Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(13):3192-3206. doi:10.7150/thno.19698 This issue Cite
Research Paper
Platelet-Targeted Delivery of Peripheral Blood Mononuclear Cells to the Ischemic Heart Restores Cardiac Function after Ischemia-Reperfusion Injury
Melanie Ziegler1, Xiaowei Wang1, 2, Bock Lim1, Ephraem Leitner1, Franco Klingberg1, Victoria Ching1, 2, 3, Yu Yao1, Dexing Huang1, Xiao-Ming Gao4, Helen Kiriazis4, Xiao-Jun Du4, Jody J. Haigh5, Alex Bobik6, Christoph E. Hagemeyer1, 2, 3#, Ingo Ahrens1*, Karlheinz Peter1, 2 ![]()
1. Atherothrombosis and Vascular Biology Laboratory, Baker Heart and Diabetes Institute, Melbourne, VIC 3004 Australia;
2. Department of Medicine, Monash University, Melbourne, VIC 3800 Australia;
3. Vascular Biotechnology Laboratory, Baker Heart and Diabetes Institute, Melbourne, VIC 3004 Australia;
4. Experimental Cardiology Laboratory, Baker Heart and Diabetes Institute, Melbourne, VIC 3004 Australia;
5. Mammalian Functional Genetics Laboratory, Division of Blood Cancers, Australian Centre for Blood Diseases, Monash University, Melbourne, VIC 3004 Australia;
6. Vascular Biology and Atherosclerosis Laboratory, Baker Heart and Diabetes Institute, Melbourne, VIC 3004 Australia.
#Present address: NanoBiotechnology Laboratory, Australian Centre for Blood Diseases, Monash University, Melbourne, VIC 3004 Australia
*Present address: Department of Cardiology and Medical Intensive Care, Augustinerinnen Hospital, Academic Teaching Hospital University of Cologne, 50678 Cologne, Germany
Received 2017-2-17; Accepted 2017-5-30; Published 2017-7-22
Abstract

One of the major hurdles in intravenous regenerative cell therapy is the low homing efficiency to the area where these cells are needed. To increase cell homing toward areas of myocardial damage, we developed a bispecific tandem single-chain antibody (Tand-scFvSca-1+GPIIb/IIIa) that binds with high affinity to activated platelets via the activated glycoprotein (GP)IIb/IIIa receptor, and to a subset of peripheral blood mononuclear cells (PBMC) which express the stem cell antigen-1 (Sca-1) receptor.
Methods: The Tand-scFvSca-1+GPIIb/IIIa was engineered, characterized and tested in a mouse model of ischemia-reperfusion (IR) injury applying left coronary artery occlusion for 60 min. Fluorescence cell tracking, cell infiltration studies, echocardiographic and histological analyses were performed.
Results: Treatment of mice undergoing myocardial infarction with targeted-PBMCs led to successful cell delivery to the ischemic-reperfused myocardium, followed by a significant decrease in infiltration of inflammatory cells. Homing of targeted-PBMCs as shown by fluorescence cell tracking ultimately decreased fibrosis, increased capillary density, and restored cardiac function 4 weeks after ischemia-reperfusion injury.
Conclusion: Tand-scFvSca-1+GPIIb/IIIa is a promising candidate to enhance therapeutic cell delivery in order to promote myocardial regeneration and thereby preventing heart failure.
Keywords: cell delivery, single-chain antibody, targeting, myocardial infarction, PBMC, regenerative cell therapy, cell tracking.
Introduction
Acute myocardial infarction (AMI) remains a leading cause of mortality and morbidity and a major economic burden worldwide [1]. Successful medical and interventional treatments mitigate the initial cardiac damage during an AMI, but the extent of this effect strongly depends on time from onset of symptoms to initial treatment. In addition, reperfusion causes further damage to the infarcted myocardial tissue. Therefore, there is an unmet need for novel therapies to minimize subsequent cardiac remodeling and preserve cardiac function.
Several cell-based therapies, in particular various stem and progenitor cell studies in preclinical animal models, have undergone intensive investigation and yielded great potential to improve tissue regeneration, enhance angiogenesis, and restore cardiac function after ischemia-reperfusion (IR) injury [2-5]. However, cell-based therapy trials for patients with AMI have shown disparate outcomes. Some trials showed improved left ventricular ejection fraction (LVEF) [6-8], but most studies did not find a significant benefit after cell therapy [9-11]. Overall, current cell therapy fails to deliver the initially anticipated clinical benefits.
Besides poor survival, low proliferation rates, insufficient engraftment, and incomplete differentiation of the transplanted cells, low cell homing efficacy to the infarcted myocardium has been one of the major limitations of cell therapy after AMI [12]. Previous studies indicated a low homing efficacy of only around 2% after intravenous cell infusion and around 5% after intracoronary transplantation. Instead, the majority of cells homed to the lung, liver, and spleen [13-15]. Despite the advantages of simplicity, cost-effectiveness, and the non-invasive nature of intravenous cell therapy, its main drawback is still poor cell retention in the ischemia-injured heart [16, 17]. Therefore, targeted cell therapy could be a promising tool to enhance cell delivery to the infarcted myocardium.
Crucial for targeted cell therapy is a highly specific target epitope to deliver cells to the specific area of myocardial injury. Activated platelets play a major role after transient myocardial ischemia, as they accumulate in the heart directly after reperfusion, and this strongly correlates with the size of the ischemic and necrotic areas [18-20]. Hence, activated platelets represent an ideal target for redirecting cells with regenerative potential into the ischemic heart. In particular, a single-chain antibody (scFv) targeted against active conformation of the major platelet integrin glycoprotein (GP)IIb/IIIa (αIIb/βIIIa; CD41/CD61) represents a unique targeting epitope [21, 22]. Specificity, sensitivity and suitability of the scFvGPIIb/IIIa as a targeting tool have been previously demonstrated [23-26], as well as specific accumulation within the ischemic myocardium [20, 27].
With the aim of increasing delivery of therapeutic cells to sites of cardiac damage and thus minimizing cardiac remodeling and restoring cardiac function after AMI, we developed a bispecific tandem single-chain antibody (Tand-scFv) that binds to activated platelets via the GPIIb/IIIa receptor, and to a subset of peripheral blood mononuclear cells (PBMCs) which express the stem cell antigen-1 (Sca-1) receptor. This bispecific Tand-scFvSca-1+GPIIb/IIIa was evaluated in functional in vitro assays and an in vivo mouse model of myocardial ischemia and reperfusion, demonstrating preservation of ventricular function and thus corroborating a new therapeutic approach for patients with AMI.
Results
Construction, expression, and purification of the bispecific Tand-scFvSca-1+GPIIb/IIIa and Tand-scFvSca-1+Mutant
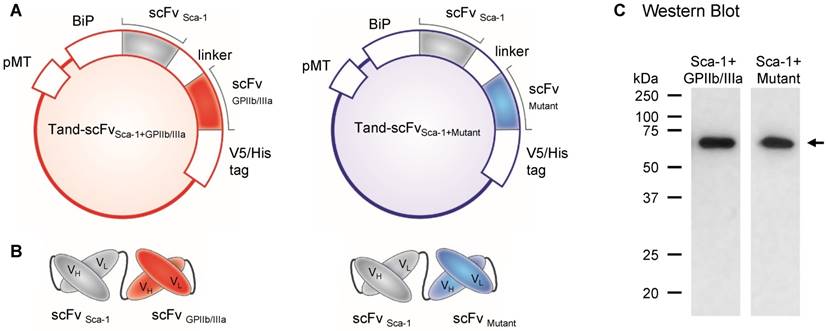
We designed and constructed two tandem scFvs, the bispecific Tand-scFvSca-1+GPIIb/IIIa and a corresponding control antibody, Tand-scFvSca-1+Mutant. Both tandem scFv fragments contain an N-terminus-located binding immunoglobulin protein (BiP) signal for secreting the antibody, followed by the scFv directed against Sca-1 to selectively home Sca-1-expressing PBMCs to the ischemic myocardium and a flexible linker sequence. At the C-terminus of the Sca-1 fragment is a linker peptide, followed by either of the targeting scFvs, directed against the active conformation of GPIIb/IIIa, or the control mutant version of this scFv (Figures 1A, B). Both proteins consist of a V5/6x-His tag at the C-terminus for purification and detection purposes. The designed constructs were cloned into the pMT expression vector in a tandem format, encoding proteins with a molecular weight of approximately 61 kDa. Purified diabodies were immunoblotted under reducing conditions using a monoclonal anti-His-HRP antibody, and the Western Blot showed a band at the predicted size of 61 kDa (Figure 1C).
Design and production of the tandem single-chain antibody (Tand-scFv)Sca-1+GPIIb/IIIa and the control Tand-scFvSca-1+Mutant. A) Plasmids of Tand-scFvs. Both proteins contain an N-terminal-located binding immunoglobulin protein (BiP) signal, followed by the single-chain antibody (scFv) against Sca-1 and a flexible linker sequence. The C-terminus of each protein is formed by a V5/His tag. Between the linker and the V5/His tag is the functional antibody, a scFv targeted against activated GPIIb/IIIa, and for the control antibody a mutant version of the scFv. B) Schematic illustration of Tand-scFvs. C) Purified Tand-scFvs, Tand-scFvSca-1+GPIIb/IIIa, and Tand-scFvSca-1+Mutant were immunoblotted under reducing conditions using an anti-His-HRP antibody and show a band at approximately 61 kDa (indicated by the arrow), which is the elxpected molecular weight.
Binding of both tandem scFvs to activated GPIIb/IIIa and Sca-1
Following the production of Tand-scFvSca-1+GPIIb/IIIa and Tand-scFvSca-1+Mutant, binding specificity was examined using flow cytometry and immunofluorescence staining. One binding site of both tandem scFvs is directed against Sca-1. Flow cytometry showed high binding affinity to Sca-1-expressing mouse PBMC for the Tand-scFvSca-1+GPIIb/IIIa as well as the corresponding control antibody Tand-scFvSca-1+Mutant (Figure 2A). The binding to Sca-1 was further confirmed by immunofluorescence staining of a novel generated Sca-1-expressing human embryonic kidney (HEK) cell line. Immunofluorescence staining of Sca-1-expressing HEK cells showed binding by both tandem scFvs as well as the commercial Sca-1 control antibody (green fluorescence), and confirmed that the scFvSca-1 is functional and binds specifically to Sca-1 (Figure 2B).
In vitro functionality of Tand-scFvSca-1+GPIIb/IIIa and Tand-scFvSca-1+Mutant. A) Representative histograms show strong binding of a commercial Sca-1 antibody (green), Tand-scFvSca-1+Mutant (blue), and Tand-scFvSca-1+GPIIb/IIIa (red) to PBMCs. B) Representative immunofluorescence images of Sca-1-expressing HEK cells showing binding by both constructs as well as by the commercial Sca-1 control antibody (green fluorescence, magnification: 400x, scale bar: 20 µm, n=3). C) Representative histograms show high affinity binding of a PAC-1 antibody (green), Tand-scFvSca-1+GPIIb/IIIa (red), but not Tand-scFvSca-1+Mutant (blue) to activated GPIIb/IIIa on human platelets. D) Representative immunofluorescence images of activated and non-activated GPIIb/IIIa-expressing CHO cells show specific binding of Tand-scFvSca-1+GPIIb/IIIa to activated GPIIb/IIIa but not to non-activated GPIIb/IIIa. Tand-scFvSca-1+Mutant binds to neither activated nor non-activated GPIIb/IIIa-expressing CHO cells (green fluorescence, magnification: 200x, scale bar: 50 µm, n=3).
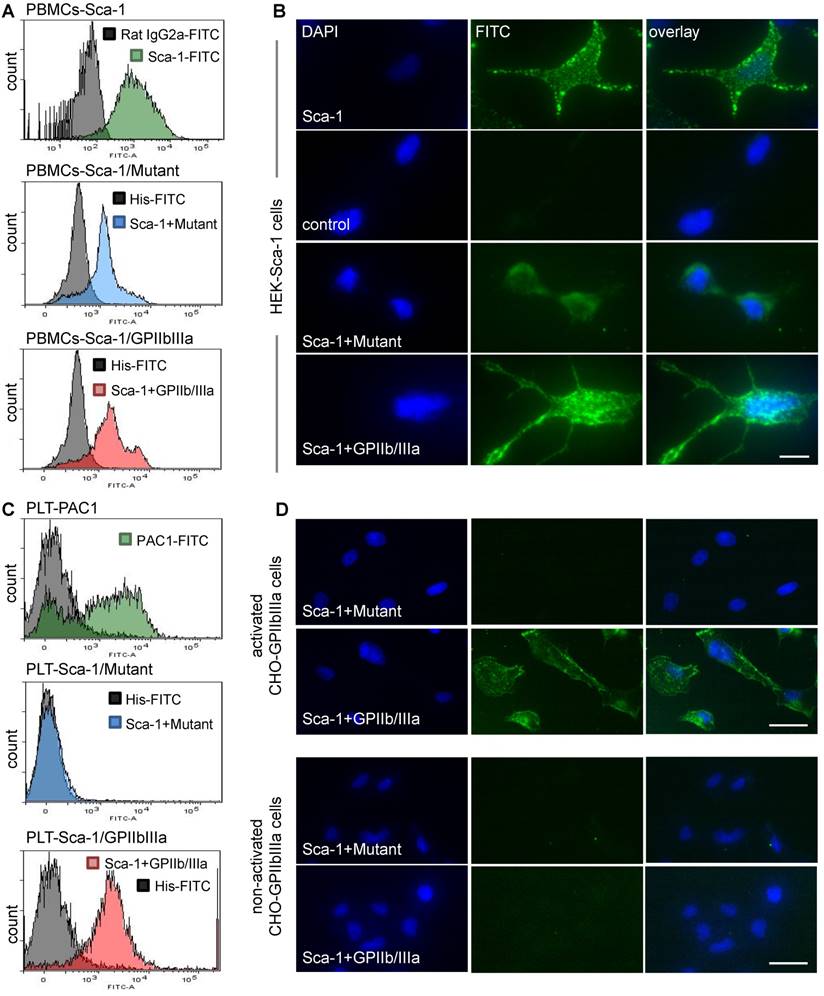
The second binding site, scFvGPIIb/IIIa, is directed against the active conformation of GPIIb/IIIa on the platelet surface. Flow cytometry displayed binding of the Tand-scFvSca-1+GPIIb/IIIa to activated human platelets, while Tand-scFvSca-1+Mutant did not bind to activated platelets. Successful platelet activation was confirmed using an anti-human PAC-1 antibody (Figure 2C). The Tand-scFvSca-1+GPIIb/IIIa preserved this activation specificity also on mouse platelets (data not shown). In addition, immunofluorescence staining was performed to visualize binding of Tand-scFvSca-1+GPIIb/IIIa to a monolayer of chinese hamster ovary (CHO) cells expressing activated GPIIb/IIIa. The control antibody Tand-scFvSca-1+Mutant did not bind to CHO cells expressing activated GPIIb/IIIa. As a negative control, neither Tand-scFvSca-1+GPIIb/IIIa nor Tand-scFvSca-1+Mutant showed significant staining of CHO cells expressing the resting, non-activated GPIIb/IIIa, and therefore confirmed specific binding of Tand-scFvSca-1+GPIIb/IIIa to activated GPIIb/IIIa (Figure 2D).
Bifunctional testing of Tand-scFvSca-1+GPIIb/IIIa and Tand-scFvSca-1+Mutant using static and dynamic adhesion assays
To examine the bifunctional character of Tand-scFvSca-1+GPIIb/IIIa, static and dynamic adhesion assays were performed using Sca-1-expressing HEK cells and a monolayer of activated platelets. Static adhesion assays showed that pre-incubation with Tand-scFvSca-1+GPIIb/IIIa significantly increased the adhesion of Sca-1-expressing HEK cells to activated platelets compared to pre-incubation with Tand-scFvSca-1+Mutant (197 ± 41 vs. 44 ± 33 cells/field of view (fov), **p<0.01) or PBS (11 ± 10 cells/fov, ***p<0.001). Control experiments provided evidence that the increase in adhesion was due to binding of Tand-scFvSca-1+GPIIb/IIIa to Sca-1 on HEK cells and activated GPIIb/IIIa on platelets, since adhesion of neither native HEK cells nor control Tand-scFvSca-1+Mutant significantly increased adhesion to the monolayer of activated platelets (Figure 3A).
In vitro bifunctionality of Tand-scFvSca-1+GPIIb/IIIa and Tand-scFvSca-1+Mutant. Tand-scFvSca-1+GPIIb/IIIa specifically increases adhesion of Sca-1-expressing HEK cells to a monolayer of ADP activated human platelets under A) static and B) physiological flow conditions (fov = field of view, n=3). Representative images show adhering Sca-1-expressing HEK cells (magnification: 400x (A) and 200x (B), scale bar: 50 µm (A) and 100 µm (B)). Data are represented as mean ± SD.
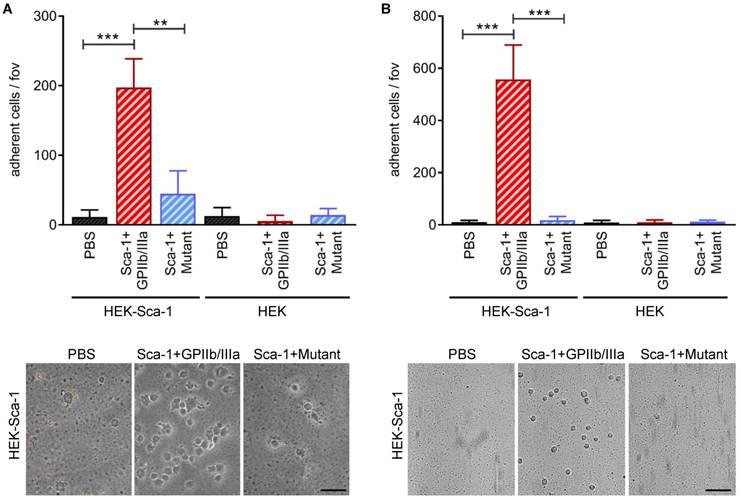
Besides these static adherence conditions, adhesion assays under physiological flow conditions were performed. Both native and Sca-1-expressing HEK cells were perfused over activated platelets pre-incubated with one of Tand-scFvSca-1+GPIIb/IIIa, Tand-scFvSca-1+Mutant, or PBS. Pre-incubation with Tand-scFvSca-1+GPIIb/IIIa showed a significant increase in firm adhesion of Sca-1-expressing HEK cells to activated platelets compared to pre-incubation with Tand-scFvSca-1+Mutant (557 ± 132 vs. 18 ± 14 cells/fov, ***p<0.001) or PBS (10 ± 7 cells/fov, ***p<0.001). As in the static adhesion assays, neither Tand-scFvSca-1+GPIIb/IIIa nor Tand-scFvSca-1+Mutant increased adhesion of native HEK cells (Figure 3B).
In summary, these data demonstrate that the novel bispecific antibody Tand-scFvSca-1+GPIIb/IIIa can direct Sca-1-expressing cells to activated GPIIb/IIIa on platelets by specifically interacting with the Sca-1 antigen and activated GPIIb/IIIa.
Characterization and accumulation of Tand-scFvSca-1+GPIIb/IIIa-targeted-PBMCs within the infarcted myocardium
Having confirmed that Tand-scFvSca-1+GPIIb/IIIa specifically binds to Sca-1 and GPIIb/IIIa and furthermore directs Sca-1-expressing cells to activated platelets in vitro, we hypothesized that Tand-scFvSca-1+GPIIb/IIIa preserves these homing properties in vivo and that directing Sca-1-expressing cells to activated platelets may have beneficial effects after myocardial infarction and reperfusion. This hypothesis was tested in a mouse model of cardiac IR injury. Mice underwent left coronary artery occlusion (CAO) for 1 h followed by reperfusion. Immediately after reperfusion, animals were injected with either PBS, PBMCs, PBMCs pre-incubated with Tand-scFvSca-1+GPIIb/IIIa, or the control antibody Tand-scFvSca-1+Mutant, respectively.
First, Tand-scFvSca-1+GPIIb/IIIa targeted PBMCs (targeted-PBMCs) were characterized using flow cytometry and it was demonstrated that 55.0 ± 4.1% of the PBMCs were Sca-1+. The major subsets of the targeted and administered Sca-1+-PBMCs were B2-cells (60%), CD4+ T-cells (24.9%), Ly6Clow-monocytes (5%), CD8+ T-cells (4.8%), and B1-cells (3.3%; Figure 4A, Supplemental Figures S1 + S2).
Enhanced delivery of targeted-PBMCs within infarcted myocardium after IR injury. A) Relative proportion of Sca-1+ PBMC subpopulations administered to infarcted mice. B) Representative IVIS image and quantitative analysis show a significant accumulation of targeted-PBMCs within the infarcted myocardium compared to non-targeted-PBMCs. C) Representative IVIS image and quantitative analysis of blood samples display comparable amounts of PBMCs throughout both treatment groups. Data are represented as mean ± SD, n=3. D) PBMCs were stained with Red CMTPX cell tracker dye and representative images of targ-PBMC within the ischemic cardiac sections are shown (magnification: 600x, scale bar: 20 µm, BF: bright field).
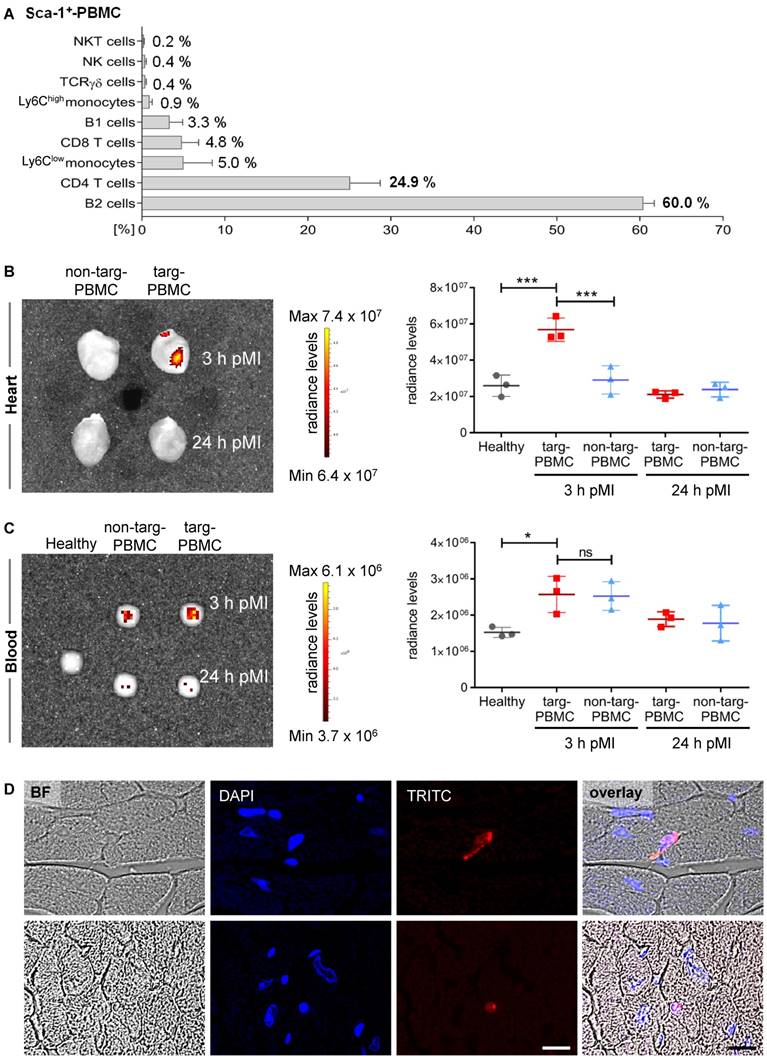
Next, we wanted to determine whether targeted-PBMCs show enhanced delivery to the infarcted myocardium. Therefore, mice after IR injury were treated with Red CMTPX cell tracker PBMCs pre-incubated with Tand-scFvSca-1+GPIIb/IIIa or the control antibody. Hearts and blood were imaged using an IVIS Lumina XRMS scanner 3 h and 24 h after cell injection. IVIS scans showed a significant accumulation of targeted-PBMCs in the myocardium 3 h after IR injury compared to the non-targeted-PBMCs and the healthy control heart (Figure 4B). No relevant fluorescence signal was detected 24 h post IR injury. A scan of whole blood displayed that both groups, targeted-PBMCs and non-targeted-PBMCs, received comparable amounts of PBMCs (Figure 4C). Furthermore, there was no significant change in radiance levels in the lung, spleen, liver, or bone throughout all groups observed. To identify the fate of targeted-PBMCs, infarcted cardiac sections were microscopically analyzed 3 h after IR injury. Targeted cells (stained with Red CMTPX cell tracker dye) could be found mainly in the microvasculature while a few cells had migrated into the infarcted myocardial tissue (Figure 4D).
Targeted-PBMCs ameliorate cardiac remodeling after IR injury
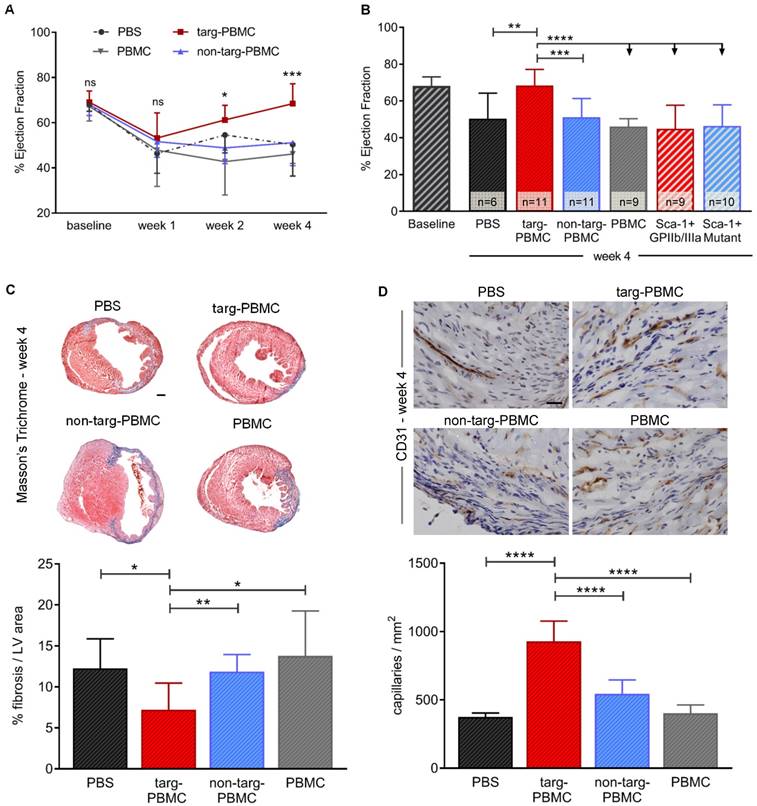
Treatment with Tand-scFvSca-1+GPIIb/IIIa-targeted-PBMCs resulted in continuous improvement in cardiac left ventricular (LV) function as demonstrated by echocardiography 1, 2, and 4 weeks after IR injury (week 1: 53.2 ± 11.1% ejection fraction (EF), week 2: 61.2 ± 6.5% EF, week 4: 68.4 ± 8.8% EF; Figure 5A). After 4 weeks, the improvement achieved was significant in the targeted-PBMC group (68.4 ± 8.8% EF) relative to the five control groups (non-targeted-PBMC: 51.1 ± 10.2% EF, PBMC: 46.2 ± 4.2% EF, PBS: 50.3 ± 14.0% EF, ScFvSca-1+GPIIb/IIIa: 50.0 ± 12.8% EF or ScFvSca-1+Mutant: 46.4 ± 11.5% EF *p<0.05, ***p<0.001, and ****p<0.0001; Figure 5B). Four weeks after IR injury, fibrotic areas in mice treated with targeted-PBMCs, as assessed by Masson's trichrome staining, were significantly reduced in comparison to mice treated with PBS, PBMCs, or non-targeted-PBMCs (Figure 5C). Furthermore, angiogenesis was analyzed by immunohistochemistry using a CD31 antibody. The average capillary density in the infarct zone was significantly enhanced in mice treated with targeted-PBMCs compared with all control groups (929 ± 148 vs. 544 ± 103 (non-targeted-PBMCs), 403 ± 61 (PBMCs), and 376 ± 29 (PBS) capillaries per square millimeter, ****p<0.0001; Figure 5D).
Treatment with PBMCs pre-incubated with Tand-scFvSca-1+GPIIb/IIIa preserves cardiac systolic function, reduces infarct size, and increases neovascularization in a mouse model of IR injury. A) Treatment with targeted-PBMCs shows a significant increase in ejection fraction from week 1 to week 4 after IR injury (significance between targ-PBMC and non-targ-PBMC in each time category). B) Four weeks post-MI, administration of targeted-PBMCs restored ejection fraction compared to administration of non-targeted-PBMCs, PBMCs, PBS, ScFvSca-1+GPIIb/IIIa or ScFvSca-1+Mutant. C) Cardiac sections were stained using Masson's Trichrome. Collagen was stained blue and muscle was counterstained (red). Quantitative analysis of fibrosis within the left ventricle (LV) is shown. Administration of targeted-PBMCs caused a significant decrease in fibrosis (scale bar 1 mm, magnification 12.5x). D) Cardiac sections were stained using a CD31 antibody. Blood vessels were stained brown and cell nuclei were counterstained with hematoxylin (blue). Quantitative analysis of capillary density in a certain tissue area (capillaries/mm2) is shown. Treatment with targeted-PBMCs significantly increased neovascularization within the infarcted tissue compared to with non-targeted-PBMCs, PBMCs, and PBS (scale bar: 25 μm, magnification: 200x). Data are represented as mean ± SD, n=6-11.
Taken together, platelet-targeted cell therapy using Tand-scFvSca-1+GPIIb/IIIa-targeted-PBMCs preserves cardiac function, decreases fibrosis, and enhances neovascularization 28 days after IR injury. To investigate the underlying cellular mechanisms of this impressive functional improvement, changes in secondary cell type infiltration were analyzed 3 days after IR injury.
Tand-scFvSca-1+GPIIb/IIIa-targeted-PBMCs reduce inflammatory response after IR injury
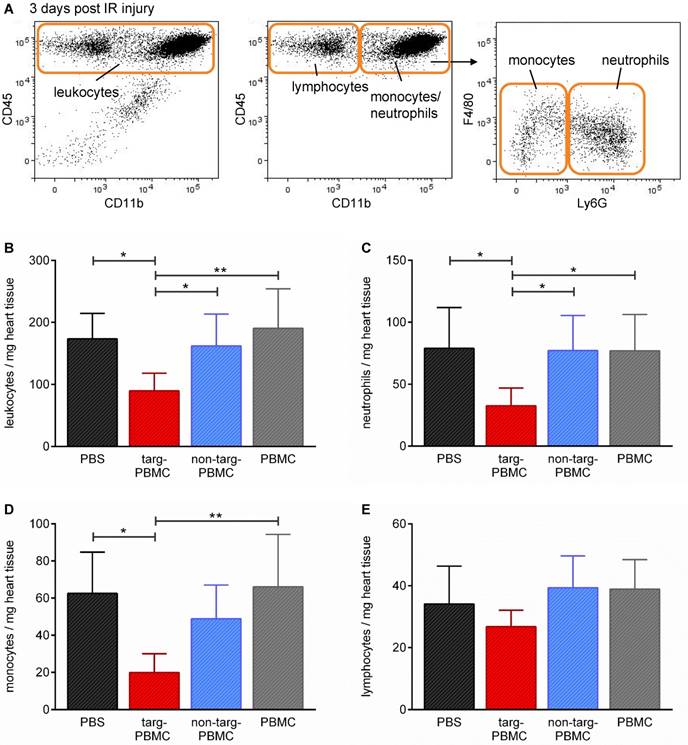
Alterations in secondary cell type infiltration due to treatment with Tand-scFvSca-1+GPIIb/IIIa-targeted-PBMCs were assessed by flow cytometric analysis 3 days after IR injury (Figure 6A). Mice treated with targeted-PBMCs demonstrated an approximately 50% reduction in the density of inflammatory cells within the infarcted myocardium, as measured by the number of total leukocytes per mg heart tissue (targeted-PBMCs: 90 ± 29 vs. non-targeted-PBMCs: 162 ± 51, PBMCs: 190 ± 64, and PBS: 173 ± 41 leukocytes/mg heart tissue, *p<0.05; Figure 6B). In particular, subtype analysis showed that in particular the numbers of neutrophils and monocytes were decreased after treatment of Tand-scFvSca-1+GPIIb/IIIa-targeted PBMCs (Figure 6C-E).
Changes in leukocytes and leukocyte subpopulations in heart tissue three days after IR injury. A) Mice were euthanized and the hearts digested and stained for flow cytometric analysis of different cell populations using the depicted gating strategy. B-E) Bar graphs show quantitative analyses of B) infiltrated leukocytes, C) neutrophils, D) monocytes, and E) lymphocytes per mg heart tissue within the infarcted myocardium. Data are represented as mean ± SD, n=6-7.
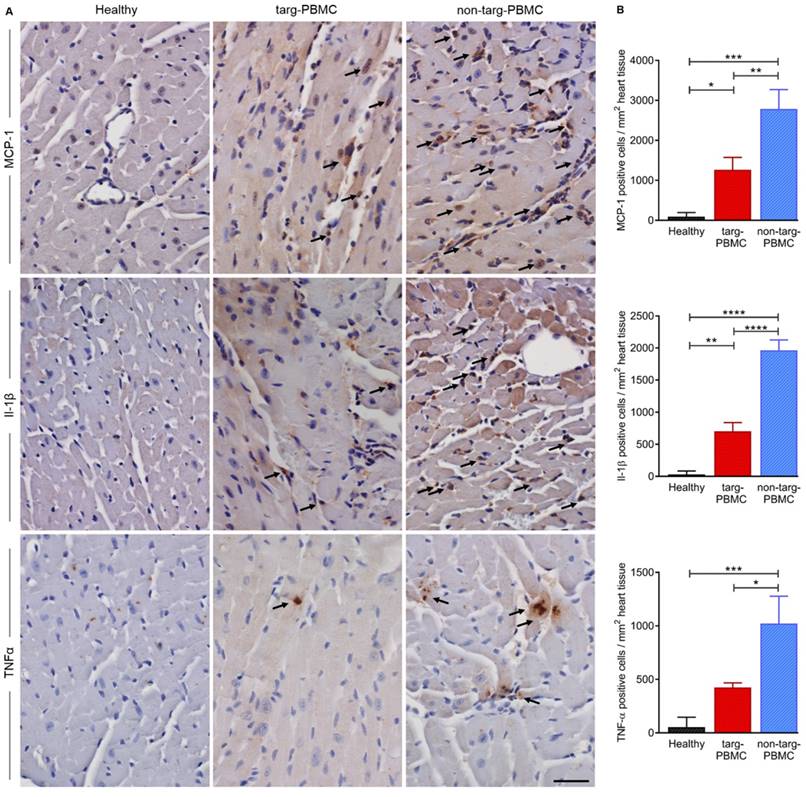
To support the reduction of inflammation within the ischemic myocardium cytokine staining for MCP-1, Il-1β, and TNFα was performed 24 h after IR injury. As expected, all cytokines were upregulated after IR injury as compared to the healthy controls. The upregulation was significantly reduced in mice treated with targ-PBMC compared to the non-targ-PBMC control group (Figure 7).
Changes in cytokine levels in cardiac tissue 24 hours after IR injury. A) Cardiac section (n=3) were stained for MCP-1, Il-1β, and TNFα. Cytokine expression was reduced in the targ-PBMC treated mice compared to non-targ-PBMC mice. Arrows indicate cytokine expressing cells (scale bar: 30 μm, magnification: 400x). B) Bar graphs show quantitative analyses of cytokine positive cells per mg heart tissue within the infarct zone. Data are represented as mean ± SD, n=3.
Taken together, these data demonstrate that treatment of Tand-scFvSca-1+GPIIb/IIIa-targeted PBMCs modulated the inflammatory profile by reducing the infiltration of inflammatory cells in the myocardium after IR injury.
Discussion
We generated the unique bivalent recombinant single-chain antibody Tand-scFvSca-1+GPIIb/IIIa, which specifically binds to activated platelets as well as to Sca-1+-PBMCs. Systemic delivery of Tand-scFvSca-1+GPIIb/IIIa pre-incubated PBMCs via intravenous injection showed enhanced delivery to the infarcted myocardium and led to reduced infiltration of inflammatory cells, reduced fibrosis, enhanced capillary density, and, most importantly, restored cardiac LV function after IR injury.
These findings strongly imply that transplantation of platelet-targeted-PBMCs might be an effective and novel therapeutic approach for AMI. Intravenous cell delivery is the least invasive and most convenient route, and leads to a uniform distribution of the cells in the infarcted area [16, 17]. Despite these advantages, the expected low homing efficiency of intravenous cell infusion has instead primarily resulted in intramyocardial or intracoronary injection being tested in clinical trials [13, 28]. However, targeted cell therapy has the potential to overcome this limitation. Recent MRI and PET/CT studies using the scFvGPIIb/IIIa, which was used in our study, showed a highly significant accumulation of the targeted antibody to activated platelets within the ischemic myocardium after IR injury, a uniform distribution throughout the ischemic region, as well as occurrence at a well-defined time window peaking 2 hours post reperfusion [20, 27]. Moreover, the scFvGPIIb/IIIa presents an excellent translational tool, as it binds to both, human and mouse activated platelets. To improve myocardial delivery of systemic cell infusion, we developed the antibody Tand-scFvSca-1+GPIIb/IIIa, which binds primarily to activated platelets and homes toward Sca-1+ cells within the ischemic myocardium. Indeed, this specific targeting by Tand-scFvSca-1+GPIIb/IIIa leads to significant enrichment of the targeted-PBMCs compared to non-targeted control PBMCs 3 h after IR injury. Therefore, targeted cell therapy is a promising approach to increasing cell retention within the infarcted myocardium.
There have been various promising candidates that showed safety and potential for cardiac cell therapy in preclinical IR injury models, including mesenchymal stem cells (MSCs), hematopoietic stem cells (HSC), tissue-resident cardiac progenitor cells (CPC), cardiac stem cells (CSC), and embryonic stem cells (ESC), induced pluripotent stem cells (iPSC), as well as bone marrow-derived or peripheral blood mononuclear cells (MNCs) [29]. However, results showed disparate outcomes in terms of efficacy and cardiovascular research still faces the challenge to define the best cell type for cardiac cell therapy in order to salvage and/or regenerate damaged myocardium after AMI [29-31]. While ongoing research aims to identify the best source of cells, PBMCs were used in this study, as a proof of concept to evaluate the potential of the targeting approach.
In general, the clinical use of PBMCs for transplantation is favorable because they can be easily, quickly and safely harvested from the peripheral blood and even from donors treated with antiplatelet agents [7]. In some cases stem cell treatment takes too long and is initiated too late to have any beneficial effect [32]. In addition, regenerative cell therapy in humans requires cell numbers in the range of billions. Importantly, this can be achieved with PBMCs, as they can be collected repeatedly and in sufficient quantities [33]. The harvesting of the PBMC population is far less invasive than the collection of stem cells, which strongly favors the use of PMBCs as cell therapeutics promoting cellular repair after AMI. A further major advantage of PBMCs as therapeutic cells is their low entrapment in the lungs, while various stem cells such as MSCs are easily entrapped in the lungs after systemic transplantation [14, 16]. This is most likely due to PBMCs' different characteristics compared to stem and progenitor cells, such as their smaller cell size, only 4-9 µm for mouse PBMCs compared to 20-45 µm for mouse MSCs [34].
Sca-1 is highly expressed not only on most PBMCs, but also on a broad variety of stem and progenitor cells, in particular on various types of promising regenerative cells including MSC [35], tissue-resident CPC [36], CSC [37], several ESCs [38], and iPSC [39]. Therefore, our newly developed biotechnological approach of delivering Sca-1+ cells represents an attractive strategy for various cell-based therapies. Sca-1 is a mouse specific antigen, which is not expressed in humans. However, the human epitope CD34 might be an equivalent antigen to choose for translation into patients. CD34 also shows a widespread distribution on various types of cells such as HPC, HSC, vascular endothelial progenitor cells (VPC), MSC, endothelial cells (EC), mast cells, interstitial dendritic cells, and fibrocytes and has been used for cell directing studies in humans before (e.g. CD34-antibody coated coronary artery stents) [40, 41]. Using CD34 in humans may allow similar targeted cell therapies as shown in our studies with Sca-1 in mice. The scFvSca-1 can be easily replaced by a scFv against a promising human analogue such as CD34. Therefore, our proof of concept study bears translational relevance for the treatment of human diseases.
Our targeted-PBMC approach differs substantially from the non-targeted PBMC approaches reported so far [7, 42]. Selecting for Sca-1+ cells changed the composition of cells delivered for cell therapies substantially. For example, the number of Ly6Chigh-monocytes decreased from approx. 7% in non-targeted-PBMCs to approx. 1% in targeted-PBMCs, while Ly6Clow-monocytes increased from approx. 2% to 5% in Sca-1+ PBMCs. This shifts the monocyte composition toward more anti-inflammatory effector cells.
Most importantly, over 63% of the delivered Sca-1+ PBMCs are B cells, mostly B2 cells, and therefore their function may be driving the paracrine-mediated cardiac-repair mechanisms when delivered immediately after AMI. While the roles of monocytes and macrophages are well characterized, there is little known about the role and underlying mechanism of B and T cells after IR injury. These cells are currently described as early modulators in myocardial healing and remodeling, and show disparate outcomes [43]. Both depletion of mature B cells using a monoclonal anti-CD20 antibody [44] and intramyocardial delivery of BMC-derived B cells [45] show therapeutic potential to preserve cardiac function. However, the comparison between these studies and our study is limited, as they focused on the role of bone marrow-derived B cells including T1 and T2 transitional (immature) B cells in permanent ligation models, while our study focuses on circulating B cells, mostly B2 cells, in a transient ischemia model of cardiac injury. Overall, B cells are described as acting by secreting antibodies, as well as by paracrine mechanisms such as T-cell activation, cytokine and chemokine secretion, monocyte mobilization, and antigen presentation [46]. Therefore, transplanted targeted-PBMCs are expected to act via paracrine effects, rather than transdifferentiation and long-term integration into the ischemic heart.
It is well-established that AMI causes an inflammatory response characterized by recruitment and activation of immune cells of the innate and adaptive immune system [47]. Cardiac injury induces chemokine and cytokine synthesis in the infarcted heart, and this plays a key role in leukocyte locomotion and trafficking to the heart [48]. Once in the heart, leukocytes contribute to inflammation, remodeling, and fibrosis within the myocardium [49]. Previous studies clearly demonstrated that depletion of neutrophils, as well as moderate reduction of monocytes, led to a decrease in IR injury and ultimately in reduction of cardiac damage [50, 51]. Therefore, a decrease in cytokines and overall leukocyte recruitment to the heart following AMI, as shown in this study, is indicative of a dampened inflammatory response as a potential mechanism for the preservation of cardiac function, as seen in mice treated with targeted-PBMCs. This study focused on the development and characterization of the bispecific antibody and the potential of such a targeted antibody approach. Further research is required to delineate the specific mechanism of the benefits achieved by PBMCs.
Furthermore, the herein developed bispecific antibody Tand-scFvSca-1+GPIIb/IIIa holds great promise for targeted cell therapy in multiple clinical conditions. Imaging of activated platelets using scFvGPIIb/IIIa demonstrated the unique potential of this tool to accumulate specifically at areas of thrombosis, ischemia, or inflammation, making this tool attractive for targeted cell delivery to tissues undergoing IR or inflammatory reactions [20, 23, 52, 53]. For example, Tand-scFvSca-1+GPIIb/IIIa could be a promising biotechnological approach to home therapeutic cells after ischemic conditions such as stroke or renal infarction, as well as inflammation-mediated diseases such as rheumatoid arthritis.
In conclusion, Tand-scFvSca-1+GPIIb/IIIa delivers a Sca-1-expressing subset of PBMCs to activated platelets within the IR myocardium and thereby preserves cardiac function after IR injury. Tand-scFvSca-1+GPIIb/IIIa-targeted intravenous delivery of PBMCs is therefore a unique and promising approach for cell-based therapy, promoting myocardial repair after AMI.
Methods
Mice
C57BL/6 and UBC-GFP/B6+/-(Tg) mice were acquired from Jackson Laboratories and bred by Alfred Medical Research and Education Precinct (AMREP) Animal Services in Melbourne, VIC. All experimental work was performed in accordance with the Australian code for the care and use of animals for scientific purposes and approved by the AMREP Animal Ethics Committee (E/1402/2013/B).
Generation, expression, and purification of bispecific Tand-scFvSca-1+GPIIb/IIIa and Tand-scFvSca-1+Mutant
Two different scFvs, activated GPIIb/IIIa-targeted scFv (scFvGPIIb/IIIa) and non-targeted scFv (scFvMutant), were fused with scFvSca-1 and cloned into the pMT vector system. A detailed description of expression and purification is given in the Supplemental Methods. Briefly, both scFvSca-1-plasmid constructs were produced using CuSO4-induced dimethyldioctadecylammonium bromide (DDAB) transfection in Drosophila melanogaster Schneider line-2 (S2) cells (Life Technologies). First, cell supernatant was applied to a chelating Sepharose fast-flow column to remove the non-specific protein and the copper. Both scFvs contain a 6xHis tag, which was used for further purification with nickel-based metal affinity chromatography (Invitrogen).
Western blotting
Purified samples were analyzed using SDS gel electrophoresis (12% acrylamide, denaturing conditions). SDS PAGE was transferred to a poly(vinylidene fluoride) membrane (Millipore) for immunodetection. The membrane was blocked with 5% skim milk in PBST and probed with an HRP-labeled anti-6xHis tag antibody (Roche). The signal was visualized using Super Signal Chemiluminescent Substrate (Thermo Scientific).
PMBC isolation
UBC-GFP/B6+/-(Tg) donor mice were anesthetized and blood was collected into 20 U of clexane (Sanofi Aventis) by cardiac puncture. The blood was then carefully layered on top of Ficoll-Paque (GE Healthcare) and centrifuged at 400 g for 30 min, acceleration set at 4 and brakes set at 1. The PBMC layer was transferred into Dulbecco's Modified Eagle Medium (DMEM, Life Technologies) and centrifuged at 250 g for 10 min. PBMCs were resuspended in DMEM and counted. GFP-PBMCs were used as donor cells for cell tracking purposes. Unfortunately, after using these mice for our study we found that the GFP signal in these cells was attenuated after isolation and injection. Thus we used the CellTrackerTM Red CMTPX Dye for the presented cell tracking experiments as described below.
PBMC characterization
Isolated PBMCs (n=4) were stained with various fluorochrome-labeled anti-mouse antibodies (Sca-1-PE or -FITC, NK1.1-PerCP-Cy5.5, CD3-APC, TCRγδ-pacific blue, CD11b-APC-Cy7, or -PerCP-Cy5.5, CD4-pacific blue, CD8-PerCP, CD5-APC, CD19-PE-Cy7, CD45-APC, Ly6C-pacific blue, and Ly6G-PerCP-Cy5.5) for 30 min at 4 °C. Samples were measured using a FACS Canto II (BD Biosciences) and Flowlogic (Inivai technologies) was used for FACS analysis. The gating strategy is presented in Supplemental Figures S1 + S2. Monocytes were gated as CD11b+Ly6G- cells and then differentiated between Ly6Chigh and Ly6Clow monocytes. CD19+CD5- as a combination of markers to identify B2 cells was previously described [54, 55] and used in our gating strategy.
Binding studies of Tand-scFvSca-1+GPIIb/IIIa and Tand-scFvSca-1+Mutant by flow cytometry
Binding capacity of scFvs against Sca-1 receptors was performed on freshly isolated mouse PBMCs. Study of binding of scFvs against activated GPIIb/IIIa was performed on human platelets. Platelets were activated using 20 µM ADP. PBMCs and activated platelets were incubated with 10 µg/ml Tand-scFvSca-1+GPIIb/IIIa and Tand-scFvSca-1+Mutant for 15 min at room temperature (RT). After washing, cells were stained using Penta-His Alexa Fluor 488 (Qiagen) directed against the 6xHis tag of the bispecific scFvs for 15 min in darkness. Binding of Tand-scFvSca-1+GPIIb/IIIa and Tand-scFvSca-1+Mutant was analyzed using a BD FACS Canto II.
Generation of Sca-1-expressing HEK cells
Sca-1-expressing HEK cells were generated by stable transfection of HEK cells with pSecTag2A-Sca-1 DNA. Control HEK cells were transfected with the plasmid only, but did not contain Sca-1. Zeocin (Life Technologies) was used as the selection reagent at 250 µg/ml.
Immunofluorescence staining
Sca-1-expressing HEK cells and CHO cells expressing either activated or non-activated GPIIb/IIIa were seeded on coverslides. Cells were grown for 2 days, and incubated with 10 µg/ml of either Tand-scFvSca-1+GPIIb/IIIa or Tand-scFvSca-1+Mutant, followed by a Penta-His Alexa Fluor 488 antibody. Cells were visualized with the IX81 Olympus microscope (Olympus, Japan) and Cell^P1692 (ANALysis Image Processing) software.
Preparation of washed platelets
Blood was collected from healthy donors into citrate and spun at 180 g for 10 min to generate platelet rich plasma (PRP). Then, PRP was acidified to pH 6.5 using Acid Citrate Dextrose solution B (ACD-B) (25 mM citric acid, 45 mM sodium citrate, 80 mM dextrose). Platelets were pelleted by centrifugation at 720 g for 10 min and the pellet was reconstituted to original PRP volume with JNL buffer pH 6.5 (6 mM dextrose, 130 mM NaCl, 9 mM Na bicarbonate, 10 mM Na citrate, 10 mM Tris base, 3 mM KCl, 2 mM HEPES, 0.81 mM KH2PO4, 0.9 mM MgCl2). Platelets were pelleted again by centrifugation at 500 g for 10 min, resuspended in 3 times the original PRP volume using JNL buffer p 7.4 and were supplemented with CaCl2 (1.8 mM).
Static adhesion assay
Washed human platelets were isolated and a 96 well plate was coated with a monolayer of activated platelets. Briefly, 100 µl of washed platelets were added per well and activated using a final concentration of 20 µM ADP. After incubation for 20 min at 37°C platelets formed a monolayer at the bottom of the wells. The platelet monolayer was rinsed twice with 200 µL of PBS and blocked with 1% BSA in Tyrode-HEPES buffer for 30 min at 37°C. In the meantime HEK cells and Sca-1 expressing HEK cells were harvested and incubated with 10 µg/ml Tand-scFvSca-1+GPIIb/IIIa, Tand-scFvSca-1+Mutant or PBS for 15 min at RT. 1x105 HEK cells per well were incubated with the platelet monolayer at 37°C for 10 min. Images were acquired using a 400x magnification and adhered HEK cells were counted manually.
Dynamic adhesion assay under flow conditions
Glass capillaries (0.2 x 2.0 x 100 mm, Vitrocom, NJ) were incubated with 100 µl washed human platelets for 20 min at 37°C. The platelet monolayer was then activated with 20 µM ADP for 15 min at 37°C, blocked with 1% BSA and incubated with 10 µg/ml Tand-scFvSca-1+GPIIb/IIIa, Tand-scFvSca-1+Mutant or PBS for 30 min at 37°C. Capillaries were connected to a programmable syringe pump (PHD 2000, Harvard Apparatus) and an inverted microscope was used for observation (Olympus IX81). 4x106/ml HEK cells were perfused over the platelet monolayer at a shear rate of 250 s-1 for 5 min. Three videos along the capillary centerline were acquired and adherent cells were manually counted offline.
Myocardial IR injury and echocardiography
Mice underwent open-chest surgery to induce left coronary artery occlusion (CAO) for 1 h, followed by reperfusion as described previously [56]. All operations were carried out in a blinded manner such that the surgeon was blinded to the treatment administered. Eight-weeks old, male C57BL/6 mice were anesthetized using a combination of ketamine HCl (100 mg/kg body weight (wt); Lyppard), xylazine HCl (5 mg/kg body wt; Lyppard), and atropine (1 mg/kg body wt; Pfizer) via intraperitoneal injection. Mice were orally intubated and ventilated using a rodent ventilator (Model 687, Harvard Apparatus), with a tidal volume of 0.18 ml at 120 breaths/min. Mice underwent open-chest surgery to induce CAO for 1 h, followed by reperfusion. Depending on treatment group, immediately after reperfusion mice were randomly injected via tail-vein with PBS, 1.5x106 GFP-PBMCs, 1.5x106 GFP-PBMCs pre-incubated with 140 µg/ml Tand-scFvSca-1+GPIIb/IIIa or Tand-scFvSca-1+Mutant, 40 µg per mouse Tand-scFvSca-1+GPIIb/IIIa or 40 µg per mouse Tand-scFvSca-1+Mutant.
Following surgery, one group of mice (n=6-7) were euthanized after 3 days for analysis of leukocyte infiltration. The other group (n=6-11) underwent serial echocardiography at 1, 2, and 4 weeks after infarction using an iE33 Ultrasound (Philips, Netherlands), equipped with a L15-7io transducer. Left ventricular ejection fraction was calculated from echocardiographic images (short-axis view). Echocardiographic imaging and the corresponding analyses were performed blinded by two different researchers independently from each other. Averages of 3 measurements per animal were used. After 28 days, mice were euthanized for histological analysis.
PBMC labelling, IVIS imaging and microscopic cell tracking
PBMCs were stained using 25 µM CellTrackerTM Red CMTPX Dye (Molecular Probes, excitation (Ex), 577 nm; emission (Em), 602 nm) for 30 min at 37 °C. Cells were then washed twice in DMEM and incubated with 140 µg/ml Tand-scFvSca-1+GPIIb/IIIa or Tand-scFvSca-1+Mutant for 15 min at RT. Mice underwent MI-inducing surgery by CAO. Immediately after reperfusion, cells were injected and mice were euthanized either 3 h or 24 h after reperfusion. The organs were perfused with PBS, collected in 10% formalin, and imaged using an IVIS Lumina XRMS system (Perkin Elmer).
After the IVIS scan the hearts were embedded in paraffin, cut into 6 µm thick slides and stained with 4',6-diamidino-2-phenylindole (DAPI). Samples were imaged using the Nikon A1r Plus Confocal Microscope, 40x objective.
Heart digestion and flow cytometry staining
Three days post-MI, the hearts were flushed with Krebs-Henseleit buffer. After weighing and mechanical disruption, hearts were digested for 2 h at 37 °C by an enzyme cocktail of 1.3 U Liberase DL Blendzyme (Roche) and 20 U DNase I (Ambion) in Tyrode-HEPES buffer. Cells were then passed through a cell strainer and stained with an antibody cocktail containing CD45-APC, CD11b-APC-Cy7, Ly6G-PerCP-Cy5.5, and F4/80-PE, for 30 min at 4 °C. Samples were measured using a BD FACS Canto II and Flowlogic (Inivai technologies) was used for FACS analysis.
Histology
Mice were euthanized after 28 days, the hearts were harvested and embedded in Tissue-Tek® OCT Compound (Sakura, Japan), followed by cutting into sections 6 µm thick. Ten defined heart sections for each mouse were stained using a Masson's Trichrome Staining Kit (Australian Biostain P/L) according to the manufacturer's protocol. Slides were imaged on an Olympus BX50F-3 microscope (Olympus Optical Co., Japan) using 12.5x magnification. Blue-stained collagen distribution was measured as the percentage area of the total left ventricle using ImageJ v1.48.
Immunohistochemistry
Three random cardiac sections for each mouse were immunostained using rat anti-mouse CD31 monoclonal antibody (0.3 µg/ml, BD Pharmingen), rabbit anti-mouse MCP-1 antibody (10 µg/ml, abcam), rabbit anti-mouse Il-1β antibody (1 µg/ml, abcam), rat anti-mouse TNFα antibody (25 µg/ml, BD Pharmingen), and a corresponding isotype control antibody. The appropriate biotinylated secondary antibody (Vector Laboratories) was applied for 30 min at RT. Immunostaining of cardiac sections was performed with a streptavidin-biotin-immunoperoxidase method (Vectostatin ABC-Peroxidase and diaminobenzidine; Vector Laboratories) per the manufacturer's protocol. Samples were imaged on an Olympus BX50F-3 microscope using 200x (for CD31) or 400x (for MCP-1, Il-1β, TNFα) magnification. The percentages of MCP-1-positive, Il-1β-positive, and TNFα-positive cells were counted from three random areas in the infarcted myocardium of each heart sample (n = 3) or the matching heart level for the healthy control and were referred to a certain tissue area (mm2).
Statistical analysis
Data were statistically analyzed using one- or two-way repeated-measures ANOVA. Post hoc, a Tukey's, Sidak's, or Bonferroni's multiple comparisons test was used. P values of less than 0.05 were considered statistically significant. All results are expressed as mean ± standard deviation (SD).
Abbreviations
AMI: acute myocardial infarction; GP: glycoprotein; PBMC: peripheral blood mononuclear cells; Sca-1: stem cell antigen-1; IR: ischemia-reperfusion; LVEF: left ventricular ejection fraction; scFv: single-chain antibody; BiP: binding immunoglobulin protein; HEK: human embryonic kidney; CHO: chinese hamster ovary; fov: field of view; CAO: coronary artery occlusion; LV: left ventricular; EF: ejection fraction; DDAB: dimethyldioctadecyl¬ammonium bromide; S2: Schneider line-2; BF: bright field
Acknowledgements
This work was supported by the National Health and Medical Research Council (GNT1070860 and GNT0472667), the German Research Foundation (MZ 1479/1-1; AH 185/1-1), and the National Heart Foundation (XW100517, CR11M6066).
Author contributions
Conceptualization, C.E.H., I.A., and K.P.; methodology, M.Z., X.W., F.K., E.L., C.E.H., I.A., and K.P.; investigation, M.Z., X.W, B.L., E.L., F.K., V.C., Y.Y., D.H., X.M.G, H.K., and X.J.D.; formal analysis, M.Z., X.W., B.L., and E.L.; resources, K.P.; writing - original draft, M.Z., and K.P.; writing - review & editing, I.A., J.J.H., and A.B.; project administration, K.P.; supervision, M.Z., X.J.D., J.J.H., A.B., C.E.H., I.A., and K.P.
Supplementary Material
Supplementary methods and figures.
Competing Interests
K. P. is an inventor on patents describing activated platelet-targeting recombinant antibodies.
References
1. World Health Organization. The top 10 causes of death in the world, 2000 and 2012. Fact sheet No. 310. http://www.who.int/mediacentre/factsheets/fs310/en/ (18 April 2017).
2. Tang XL, Li Q, Rokosh G, Sanganalmath SK, Chen N, Ou Q. et al. Long-Term Outcome of Administration of c-kitPOS Cardiac Progenitor Cells After Acute Myocardial Infarction: Transplanted Cells Do not Become Cardiomyocytes, but Structural and Functional Improvement and Proliferation of Endogenous Cells Persist for at Least One Year. Circ Res. 2016;118:1091-105
3. Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B. et al. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701-5
4. Ziegler M, Elvers M, Baumer Y, Leder C, Ochmann C, Schonberger T. et al. The bispecific SDF1-GPVI fusion protein preserves myocardial function after transient ischemia in mice. Circulation. 2012;125:685-96
5. Chong JJ, Yang X, Don CW, Minami E, Liu YW, Weyers JJ. et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature. 2014;510:273-7
6. Schachinger V, Erbs S, Elsasser A, Haberbosch W, Hambrecht R, Holschermann H. et al. Improved clinical outcome after intracoronary administration of bone-marrow-derived progenitor cells in acute myocardial infarction: final 1-year results of the REPAIR-AMI trial. European heart journal. 2006;27:2775-83
7. Tatsumi T, Ashihara E, Yasui T, Matsunaga S, Kido A, Sasada Y. et al. Intracoronary transplantation of non-expanded peripheral blood-derived mononuclear cells promotes improvement of cardiac function in patients with acute myocardial infarction. Circ J. 2007;71:1199-207
8. Bolli R, Chugh AR, D'Amario D, Loughran JH, Stoddard MF, Ikram S. et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet. 2011;378:1847-57
9. Schaefer A, Zwadlo C, Fuchs M, Meyer GP, Lippolt P, Wollert KC. et al. Long-term effects of intracoronary bone marrow cell transfer on diastolic function in patients after acute myocardial infarction: 5-year results from the randomized-controlled BOOST trial-an echocardiographic study. Eur J Echocardiogr. 2010;11:165-71
10. Lunde K, Solheim S, Aakhus S, Arnesen H, Abdelnoor M, Egeland T. et al. Intracoronary injection of mononuclear bone marrow cells in acute myocardial infarction. The New England journal of medicine. 2006;355:1199-209
11. Surder D, Manka R, Lo Cicero V, Moccetti T, Rufibach K, Soncin S. et al. Intracoronary injection of bone marrow-derived mononuclear cells early or late after acute myocardial infarction: effects on global left ventricular function. Circulation. 2013;127:1968-79
12. Terrovitis JV, Smith RR, Marban E. Assessment and optimization of cell engraftment after transplantation into the heart. Circ Res. 2010;106:479-94
13. Hofmann M, Wollert KC, Meyer GP, Menke A, Arseniev L, Hertenstein B. et al. Monitoring of bone marrow cell homing into the infarcted human myocardium. Circulation. 2005;111:2198-202
14. Barbash IM, Chouraqui P, Baron J, Feinberg MS, Etzion S, Tessone A. et al. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: feasibility, cell migration, and body distribution. Circulation. 2003;108:863-8
15. Aicher A, Brenner W, Zuhayra M, Badorff C, Massoudi S, Assmus B. et al. Assessment of the tissue distribution of transplanted human endothelial progenitor cells by radioactive labeling. Circulation. 2003;107:2134-9
16. Kanelidis A, Premer C, Lopez JG, Balkan W, Hare JM. Route of Delivery Modulates the Efficacy of Mesenchymal Stem Cell Therapy for Myocardial Infarction: A Meta-Analysis of Preclinical Studies and Clinical Trials. Circ Res. 2017;120:1139-1150
17. Lalit PA, Hei DJ, Raval AN, Kamp TJ. Induced pluripotent stem cells for post-myocardial infarction repair: remarkable opportunities and challenges. Circ Res. 2014;114:1328-45
18. Xu Y, Huo Y, Toufektsian MC, Ramos SI, Ma Y, Tejani AD. et al. Activated platelets contribute importantly to myocardial reperfusion injury. American journal of physiology Heart and circulatory physiology. 2006;290:H692-9
19. Liu Y, Gao XM, Fang L, Jennings NL, Su Y, Q X. et al. Novel role of platelets in mediating inflammatory responses and ventricular rupture or remodeling following myocardial infarction. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:834-41
20. von Elverfeldt D, Maier A, Duerschmied D, Braig M, Witsch T, Wang X. et al. Dual-contrast molecular imaging allows noninvasive characterization of myocardial ischemia/reperfusion injury after coronary vessel occlusion in mice by magnetic resonance imaging. Circulation. 2014;130:676-87
21. Armstrong PC, Peter K. GPIIb/IIIa inhibitors: from bench to bedside and back to bench again. Thrombosis and haemostasis. 2012;107:808-14
22. Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9:182-212
23. Wang X, Hagemeyer CE, Hohmann JD, Leitner E, Armstrong PC, Jia F. et al. Novel single-chain antibody-targeted microbubbles for molecular ultrasound imaging of thrombosis: validation of a unique noninvasive method for rapid and sensitive detection of thrombi and monitoring of success or failure of thrombolysis in mice. Circulation. 2012;125:3117-26
24. Ziegler M, Hohmann JD, Searle AK, Abraham MK, Nandurkar HH, Wang X. et al. A single-chain antibody-CD39 fusion protein targeting activated platelets protects from cardiac ischaemia/reperfusion injury. European heart journal. 2017 [Epub ahead of print]
25. Stoll P, Bassler N, Hagemeyer CE, Eisenhardt SU, Chen YC, Schmidt R. et al. Targeting ligand-induced binding sites on GPIIb/IIIa via single-chain antibody allows effective anticoagulation without bleeding time prolongation. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:1206-12
26. Wang X, Gkanatsas Y, Palasubramaniam J, Hohmann JD, Chen YC, Lim B. et al. Thrombus-Targeted Theranostic Microbubbles: A New Technology towards Concurrent Rapid Ultrasound Diagnosis and Bleeding-free Fibrinolytic Treatment of Thrombosis. Theranostics. 2016;6:726-38
27. Ziegler M, Alt K, Paterson BM, Kanellakis P, Bobik A, Donnelly PS. et al. Highly Sensitive Detection of Minimal Cardiac Ischemia using Positron Emission Tomography Imaging of Activated Platelets. Scientific reports. 2016;6:38161
28. Fisher SA, Doree C, Mathur A, Martin-Rendon E. Meta-analysis of cell therapy trials for patients with heart failure. Circ Res. 2015;116:1361-77
29. Dixit P, Katare R. Challenges in identifying the best source of stem cells for cardiac regeneration therapy. Stem cell research & therapy. 2015;6:26
30. Faiella W, Atoui R. Therapeutic use of stem cells for cardiovascular disease. Clin Transl Med. 2016;5:34
31. Bolli R, Ghafghazi S. Stem cells: Cell therapy for cardiac repair: what is needed to move forward? Nat Rev Cardiol. 2017;14:257-8
32. Kolvenbach R, Kreissig C, Cagiannos C, Afifi R, Schmaltz E. Intraoperative adjunctive stem cell treatment in patients with critical limb ischemia using a novel point-of-care device. Ann Vasc Surg. 2010;24:367-72
33. de Mello VD, Kolehmanien M, Schwab U, Pulkkinen L, Uusitupa M. Gene expression of peripheral blood mononuclear cells as a tool in dietary intervention studies: What do we know so far? Mol Nutr Food Res. 2012;56:1160-72
34. Hoogduijn MJ, van den Beukel JC, Wiersma LC, Ijzer J. Morphology and size of stem cells from mouse and whale: observational study. Bmj. 2013;347:f6833
35. Meirelles Lda S, Nardi NB. Murine marrow-derived mesenchymal stem cell: isolation, in vitro expansion, and characterization. Br J Haematol. 2003;123:702-11
36. Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y. et al. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12313-8
37. Zwetsloot PP, Vegh AM, Jansen of Lorkeers SJ, van Hout GP, Currie GL, Sena ES. et al. Cardiac Stem Cell Treatment in Myocardial Infarction: A Systematic Review and Meta-Analysis of Preclinical Studies. Circ Res. 2016;118:1223-32
38. Miles C, Sanchez MJ, Sinclair A, Dzierzak E. Expression of the Ly-6E.1 (Sca-1) transgene in adult hematopoietic stem cells and the developing mouse embryo. Development. 1997;124:537-47
39. Suzuki H, Shibata R, Kito T, Ishii M, Li P, Yoshikai T. et al. Therapeutic angiogenesis by transplantation of induced pluripotent stem cell-derived Flk-1 positive cells. BMC Cell Biol. 2010;11:72
40. Sidney LE, Branch MJ, Dunphy SE, Dua HS, Hopkinson A. Concise review: evidence for CD34 as a common marker for diverse progenitors. Stem Cells. 2014;32:1380-9
41. Klomp M, Beijk MA, Varma C, Koolen JJ, Teiger E, Richardt G. et al. 1-year outcome of TRIAS HR (TRI-stent adjudication study-high risk of restenosis) a multicenter, randomized trial comparing genous endothelial progenitor cell capturing stents with drug-eluting stents. JACC Cardiovasc Interv. 2011;4:896-904
42. Kamihata H, Matsubara H, Nishiue T, Fujiyama S, Amano K, Iba O. et al. Improvement of collateral perfusion and regional function by implantation of peripheral blood mononuclear cells into ischemic hibernating myocardium. Arteriosclerosis, thrombosis, and vascular biology. 2002;22:1804-10
43. Hofmann U, Frantz S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ Res. 2015;116:354-67
44. Zouggari Y, Ait-Oufella H, Bonnin P, Simon T, Sage AP, Guerin C. et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. 2013;19:1273-80
45. Goodchild TT, Robinson KA, Pang W, Tondato F, Cui J, Arrington J. et al. Bone marrow-derived B cells preserve ventricular function after acute myocardial infarction. JACC Cardiovasc Interv. 2009;2:1005-16
46. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221-41
47. Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J. et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. Journal of molecular and cellular cardiology. 2013;62:24-35
48. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159-73
49. Ruparelia N, Digby JE, Jefferson A, Medway DJ, Neubauer S, Lygate CA. et al. Myocardial infarction causes inflammation and leukocyte recruitment at remote sites in the myocardium and in the renal glomerulus. Inflamm Res. 2013;62:515-25
50. Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437-45
51. Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA, Lucchesi BR. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation. 1983;67:1016-23
52. Heidt T, Ehrismann S, Hovener JB, Neudorfer I, Hilgendorf I, Reisert M. et al. Molecular Imaging of Activated Platelets Allows the Detection of Pulmonary Embolism with Magnetic Resonance Imaging. Scientific reports. 2016;6:25044
53. von zur Muhlen C, von Elverfeldt D, Moeller JA, Choudhury RP, Paul D, Hagemeyer CE. et al. Magnetic resonance imaging contrast agent targeted toward activated platelets allows in vivo detection of thrombosis and monitoring of thrombolysis. Circulation. 2008;118:258-67
54. Tay C, Liu YH, Hosseini H, Kanellakis P, Cao A, Peter K. et al. B-cell-specific depletion of tumour necrosis factor alpha inhibits atherosclerosis development and plaque vulnerability to rupture by reducing cell death and inflammation. Cardiovasc Res. 2016;111:385-97
55. Ying W, Wollam J, Ofrecio JM, Bandyopadhyay G, El Ouarrat D, Lee YS. et al. Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. The Journal of clinical investigation. 2017;127:1019-30
56. Gao XM, Xu Q, Kiriazis H, Dart AM, Du XJ. Mouse model of post-infarct ventricular rupture: time course, strain- and gender-dependency, tensile strength, and histopathology. Cardiovasc Res. 2005;65:469-77
Author contact
![]() Corresponding author: karlheinz.peteredu.au
Corresponding author: karlheinz.peteredu.au