Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(12):3090-3105. doi:10.7150/thno.18114 This issue Cite
Research Paper
The gRNA-miRNA-gRNA Ternary Cassette Combining CRISPR/Cas9 with RNAi Approach Strongly Inhibits Hepatitis B Virus Replication
Jie Wang1*, Ran Chen1*, Ruiyang Zhang1*, Shanlong Ding1, Tianying Zhang2, Quan Yuan2, Guiwen Guan1, Xiangmei Chen1, Ting Zhang1, Hui Zhuang1, Frederick Nunes3, Timothy Block4, Shuang Liu5, Zhongping Duan5, Ningshao Xia2 ![]() , Zhongwei Xu3
, Zhongwei Xu3 ![]() , Fengmin Lu1
, Fengmin Lu1 ![]()
1. State Key Laboratory of Natural and Biomimetic Drugs, Department of Microbiology & Infectious Disease Center, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, China;
2. State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics, School of Public Health, Xiamen University, Xiamen, China;
3. Department of Gastroenterology, Pennsylvania Hospital, University of Pennsylvania, Philadelphia, Pennsylvania, USA;
4. Baruch S. Blumberg Institute, Doylestown, Pennsylvania, USA;
5. Beijing Artificial Liver Treatment & Training Center, Beijing Youan Hospital, Capital Medical University, Beijing, China.
* These authors contributed equally to this work.
Received 2016-10-28; Accepted 2017-6-8; Published 2017-7-22
Abstract

The CRISPR/Cas9 system is a novel genome editing technology which has been successfully used to inhibit HBV replication. Here, we described a novel gRNA-microRNA (miRNA)-gRNA ternary cassette driven by a single U6 promoter. With an anti-HBV pri-miR31 mimic integrated between two HBV-specific gRNAs, both gRNAs could be separated from the long transcript of gRNA-miR-HBV-gRNA ternary cassette through Drosha/DGCR8 processing. The results showed that the gRNA-miR-HBV-gRNA ternary cassette could efficiently express two gRNAs and miR-HBV. The optimal length of pri-miRNA flanking sequence in our ternary cassette was determined to be 38 base pairs (bp). Besides, HBV-specific gRNAs and miR-HBV in gRNA-miR-HBV-gRNA ternary cassette could exert a synergistic effect in inhibiting HBV replication and destroying HBV genome in vitro and in vivo. Most importantly, together with RNA interference (RNAi) approach, the HBV-specific gRNAs showed the potent activity on the destruction of HBV covalently closed circular DNA (cccDNA). Since HBV cccDNA is an obstacle for the elimination of chronic HBV infection, the gRNA-miR-HBV-gRNA ternary cassette may be a potential tool for the clearance of HBV cccDNA.
Keywords: CRISPR/Cas9, miRNA, gRNA-miRNA-gRNA ternary cassette, HBV, cccDNA.
Introduction
Hepatitis B virus (HBV) infection is an important public health problem worldwide. It has been estimated that as much as 40% of men and 15% of women with perinatally acquired chronic HBV infection will die of liver cirrhosis or hepatocellular carcinoma [1].
HBV is a small enveloped hepatotropic DNA virus, with a ~3.2 kb long, partially double-stranded relaxed circular DNA (rcDNA) genome, which is reversely transcribed from the pregenomic RNA (pgRNA). Upon infection, rcDNA is transported into nucleus where it is converted into covalently closed circular DNA (cccDNA). As a template for HBV replication, cccDNA is responsible for all viral transcripts including pgRNA [2, 3]. Chronic HBV infection is characterized by the persistence of HBV cccDNA in the nucleus of the infected hepatocyte. Unfortunately, none of the currently used antiviral agent could efficiently eradicate cccDNA, so it is still difficult to achieve clinical cure of chronic hepatitis B (CHB) [4-6]. Besides, we and other groups have proved that the encapsidated HBV pgRNAs can be released as extracellular HBV pgRNA virions, which may be associated with persistence of viral infection and rebound [7, 8]. As we know, HBV pgRNA is directly transcribed from cccDNA. Therefore, a new strategy aimed to eradicate cccDNA is of clinical significance.
Currently, genome editing technologies bring light to the antiviral therapy of HBV, including zinc finger nucleases (ZFNs) [9, 10], transcription activator-like effector nucleases (TALENs) [11, 12], and the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) system [13-16]. Compared with ZFNs and TALENs, CRISPR/Cas system is superior for its easier genome editing [17-19]. Type II CRISPR/Cas system consisting of DNA endonuclease Cas9, CRISPR RNAs (crRNAs) and trans-activating crRNAs (tracrRNAs) is the simplest CRISPR/Cas system, particularly after fusing the crRNA and tracrRNA into a single chimeric guide RNA (gRNA) [20, 21]. The specific gRNA directs Cas9 to the target DNA sequence called the protospacer on the target DNA next to the protospacer adjacent motif (PAM) for site-specific cleavage [22, 23]. For anti-HBV therapy, we have proved that dual gRNAs guided CRISPR/Cas9 system showed a synergistic effect in suppressing HBV replication [24]. Besides, a lot of miRNAs have been proved to be involved in the control of HBV replication by directly targeting to HBV mRNAs or by indirectly regulating cellular transcription factors [25]. Recently, TALENs or CSRISPR/Cas9 nucleases were used to site-specifically integrate an anti-hepatitis C virus (HCV) shmiRNA into an engineered genomic miRNA (miR-122/hcr) locus in hepatoma cells, and successfully impair HCV replication, which was an original combination of DNA engineering and RNAi expression technologies [26].
There are several approaches to enable simultaneous expression of two gRNAs, including the crRNA array [27], U6-gRNA cassette in tandem [28], dimeric RNA-guided FokI nucleases (RFNs) [29] and Cas6/Csy4-based RNA processing strategies [29, 30]. Here, we developed a powerful approach in inhibiting HBV replication, which combined the CRISPR/Cas9 and RNAi approaches together. In this approach, a gRNA-microRNA (miRNA)-gRNA ternary cassette was designed, which used the endogenous nuclear RNaseIII enzyme Drosha/DGCR8 to process pri-miRNA and produce a pre-miRNA and two HBV-specific gRNAs. To increase the antiviral efficiency, the anti-HBV pri-miR-31 mimic was used to produce miR-HBV, which had a similar structure to that of naturally occurring pri-miR-31 [31, 32]. Moreover, we evaluated the potential value of using the gRNA-miR-HBV-gRNA ternary cassette to clear HBV cccDNA.
Materials and Methods
Plasmids
The gRNA/Cas9 dual expression vector pSpCas9 (BB)-2A-GFP (PX458) was obtained from Addgene (Addgene, Cambridge, MA). The pSpCas9(BB)-2A-GFP-XhoI/HindIII (PX458M) plasmid was constructed by inserting XhoI and HindIII restriction endonucleases cutting sites into PX458 plasmid, using QuikChange site-directed mutagenesis method (Agilent technologies, Santa Clara, CA). The primers were 5'- TGGCACCGAGTCGGTGCCTCGAGTCCAAGCTTAGGCTAGTCCGTTTTTAGCGC-3' (Forward) and 5'- GCGCTAAAAACGGACTAGCCTAAGCTTGGACTCGAGGCACCGACTCGGTGCCA-3' (Reverse). The pSpgRNA3-Pri-miR31(51+51)/miR-HBV-gRNA2-Cas9(BB)-2A-GFP (3-H51-2) vector was constructed by ligating the annealed oligo-nucleotides of gRNA3 into the PX458M plasmid digested with Bbsl enzyme, then the plasmid was digested with XhoI and HindIII enzymes (New England Biolabs, Ipswich, MA), and ligated with synthesized gene fragment pri-miR-31(51+51)/miR-HBV-gRNA2. The gRNA3-H22-gRNA2 (3-H22-2), gRNA3-H30-gRNA2 (3-H30-2), gRNA3-H38-gRNA2 (3-H38-2) and gRNA3-mutant miR-HBV (38 bp flanking sequence)-gRNA2 (3-M38-2) plasmids were constructed by ligating the annealed oligo-nucleotides (Supplementary Table S1) or synthesized gene fragment (Supplementary Table S2) into the 3-H51-2 plasmid digested with XhoI and HindIII. The pSpgRNA3-gRNA2-Cas9(BB)-2A-GFP (gRNA3-gRNA2) plasmid was constructed by ligating the synthesized gene fragment (Supplementary Table S2) into the PX458M plasmid digested with XhoI and HindIII. The pGL3-HBV (1575-1604) plasmid was constructed by ligating the annealed oligo-nucleotides containing the miR-HBV target sequence [nt 1575-1595: CCGTGTGCACTTCGCTTCACC (Genebank accession AB014381)] and the pGL3-control plasmid digested with XbaI enzyme. The oligo-nucleotides were 5'- CTAGCCGTGTGCACTTCGCTTCACCTCTGCACGT-3' (Top) and 5'- CTAGACGTGCAGAGGTGAAGCGAAGTGCACACGG-3' (Bottom). The expression plasmids of PX458-miR-HBV and PX458-mutant miR-HBV was constructed by PCR from the expression plasmids of 3-H38-2 and 3-M38-2 ternary cassette respectively, and subsequently ligated PCR products and PX458M plasmid digested with Bbsl and HindIII. The sequences of oligo-nucleotides and synthesized gene fragment for constructing the HBV-specific double gRNAs/Cas9 dual expression vectors were listed in Table S2 and S3. The 1.2×HBV construct (pBB4.5-HBV1.2, genotype C) was constructed using a 1.2-fold length genome of genotype C HBV DNA sequence, based on pBB4.5-HBV1.3 (genotype D, G1896A mutation) which was kindly provided by professor Locarnini SA from the Victorian Infectious Diseases Reference Laboratory, Australia [33]. The HBV-expression vectors pGEM-HBV1.3A (genotype A) and pGEM-HBV1.3B (genotype B) were kindly provided by Professor Ningshao Xia from School of Public Health, Xiamen University, China.
Cell transfection
HepG2-NTCP-tet cells, in which the expression of sodium taurocholate cotransporting polypeptide (NTCP) gene was tetracycline inducible, was kindly provided by State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics, School of Public Health, Xiamen University. Xiamen, Fujian, China. HepG2-NTCP-tet cells, human liver cancer cell lines HuH7 and HepAD38 (stable expression of HBV) were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA). HuH7 and HepAD38 cells were seeded in a 12-well plate at 2×105 cells/well. HuH7 cells were co-transfected with HBV expression vectors (HBV of genotype A, B or C) and HBV-specific gRNA/Cas9 dual expression vector (pSpCas9(BB)-2A-GFP) by lipofectamain 2000 (Life technologies, Carlsbad, CA). HepAD38 cells and HepG2-NTCP-tet cells were transfected with HBV specific gRNA/Cas9 dual expression vector by lipofectamain 3000 (Life technologies).
Dual luciferase reporter assay
HuH7 cells were seeded in a 12-well plate at 2×105 cells/well and allowed to grow for 16-24 h, then transiently co-transfected with the pGL3-HBV (1575-1604) or pGL3-control, PRL-TK and gRNA3-H22-gRNA2 or gRNA3-H30-gRNA2 or gRNA3-H38-gRNA2 or gRNA3-H51-gRNA2 plasmids. The luciferase activity in each well was quantified using a dual luciferase reporter kit (Promega, Madison, WI) at 36 hours post transfection, and was detected by EnSpire multimode plate reader (PerlinElmer, Waltham, MA) following the manufacturer's protocol.
RNA extraction, PolyA tailing, Reverse transcription and PCR amplification
Small RNA was extracted by mirVanaTM miRNA Isolation Kit (Life technologies), and the RNA was added polyA using Poly(A) Tailing Kit (Life technologies) in accordance with manufacturer's instructions. The reverse transcription was performed using Transcriptor First Strand cDNA Synthesis Kit (Roche, Basel, Switzerland) in accordance with manufacturer's instructions. The reverse transcription primers RT-miR-HBV (5'-ACCACGCTATCGCTACTCACTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTCCGTGTGC-3'), RT-mutant miR-HBV (5'- ACCACGCTATCGCTACTCACTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTAGCTATGC-3') and RT-gRNA3 (5'-ACCACGCTATCGCTACTCACTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTCCTCTCCAG-3') were used for the detection of mature miR-HBV, mutant miR-HBV and gRNA3 carrying the flanking sequence of anti-HBV pri-miR31 mimic separated by the endogenous Drosha/DGCR8 system. The reverse transcription primer RT-random (5'-ACCACGCTATCGCTACTCACTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-3') was used for the detection of gRNA2 and 5S rRNA. PCR or qPCR amplification was performed using the following primers to detect the small RNAs. Primers miR-HBVF: 5'-GGTGAAGCGAAGTGCACACG-3', miR-HBVmF: 5'-GGTGAAGCGAATGGCATAGC-3', 5'-CAAGCCTCCAAGCTGTGCCT-3', 5'-GGTTGCGTCAGCAAACACT-3' and AnchoredR: 5'-ACCACGCTATCGCTACTCAC-3' were used to detect mature miR-HBV, mutant miR-HBV, gRNA3 and gRNA2, respectively. Primers 5SF: 5'-5SF: CCATACCACCCTGAACGC-3' and 5SR: 5'-AGCCTACAGCACCCGGTAT-3' were used to detect 5S rRNA.
HBV infection
HBV infection of HepG2-NTCP-tet cells was conducted as previously described [34]. Briefly, HepG2-NTCP-tet cells which were maintained in DMEM medium containing 2μg/ml Doxycycline (Merk, Kenilworth, NJ) for 4 days were seeded in a 24-well plate (2×105 cells/well), and were inoculated with 2×107 copies of genome equivalent HBV in the presence of 4% PEG 8000 for 20 hr. HepG2-NTCP cells cells were then washed with PBS six times and maintained in fresh DMEM medium.
Detection of HBsAg and HBeAg
Cell pellet and culture supernatant were harvested at 72 hours post transfection, and the levels of HBsAg and HBeAg were detected by a time-resolved fluoroimmunoassay (TRFIA) according to manufacturer's instructions (PerkinElmer). In brief, culture supernatant (100 μl) was added into a microtiter plate coated with anti-HBs or anti-HBe and shaked for 40 min in room temperature, then washed for 4 times. Europium-labeled anti-HBsAg or anti-HBeAg was diluted 1:50 by HBsAg or HBeAg dilution buffer and added at 100 μl per well, shaked for 40 min in room temperature, then washed for 6 times. At last, after incubation with enhancement solution (100 μl) for 5 min, the plates were read using Anytest reader (PerkinElmer).
Detection of HBV DNA fragments cleaved by dual gRNAs
DNA was extracted from cells using QIAGEN DNA mini kit (Qiagen, Hilden, Germany) in accordance with the manufacturer's instruction. Specific primers listed below were used to PCR amplify the cleaved HBV DNA fragments. HBV1856F: 5'-CCTACTGTTCAAGCCTCCAAGC-3'; HBV434R: 5'-AGAAGATGAGGCATAGCAGC-3'; HBV321F: 5'-CAACCTCCAATCACTCACCAAC-3'; HBV2006R: 5'-CTAGGAGTTCCGCAGTATGGAT-3'. HBV1856F and HBV434R were used for detection the fragment cleaved by dual gRNAs of gRNA1+4; HBV321F and HBV2006R were used for detection the fragment cleaved by dual gRNAs of gRNA2+3, respectively.
PCR reaction mixture (20 μl) contained 2×Taq mix 10 μl, forward primer (10 μM) 1 μl, reverse primer (10 μM) 1 μl, DNA template (200 ng/μl) 1 μl and double distilled water 7 μl. The reaction mixture was denatured at 95°C for 5 min, followed by 35 cycles at 95°C for 30 s, 60°C for 30 s, 72°C for 1 min, and at last 72°C for 5 min. Agarose gel (1.5%) was used for separation of DNA fragments with different length. The DNA fragment of expected length was sequenced.
Southern Blot
Total cellular DNA was extracted from the transfected HuH7 cells (3 × 106 cells each group) using QIAGEN DNA mini kit (Qiagen). DNA samples were resolved on a 1.0% agarose gel, transferred to a nylon membrane (Bio-Rad, Hercules, CA, USA), and hybridized with a digoxin-labeled DNA fragment covering the whole HBV genome. The probe was amplified from pGEM-HBV1.3A and pGEM-HBV1.3B plasmid, and labeled with PCR DIG Probe Synthesis Kit (Roche, Mannheim, Germany), respectively. The primers were 5'-TTTGCCTTCTGACTTCTTTC-3' (1954-1973 nt) and 5'-CCAAATTCTTTATAMGGGTC-3' (1929-1910 nt).
Western Blot
Western Blot analysis was performed as previously described [7]. Briefly, the protein lysates (total 60 μg) dissolved in 1×laemmli buffer (with 5% 2-mercaptoethanol), boiled for 10 min and resolved on 4-12% gradient SDS-PAGE gel (BioRad). Subsequently, the proteins were transferred to a nitrocellulose membrane and incubated with mouse anti-core protein antibody (1μg/mL, a gift from State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics, School of Public Health, Xiamen University. Xiamen, Fujian, China) and rabbit anti-β-actin antibody (1μg/mL, Santa Cruz Biotech, Dallas, TX, USA) overnight at 4℃. Reactive proteins were developed with anti-mouse and anti-rabbit antibodies conjugated to horseradish peroxidase (Zhongshan Golden Bridge, Beijing, China), and visualized using SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol.
Detection of HBV cccDNA
Two gRNA/Cas9 dual expression vectors were co-transfected into HepAD38 cells at least 7 days after tetracycline removed, and the cells were harvested at 72 hours post of transfection. The DNA was extracted from cells using QIAGEN DNA mini kit (Qiagen) in accordance with the manufacturer's instruction. To obtain cccDNA, KCl precipitation was used to isolate HBV cccDNA [6, 35], and plasmid-safeTM ATP-dependent DNase (PSAD) (Epicentre, Madison, WI) were used to enhance the specificity of cccDNA detection, for it can digest the integrated linear HBV DNA, the residue HBV rcDNA, double-strand linear (DSL) and single-strand (SS) DNA [36, 37]. Afterwards, rolling Circle Amplification (RCA) was conducted to selectively amplify cccDNA. Finally, PCR was performed using RCA products as template, and using cccDNA specific primers which target the gap region of HBV genome.
Hydrodynamic injection (HDI)
The hydrodynamic injection was performed as previously described [38]. Briefly, 8 µg 1.2×HBV expression and 24µg control (vector), 3-2 binary or 3-H38-2 ternary cassette expression plasmids were injected into the tail vein of C57BL/6 mice (5-7 weeks old) in 2 ml PBS to each mouse (almost equivalent to 8% of the mouse body weight) within 5 seconds. There were 8 mice in each group. Sera were collected at 3, 5 and 7 days post-injection. Two mice were sacrificed at 72 hours post-injection, and the liver was removed. One part of liver was frozen in liquid nitrogen and grinded for DNA extraction, the other part of liver was fixed for immunohistochemistry analysis. The HBsAg levels in sera and HBV DNA detection were performed as above description. All mouse care and experiments were carried out according to the guidelines established by the Institutional Animal Care and Use Committee at the Peking University Health Science Center. All mouse experiments were approved by the Institutional Animal Care and Use Committee at the Peking University Health Science Center.
Immunohistochemistry (IHC)
The mice were sacrificed at 72 hours post-injection, and the liver was removed and fixed in formalin (3.7% Formaldehyde in PBS) for immunohistochemistry analysis, HBcAg was detected by immunohistochemical staining by rabbit anti-HBc (DAKO, Carpinteria, CA; Biomeda, Foster City, CA) and MaxVision TM HRP-Polymer anti-Rabbit IHC Kit (Maixin, Fuzhou, China).
Statistical analysis
For statistical analyses, Mann-Whitney U test and Kruskal-Wallis H (K) test were conducted using (SPSS Statistics 21.0). In all cases, a P value of less than 0.05 was considered significant.
Results
Construction of HBV-specific gRNA-miR-HBV-gRNA ternary cassette
Structure of the ternary cassette producing two HBV-specific gRNAs and a miR-HBV is depicted schematically in Figure 1A. This gRNA-miR-HBV-gRNA ternary cassette should express a long RNA transcript containing two HBV-specific gRNAs and an anti-HBV pri-miR-31 mimic under the control of a U6 RNA polymerase (Pol) III promoter. Subsequently, the anti-HBV pri-miR-31 mimic would be processed by the endogenous Drosha/DGCR8, thus a pre-miR-HBV and two HBV-specific gRNAs containing part of pri-miR-31 flanking sequence would be ultimately separated (Figure 1A). To design the HBV-specific gRNA, the target sites of HBV-specific gRNAs were searched at different regions of HBV genome, which were conserved among genotypes A-D (Figure 1B). The target sequences of HBV-specific gRNAs were shown in Supplementary Table S3.
The schematic diagram of the HBV-specific gRNA-miR-HBV-gRNA cassette. (A) Schematic illustration of gRNA-miR-HBV-gRNA cassette. (B) The locations of gRNA and miR-HBV target sites in HBV genome. nt: nucleotide. (C) The sequences of pri-miR31 and anti-HBV pri-miR-31 mimics with different lengths (base pairs, bp) of flanking sequences.
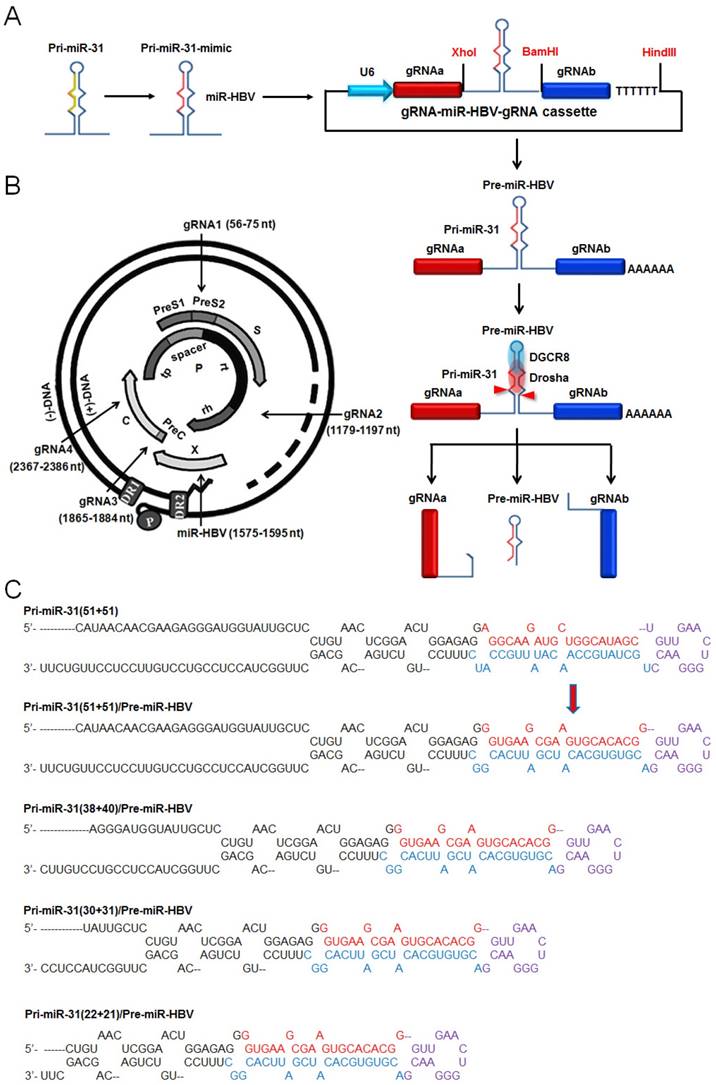
The anti-HBV pri-miR-31 mimic has a similar structure to that of naturally occurring pri-miR-31, which was designed by replacing the mature miR-31 with miR-HBV sequence predicted by the MFold algorithm [32]. The target site of miR-HBV was also shown in Figure 1B. The length of pri-miRNA backbones is considered as a variable affecting the recognition of Drosha/DGCR8 and the flanking sequences of gRNA may interfere with the gRNA-mediated cleavage of target sequences. Therefore, to optimize the anti-HBV efficiency of gRNA-miR-HBV-gRNA cassette, different lengths of anti-HBV pri-miR-31 mimics were designed (Figure 1C). The full DNA sequences of all gRNA-miR-HBV-gRNA cassettes were shown in Supplementary Table S4.
The gRNA-miR-HBV-gRNA ternary cassette can efficiently produce HBV-specific gRNAs and miR-HBV
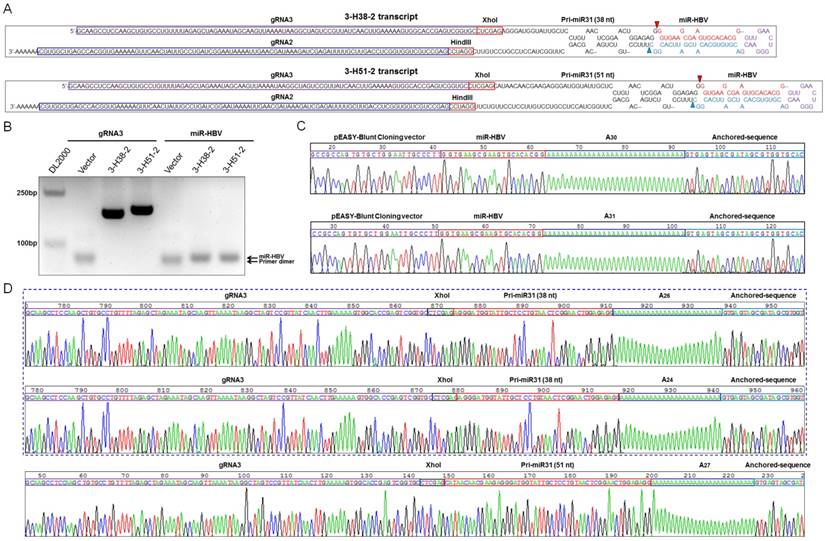
Based on the length of anti-HBV pri-miR-31 mimics, we constructed a series of gRNA3-miR-HBV-gRNA2 (3-Hn-2) cassettes including 3-H22-2, 3-H30-2, 3-H38-2 and 3-H51-2, which were inserted into pSpCas(BB)-2A-GFP (PX458) plasmid respectively. Two gRNA3-miR-HBV-gRNA2 cassettes (3-H38-2 and 3-H51-2) were chosen to confirm the expression of individual gRNAs and miR-HBV (Figure 2A). To specifically amplify the separated HBV-specific gRNA and miR-HBV, a polyA tailing method combined with anchoring random sequence through reverse transcription and PCR amplification was employed. Since gRNA2 was located at the 3' end of gRNA3-miR-HBV-gRNA2 ternary cassette, both gRNA2 in the long gRNA3-miR-HBV-gRNA2 transcript and gRNA2 separated by endogenous Drosha/DGCR8 could be added polyA. Consequently, polyA-tailed gRNA2 could not specifically reflect the formation of separated gRNA2. Therefore, gRNA3 and miR-HBV were detected in this study. As shown in Figure 2B, HBV-specific gRNA3 and miR-HBV were successfully expressed and separated from 3-H38-2 and 3-H51-2 ternary cassettes. Moreover, these PCR products were purified and inserted into pEASY-Blunt Cloning vector, and the expression of mature miR-HBV and the polyA-tailed gRNA3 carrying different flanking sequences of anti-HBV pri-miR-31 mimics were further confirmed by the clone-based sequencing analyses (Figure 2C and 2D).
Detection on the expression of functional HBV-specific gRNAs and miR-HBV from gRNA3-miR-HBV-gRNA2 cassette. (A) The sequences of 3-H38-2 and 3-H51-2 ternary cassettes. The triangle indicates the cleavage site of endogenous Drosha/DGCR8. (B) The expression plasmid containing 3-H38-2 or 3-H51-2 ternary cassette was transfected into HuH7 cells. Cellular small RNA (<200 bp) was harvested at 48 hours after transfection, then polyA tailing, anchoring random sequence through reverse transcription and PCR amplification were performed to detect the expression of separated HBV-specific gRNA3 and miR-HBV. Clone-based sequencing analyses of the above PCR products were conducted to confirm the expression of mature miR-HBV (C) and HBV-specific gRNA3 with flanking sequence (D) of anti-HBV pri-miR-31 mimics.
The flanking sequence of anti-HBV pri-miR-31-mimic can influence the efficiency of ternary cassette in suppressing HBV replication
To further demonstrate the functional miR-HBV was expressed from gRNA-miR-HBV-gRNA ternary cassette, the HBV specific sequence (1575-1604 nucleotides in HBV genome) containing the target sequence (1575-1595 nucleotides in HBV genome) of miR-HBV was inserted into the 3'UTR region of luciferase gene in pGL3-control vector, and the production of functional miR-HBV was assessed by a dual luciferase assay. The results revealed that all gRNA3-miR-HBV-gRNA2 ternary cassettes with different lengths of anti-HBV pri-miR-31-mimic could efficiently suppress the activity of luciferase gene which carries the target sequence of miR-HBV at the 3'UTR region, and the efficiency was increased along with the increase in the length of anti-HBV pri-miR-31 mimic (Figure 3A). Consistently, the level of mature miR-HBV expressed from gRNA3-miR-HBV-gRNA2 ternary cassette was also increased along with the increase in the length of anti-HBV pri-miR-31 mimic, and the longest anti-HBV pri-miR-31 mimic carrying 51 bp flanking sequences could produce the most mature miR-HBV in this study (Figure 3B).
The effect of anti-HBV pri-miR-31-mimic flanking sequence in ternary cassette. (A) A dual-luciferase assay was conducted to detect the production of functional miR-HBV from gRNA3-miR-HBV-gRNA2 cassettes with different lengths of anti-HBV pri-miR-31 mimic in HuH7 cells co-transfected with pGL3-HBV (1575-1604) or pGL3-control, PRL-TK and each of the plasmids containing gRNA3-miR-HBV-gRNA2 cassettes with different length of anti-HBV pri-miR-31 mimics. The vectors pGL3-control and PRL-TK were used as negative control and internal control, respectively. Data was shown as mean±SD of 4 independent experiments. (B) A polyA tailing reaction and a quantitative reverse transcription-PCR (qRT-PCR) were conducted to detect the production of mature miR-HBV in HuH7 cells transfected with the plasmids containing gRNA3-miR-HBV-gRNA2 cassettes with different length of anti-HBV pri-miR-31 mimics. (C) Schematic illustration of gRNA-gRNA binary cassette. (D) The expression plasmid of 1.2×HBV was co-transfected with the expression plasmid containing 3-2 binary cassette or gRNA3-miR-HBV-gRNA2 ternary cassette with different length of anti-HBV pri-miR-31 mimic flanking sequence in HuH7 cells at the ratio of 3:1 and 1:3. HBsAg levels in the cell culture supernatant were measured using a time-resolved fluoroimmunoassay at 72 hours after transfection. Data was shown as mean±SD of 5 independent experiments. (* indicated P<0.05, Mann-Whitney U test). PX458 plasmid was used as a vector control. 5S rRNA was used as the internal control.
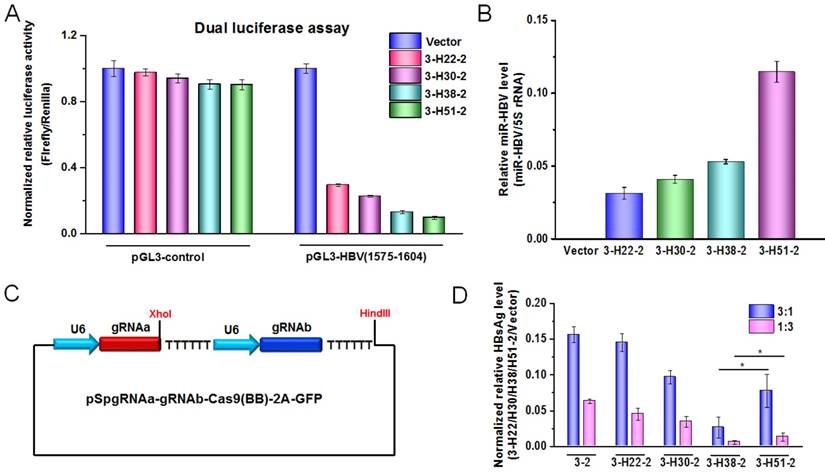
To assess the antiviral efficiency of gRNA-miR-HBV-gRNA ternary cassette, the gRNA3-gRNA2 binary cassette (3-2) in which two gRNAs were transcribed from different U6 promoters was constructed (Figure 3C). As shown in Figure 3D, all of the gRNA3-miR-HBV-gRNA2 ternary cassettes exhibited higher efficiency in suppressing HBV replication than that of gRNA-gRNA binary cassette. Whereas, not the 3-H51-2 ternary cassette carrying the longest (51 bp) flanking sequence of anti-HBV pri-miR-31 mimic but the 3-H38-2 ternary cassette showed the highest efficiency in suppressing HBV replication.
The miR-HBV and two HBV-specific gRNAs in gRNA-miR-HBV-gRNA ternary cassette can get a synergistic effect in suppressing HBV replication
To explore the effect of miR-HBV and two HBV-specific gRNAs expressed from gRNA-miR-HBV-gRNA ternary cassette in suppressing HBV replication, the sequence of miR-HBV in 3-H38-2 ternary cassette was mutated to lose the recognition ability of its target sequence, which was named as gRNA3-miR-HBVm-gRNA2 (3-M38-2) ternary cassette (Figure 4A). Meanwhile, the mutant miR-HBV was confirmed to lose the ability in suppressing HBV replication, which could be used as a negative control of miR-HBV (Figure 4B). Besides, the vector control plasmid used in each experiment was pSpCas9 (BB)-2A-GFP (PX458) which expressed a nonsense gRNA (GGGTCTTCGAGAAGACCT). Thus, the vector control could be used as a negative control of two gRNAs.
The function of miR-HBV in gRNA-miR-HBV-gRNA ternary cassettes. (A) A dual-luciferase assay was conducted to analyze the effect of 3-M38-2 ternary cassette in the target sequence of miR-HBV. The 3-M38-2 ternary cassette was used as a positive control. Data was shown as mean±SD of 5 independent experiments. (B) The expression plasmids of 1.2×HBV (genotype C) and miR-HBV or mutant miR-HBV (miR-HBVm) were co-transfected into HuH7 cells. The HBsAg levels in the cell and culture supernatant of HuH7 cells were measured using a time-resolved fluoroimmunoassay at 48 hours after transfection. Data was shown as mean±SD of 4 independent experiments. (C) The expression plasmids of 1.2×HBV and 3-2 binary cassette, 3-M38-3 or 3-H38-2 ternary cassette were co-transfected into HuH7 cells. The levels of HBsAg and HBeAg in the culture supernatant were measured using a time-resolved fluoroimmunoassay at 72 hours after transfection. Data was shown as mean±SD of 5 independent experiments. (D) The level of gRNA2 in the HuH7 cells transfected with the expression plasmid of vector control, 3-2 binary or 3-H38-2 ternary cassette was detected by qRT-PCR (SYBR Green). The level of (E) gRNA3 carrying flanking sequence of anti-HBV pri-miR-31 mimic and (F) mature miRNA (miR-HBV or mutant miR-HBV) in the HuH7 cells transfected with the expression plasmid of vector control, 3-2 binary or 3-H38-2 ternary cassette was detected by qRT-PCR (SYBR Green). Data was shown as mean±SD of 4 independent experiments. (** indicated P<0.01, ***indicated P<0.001, Mann-Whitney U test). PX458 plasmid was used as a vector control.
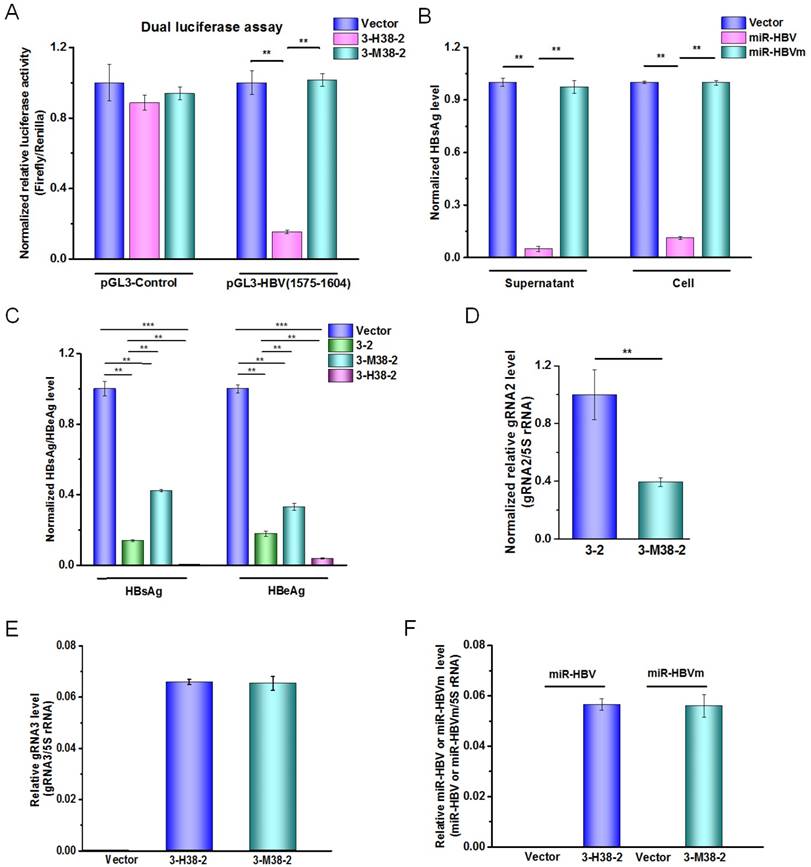
As shown in Figure 4C, both 3-2 binary cassette and 3-M38-2 ternary cassette could significantly inhibit HBV replication, suggesting that the combination of two HBV-specific gRNAs exhibited a potent ability in suppressing HBV replication. It was consistent with our previous report in which gRNA5 and gRNA12 were corresponding to gRNA2 and gRNA3, respectively [24]. However, 3-2 binary cassette showed higher efficiency in suppressing HBV replication than that of 3-M38-2 ternary cassette, which might be due to the possibility of the longer RNA the lower transcription efficiency of U6 promoter. To test this possibility, the expression levels of gRNA2 in 3-2 binary cassette and 3-M38-2 ternary cassette was measured by qRT-PCR. The result revealed that the level of gRNA2 in 3-2 binary cassette was significantly higher than that of 3-M38-2 ternary cassette, which supported the above possibility (Figure 4D). Since the levels of mature miRNA (miR-HBV or mutant miR-HBV) and gRNA3 carrying flanking sequence of anti-HBV pri-miR-31 mimic were almost equal between 3-H38-2 and 3-M38-2 ternary cassettes, the transcription efficiencies of ternary cassette and the processing of pri-miRNA were similar between these two ternary cassettes (Figures 4E and 4F). While, the efficiency of 3-H38-2 ternary cassette in suppressing HBV replication was significantly higher than that of 3-M38-2 ternary cassette (Figure 4C). The above results suggested that miR-HBV and two HBV-specific gRNAs in gRNA-miR-HBV-gRNA ternary cassette can get a synergistic effect in suppressing HBV replication.
The gRNA-miR-HBV-gRNA ternary cassette can efficiently suppress the replication of multi-genotypes HBV in vitro
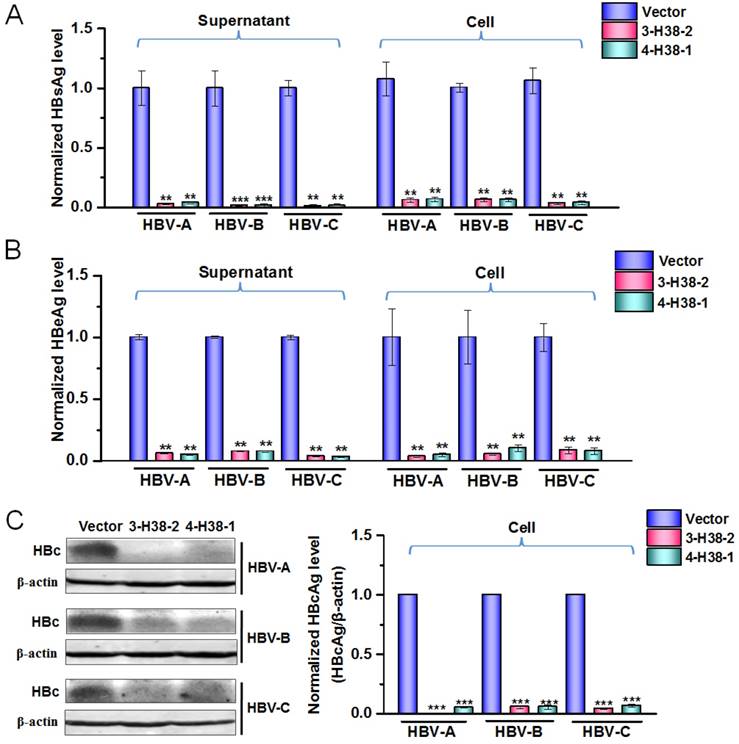
To further show the efficiency of gRNA-miR-HBV-gRNA ternary cassette in suppressing the replication of multi-genotypes HBV, another ternary cassette composed by gRNA4, gRNA1 and an anti-HBV pri-miR-31 mimic carrying 38 bp flanking sequences was constructed, which was named as 4-H38-1 ternary cassette. As shown by a dual luciferase assay, 4-H38-1 could also efficiently produce miR-HBV (Supplementary Figure S1). Since the target sequences of gRNAs 1-4 and miR-HBV were conserved in HBV genome of genotypes A-D, the consistent effect in suppressing the replication of multiple-genotypes HBV should be observed. The inhibitory effects of the individual gRNAs 1-4 in HBV replication have been confirmed in our previous study [24]. Consistently, both 3-H38-2 and 4-H38-1 ternary cassettes could significantly suppress the HBsAg, HBeAg and HBcAg levels of genotypes A to C HBV (Figure 5A-5C). No significant effect of either gRNAs 1-4 or miR-HBV on cell proliferation was observed using an MTT assay, which suggested that miR-HBV and each gRNA were free of cytotoxicity (Supplementary Figure S2).
The efficiency of gRNA-miR-HBV-gRNA ternary cassettes in suppressing HBV replication. The expression plasmids of each genotype (A, B or C) HBV and 3-H38-2 or 4-H38-1 cassette were co-transfected into HuH7 cells, and the levels of HBsAg (A) and HBeAg (B) in the cell culture supernatant or cells were measured using a time-resolved fluoroimmunoassay at 72 hours after transfection. Data was shown as mean±SD of 5 independent experiments. (C) The level of HBcAg in the HuH7 cells co-transfected with the expression plasmids of each genotype (A, B or C) HBV and 3-H38-2 or 4-H38-1 cassette were measured by Western Blot. The right figures were the statistical graphs of Western Blot, which was analyzed by Image J software (NIH, Bethesda, USA) at least in triplicate. HBc: HBcAg. PX458 plasmid was used as a vector control. (** indicated P<0.01, ***indicated P<0.001, Mann-Whitney U test).
The gRNA-miR-HBV-gRNA ternary cassette can efficiently destroy HBV genome
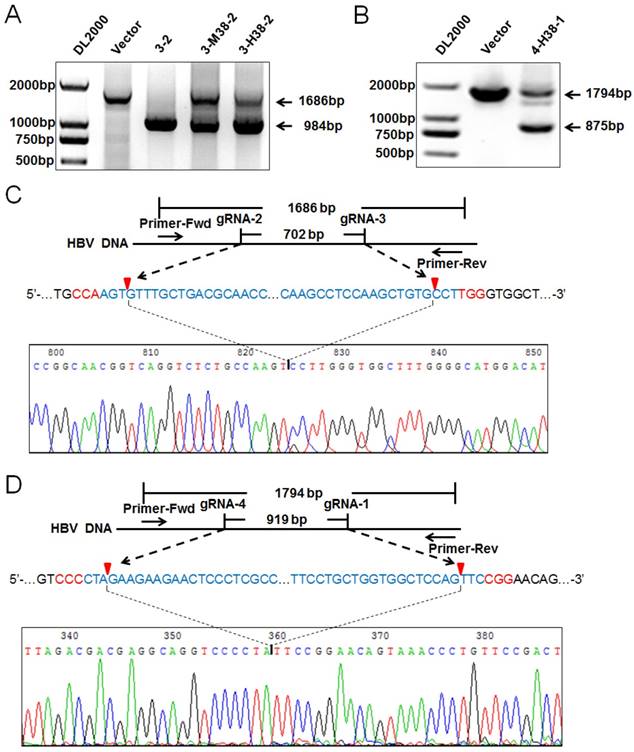
To confirm the HBV-specific gRNAs-mediated suppression of HBV replication was through genome cleavage, the expression plasmids of 1.2×HBV and 3-2 binary, 3-M38-2 or 3-H38-2 ternary cassette were co-transfected into HuH7 cells, and the HBV DNA genome region covering the cleavage sites of gRNA2 and gRNA3 was amplified by PCR. Once two gRNAs mediated the double-strand break (DSB) of target DNA, the fragment between the cleavage sites would be removed, and a smaller PCR product would be detected. Consistent with our previous report [24], the smaller fragment was detected, especially for 3-2 binary cassette which cleft most HBV genome, indicating that the cleavage efficiency was extremely high (Figure 6A). Similarly to gRNA2 and gRNA3, gRNA1 and gRNA4 could also efficiently destroy HBV genome (Figure 6B). Furthermore, the Cas9/gRNA-mediated destruction of HBV genome was demonstrated by Southern Blot analyses (Supplementary Figures S2A and S2B). To confirm the cleavage specificity of two HBV-specific gRNAs, the smaller fragments of PCR products were sequenced directly. The results revealed that most rejoining sites were the gRNA-cleavage sites of two HBV-specific gRNAs (Figure 6C and 6D).
Detection on the HBV-specific gRNAs-mediated destruction of HBV genome. (A) The expression plasmids of 1.2×HBV and 3-2 binary, 3-M38-2, 3-H38-2 or 4-H38-1 (B) ternary cassette were co-transfected into HuH7 cells. Cellular DNA was extracted at 72 hours after transfection, and PCR amplifications were performed using the primers beyond the cleavage sites of two gRNAs. (C) Sequencing analysis of the smaller fragment cleft by gRNA2 and 3. (D) Sequencing analysis of the smaller fragment cleft by gRNA1 and 4. PX458 plasmid was used as a vector control.
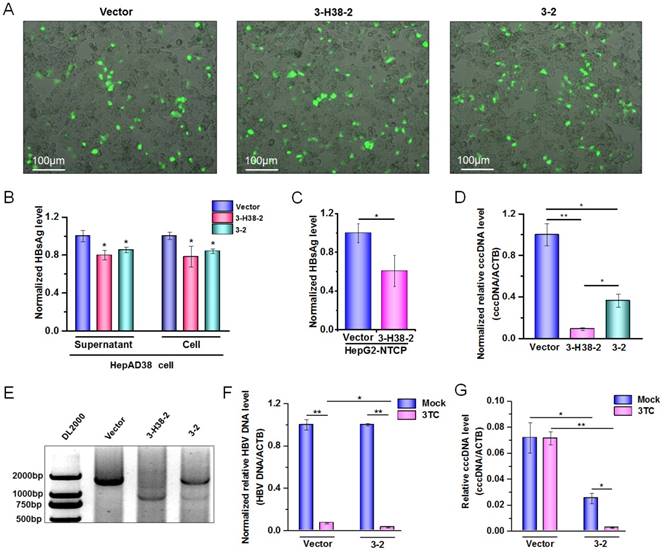
Since HBV-specific gRNA could destroy HBV DNA, theoretically, it could also destroy HBV cccDNA. To confirm this, the expression plasmid containing 3-2 binary cassette or 3-H38-2 ternary cassette was transfected into HepAD38 cells in which the HBV of genotype D was produced under the control of tet-off system [39]. Firstly, we demonstrated that 3-2 binary or 3-H38-2 ternary cassette in HepAD38 cells could also significantly reduce the HBsAg levels either in the cell culture supernatant or in cells, even the transfection efficiency was low (Figure 7A and 7B). Consistently, in the case of low transfection efficiency, 3-H38-2 ternary cassette could also significantly reduce the HBsAg levels in the culture supernatant of HepG2-NTCP-tet cells infected with HBV (Figure 7C). To overcome the interference of low transfection efficiency, the enhanced green fluorescence protein (EGFP) positive HepAD38 cells were selected by Flow Cytometry to enrich the successfully transfected cells which could express Cas9, EGFP and 3-2 binary or 3-H38-2 ternary cassette together. Since cccDNA reservoirs are the cause of chronic HBV infection, the level of cccDNA in the EGFP positive HepAD38 cells was measured. To make sure the quantitative measurement of the HBV cccDNA was reliable, KCl precipitation, plasmid-safe ATP-dependent DNase (PSAD) digestion and rolling circle amplification were employed, followed by the quantitative PCR with cccDNA specific primers (sense primer covers the cleavage site of gRNA3) that target the gap region of HBV genome. As expected, both 3-2 binary and 3-H38-2 ternary cassette could significantly decrease cccDNA level in HepAD38 cells (Figure 7D). Besides, HBV cccDNA could also be destroyed by both 3-2 binary and 3-H38-2 ternary cassette (Figure 7E). Intriguingly, the efficiency of 3-H38-2 ternary cassette on cccDNA destruction was significantly higher than that of 3-2 binary cassette, which was opposite to their effects on HBV DNA destruction (Figure 6A and Figure 7D). For this phenomenon, an explanation was that except for the direct cccDNA cleavage mediated by two gRNAs, miR-HBV might indirectly increase the percentage of destroyed cccDNA through suppressing HBV replication and subsequently inhibiting the supplement of cccDNA pool. To confirm this postulation, we used lamivudine (3TC) to treat HepAD38 cells transfected with the expression plasmid of 3-2 binary cassette and inhibit the supplement of cccDNA pool through suppressing HBV replication. Firstly, the synergistic effect of HBV-specific gRNA and 3TC was found in suppressing HBV replication (Figure 7F). Next, the gRNA-mediated cccDNA destruction was confirmed to be enhanced by 3TC treatment (Figure 7G), which supported the above hypothesis that miR-HBV might indirectly increase the proportion of destroyed cccDNA through inhibiting the supplement of cccDNA pool.
Detection on the HBV-specific gRNAs-mediated destruction of HBV cccDNA. (A) HepAD38 cells were seeded into 10 cm dish. The expression plasmid of gRNA3-RNA2 binary or gRNA-miRNA-gRNA ternary cassette was co-transfected into HepAD38 cells. Transfection efficiency of HepAD38 cells was evaluated by observing the enhanced green fluorescent protein (EGFP) expressing cells under immunofluorescence microscopy. (B) HBsAg and HBeAg levels in the cell culture supernatant of HepAD38 cells were measured using a time-resolved fluoroimmunoassay. Data was shown as mean±SD of 6 independent experiments. (C) HBsAg levels in the cell culture supernatant of HepG2-NTCP-tet cells were measured using a time-resolved fluoroimmunoassay. Data was shown as mean±SD of 5 independent experiments. (D) HBV cccDNA levels in EGFP positive HepAD38 cells selected by Flow cytometry were measured using a KCl precipitation, plasmid-safe ATP-dependent DNase (PSAD) digestion, rolling circle amplification and quantitative PCR (SYBR green) combined method. Data was shown as mean±SD of 4 independent experiments. (E) PCR amplification of cccDNA was performed using the primers beyond the cleavage sites of two gRNAs following above rolling circle amplification. HepAD38 cells were transfected with 3-2 binary cassette expression plasmid, and then treated with or without lamivudine (3TC). The HBV DNA levels (F) in HepAD38 cells were detected by quantitative PCR, and HBV cccDNA levels (G) in EGFP positive HepAD38 cells selected by Flow cytometry were measured as above. Data was shown as mean±SD of 4 independent experiments. (* indicated P<0.05, ** indicated P<0.01, Mann-Whitney U test). PX458 plasmid was used as a vector control.
The gRNA-miR-HBV-gRNA ternary cassette can efficiently suppress HBV replication in vivo
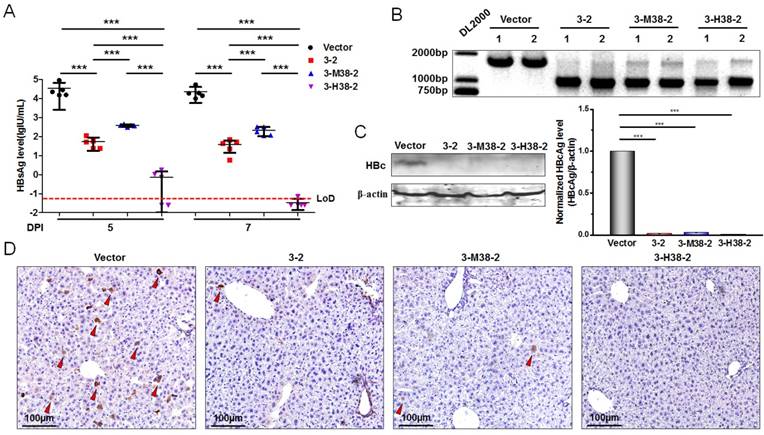
To further demonstrate the antiviral advantage of gRNA-miRNA-gRNA ternary cassette, hydrodynamic injection of the expression plasmids of 1.2×HBV and 3-2 binary or 3-H38-2 ternary cassette was conducted in C57BL/6 mice (5-7 weeks old). The HBsAg levels in serum were detected at 5 and 7 days post-injection. Intriguingly, 3-2 binary, 3-M38-2 and 3-H38-2 ternary cassettes could efficiently reduce the levels of HBsAg. The 3-H38-2 ternary cassette showed higher efficiency in inhibiting HBV replication, in which the HBsAg levels were below the lower limit of detection (Figure 8A). Furthermore, two mice of each group were sacrificed under anesthesia at 7 days post injection. The HBV DNA in the liver tissue was extracted and amplified by PCR. As expected, HBV-specific gRNA could also efficiently destroy HBV genome in vivo, and the cleavage efficiency was so high that most HBV genome was cleft (Figure 8B). Besides, the level of HBcAg in the liver tissue was measured by Western Blot and Immunohistochemistry assay (IHC) at 7 days post injection. The results of Western Blot analyses revealed that the levels of HBcAg were significantly reduced to the level that were undetectable with our relatively low-sensitive Western Blot analyses in the 3-2, 3-M38-2 and 3-H38-2 injected mice (Figure 8C). Consistently, compared to vector control group, the number of HBcAg-positive hepatocytes decreased significantly in the 3-2, 3-M38-2 and 3-H38-2 injected mice, even no HBcAg-positive hepatocyte was found in the 3-H38-2 injected mice (Figure 8D).
The efficiency of the gRNA-miR-HBV-gRNA ternary cassettes in suppressing HBV replication in vivo. (A) Hydrodynamic injection of the 1.2×HBV (8µg) and gRNA3-RNA2 binary or gRNA-miRNA-gRNA ternary cassette expression plasmids (24µg) in C57BL/6 mice. The HBsAg levels (lgIU/mL) in sera were detected at 5 and 7 days post-injection (DPI) [*** indicated P<0.001, Kruskal-Wallis H (K) test]. (B) Liver tissue DNA was extracted at 7 days post-injection, and PCR amplifications were performed using the primers beyond the cleavage sites of each gRNAs. 1: liver tissue from 1# mice. 2: liver tissue from 2# mice. (C) The levels of HBcAg in the liver tissues of mice at 7 days post-injection were detected by Western Blot. The right figures were the statistical graphs of Western Blot, which was analyzed by Image J software (NIH, Bethesda, USA) at least in triplicate. HBc: HBcAg. (** indicated P<0.01, ***indicated P<0.001, Mann-Whitney U test). HBcAg: HBc. (D) Representative micrographs showing the levels of HBcAg in the liver tissues measured by IHC at 7 days post-injection. The red triangle indicates the HBcAg positive hepatocytes.
Discussion
Clinically, the nucleos(t)ide analogues (NAs) and pegylated-interferon-α (PEG-IFN-α) are two kinds of antiviral agents commonly used to treat CHB. NAs block HBV replication mainly through inhibiting the reverse transcription of HBV pre-genome RNA, which have little or no direct effect on cccDNA. Since cccDNA is noticeably stable and declines slowly under NAs therapy, the recurrence of hepatitis B often occurs after the cessation of NAs treatment [4, 5, 40]. Although IFN-α can inhibit HBV replication through degradation of cccDNA, its efficiency is limited due to systemic side effect [6, 41]. Therefore, clinical cure of CHB is still an unresolved problem and a long-term treatment of CHB is required, which is expensive and may lead to concomitant resistant [40]. It is an urgent necessary to develop new strategies of clearing the persistent HBV cccDNA in hepatocytes.
In this report, a combined approach of CRISPR/Cas9 system and RNAi was developed. The HBV-specific gRNAs and miR-HBV were designed based on the conserved sequences among HBV genome of genotypes A-D which are the most predominant genotypes in the world [42]. Naturally, pri-miRNAs are transcribed from miRNA genes by RNA polymerase (Pol) II, and subsequently processed by nuclear RNaseIII enzyme Drosha together with DGCR8 to form pre-miRNAs in the nucleus, pre-miRNAs are ultimately processed by Dicer in the cytoplasm to form the resulting 19-24 nucleotides mature miRNAs [43]. Pri-miRNA mimic is the analogue of nascent miRNA, and can get silencing of the target gene by simulating natural miRNA processing [31, 32, 44]. To take advantage of this feature, we inserted an anti-HBV pri-miR-31 mimic between two HBV-specific gRNAs to construct a gRNA-miR-HBV-gRNA ternary cassette. Although a modest flanking sequence (≥8 bp) beyond the 5' end of a pre-miRNA is required for the recognition of Drosha/DGCR8 and subsequent cleavage of pri-miRNA [45], the flanking sequence of anti-HBV pri-miR-31 mimic may interfere with the activity of gRNA. Therefore, to get the maximal anti-HBV efficiency, a series of anti-HBV pri-miR-31 mimics carrying different lengths of flanking sequences were designed based on the pri-miR-31/5 sequence which was successfully applied for inhibiting HBV replication [32]. As expected, HBV-specific gRNAs and miR-HBV could be efficiently expressed and separated in gRNA-miR-HBV-gRNA ternary cassette. Although the production of the artificially designed miR-HBV was the same with miRNA, its activity should be similar with the short interference RNA (siRNA), because its sequence was completely matched with the target sequence. Since the longer flanking sequence of anti-HBV pri-miR-31 mimic the stronger ability in suppressing luciferase activity in which miR-HBV-target sequence (HBV 1575-1595) was located at 3' UTR region of luciferase gene, the expression level of mature miR-HBV should be increased with the increase in length of flanking sequence. Whereas, a 38 bp flanking sequence of anti-HBV pri-miR-31 mimic in the gRNA-miR-HBV-gRNA ternary cassette was the optimal length in suppressing HBV replication, but not a 51 bp flanking sequence, which suggested that the 5' end and 3' end extension of two resulting gRNAs might affect their cleavage efficiencies.
Next, the in vitro and in vivo experiments were conducted to evaluate the antiviral effect of gRNA-miR-HBV-gRNA ternary cassette, and showed that this ternary cassette could efficiently suppress the replication of genotype A to D HBV, including the levels of HBsAg, HBeAg and HBcAg. Further, PCR, Southern Blot or direct sequencing confirmed that two HBV-specific gRNAs expressed from gRNA-miR-HBV-gRNA ternary cassette could efficiently and specifically destroy HBV genome in vitro and in vivo, suggesting that the anti-HBV effect of CRISPR/Cas9 was due to the cleavage of HBV genome. However, the cleavage efficiency of gRNA-miR-HBV-gRNA ternary cassette was lower than that of gRNA-gRNA binary cassette. For this phenomenon, one reason was that the transcription efficiency of U6 promoter for the long gRNA-miR-HBV-gRNA ternary cassette (about 340 bp) was lower than that of gRNA-gRNA binary cassette (about 100 bp), another reason might be that the flanking sequences of anti-HBV pri-miR-31 mimic interfered with the activity of gRNA. When targeted by a single gRNA, the DNA cleft by Cas9 usually underwent the non-homologus end joining (NHEJ) repair, even for the HBV genome, which introduces the indel mutation and inhibits HBV replication [46, 47]. Since dual gRNAs can remove the DNA fragment between two gRNAs, the damage in HBV genome should be greater than that of single gRNA. Interestingly, the indel mutation was not found in the sequencing map of the smaller PCR product. It was suggested that the DNA fragment between two gRNAs was precisely removed, which was consistent with our and other's previous reports [24, 27]. It was indicated that two cleavage sites might rapidly ligate without forming indel mutation when a fragment of HBV genome was removed by two gRNAs.
Intriguingly, we found that the gRNA-miR-HBV-gRNA ternary cassette could get higher efficiency in suppressing HBV replication than that of the gRNA-gRNA binary cassette, suggesting a synergistic effect of CRISPR/Cas9 system and RNAi approach. It has been reported that HBV cccDNA can be cleft by CRISPR/Cas9 system in HuH7 and HepG2 cells after transfection [46-49]. Consistently, we found that gRNA-miR-HBV-gRNA ternary cassette could also efficiently destroy HBV cccDNA in HepAD38 cells, and the efficiency was higher than that of gRNA-gRNA binary cassette, which was different from the observation in HBV genome of 1.2×HBV expression plasmid. For this phenomenon, we postulated that miR-HBV might additionally inhibit the supplement of cccDNA pool in HepAD38 cells by suppressing HBV replication, and thus indirectly promoted the CRISPR/Cas9 system-mediated destruction of cccDNA. To confirm the above postulation, lamivudine (3TC) treatment was used to suppress HBV replication and subsequently inhibit the supplement of cccDNA pool. Consistently, 3TC treatment indeed could promote the gRNA-gRNA binary cassette-mediated destruction of cccDNA. These results exhibited the superiority of gRNA-miR-HBV-gRNA ternary cassette in suppressing HBV replication and destroying HBV cccDNA.
As we know, the off-target effect of CRISPR/Cas9 system is a common problem and poses a major challenge for its clinical application. Since the possibility of non-specific effects could not be completely addressed using wild-type SpCas9, and it is likely to be higher when the number of gRNAs introduced into cell is increased. Fortunately, some SpCas9 variants can reduce off-target effects to undetectable level [50, 51]. Therefore, it is possible for the ultimate therapeutic utility of this technology. More excitingly, Staphylococcus aureus Cas9 (SaCas9) and its gRNA expression cassette can be packaged into a single adeno-associated virus (AAV) vector, which make a step further in the clinical application of CRISPR/Cas9 system [52]. Therefore, the gRNA-miR-HBV-gRNA ternary cassette can be inserted into the SaCas9-AAV expression vector to promote the clinical application of the gRNA-miRNA-gRNA ternary cassette. Besides, based on the gRNA-miRNA-gRNA ternary cassette, we can get the gRNA-miRNA-gRNA-miRNA-gRNA……multiplex gRNAs and miRNAs expression cassette for multiplex genes editing.
In conclusion, our results proved that gRNA-miR-HBV-gRNA ternary cassette could efficiently suppress the replication of multi-genotypes HBV through RNAi and destroying HBV genome, which showed the synergistic or additional effect of CRISPR/Cas9 system and RNAi approach. The development of gRNA-miRNA-gRNA ternary cassette should make them broadly useful in research and therapeutic applications for genome editing, particularly for antiviral therapy.
Acknowledgements
This work was supported by grants from the Natural Science Foundation of China (No. 81471938 and 81672013), the National S & T Major Project for Infectious Diseases (No.2013ZX10002-002 and 2012ZX10002-005), and 111 project (B07001). The authors declare no conflicts of interest.
Author contributions
Lu FM, Xu ZW, Xia NS and Wang J designed the research; Wang J, Chen R, Zhang RY, Ding SL, Zhang TY, Yuan Q and Zhang T performed the research; Wang J and Chen R supplemented the experiments; Guan GW drew the graphical abstract; Lu FM, Xu ZW, Xia NS, Wang J, Chen R and Zhang RY analyzed the data; Wang J and Lu FM wrote the paper; Chen XM, Zhuang H, Nunes F, Block T, Liu S and Duan ZP revised the paper.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Trepo C, Chan HL, Lok A. Hepatitis B virus infection. Lancet. 2014;384:2053-63
2. Ganem D, Prince AM. Hepatitis B virus infection-natural history and clinical consequences. N Engl J Med. 2004;350:1118-29
3. Gao W, Hu J. Formation of hepatitis B virus covalently closed circular DNA: removal of genome linked protein. J Virol. 2007;81:6164-74
4. Dandri M, Burda MR, Will H. et al. Increased hepatocyte turnover and inhibition of woodchuck hepatitis B virus replication by adefovir in vitro do not lead to reduction of the closed circular DNA. Hepatology. 2000;32:139-46
5. Wursthorn K, Lutgehetmann M, Dandri M. et al. Peginterferon alpha-2b plus adefovir induce strong cccDNA decline and HBsAg reduction in patients with chronic hepatitis B. Hepatology. 2006;44:675-84
6. Lucifora J, Xia Y, Reisinger F. et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343:1221-8
7. Wang J, Shen T, Huang X. et al. Serum hepatitis B virus RNA is encapsidated pregenome RNA that may be associated with persistence of viral infection and rebound. J Hepatol. 2016;65:700-10
8. Jansen L, Kootstra NA, van Dort KA. et al. Hepatitis B virus pregenomic RNA is present in virions in plasma and as associated with a response to pegylated interferon alfa-2a and nucleos(t)ide analogues. J Infect Dis. 2016;213:224-32
9. Zimmerman KA, Fischer KP, Joyce MA. et al. Zinc finger proteins designed to specifically target duck hepatitis B virus covalently closed circular DNA inhibit viral transcription in tissue culture. J Virol. 2008;82:8013-21
10. Cradick TJ, Keck K, Bradshaw S. et al. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol Ther. 2010;18:947-54
11. Bloom K, Ely A, Mussolino C. et al. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol Ther. 2013;21:1889-97
12. Chen J, Zhang W, Lin J. et al. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol Ther. 2014;22:303-11
13. Lin SR, Yang HC, Kuo YT. et al. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates In Vivo. Mol Ther Nucleic Acids. 2014;3:e186
14. Seeger C, Sohn JA. Targeting hepatitis B virus with CRISPR/Cas9. Mol Ther Nucleic Acids. 2014;3:e216
15. Kennedy EM, Bassit LC, Mueller H. et al. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology. 2015;476:196-205
16. Zhen S, Hua L, Liu YH. et al. Harnessing the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated Cas9 system to disrupt the hepatitis B virus. Gene Ther. 2015;22:404-12
17. Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167-70
18. Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49-55
19. Urnov FD, Rebar EJ, Holmes MC. et al. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636-46
20. Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347-55
21. Cong L, Zhang F. Genome engineering using CRISPR-Cas9 system. Methods Mol Biol. 2015;1239:197-217
22. Mali P, Yang L, Esvelt KM. et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823-6
23. Ran FA, Hsu PD, Wright J. et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281-308
24. Wang J, Xu ZW, Liu S. et al. Dual gRNAs guided CRISPR/Cas9 system inhibits hepatitis B virus replication. World J Gastroenterol. 2015;21:9554-65
25. Deng W, Lu M. The role of microRNAs in hepatocyte metabolism and hepatitis B virus replication. Vriol Sin. 2016;31:472-79
26. Senis E, Mockenhaupt S, Rupp D. et al. TALEN/CRISPR-mediated engineering of a promoterless anti-viral RNAi hairpin into an endogenous miRNA locus. Nucleic Acids Res. 2017;45:e3
27. Cong L, Ran FA, Cox D. et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819-23
28. Platt RJ, Chen S, Zhou Y. et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440-55
29. Tsai SQ, Wyvekens N, Khayter C. et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. 2014;32:569-76
30. Nissim L, Perli SD, Fridkin A. et al. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol Cell. 2014;54:698-710
31. Ely A, Naidoo T, Mufamadi S. et al. Expressed anti-HBV primary microRNA shuttles inhibit viral replication efficiently in vitro and in vivo. Mol Ther. 2008;16:1105-12
32. Ely A, Naidoo T, Arbuthnot P. Efficient silencing of gene expression with modular trimeric Pol II expression cassettes comprising microRNA shuttles. Nucleic Acids Res. 2009;37:e91
33. Chen RY, Edwards R, Shaw T. et al. Effect of the G1896A precore mutation on drug sensitivity and replication yield of lamivudine-resistant HBV in vitro. Hepatology. 2003;37:27-35
34. Yan H, Zhong G, Xu G. et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;1:e00049
35. Summers J, Smith PM, Horwich AL. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J Virol. 1990;64:2819-24
36. Zhong Y, Han J, Zou Z. et al. Quantitation of HBV covalently closed circular DNA in micro formalin fixed paraffin-embedded liver tissue using rolling circle amplification in combination with real-time PCR. Clin Chim Acta. 2011;412:1905-11
37. Li W, Zhao J, Zou Z. et al. Analysis of hepatitis B virus intrahepatic covalently closed circular DNA and serum viral markers in treatment-naive patients with acute and chronic HBV infection. PLoS One. 2014;9:e89046
38. Zhang TY, Yuan Q, Zhao JH. et al. Prolonged suppression of HBV in mice by a novel antibody that targets a unique epitode on hepatitis B surface antigen. Gut. 2016;65:658-71
39. Ladner SK, Otto MJ, Barker CS. et al. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother. 1997;41:1715-20
40. Zoulim F. Hepatitis B virus resistance to antiviral drugs: where are we going? Liver Int. 2011;31(Suppl 1):S111-S116
41. Belloni L, Allweiss L, Guerrieri F. et al. IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest. 2012;122:529-37
42. Schaefer S. Hepatitis B virus taxonomy and hepatitis B virus genotypes. World J Gastroenterol. 2007;13:14-21
43. Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594-6
44. Boudreau RL, Martins I, Davidson BL. Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol Ther. 2009;17:169-75
45. Zeng Y, Yi R, Cullen BR. Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. Embo J. 2005;24:138-48
46. Dong C, Qu L, Wang H. et al. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antiviral Res. 2015;118:110-7
47. Zhu W, Xie K, Xu Y. et al. CRISPR/Cas9 produces anti-hepatitis B virus effect in hepatoma cells and transgenic mouse. Virus Res. 2016;217:125-32
48. Ramanan V, Shlomai A, Cox DB. et al. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci Rep. 2015;5:10833-41
49. Guo X, Chen P, Hou X. et al. The recombined cccDNA produced using minicircle technology mimicked HBV genome in structure and function closely. Sci Rep. 2016;6:25552-61
50. Slaymaker IM, Gao L, Zetsche B. et al. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84-8
51. Kleinstiver BP, Pattanayak V, Prew MS. et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490-5
52. Ran FA, Cong L, Yan WX. et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186-91
Author contact
![]() Corresponding authors: Fengmin Lu, MD, State Key Laboratory of Natural and Biomimetic Drugs, Department of Microbiology & Infectious Disease Center, School of Basic Medical Sciences, Peking University Health Science Center, 38 Xueyuan Road, Beijing 100191, P.R. China. Fax: +86-10-82805136; lu.fengminpku.edu.cn (F Lu); Zhongwei Xu, MD, PhD, Department of Gastroenterology, Pennsylvania Hospital, University of Pennsylvania, 203 West Washington Square, Philadelphia, PA, 19106, USA. zhongwei.xuupenn.edu (Z Xu); Ningshao Xia, State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics, School of Public Health, Xiamen University, Xiamen, P.R. China. nsxiaedu.cn (N Xia).
Corresponding authors: Fengmin Lu, MD, State Key Laboratory of Natural and Biomimetic Drugs, Department of Microbiology & Infectious Disease Center, School of Basic Medical Sciences, Peking University Health Science Center, 38 Xueyuan Road, Beijing 100191, P.R. China. Fax: +86-10-82805136; lu.fengminpku.edu.cn (F Lu); Zhongwei Xu, MD, PhD, Department of Gastroenterology, Pennsylvania Hospital, University of Pennsylvania, 203 West Washington Square, Philadelphia, PA, 19106, USA. zhongwei.xuupenn.edu (Z Xu); Ningshao Xia, State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics, School of Public Health, Xiamen University, Xiamen, P.R. China. nsxiaedu.cn (N Xia).