatment (Figure 4G). The enrichment of WDR76 to the indicated genes decreased with the treatment of ATRA in the A549 cells whereas LSH expression decreased (Figure 4H). A549 cells were forced expression of LSH after the treatment of ATRA, then cell viability decreased in response to erastin, indicating that the regulation of LSH is related to the cellular effects of ATRA (Supplementary Figure S6C). Moreover, we found that the iron level and lipid ROS levels increased in the A549 cells after ATRA treatment (Figure 4I-K). These results are consistent with the model that ATRA suppresses tumor growth by decreasing LSH expression and promoting ferroptosis.
Retinoic acid promotes ferroptosis by attenuating LSH expression. (A, B) western blot analysis of LSH in A549 (A) and H522 (B) cells after the treatment of indicated chemicals. (C-E) Volume (C), representative images (D) and weight (E) of A549 tumors in nude mice after treatment with ATRA. (F, G) RT-qPCR analysis of ferroptosis-associated genes in ATRA-treated A549 cells in culture (F) or in ATRA-treated xenograft tumor samples (G). (H) ChIP analysis of WDR76 at the promoters of GLUT1, SCD1 and FADS2 in ATRA-treated A549 cells. (I-K) The total iron (I) and ferrous iron (J) and lips ROS (K) levels in ATRA-treated A549 cells. * P<0.05, ** P<0.01, *** P<0.001.
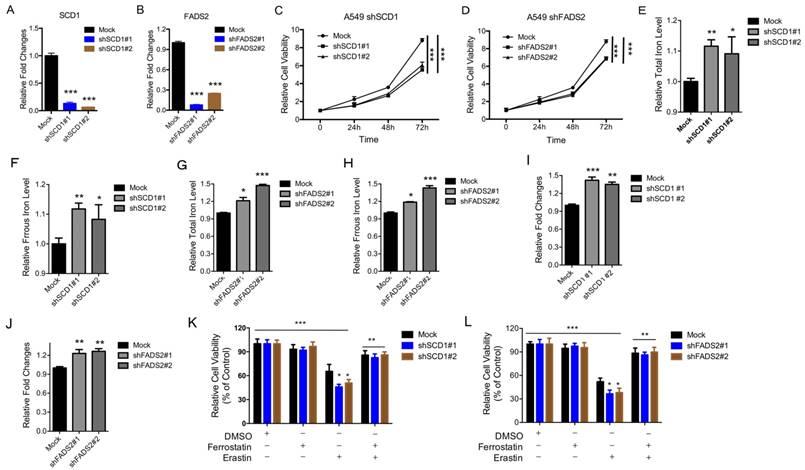
Depletion of the SCD1 and FADS2 metabolic genes induces ferroptosis
To determine the physiological roles of the LSH target genes (i.e., SCD1 and FADS2) in lung carcinogenesis and erastin-induced cell death, we stably knocked down SCD1 and FADS2 in A549 cancer cells. The knockdown approach successfully reduced the SCD1 and FADS2 mRNA levels to less than 15% and 20% respectively (Figure 5A-B). The ratio of cell growth was significantly reduced after the SCD1 (Figure 5C) and FADS2 (Figure 5D) depletion. Furthermore, the iron levels and lipid ROS increased when SCD1 and FADS2, respectively, were depleted (Figure 5E-J). We also found that erastin-induced cell death increased after the knockdown of SCD1 and FADS2 (Figure 5K-L). Moreover, knockdown of SCD1 and FADS2 decreased the ferroptosis-associated regulators, such as SOD2, SLC2A4, SLC2A14, SLC7A11, SLC1A5, SLC3A2, CARS, EMC2, RPL8 and GLUD1, at the mRNA level (Supplementary Figure S7A-B). In summary, these findings suggest that the LSH target genes SCD1 and FADS2 contribute to ferroptosis.
Inhibition of the SCD1 and FADS2 metabolic genes promotes ferroptosis. (A, B) RT-qPCR analysis was conducted to detect SCD1 (A) and FADS2 (B) in A549 cells that were stably transfected with two distinct target gene shRNA expression vectors and in control cells (siCTRL). (C, D) The MTT assay was performed to assess A549 cell viability after stable depletions of SCD1 (C) or FADS2 (D). (E-H) The Total iron and ferrous iron levels were analyzed in A549 cells in the depletion of SCD1 (E, F) and FADS2 (G, H). (I-L) Lipid ROS was measured by C11-BODIPY staining coupled to flow cytometry in A549 cells after the depletion of SCD1 (I) and FADS2 (J). A549 cells responses to erastin (5μM) ± Ferrostatin(1μM) after depletion of SCD1 (K) and FADS2 (L). * P<0.05, ** P<0.01, *** P<0.001.
EGLN1 induces LSH expression by repressing HIF1α
The iron- and oxygen- dependent, 2-oxoglutarate-dependent dioxygenase family includes EGLN-1, -2 and -3[37]. To identify the potential link between LSH and EGLN1, we analyzed the mRNA levels of EGLNs and LSH in an independent panel of 47 primary lung tumors. Compared to the adjacent normal tissues, we found that the LSH, EGLN1 and EGLN3 mRNA levels were highly expressed in the lung cancer tumors (Figure 6A-C), while the mRNA levels of EGLN2 were unchanged (Data not shown). Furthermore, LSH expression was higher in both lung ADC (Supplementary Figure S8A) and SCC (Supplementary Figure S8B). The correlation between LSH and EGLN1 or EGLN3 was further analyzed, and we found that there was a strong correlation between LSH and the EGLNs (Figure 6D-E). Moreover, we detected the LSH, EGLN1 and EGLN3 protein levels in four lung cancer cases (case 1: lung ADC, case 2: lung adenosquamous cancer, and case 3 and 4 : lung SCC); notably, EGLN1, EGLN3 and LSH increased in all lung cancers (Figure 6F), further indicating that a strong correlation between LSH and EGLN1 exists. Furthermore, we found that the protein expression of EGLN1 was strongly associated with the expression of LSH in a panel of lung cell lines (Figure 6G).
The EGLN1/c-Myc axis activates LSH expression. (A-E) RT-qPCR analysis of LSH (A), EGLN1 (B) and EGLN3 (C) expression in 47 lung cancer samples paired with corresponding normal lung tissues. The correlations between LSH and EGLN1 (D), EGLN3 (E) were analyzed. (F) (F, G) Western blot analysis of LSH, EGLN1 and EGLN3 in lung cancer tissues and normal lung tissues (F) and a panel of lung cancer cells (G). (H) Western blot analysis of LSH, EGLN1 and EGLN3 in H358 cells overexpressing LSH or EGLN1. (I) Determination of LSH mRNA level by qRT-PCR after depletion of EGLN1 in A549 cells. (J) MTT assays to assess cell viability in A549 cells after depletion of EGLN1. (K-M) Western blot analysis of the indicated proteins after the treatment with DMOG (K), BAY 85-394 (L) and CoCL2 (M). (N) Western blot analysis of the indicated proteins after ATRA treatment. * P<0.05, ** P<0.01, *** P<0.001.
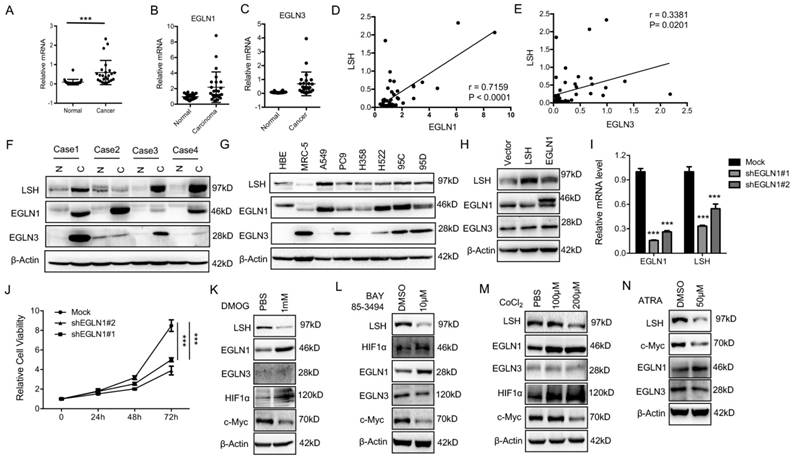
To further confirm the link between EGLN1 and LSH, we analyzed the EGLN1 protein level after LSH was overexpressed (Supplementary Figure S9A) or knocked down in lung cancer cells (Supplementary Figure S9B), we did not find that LSH altered EGLN1 expression. However, EGLN1 was overexpressed in the cells, and we found that the expression of LSH was increased (Figure 6H). The knockdown of EGLN1 decreased the mRNA expression of LSH and cell growth (Figure 6I-J), indicating EGLN1 regulates LSH expression. We treated the A549 cells with the following EGLN/PHD inhibitors: dimethyloxalyglycine (DMOG), CoCl2, inhibitors of dioxygenases, and BAY 85-3494 (BAY), a specific inhibitor of EGLNs with highest potency against EGLN1. We found that inhibition of EGLNs increased HIF1α and decreased LSH expression at the protein levels (Figure 6K-M), whereas inhibiting of EGLN/PHD further decreased ferroptosis in response to erastin (Supplementary Figure S9C). In summary, these data indicate that HIF-1α might function as a repressor of LSH expression.
When HIF-1α is induced, it immediately disrupts c-Myc complexes [38-40], indicating that inhibition of HIF-1α and EGLN1 might induce the expression of c-Myc. As expected, we found that the expression of c-Myc decreased after both the inhibition of the EGLNs and the induction of HIF-1α (Figure 6K-M). Moreover, we showed that EGLN and c-Myc were recruited to the promoter of LSH at two HIF-1α binding sites in the lung cancer cells (Supplementary Figure S10A-B), whereas inhibition of EGLN activity after the treatment of BAY decreased the binding of EGLN1 and c-Myc to the LSH promoter region (Supplementary Figure S10C-D). After the treatment of CoCl2 in A549 cells, both EGLN1 and c-Myc was recruited to the LSH promoter region (Supplementary Figure S10E-F) Lastly, ATRA decreased LSH and c-Myc protein expression levels in A549 cells (Figure 6N), which further support the hypothesis that the c-Myc/EGLN1 axis upregulated the expression of LSH expression by recruiting c-Myc to the LSH promoter.
LSH functions as an oncogene in lung cancer
To determine the physiological role of LSH in lung cancer, we analyzed the role of LSH in cancer in vitro and in vivo. The overexpression of LSH significantly increased the growth of all cell lines in vitro (Figure 7A, B), and increased migration and invasion in an in vitro assay (supplementary Figure S11A). To address whether LSH plays a role in lung cancer in vivo, we utilized a xenograft model to assess tumor formation in nude mice. We found that the injection of H358-LSH cells (2×106) significantly increased the tumor size (supplementary Figure S11B), tumor volume (Figure 7C) and tumor weight (Figure 7D), while the whole-body weight remained unchanged (supplementary Figure S11C). The expression levels of metabolism-associated genes, including SCD1 and FADS2 were increased by LSH in the xenograft tumors (Figure 7E).
LSH functions in vitro and in vivo as an oncogene in lung cancer. (A) The MTT assay was used to assess the cell viability of PC9 lung cancer cells that were stably transfected with an LSH expression vector. (B) Growth in soft agar was measured for PC9 cells that stably overexpressed LSH with quantitative analysis. (C) Tumor volumes in nude mice are shown after injection of PC9 cells stably expressing control vector or LSH expression plasmids as indicated time. (D) Tumor weights were recorded. (E) mRNA expression of the indicated genes measured by qPCR in xenograft tumors derived from PC9 cells with highly expression of LSH. (F) The MTT assay was performed to assess the cell viability of A549 cells that were stably transfected with two distinct LSH shRNA expression vectors (siLSH#1 and siLSH#2) and of control cells (Mock). (G) Growth in soft agar was measured for A549 cells that LSH was stably knocked down with quantitative analysis. (H) Tumor volumes in nude mice are shown after injection of A549 cells stably depletion of LSH as indicated time and (I) Tumor weights were recorded. (J) mRNA expression of the indicated genes measured by qPCR in xenograft tumors derived from A549 cells in the depletion of LSH. (K) The LSH protein levels in lung cancer patients were determined by immunohistochemistry. (L) The lung cancer tissue-associated LSH expression levels are significantly elevated. Kaplan-Meier curves for the overall survival rates that are associated with samples measured here for lung cancer (M) and lung ADCs (N). * P<0.05, ** P<0.01, *** P<0.001.
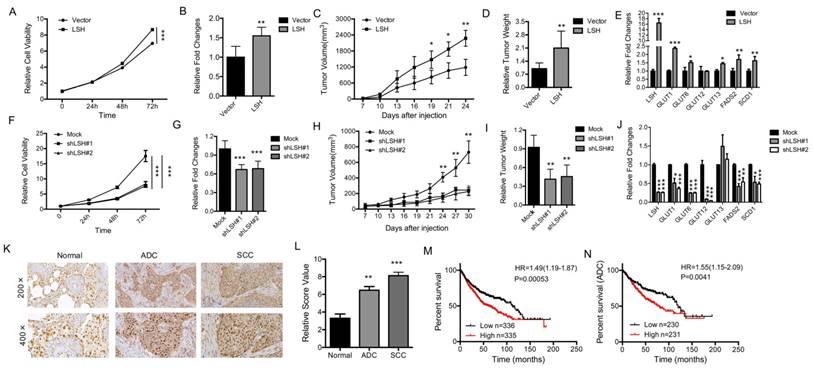
Conversely, LSH knockdown resulted in significantly reduced cell growth rate (Figure 7F) and impaired the formation of colonies (Figure 7G and supplementary Figure 11D). When we injected 3 × 106 A549 cells into nude mice, we observed that LSH depletion significantly reduced the tumor volume (Figure 7H), tumor formation and tumor weight (supplementary Figure S11E and Figure 7I), while the body weight did not significantly change in either groups (supplementary Figure S11F). The expression levels of metabolism-associated genes, including SCD1 and FADS2 were decreased after depletion of LSH in the xenograft tumors (Figure 7J).
We observed the increase of LSH mRNA increased in lung tumors (Fig 6A) and the increase of LSH protein in limited lung tumor samples (Fig 6F). To further determine the role of LSH in lung cancer, we performed immunohistochemical analysis on tissues from lung cancer patients. The LSH protein was present in the nuclei of the normal lung tissues, and its expression was greatly increased in the lung ADC and SCC tissues (Figure 7K-L). However, we did not detect any point mutations in LSH by sequencing in the 10 lung cancer tissues (data not shown). Notably, a Kaplan-Meier analysis was performed on a cohort of patients with these lung cancers and showed that lower LSH expression was associated with the overall survival in all lung cancers (Figure 7M), and ADCs (Figure 7N), but not in lung SCCs (supplementary Figure S11G). Taken together, LSH functioned as an oncogene in lung cancer progression.
Based on our observations, we propose a model for LSH-mediated inhibition of ferroptosis and enhancement of lung tumorigenesis (supplementary Figure S12). In this model, LSH acts as a novel inhibitor of ferroptosis by regulating several metabolism-related genes. LSH is upregulated by c-Myc, which is enriched at the LSH promoter by the EGLN1-mediated repression of HIF-1α. The induced LSH interacts with WDR76, which, in turn, upregulates the lipid metabolic genes, including GLUT1, SCD1 and FADS2. These metabolic genes inhibit the accumulation of lipid ROS and intracellular iron, which are required for ferroptosis, and inhibition of ferroptosis by LSH ultimately promotes cancer progression.
Discussion
Our findings provide evidence that LSH plays an important role in lung carcinogenesis. Our findings are the first to suggest that LSH acts as an important inhibitor of ferroptosis in carcinogenesis by promoting lipid metabolism-related genes, which act as novel key regulators of ferroptosis; moreover, LSH is upregulated by the EGLN1/c-Myc axis, whereas HIF-1α functions as a repressor of LSH expression.
A large-scale genome-wide analysis of various cancer model systems has revealed a strong correlation between aberrant epigenetic modifications and tumor progression, thereby highlighting the importance of chromatin-modifying enzymes in cancer [41, 42]. Interestingly, chromatin-modifying enzymes mediate sensing of the intermediary metabolism products to modulate gene regulation and disease progression [43]. LSH is critical for chromatin function and the establishment of DNA methylation [20, 21, 32, 35]. Several reports have shown that LSH contributes to the malignant progression of prostate cancer, melanoma, head and neck cancer, nasopharyngeal carcinoma and glioma [44-47], the mechanism for this remains less known. We recently indicate that LSH is shown to co-operate with partners, such as G9a to drive cancer progression [33, 44, 48]. However, the molecular mechanisms, particularly in lung cancer, are not well understood. Here, we demonstrate that LSH contributes to lung cancer progression by directly upregulating metabolic genes including SCD1 and FADS2. LSH-mediated increases in metabolic gene expression may occur through a DNA methylation-independent mechanism rather than through chromatin regulation [33, 49, 50]. Here, we provide evidence for an interaction between LSH and WDR76, which is a nuclear WD40 protein of unknown function in mammals. The LSH-dependent recruitment of WDR76 to the metabolic gene promoters and the subsequent chromatin modification that leads to metabolic gene activation, links epigenetic regulation by LSH to upregulation of the emerging metabolic genes.
The ferroptotic mode of programmed necrosis was recently discovered as an apoptosis-independent form of cell death in Ras-transformed cells [51]; the K-ras mutant is common in lung cancer. Ferroptotic death is morphologically, biochemically, and genetically distinct from apoptosis, necrosis (various forms), and autophagy. This process is characterized by an overwhelming, iron-dependent accumulation of lethal lipid ROS [7, 8]. Here we demonstrate that LSH decreases the lipid ROS and iron concentrations, which supports an inhibitory role for LSH in ferroptosis. Ferroptosis is characterized by increased levels of lipid peroxidation, which can be caused by compound-mediated inhibition of GPX4, or by indirectly targeting GPX4 through glutathione depletion [5]. Erastin, a ferroptosis-inducer, inhibits ferroptosis by inhibiting the cysteine/glutamate antiporter system [52], but the regulatory mechanism for this remains unclear. We demonstrated here that LSH is resistant to ferroptotic cell death in cancer cells. RNA sequencing analysis indicates that LSH is closely associated with the metabolic process, which suggests that LSH inhibits ferroptosis by affecting these metabolic genes. Interestingly, antioxidant reagents, vitamin C and aspirin, do not affect the expression of LSH or of mitochondria related genes. Vitamin E is regarded as a high efficient ferroptosis inhibitor[53, 54]. However, vitamin E did not affect LSH expression, indicating that types of cells and diseases might affect the efficiency of ferroptosis inhibitors. However, inducing ferroptosis including well-designed nanomedicines might provide a new insight to treat cancer [54, 55].
The iron dependent enzymes EGLNs catalyze HIF prolyl hydroxylation, which leads to HIF-1α and HIF-2α degradation. HIF-1α regulates oxygen-dependent glucose and glutamine metabolism, playing a critical role in cancer progression [12, 13, 56, 57]. In fact, EGLN1 inhibition causes accumulation of circulating metabolites [58]. Interestingly, some oncometabolites stimulate EGLN activity, which leads to diminished HIF levels [14]. For example, high extracellular glutamate levels inhibit the xCT glutamate-cysteine antiporter and thereby interfere with cysteine uptake, which results in lower intracellular cysteine levels. Decreased intracellular cysteine levels inhibit EGLN activity and stabilize HIF-1α [59]. We have recently shown that oncometabolites also activate LSH expression [33]. Here, we have demonstrated that EGLN1 upregulates LSH expression by inhibiting HIF‐1α, which highlights HIF‐1α as a key repressor of LSH expression. EGLN2 is to be essential for cell death and is a candidate driver of iron chelation-mediated inhibition of cell death [9]. Interestingly, HIF-1α and c-Myc counteract each other [38-40]. We found that c-Myc is recruited to the HIF-1α binding site on the LSH promoter in the normoxic state. Lipid ROS accumulation is a key characteristic of ferrotosis; we have shown that several lipid metabolism genes influence ferroptosis by affecting the lipid ROS and iron levels.
In summary, we demonstrated that LSH is involved in ferroptosis and is a potential therapeutic target in cancer because of its crucial role in ferroptosis. Previous studies have linked ferroptosis with oncogenic Ras [4], and more recently, the tumor suppressor p53 was demonstrated to positively regulate ferroptosis by transcriptionally inhibiting the expression of the cysteine/glutamate antiporter SLC7A11 [4, 6]; our findings demonstrate that LSH promotes the lipid metabolic genes, including SCD1 and FADS2. Given our finding that ferroptosis is epigenetically regulated by the chromatin-remodeling factor LSH, which is highly expressed in cancer tissues as an oncogene, ferroptosis could be preferentially triggered in cancer cells as a therapeutic option.
Abbreviations
ADC: Adenocarcinoma; BAY: BAY 85-3494; ChIP: Chromatin immunoprecipitation; Co-IP: Co- Immunoprecipitation; ESCs: embryonic stem cells; FPKM: Fragments Per Kilobase of exon per million reads; GLUTs: glucose transporters; FADS2: fatty acid desaturase 2; hMeDIP: hydroxymethylated DNA Immunoprecipitation; 5-hmC: 5-(hydroxyl) methylcytosine; GPX4: glutathione peroxidase 4; HIFα: hypoxia inducible factor α; IHC: Immunohistochemical; LSH: lymphoid-specific helicase; DMOG: dimethyloxalyglycine; NSCLC: non-small cell lung cancer; ROS: reactive oxygen species; PHD: prolyl hydroxylase domain; ATRA: Retinoic acid; SCC: squamous cell carcinoma; SCD1: sterol-CoA desaturase 1; TSSs: transcription start sites.
Acknowledgements
This work was supported by the National Basic Research Program of China [2015CB553903(Y.Tao)], and the National Natural Science Foundation of China [81372427 and 81672787(Y.Tao), 81271763 and 81672991(S.Liu), 81302354(Y.Shi), 81672307(X.Wang), 81422051 and 81472593 (Y.Cheng)].
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Green DR, Galluzzi L, Kroemer G. Cell biology. Metabolic control of cell death. Science. 2014;345:1250256
2. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27-47
3. Martinho O, Silva-Oliveira R, Cury FP, Barbosa AM, Granja S, Evangelista AF. et al. HER Family Receptors are Important Theranostic Biomarkers for Cervical Cancer: Blocking Glucose Metabolism Enhances the Therapeutic Effect of HER Inhibitors. Theranostics. 2017;7:717-32
4. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72
5. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317-31
6. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57-62
7. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Molecular cell. 2015;59:298-308
8. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9-17
9. Siddiq A, Aminova LR, Troy CM, Suh K, Messer Z, Semenza GL. et al. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J Neurosci. 2009;29:8828-38
10. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M. et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464-8
11. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR. et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43-54
12. LaGory EL, Giaccia AJ. The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol. 2016;18:356-65
13. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123:3664-71
14. Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S. et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484-8
15. Lee DC, Sohn HA, Park ZY, Oh S, Kang YK, Lee KM. et al. A lactate-induced response to hypoxia. Cell. 2015;161:595-609
16. Katada S, Imhof A, Sassone-Corsi P. Connecting threads: epigenetics and metabolism. Cell. 2012;148:24-8
17. Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell metabolism. 2012;16:9-17
18. Badeaux AI, Shi Y. Emerging roles for chromatin as a signal integration and storage platform. Nat Rev Mol Cell Biol. 2013;14:211-24
19. Zemach A, Kim MY, Hsieh PH, Coleman-Derr D, Eshed-Williams L, Thao K. et al. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell. 2013;153:193-205
20. Yu W, McIntosh C, Lister R, Zhu I, Han Y, Ren J. et al. Genome-wide DNA methylation patterns in LSH mutant reveals de-repression of repeat elements and redundant epigenetic silencing pathways. Genome Res. 2014;24:1613-23
21. Tao Y, Xi S, Shan J, Maunakea A, Che A, Briones V. et al. Lsh, chromatin remodeling family member, modulates genome-wide cytosine methylation patterns at nonrepeat sequences. Proc Natl Acad Sci U S A. 2011;108:5626-31
22. Myant K, Termanis A, Sundaram AY, Boe T, Li C, Merusi C. et al. LSH and G9a/GLP complex are required for developmentally programmed DNA methylation. Genome Res. 2011;21:83-94
23. Fan T, Yan Q, Huang J, Austin S, Cho E, Ferris D. et al. Lsh-deficient murine embryonal fibroblasts show reduced proliferation with signs of abnormal mitosis. Cancer Res. 2003;63:4677-83
24. Burrage J, Termanis A, Geissner A, Myant K, Gordon K, Stancheva I. The SNF2 family ATPase LSH promotes phosphorylation of H2AX and efficient repair of DNA double-strand breaks in mammalian cells. Journal of cell science. 2012;125:5524-34
25. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
26. Reck M, Heigener DF, Mok T, Soria JC, Rabe KF. Management of non-small-cell lung cancer: recent developments. Lancet. 2013;382:709-19
27. Forde PM, Brahmer JR, Kelly RJ. New strategies in lung cancer: epigenetic therapy for non-small cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:2244-8
28. Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17:284-99
29. Shi Y, Tao Y, Jiang Y, Xu Y, Yan B, Chen X. et al. Nuclear epidermal growth factor receptor interacts with transcriptional intermediary factor 2 to activate cyclin D1 gene expression triggered by the oncoprotein latent membrane protein 1. Carcinogenesis. 2012;33:1468-78
30. Jiang Y, Yan B, Lai W, Shi Y, Xiao D, Jia J. et al. Repression of Hox genes by LMP1 in nasopharyngeal carcinoma and modulation of glycolytic pathway genes by HoxC8. Oncogene. 2015;34:6079-91
31. Peck B, Schulze A. Lipid desaturation - the next step in targeting lipogenesis in cancer? FEBS J. 2016;283:2767-78
32. Ren J, Briones V, Barbour S, Yu W, Han Y, Terashima M. et al. The ATP binding site of the chromatin remodeling homolog Lsh is required for nucleosome density and de novo DNA methylation at repeat sequences. Nucleic Acids Res. 2015;43:1444-55
33. He X, Yan B, Liu S, Jia J, Lai W, Xin X. et al. Chromatin Remodeling Factor LSH Drives Cancer Progression by Suppressing the Activity of Fumarate Hydratase. Cancer research. 2016;76:5743-55
34. Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C. et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146-59
35. Yu W, Briones V, Lister R, McIntosh C, Han Y, Lee EY. et al. CG hypomethylation in Lsh-/- mouse embryonic fibroblasts is associated with de novo H3K4me1 formation and altered cellular plasticity. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:5890-5
36. Xi S, Geiman TM, Briones V, Guang Tao Y, Xu H, Muegge K. Lsh participates in DNA methylation and silencing of stem cell genes. Stem Cells. 2009;27:2691-702
37. Losman JA, Kaelin WG Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836-52
38. Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108-13
39. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI. et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407-20
40. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and Cancer. Cancer Discov. 2015;5:1024-39
41. Etchegaray JP, Mostoslavsky R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Molecular cell. 2016;62:695-711
42. Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502:480-8
43. Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489-98
44. von Eyss B, Maaskola J, Memczak S, Mollmann K, Schuetz A, Loddenkemper C. et al. The SNF2-like helicase HELLS mediates E2F3-dependent transcription and cellular transformation. EMBO J. 2012;31:972-85
45. Waseem A, Ali M, Odell EW, Fortune F, Teh MT. Downstream targets of FOXM1: CEP55 and HELLS are cancer progression markers of head and neck squamous cell carcinoma. Oral oncology. 2010;46:536-42
46. Ryu B, Kim DS, Deluca AM, Alani RM. Comprehensive expression profiling of tumor cell lines identifies molecular signatures of melanoma progression. PLoS One. 2007;2:e594
47. Xiao D, Huang J, Pan Y, Li H, Fu C, Mao C. et al. Chromatin Remodeling Factor LSH is Upregulated by the LRP6-GSK3beta-E2F1 Axis Linking Reversely with Survival in Gliomas. Theranostics. 2017;7:132-43
48. Keyes WM, Pecoraro M, Aranda V, Vernersson-Lindahl E, Li W, Vogel H. et al. DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell stem cell. 2011;8:164-76
49. Jiang Y, Liu S, Chen X, Cao Y, Tao Y. Genome-wide distribution of DNA methylation and DNA demethylation and related chromatin regulators in cancer. Biochimica et biophysica acta. 2013;1835:155-63
50. Liu S, Tao Y. Interplay between chromatin modifications and paused RNA polymerase II in dynamic transition between stalled and activated genes. Biological reviews of the Cambridge Philosophical Society. 2013;88:40-8
51. Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A. et al. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:8773-8
52. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M. et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523
53. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212:555-68
54. Zheng DW, Lei Q, Zhu JY, Fan JX, Li CX, Li C. et al. Switching Apoptosis to Ferroptosis: Metal-Organic Network for High-Efficiency Anticancer Therapy. Nano Lett. 2017;17:284-91
55. Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I. et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. 2016;11:977-85
56. Lin YJ, Shyu WC, Chang CW, Wang CC, Wu CP, Lee HT. et al. Tumor Hypoxia Regulates Forkhead Box C1 to Promote Lung Cancer Progression. Theranostics. 2017;7:1177-91
57. Lo Dico A, Costa V, Martelli C, Diceglie C, Rajata F, Rizzo A. et al. MiR675-5p Acts on HIF-1alpha to Sustain Hypoxic Responses: A New Therapeutic Strategy for Glioma. Theranostics. 2016;6:1105-18
58. Olenchock BA, Moslehi J, Baik AH, Davidson SM, Williams J, Gibson WJ. et al. EGLN1 Inhibition and Rerouting of alpha-Ketoglutarate Suffice for Remote Ischemic Protection. Cell. 2016;164:884-95
59. Briggs KJ, Koivunen P, Cao S, Backus KM, Olenchock BA, Patel H. et al. Paracrine Induction of HIF by Glutamate in Breast Cancer: EglN1 Senses Cysteine. Cell. 2016;166:126-39
Author contact
![]() Corresponding author: Y.T. Email: taoyongedu.cn; Tel. +(86) 731-84805448; Fax. +(86) 731-84470589.
Corresponding author: Y.T. Email: taoyongedu.cn; Tel. +(86) 731-84805448; Fax. +(86) 731-84470589.