Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and Discussions
Conclusions
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(12):3034-3052. doi:10.7150/thno.19279 This issue Cite
Research Paper
Albumin-Gold Nanorod Nanoplatform for Cell-Mediated Tumoritropic Delivery with Homogenous ChemoDrug Distribution and Enhanced Retention Ability
Hsien-Ting Chiu1, Cheng-Kuan Su2, Yuh-Chang Sun1, Chi-Shiun Chiang1, Yu-Fen Huang1 ![]()
1. Department of Biomedical Engineering and Environmental Sciences, National Tsing Hua University, Hsinchu 30013, Taiwan, ROC;
2. Department of Bioscience and Biotechnology, National Taiwan Ocean University, Keelung, 20224, Taiwan, ROC.
Received 2017-1-20; Accepted 2017-6-5; Published 2017-7-22
Abstract

Recently, living cells with tumor-homing properties have provided an exciting opportunity to achieve optimal delivery of nanotherapeutic agents. However, premature payload leakage may impair the host cells, often leading to inadequate in vivo investigations or therapeutic efficacy. Therefore, a nanoplatform that provides a high drug-loading capacity and the precise control of drug release is required. In the present study, a robust one-step synthesis of a doxorubicin (DOX)-loaded gold nanorod/albumin core-shell nanoplatform (NR@DOX:SA) was designed for effective macrophage-mediated delivery to demonstrate how nanoparticle-loaded macrophages improve photothermal/chemodrug distribution and retention ability to achieve enhanced antitumor effects. The serum albumin shell of these nanoagents served as a drug reservoir to delay the intracellular DOX release and drug-related toxicity that impairs the host cell carriers. Near-infrared laser irradiation enabled on-demand payload release to destroy neighboring tumor cells. A series of in vivo quantitative analyses demonstrated that the nanoengineered macrophages delivered the nanodrugs through tumor-tropic migration to tumor tissues, resulting in the twice homogenous and efficient photothermal activations of drug release to treat prostate cancer. By contrast, localized pristine NR@DOX:SAs exhibit limited photothermal drug delivery that further reduces their retention ability and therapeutic efficacy after second combinational treatment, leading to a failure of cancer therapy. Moreover, the resultant unhealable wounds impair quality of life. Free DOX has rapid clearance and therefore exhibits limited antitumor effects. Our findings suggest that in comparison with pristine nanoparticles or free DOX, the nanoengineered macrophages effectively demonstrate the importance and effect of homogeneous drug distribution and retention ability in cancer therapy.
Keywords: albumin, gold nanorod, core-shell nanoparticle, cell-mediated drug delivery, homogenous drug distribution, drug retention ability.
Introduction
Nanotechnology plays a prominent role in translating cancer research into clinical practice. Nanomaterials have promising applications depending on their features and are currently used as nanotherapeutic agents. However, systemic and local administration of these nanoagents have revealed that these materials have disadvantages that limit their applicability in cancer therapy, namely the common occurrence of elevated intratumoral interstitial fluid pressure, a dense extracellular matrix with impaired perfusion, and a hypoxic area in the tumor microenvironment [1, 2]. Furthermore, additional investigations are required to explore the systemic administration. During circulation, injected nanoparticles (NPs) are sequestered by the reticuloendothelial system [3].
Engineering of natural cells may be a promising solution to fulfill the criteria for next-generation personalized medicine [4, 5]. Nanodrugs that can be incorporated into neutral cells provide advantages such as prolonged circulation time [6], tumor-tropic migration [7], and improved drug distribution [8] for cancer therapy. However, of the studies that aimed to develop nanosystems for enhancing therapeutic efficacy, relatively few have utilized or designed nanoparticles by conducting fundamental in vivo studies. Until now, few in vivo investigations have been conducted regarding the mechanism through which the nanomaterials precisely control the behaviors of host cells, influence payload efficacy and release, and enable the long-term monitoring of cellular behaviors in tumor microenvironments and conduct a fair comparison with pristine nanoparticles or conventional drug therapy in the resultant therapeutic efficacy and biological effect [5, 9-11].
Gold (Au) NPs are easily functionalized with biomolecules, which facilitate their integration into biological systems. When Au NPs are anchored to targeted cell carriers, they tend to remain essentially inert and nontoxic, thus maintaining the basic functions of the host cell carriers, including migration and immunotropic activities. Numerous Au NPs with spectral absorbance in the near-infrared (NIR) wavelength range have demonstrated the criteria necessary for serving as cell-based nanoplatforms for photoablation because NIR light is weakly absorbed by tissues and water [12]. Au nanoshell-laden macrophages have been reported to infiltrate tumor spheroids and are highly successful in photothermal therapy [13-15]. Gold nanorods (Au NRs), which offer a greater optical cross section than do silica-Au nanoshells and nanocages [16, 17], are promising candidates for incorporation into a cell-based drug delivery system as photothermal [8, 18-20] and imaging agents [21, 22]. However, to date, most Au NP-anchored cancer therapies have been solely based on the photothermal effect. Because the combinational therapy with chemotherapeutic agents is an emerging treatment strategy against cancer, a novel drug carrier that is compatible with both chemotherapy and photothermal therapy is required.
Serum albumin (SA) has emerged as a versatile carrier in the drug delivery regime because it is intrinsically nontoxic, biocompatible, and biodegradable; has strong potential in various drug-loading capacities; and can be combined with other nanomaterials [23]. Studies have reported that SA-covered Au NRs evade cetyltrimethylammonium bromide (CTAB)-triggered cytotoxic effects [24], and the SA absorbed on the NPs can consequently induce macrophage phagocytosis [25]. However, covering an individual Au NR with a uniform and complete protein shell in a controlled manner poses a major challenge, because SA tends to induce slight clustering [26, 27] and unequivalent encapsulation [28, 29] of Au NRs during the fabrication of NR-embedded albumin NPs. This may cause cell-mediated drug delivery to have diverse therapeutic effects in cancer treatment. In the present study, we developed homogeneous core-shell nanoplaforms, NR@SAs, for efficient cellular uptake and effective drug loading (Figure 1A). To enhance therapeutic efficacy, a chemotherapeutic drug, doxorubicin (DOX), was incorporated during the synthesis of NR@DOX:SAs by using a novel modified desolvation protocol. After the fabricated NR@DOX:SAs were internalized by a model cell carrier (RAW264.7, macrophage), the SA shell played a critical role in delaying the release of encapsulated DOX to temporarily protect the cell carriers. The NIR laser-induced photothermal effect damaged the host cells and facilitated the release of drug payload into the surrounding media. The combinational photothermal treatment and chemotherapy showed a synergistic effect against the in vitro and in vivo growth of neighboring tumor cells.
Core-shell nanoplatform (NR@DOX:SA) demonstrating the distinctive behaviors of a cell-mediated drug delivery system. (A) Preparation of the NR@DOX:SAs programmed for cell-mediated delivery. The precise and efficient activation of controlled drug release was modulated through NIR light irradiation for in vitro and in vivo studies. (B) The key concepts of homogenous drug distribution and retention ability as demonstrated by three therapeutic drugs: NP-laden macrophages, pristine NPs, and free DOX in tumor-bearing mice.
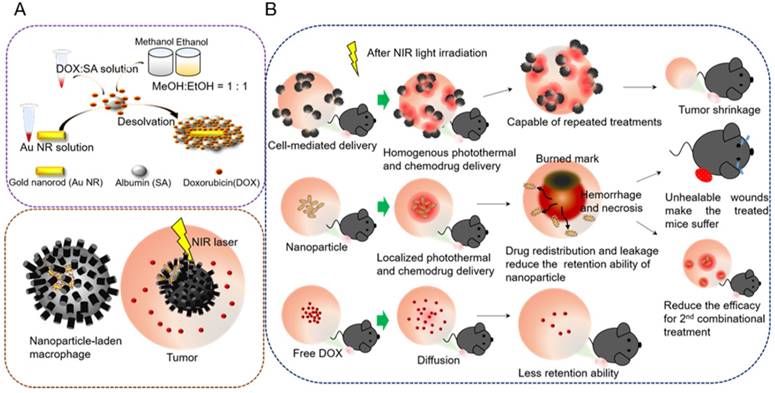
In addition, localized preoperative chemotherapy or hyperthermia treatment with subsequent surgical resection or irradiation have been the established practices for limiting the adverse systemic events of cancer progression. However, additional investigations are required to overcome the negative effects of this strategy, including technical control injection site [30, 31] and tumor microenvironment heterogeneity [1, 32-34], and to elucidate the resultant biological effects after cancer therapy [35-38]. Furthermore, tumor-tropic delivery vesicles, which can automatically enhance drug coverage [8, 39, 40] and prolong drug activity [32, 37] in preoperative intratumoral photothermal therapy and chemotherapy, should be developed. Our findings revealed that NR@DOX:SA-laden macrophages had consistently homogenous photothermal and drug distribution as well as prolonged retention of cytotoxic nanoagents, demonstrating optimal tumor elimination after multiple injections. By contrast, pristine NR@DOX:SAs were limited in their spatial distribution, leading to a severe and unhealable wound that reduced the retention ability of NR@DOX:SAs and impaired quality of life. Although free DOX diffused readily, it was rapidly washed out after direct intratumoral injection. Eventually, both pristine NR@DOX:SAs and free DOX did not exert antitumor effects against prostate cancer in tumor-bearing mice. These findings strongly suggest that homogeneous drug distribution and retention ability are determinants of the outcomes of cancer therapy (Figure 1B). Furthermore, preliminary findings indicated that compared with pristine NR@DOX:SAs, NR@DOX:SA-laden macrophages demonstrated greater tumor-tropic migration and specific accumulation in the hypoxic region, which might influence the final outcomes of drug delivery, distribution, and retention ability and the resultant therapeutic effects. Altogether, this study provides a rational design concept of a programmable nanoplatform for cell-mediated drug delivery to illustrate the importance of a more homogenous and long-lasting therapeutic effect in cancer treatment.
Materials and Methods
Materials
Sodium tetrachloroaurate(III) dihydrate (99%), bovine serum albumin (98%), cetyltrimethylammonium bromide (CTAB), methanol (99.5%), ethanol (99.5%), paraformaldehyde, phosphotungstic acid hydrate, pimonidazole HCl, doxorubicin, hematoxylin and eosin were obtained from Sigma-Aldrich (St. Louis, MO, USA). Phosphate buffered saline (PBS), fetal bovine serum, RPMI 1640 medium, Dulbecco's modified Eagle's medium (DMEM) were obtained from GIBCO (Grand Island, NY, USA). Penicillin-streptomycin, trypsin-ethylenediaminetetraacetic acid, 4',6-diamidino-2-phenylindole, dihydrochloride (DAPI), Hochest 33342, transferrin-Alexa 633 and tetramethylrhodamine isothiocyanate (TRITC) were bought from invitrogen (Invitrogen, Carlsbad, CA, USA). Alamar blue® was purchased from AbD Serotec (Oxford, UK). Deionized water (18.2 MΩ. cm) was used to prepare all of the aqueous solutions. For the cellular experiments, all of the equipment and deionized water was steamed at 134 ℃ in 15 min by autoclave (TAT-202A20, EQUS, Taiwan) before any use.
Preparation and characterization of NR@DOX:SAs
The seed and growth method [41] was used in this study to fabricate Au NR. The produced Au NR solution was followed by centrifugation at 12000 rpm for 15 min until the CTAB solution was reduced to 5×10-4 M. After analyzing by inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7700 Series ICP-MS, USA), the final concentration of the resultant Au NR in 0.3 nM was denoted as 1× and the solution was further concentrated to 50× to preserve as the stock solution. To prepare DOX loaded NR@SAs, DOX solution (160 μL) was pre-mixed with SA (0.125 wt%) for 2 h, followed by a gently mixing and incubation with the as-prepared Au NR stock solution (40 μL). 800 μL of EM solution (50% methanol+50% ethanol) was added immediately and left undisturbed overnight. The resultant product was washed three times with 1% SA by centrifugation at 6500 rpm for 20 min and redispersed in DI water, PBS or culture medium for further application. UV/Vis spectra were acquired using a Cary 100 UV-Vis spectrometer (Varian, Palo Alto, CA, USA) while the hydrodynamic size and zeta potential were measured by a Zetasizer Nano ZS (Malvern Instruments Ltd.). NR@SAs stained with 4% phosphotungstic acid were verified by transition electron microscopy (TEM, H-7100, Hitachi, Tokyo, Japan). To determine the drug loading capacity of NR@DOX:SAs, the absorbance of the residual DOX in supernatant was recorded at 490 nm. All the drug molecules unentrapped in the supernatants during multiple washing steps have been collected and quantified. DOX encapsulation efficacy (EE) was calculated using the following equation: %EE = [(DOX in feed -unentrapped DOX) / DOX in feed] × 100%. Drug loading (or drug content, %) is defined as: (mass of DOX loaded to the NPs/mass of DOX-loaded NPs) × 100%.
Thermal heating and drug release via NIR-activation of NR@DOX:SAs
150 μL per well of 0.6 nM NR@DOX32:SA (in PBS), 0.6 nM NR@SA (in PBS) and PBS was added into 96 well-plate at room temperature. Samples were then irradiated with an 808-nm NIR laser (DPSS laser, Unice E-O Services Inc., Taiwan) for 8 min at various power densities of 0.25 W/cm2, 0.75 W/cm2 or 2.65 W/cm2, respectively. The temperature change curves were recorded using an IR camera (Thermo Shot F30, NFC Avio Infrared Technologies Co., Ltd). Payload release of DOX or tetramethylrhodamine isothiocyanate (TRITC) labeled-SA from the suspension of NR@DOX:SAs or NR@SA-TRITC in PBS was measured at 20 min time intervals for 1 h under NIR light exposure. The fluorescence of DOX or SA-TRITC was measured by the plate reader to determine the released amount of DOX or SA-TRITC (excitation/emission for DOX: 480/596 nm; excitation/emission for TRITC: 535/572 nm.). For all the NIR-activated drug release tests, a stir bar stirring at 500 rpm was used to fasten the thermal homogeneity. Each well was sealed with transparent tape to prevent vaporization especially for those groups with long time laser irradiation (> 20 min).
Cell culture
Murine macrophage cell line (RAW 264.7) and murine prostate cancer cell line (Tramp-C1) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Both Cell lines were cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin and maintained at 37 °C, 5% CO2 in a humidified incubator. Cells were always subcultured and splitted when they reached approximately 70% confluence.
In vitro fluorescence images and cytotoxicity analyses of nanoparticle loaded macrophages
A laser scanning confocal microscopy (Zeiss LSM 510, Zeiss, Jena, Germany) was used to study the intracellular drug transport in treated macrophages. Macrophages (2×104) were seeded onto 10 mm round glass coverslips placed in a 48-well plate and cultured overnight. Cells were then exposed to NR@DOX:32:SAs and free DOX at the corresponding concentration for 2 h, respectively. After treatment, cells were washed with PBS twice and maintained in fresh culture medium for indicated time intervals. Cells were fixed with 4% paraformaldehyde and stained with DAPI (1.5 μM) for 15 min before microscopic examination. Macrophages (2×104) were plated into 96-well and allowed to adhere for 12 h prior to treatment with 8× NR@SAs, NR@DOX32:SAs, NR@DOX128:SAs and free DOX at the corresponding concentration for 2 h, respectively. Cell viability was measured using AlamarBlue assay after recovery at 2, 6, 12 and 24 h. The absorbance of each sample well was measured at 570 nm and 600 nm using a plate reader (Tecan Infinite 200, Tecan Group AG, Basel, Switzerland). The intracellular DOX concentration of therapeutic macrophages was also accessed according to a modified method from published protocols.[42, 43] Treated cells were washed with PBS and lysed in CelLytic M (Sigma-Aldrich) for 15 min. DOX of various concentration was added to aliquots of untreated cell lysates to construct a calibration curve. Prior to fluorescence spectrometer measurement, each sample was incubated with 10 M HCl for 90 min (volume of sample: HCl = 9:1). The intracellular drug concentration in macrophages after 2 h exposure of 1.5 μM and 4.8 μM free DOX was found to be approximately 0.47 (±0.02) μM and 1.35 (±0.24) μM, respectively.
In vitro drug release and cytotoxicity analyses of therapeutic macrophage against tumor cells
To analyze the cytotoxicity, 100 μL of macrophage (5×103) cell suspension was seeded into the 96-well plate and left to adhere for 12 h. Cells were then exposed to 2.4 nM NR@SAs, NR@DOX:SAs and free DOX for 2 h, washed with PBS twice and resuspended in fresh culture medium at room temperature. After 1 h of exposure to an 808-nm NIR laser at a power density of 2.65 W/cm2, the treated cells were cultured for additional 48 h prior to AlamarBlue assay. In addition, the practicability of drug release from different therapeutic macrophages was further evaluated. Similarly, seeded macrophages (5×103) in 96-well plate were incubated with 2.4 nM NR@SAs, 2.4 nM NR@DOX:SAs and free DOX at the corresponding concentration (47.6 μM) for 2 h, cleaned with PBS twice and re-cultured in fresh culture medium at room temperature. Cells were then exposed to 2.65 W/cm2 NIR laser irradiation at 808 nm for 1 h. After additional 24 h incubation that was given to allow drug release, the conditioned medium (CM) taken from the treated macrophages were either transferred to the cultivation of Tramp-C1 cells (5×103 cells per well in 96-well plate) or submitted to drug payloads analysis. For therapeutic efficacy assessment, Tramp-C1 cells were incubated with various CM for 48 h and the viability was analyzed through Alamarblue assay. As for drug release assessment, each CM was either added with 20 μL 12 M HCl for 1 day to measure the DOX fluorescence signal or diluted with 500 μL aqua regia for 12 h to analyze the gold content by ICP-MS.
In vitro tumortropic studies of therapeutic macrophages
The migration assay was performed using Boyden transwell chamber for 24-well plate (8 μm-pore size, Corning Inc., NY, USA). Various payload-containing macrophages in 300 μL culture medium were seeded in the top of the insert at 6×104 cells/well. The bottom chambers were filled with either traditional cell growth medium (5% FBS) or tumor conditioned medium (TCM) of 600 μL. The migration was proceeded in 4 h at 37 °C, 5% CO2 in a humidified incubator. The migratory cells were stained with 2 μM DAPI for 20 min before being monitored and quantified by cell counting under a fluorescence microscopy (IX-71, Olympus, Center Valley, PA, USA). The data was obtained at least 3 independent experiments and statistically analyzed.
In vivo combinational therapy of therapeutic macrophage in a mouse xenograft model
Six week-old male C57BL/6J mice were purchased from National Laboratory Animal Center (Taiwan) for all in vivo experiments. 3×106 Tramp-C1 cells were subcutaneously injected into both the right and left hind limbs. As the tumor volume was reached to ~150 mm3 in 3 weeks, mice were randomly divided into 6 groups (4-5 mice per group) to analyze the tumor growth rate. 100 μL of PBS, RAW 264.7, RAW-NR@SAs, RAW-NR@DOX128:SAs, NR@DOX:SAs and free DOX (macrophage: 5×106 cells/agent, Au: 3.1 mg/kg, DOX: 3.3 mg/kg) was administrated intratumorally at day 0 and day 3 for comparison. NIR laser irradiation (808 nm, 0.75 W/cm2, 3 min) of tumor on the left limb was subsequently commenced at day 1 and day 4. Tumor size was measured via a caliper every day until day 11 and the volume (V) was calculated using the following formula: V = 1/2 × L × W2, where L and W are the longest and shortest dimension of tumor. To access in vivo targeted therapy of therapeutic macrophage, mice were furthermore received therapeutic macrophage (macrophage: 1×107 cells/agent, Au: 6.2 mg/kg, DOX: 6.6 mg/kg) through tail vein administration (two dose on day 0 and 3) when the tumor volume on the left limb was reached to 80-100 mm3 in a subcutaneous model mouse. NIR laser irradiation was subsequently performed on day 1 and day 4 and tumor size was recorded daily until day 18.
In vivo imaging system (IVIS) analysis
SA was labeled with near-IR fluorophore, Cy5.5, for the IVIS fluorescence detection. After the single intratumoral injection of free SA, NR@DOX:SAs and RAW-NR@DOX:SAs, respectively, tumor-bearing mice were imaged by IVIS (Cy5.5 filter: excitation/emission = 675/720 nm) at day 0, 1, 4 and 7 post-treatment. Region of interest of each fluorescence image was quantified and further analyzed by the Living Image® 3.1 software (Xenogen, Alameda, CA).
In vivo histological studies
To identify the hypoxia and normoxia region, 100 μL pimonidazole HCl (PIMO, 40 mg/kg) and 10 mg/ml Hochest 33342 in saline buffer was respectively intraperitoneal and intravenous administrated to the mice before 1 h sacrifice if the experiment was required. Tumors collected from mice at indicated time point were embedded in OCT (Sakura Finetek, Torrance, CA, USA), sectioned (10 μm) and mounted onto slides. Tissue slides were fixed in the ice-cold methanol for 5 min and rinsed three times in PBS. The tumor tissues for necrosis analysis were explored through hematoxylin and eosin stain. Apoptosis induced by chemo-, photo-, or combinational treatment was further evaluated if required. Tissue slides were first blocked with blocking buffer (4% FBS, 1% goat serum and 0.01% Tween-20 in PBS) for 1 h and incubated with anti-PIMO, anti-rabbit CD68 or anti-rabbit caspase 3 (Abcam, Cambridge, MA, USA) at 4 °C. After 24 h reaction, slides were probed with Alexa Fluor 488-conjugaed secondary antibody (Abcam, Cambridge, MA, USA) for 1 h and washed three times with saline buffer. Cell nuclei were identified by DAPI staining (5 μg/ml) using fluorescence microscopy. The fluorescence intensities, determined as the pixel numbers within the selected field were analyzed by Image J software (National Institutes of Health, Bethesda, Maryland, USA) and the average pixel density (mean fluorescence intensity) for the corresponding tumor section was carried out on five randomly selected fields. Similarly, the dark-field images were obtained from (IX-71, Olympus, Center Valley, PA, USA). The region of interest was determined by the pixel numbers through Image J software and the average intensity was obtained from at least nine selected area, which crossed the entire tumor section.
Statistical Analysis
Experimental data was quantitatively presented as the mean ± standard deviation. Statistical significance by a two tailed student ′s test (P < 0.05) was determined unless otherwise stated.
Results and Discussions
Preparation and characterization of core-shell nanoplatform
CTAB-capped Au NRs fabricated via a seed-mediated method [41] in this study, had an aspect ratio of 4.7, with a length and width of 56.4 (±3.5) nm and 11.9 (±1.8) nm, respectively, as determined by transmission electron microscopy (TEM) examination. To facilitate bioconjugation and DOX loading, the Au NRs were constructed with a serum albumin (SA) shell using a new desolvation method, as shown in Figure 1A. SA was initially attracted to the Au NR surface through electrostatic force with CTAB bearing the opposite charge. Coacervation, mostly driven by hydrophobic interactions, occurred rapidly between SA molecules after the addition of a denaturing agent.[29] A homogenous colloidal suspension of NR@SAs was therefore obtained and a definite core-shell morphology was observed in TEM images by negative staining (Figure 2A). To elucidate the role of the denaturation process of SA in forming the protein corona, solutions containing different percentages of the denaturing agent, e.g., ethanol, were added to facilitate particle fabrication. Incubation in an ethanol solution with an ethanol concentration lower than 60% led to lower colloidal stability, as shown in Figure S1A, indicating insufficient denaturation of SA to protect the Au NRs against aggregation. However, SA incubated in 80% ethanol solution formed a more complete coacervate with a higher SA loading (Figure S1B), resulting in less nanoparticle aggregation compared with the samples prepared using other ethanol concentrations. Moreover, ethanol solution was premixed with a more polar solvent, methanol, as an alternative denaturing agent [44]. A solution with a 1:1 ratio of ethanol to methanol (named 80% EM solution) was found to provide both an optimal capacity of SA loading as well as colloidal stability. In subsequent experiments, 80% EM solution was used to prepare drug-loaded NR@SAs.
Synthesis and the characterization of NR@DOX:SAs. (A) TEM images of NR@SAs. Scale bar: (a) 200 nm and (b) 50 nm. (B) Drug loading study of DOX to NR@SAs (2×, 0.6 nM). Aliquots of SA solution (3.03 μM) were pre-mixed with a serial concentration of DOX (4 to 256 μM) and used for NR@DOX:SAs construction. (C) Encapsulation efficiency (EE%) of DOX into NR@SAs (0.6 nM). (D) UV-Vis spectra of Au NRs, NR@SAs, and NR@DOX:SAs in phosphate buffered saline (PBS). (E) Hydrodynamic size and zeta potential of different nanoconjugates in PBS.
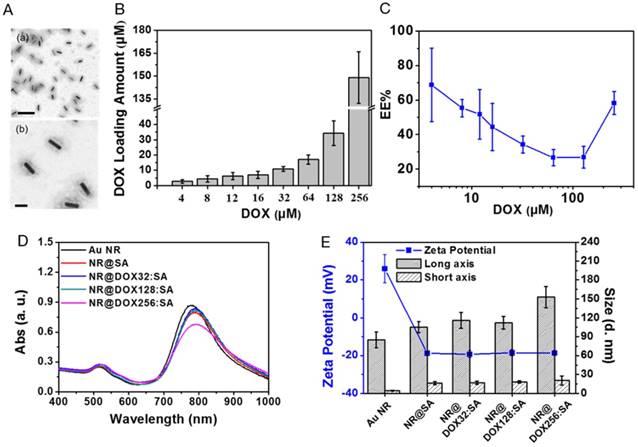
DOX was selected as a representative antineoplastic agent to be incorporated into the protein matrix of NR@SAs. An aliquot of the stock solution containing the desired amount of DOX was pre-equilibrated with SA at molar ratios ranging from 1.33 to 85.33, followed by the previously described desolvation process. To evaluate the loading capacity of DOX into NR@SAs, the supernatant removed from individual suspensions was analyzed by UV-Vis spectroscopy. As shown in Figure 2B, DOX was effectively encapsulated by NR@SAs and the loading amount increased gradually as the initial drug input feeding to the NR@SAs (0.6 nM of Au NRs and 3.03 μM of SA) was increased to 128 μM. At higher concentrations, the encapsulation efficiency increased markedly (Figure 2C). Approximately 4, 13, and 58 DOX molecules per SA were loaded for NR@DOX32:SAs, NR@DOX128:SAs, and NR@DOX256:SAs, respectively. In addition, UV-Vis spectra in Figure 2D showed that the absorption profile of NR@DOX32:SAs and NR@DOX128:SAs was similar to that of NR@SAs, whereas the optical intensity of NR@DOX256:SAs was obviously reduced in the NIR region; no distinguished difference in size and zeta potential was observed after the drug loading except for the NR@DOX256:SAs (Figure 2E). This result was in agreement with a previous report showing that the cationic properties of DOX can shield the surface charges and thus reduce the repulsion forces between the SA components during the desolvation process [45]. Monodisperse nanoparticles were therefore only obtained for drug inputs less than 128 μM because higher drug concentrations led to NR@DOX:SAs with slightly increased and broader size distributions. Eventually, initial drug inputs equal to 32 and 128 μM were selected and encapsulated into NR@SAs for subsequent cellular delivery. The loading content of DOX entrapped inside the albumin shell of NR@DOX32:SAs and NR@DOX128:SAs was 3.4 (±1.0)% and 10.6 (±2.2)%. The corresponding mass ratio (μg/μg) of Au to DOX was 3.07 and 0.94, respectively. Notably, the dense albumin shell of NR@SAs exhibited excellent performance in cargo confinement; only minute DOX leakage (< 3%) was detected for NR@DOX:SAs in phosphate buffered saline (PBS, pH 7.4) within 6 days (Figure S2A). The release kinetics of drug molecules from NR@DOX:SAs was further assessed by means of a dynamic dialysis technique.[46] In contrast to free DOX solution, a negligible drug diffusion was observed for the colloidal suspension when dialyzing against PBS for 48 h (Figure S2B), indicating the albumin shell coating of NR@SAs was effective for delayed drug release.
SA shell delays drug-related toxicity in the host cell carriers
To confirm that the resultant nanodrugs exhibited low cytotoxicity against the cellular host in the absence of external triggers, the time-dependent cell viability of the DOX-loaded NR@SAs for RAW 264.7 macrophages was investigated with free DOX molecules serving as the control sample. As shown in Figure 3A, DOX-loaded NR@SA nanoconjugates delayed the drug-related toxicity to the cellular carrier over the first 24 h; 70% of the NR@DOX128:SA-laden macrophages remained alive. However, less than 30% of the exposed cells survived after treatment with free DOX at 4.8 μM, which was the concentration of NR@DOX128:SAs uptaken by the cell carrier. Moreover, the percentage of viable cell carriers pre-treated with NR@DOX128:SAs was comparable to that of NR@DOX32:SAs over a 24-h period, demonstrating that the albumin shell coating effectively postponed both DOX release and the short-term cytotoxic effect. We also noted that the developed nanoagents were taken up efficiently by phagocytic cells. Compared to the non-treated cell carriers, a strong yellowish scattering light was observed to originate from nanoparticles in both the NR@SA- and NR@DOX:SA-laden macrophages by dark-field microscopy (Figure S3). These observations are also supported by ICP-MS results indicating that approximately 13.2 (±0.1) and 13.8 (±2.2) pg Au/cell (referring to NR@SAs and NR@DOX:SAs, respectively) was taken up by the host cells. The Au NR encapsulation efficiency of therapeutic macrophages was determined as 3%. Accordingly, the loading amount of DOX inside macrophages treated with NR@DOX32:SA and NR@DOX128:SA was found to be 4.5 (±0.6) pg/cell and 14.5 (±3.1) pg/cell, respectively.
The evaluation of macrophages as a drug biocarrier. (A) After the uptake of NR@SAs, NR@DOX32:SAs, NR@DOX128:SAs, and free DOX at the corresponding concentration, macrophages (RAW 264.7) were recovered at various time intervals for viability assay. The Au NR concentration of all samples was maintained at 0.15 nM. (B) The intracellular drug transport within macrophages incubated with NR@DOX:SAs and DOX was visualized at different recovery times by laser scanning confocal microscopy. Nuclei were stained with DAPI (blue), and the acidic endolysosomal compartments were stained with transferrin-Alexa 633 (green). Scale bar: 5 μm.
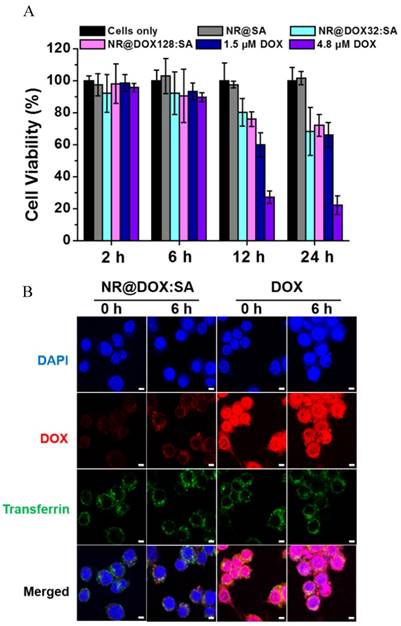
The mechanism of the postponed drug release from NR@DOX:SAs was further investigated by laser scanning confocal microscopy. As shown in Figure 3B, a weak DOX signal was initially detected in NR@DOX:SA-treated macrophages and a subsequent recovery of the fluorescence signal was monitored for cells subjected to additional incubation. The spots of DOX signals were essentially overlapped with the fluorescently labeled transferrin with Alexa 633 as a marker, indicating that NR@DOX:SAs could be successfully delivered into lysosome after internalization. However, in the case of cells incubated with the same amount of DOX, strong emission intensities were observed, located mainly in the cell nuclei and perinuclear region. These results support the efficient shielding of the albumin shell on NR@DOX:SAs, which could prohibit premature drug leakage in vitro, thereby minimizing the associated toxicity. Similar results have also been reported for host cells pre-incubated with NR@SAs. Because the adsorbed albumin was labeled with TRITC, the fluorescent NR@SAs could be trafficked to the lysosomes as they enter cells (Figure S4) [47]. The appearance of an unremarkable fluorescence recovery over 6 h [48] indicates that the dense corona on the NR@SA surface was slowly degraded by the lysosomal enzymes, leading to a delayed cytosolic release of lysosomal content.
NIR light-activated drug release from payload-laden macrophages
A major impediment in cell-based delivery systems is the inefficient liberation of drug payloads from host cell carriers [9, 10]. We therefore investigated the capability of on-demand drug release by NIR light irradiation using serial analysis, as illustrated in Figure 4A. Briefly, the conditioned medium of macrophages was obtained 24 h post NIR laser irradiation and transferred to cultivation of Tramp-C1 tumor cells. The viabilities of both macrophages and tumor cells were analyzed collaterally. As displayed in Figure 4B, 58.8 (±6.8)% of macrophages remained alive after treatment with NR@DOX:SAs, whereas only 23.9 (±7.4)% of the cells survived following exposure to NIR light. By contrast, a slight cytotoxicity (17.7±11.8%) was observed for RAW-NR@SAs submitted to additional NIR irradiation. Additionally, a live/dead cell double staining assay (Figure S5) further confirmed that a prompt cell disruption was achievable by laser irradiation of NR@DOX:SA-treated macrophages. These results suggest that the therapeutic cargo NR@DOX:SAs was effective in damaging the cellular host in response to NIR activation.
Evaluation of the in vitro therapeutic efficacy of macrophage-NR@DOX:SA against tumor cells. (A) Schematic of the experimental design of the pharmacokinetic bioassay. (B) Relative cell viability of macrophages (RAW 264.7) treated with NR@SAs, NR@DOX:SAs, and free DOX at the corresponding concentration. Cells were irradiated with NIR light (808 nm, 2.65 W/cm2, 1 h) after 2 h cellular uptake and were then recovered for an additional 48 h prior to AlamarBlue assay. **p < 0.01. #p < 0.001 versus cells only (-). (C) Tramp-C1 cell viability at the end point after 48 h of incubation with a conditioned medium (CM). **p < 0.01. #p < 0.001 versus control RAW 264.7 (-) group. (D) Fluorescence measurement was used to assess the drug release in CM from therapeutic macrophages. (E) Microscopic images collected to determine the intracellular DOX fluorescence intensity within Tramp-C1 cancer cells incubated for 12 h with conditioned medium of RAW-NR@DOX:SAs with (+) and without (-) NIR treatment. The fluorescence intensity ratio of DOX/DAPI from each image was quantified using the Image J software. (n = 6, *p < 0.05.) Scale bar: 20 μm.
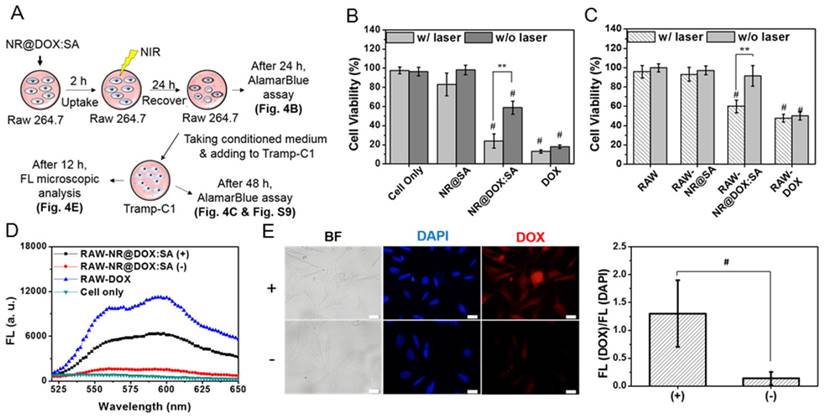
Next, we verified the NIR-mediated drug release by monitoring the DOX fluorescence of the supernatant removed from the NR@DOX:SA suspension. As shown in Figure S6A, the temperature of the colloidal suspension (100 μL of 0.6 nM NR@DOX:SA) rapidly increases and reaches a plateau under NIR light exposure for approximately 6 min. The maximum temperature increases from 35.8 (±2.1)°C to 76.7 (±5.9)°C as the irradiation power was increased from 0.75 W/cm2 to 2.65 W/cm2. Approximately 13% of drug release was also observed for NR@DOX:SA (Figure S6B) in response to NIR exposure (2.65 W/cm2) for 1 h. In accordance with a 29% release of the loaded SA-TRITC from the same carrier upon NIR activation, the subsequent payload release was attributed to the reorganization and denaturation of the adsorbed protein corona in proximity to the heated NRs [26]. It should also be noted that the optical intensity of NR@DOX:SAs was only reduced slightly (9.8% in Figure S6C); no distinguished difference in hydrodynamic size was observed after NIR irradiation (Figure S6D). Additionally, NIR light-mediated DOX release was further improved for NR@DOX:SAs pre-treated with cathepsin B, a lysosomal cysteine protease (Figure S7).[49] This result suggests that the lysozyme enzymatic activity within cells can accelerate the drug release from NR@DOX:SAs via a photothermal effect upon NIR irradiation. In confocal fluorescence images (Figure S8), the fluorescence from TRITC-labeled NR@SAs inside cells after NIR-treatment was also visualized as more diffused orange spots in the cytosol (white arrows). This fluorescence was clearly distinguishable from the fluorescence signals detected from cells prior to irradiation, which were mostly co-localized with green spots emanating from transferrin-Alexa 633. These results again indicate that the photothermal effect on NR@DOX:SAs triggered the disruption of the endo/lysosomal membranes after cellular uptake and further accelerated payload release by local heat generation.
To examine the release of drug from RAW-NR@DOX:SAs followed by NIR irradiation (+) and the subsequent drug cytotoxic activity toward tumor cells, we analyzed the viability of Tramp-C1 cells (Figure 4C). As expected, the survival rate of Tramp-C1 cells decreased substantially (60.0±6.7%) after incubation with conditioned medium (CM) taken from RAW-NR@DOX:SA (+). By contrast, cancer cells survived well (91.6±10.8%) when exposed to CM from non-irradiated RAW-NR@DOX:SA (-). Coordinately, a statistically significant decrease in cell numbers was observed in living Tramp-C1 cells after treatment with CM from RAW-NR@DOX:SA (+) again confirmed that the pronounced drug activity of RAW-NR@DOX:SAs in killing cancer cells was activatable by NIR irradiation (Figure S9). Additionally, the intracellular DOX accumulation of cancer cells was further investigated by fluorescence microscopy. In agreement with the detection of an increasing fluorescence signal in CM from irradiated RAW-NR@DOX:SA (+) than its non-irradiated counterpart (Figure 4D), a dramatic increase in the DOX fluorescence intensity was further observed for Tramp-C1 cells incubated with CM from RAW-NR@DOX:SA (+) as compared to that from RAW-NR@DOX:SA (-) (Figure 4E). Quantitative measurement of the average fluorescence DOX/DAPI ratio indicated an approximate 9.5-fold increase in DOX accumulation level. As evidenced by a non-negligible increase in the concentration of gold ions detected by ICP-MS in the CM from RAW-NR@DOX:SAs post NIR treatment (p < 0.01), the results of the aforementioned analyses indicate that both free drugs and drug-containing nanoconjugates were liberated upon input of an external trigger. NIR activation on therapeutic macrophages led to payload release in large quantities, thus exerting a cytotoxic action toward tumor cells similar to that of RAW-DOX (+/-), as displayed in Figure 4C.
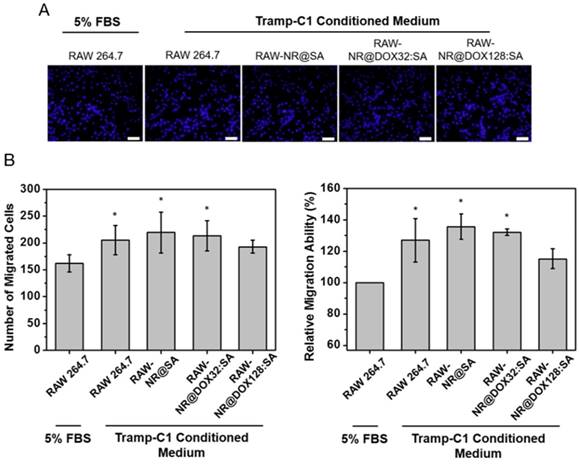
In vitro tumoritropic migratory properties of payload-laden macrophages
Prior to conducting in vivo studies, we evaluated the tumoritropic migratory potential of therapeutic macrophages using the Boyden chamber assay. The migratory cells that move toward the chemoattractant (Tramp-C1 tumor-conditioned medium, TCM) or traditional cell growth medium (5% FBS) were stained and quantified by cell counting, respectively (Figure 5). In our studies, payload-free macrophages exhibited positive chemotaxis to a simulated tumor microenvironment and migrated ca 1.3-fold faster toward TCM compared to the 5% FBS control (p = 0.03). The NR@SA- and NR@DOX32:SA-laden macrophages exhibited similar migration capacities toward TCM, leading to 1.4-fold and 1.3-fold increases in migrated cell number compared to the control, respectively. As evidenced by the observation that chemotactic migration decreased only slightly for macrophages loaded with NR@DOX128:SAs, the therapeutic cargo meets the cell-based delivery requirement that payloads remain essentially harmless to the cellular host on its tumor tropism.
Relative tumortropism of untreated macrophages (RAW 264.7) and NR@SA- and NR@DOX:SA-treated macrophages (RAW-NR@SAs and RAW-NR@DOX:SAs) that migrated toward Tramp-C1 tumor-conditioned medium or traditional cell growth medium (5% FBS). (A) Cells attached onto the backside of a Transwell membrane were stained with DAPI and visualized by fluorescence microscopy. (B) Cell migration, referred to as the cell counts per view of Transwell membrane as well as the relative turmotropic migration ability (%) of therapeutic macrophages over the control (Raw 264.7 in 5% FBS) were quantitatively analyzed. *p < 0.05 versus control RAW 264.7 (5% FBS). Scale bar: 50 μm.
Cell-mediated drug delivery system improves drug coverage and distribution and enhances retention ability
To illustrate the in vivo performance of various agents, the cell-mediated delivery system, pristine conjugates, and free drug, in vivo NIR fluorescence imaging was performed to evaluate the delivery process of pristine conjugates and RAW-NR@DOX:SAs after intratumoral administration (Figure 6A). A relatively low amount of chemodrug (3.3 mg/kg) [50-52] was used for the experiments to ensure the groups had similar tumor sizes at different growth stages over 11 d. Because the visible light emitted by DOX was limited by its poor penetration into tissue, the SA of different conjugates was covalently linked with a NIR fluorophore, Cy5.5, for clear visualization of the entire mouse tumor. After the instantaneous injection, little or no fluorescence signal was observed from any mice tumor at day 0 (Figure 6B). This lack of signal is attributed to an extensive aggregation-induced quenching effect of therapeutic agents with a high localized concentration in the central core of the tumor. However, the fluorescence signal of mouse tumors injected with RAW-NR@DOX:SAs increased approximately 6.5-fold at day 1 and continued to increase (9.6-fold at day 4 and 11.6-fold at day 7). The fairly strong fluorescence recovery of RAW-NR@DOX:SAs compared with that of pristine conjugates (maximum change: 6.8-fold) indicates that a greater drug coverage of the tumor was achieved via macrophage vehicles. Additionally, Cy5.5-labeled SA exhibited the highest fluorescence recovery (2.8-fold) at 1 d post-injection; its recovery decreased significantly over time (2.0-fold at day 4 and 1.6-fold at day 7). These data suggest that free molecules, although diffused readily, could be eliminated rapidly from the tumor mass, resulting in an insufficient intratumoral drug concentration to inhibit subsequent tumor growth.
The evaluation of chemodrug retention and distribution. (A) In vivo NIR fluorescence images of tumor-bearing mice; the images were acquired after intratumoral administration of SA, NR@DOX:SAs, and RAW-NR@DOX:SAs on day 0 to day 7. SA was labeled with Cy5.5 as an NIR fluorophore. (B) The total intensity fluorescence values and corresponding ratio (Fx/F0, Fx is the fluorescence value at a defined date and F0 represents fluorescence signal obtained immediately after injection) in the region of interest for each group were analyzed quantitatively (n = 3). On day 11, intratumoral drug distribution of the tumor harvested from mice injected intratumorally with RAW-NR@SAs, NR@SAs, and DOX. (C) Schematic diagram of respectively analyzing x-y plane and z direction of the harvested tumor. D) Fluorescence microscopic images and the corresponding intensity of DOX signals were observed within the largest cross section of tumor harvested individually. White arrows indicate the needle path of each insertion site. Scale bar: 5 mm.
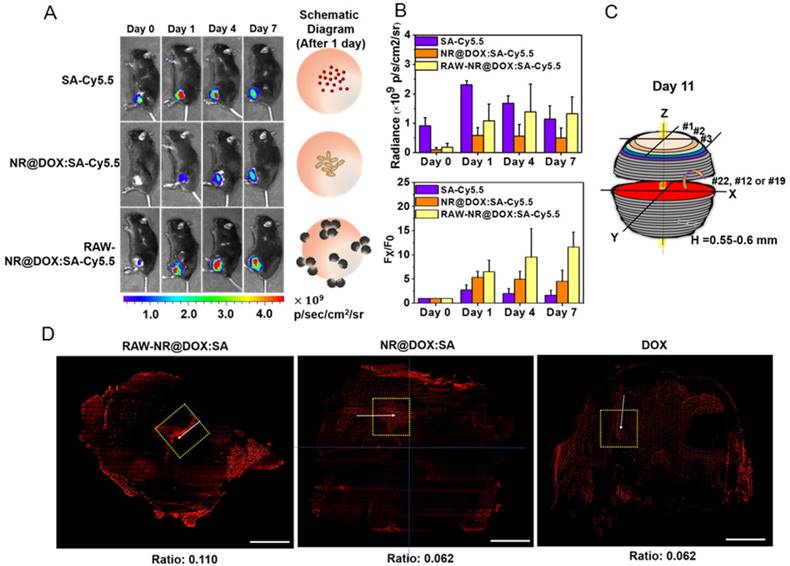
For a detailed investigation of the intratumoral drug distribution, we harvested the three major groups of tumors at 11 d after injection and obtained approximately 25 sections at 550-600-μm intervals (Figure 6C). On the basis of the observation of the maximum tumor section (x-y plane) in Figure 6D, patches of bright-red patterns originated from DOX signals were predominantly observed in the border region of tumors from mice treated with RAW-NR@DOX:SAs. By contrast, NR@DOX:SAs remained more localized near the needle insertion site (the white arrows indicate the needle path of the tumor). Although free DOX appeared to be homogeneously distributed throughout the tumor, the emitted fluorescent signal was considerably less intense than that of RAW-NR@DOX:SAs. After quantitative analysis, the average fluorescence DOX/DAPI ratio of RAW-NR@DOX:SAs was nearly 2-fold larger than the other groups. Moreover, the total area consisting of DOX fluorescence from RAW-NR@DOX:SAs accounted for 27.5 % of entire tumor section. It was obviously larger than NR@DOX:SAs (14.5%) and free DOX (18.5 %). In addition, a 2-D horizontal scan was performed in each fault plane along the vertical axis (Z axis) to examine the drug distribution within the 5 × 5 mm2 area (yellow dashed rectangle) across the center of the intratumoral injection site. Fluorescence images (Figure S10A) from the 25 section planes again confirmed that RAW-NR@DOX:SAs exhibited a much stronger signal and a remarkably broader distribution compared to the other groups. Together with the quantitative analysis results in Figure S10B that indicate 25 consecutive sections of tumor-bearing mice injected with RAW-NR@DOX:SAs exhibit the highest average DOX/DAPI intensity ratio and more distributed profile, these findings demonstrate the potential benefits of long-lasting DOX preservation by macrophages. Meanwhile, their tumor-tropic migration improve spatial positioning of drug distribution and penetration.
Homogenous photothermal drug delivery was achieved by combinational therapy through multiple intratumoral injections
Because the spatial distribution of both Au NRs and chemodrug were altered, the photothermal effects, particularly of the cell-mediated delivery system and pristine conjugates, were investigated. Compared with other Au NR-based delivery systems, the present delivery system had a relatively high amount of Au NRs (3.1 mg/kg) but low laser power and irradiation time (0.75 W/cm2 and 3 min, respectively) [8, 36] to provide a detailed demonstration of the intratumoral delivery profile of each therapeutic agent. The NIR images indicated that the highest corresponding temperature of the exposed region of the tumor after two treatments of NR@DOX:SAs (Figure 7A) were approximately 70 °C and 49 °C, respectively, and the maximum temperatures for both the RAW-NR@DOX:SA and RAW-NR@SA groups were approximately 44 °C and 46 °C during the first and second treatment, respectively. Under the same irradiation conditions, the temperature of all remaining groups was 37-38 °C. Although a relatively high temperature was obtained by the pristine NPs in the first treatment, the average temperature within the laser spot was approximately 40.5 °C, which was 2 °C higher than that of the cell-mediated delivery system (Figure 7B). In the PBS control, the temperature was approximately 33-34 °C. Furthermore, a significant temperature gradient was observed with the pristine NR@DOX:SAs (Figure 7C). Only approximately 20% of the area under the laser spot was >50 °C, and approximately 10% of the area was distributed at each interval (36-50 °C). Furthermore, approximately 40% of the area remained completely unaffected (<36 °C) by the NIR laser irradiation. These results suggested that the localized distribution of pristine NR@DOX:SAs followed by direct intratumoral injection, leads to an inefficient heat transfer in the tumor microenvironment. By contrast, heat (>38 °C) generated through a cell-mediated delivery system covered 50%-60% of the area, and only 10%-20% of the area remained completely unaffected, indicating that the cell-mediated delivery system had more efficient photothermal distribution for intratumoral coverage. Furthermore, the skewness test (Figure 7D) showed that both pristine NPs and cell-mediated delivery system skewed right (>0); however, the skewness of the cell-mediated delivery system was closer to 0 (Figure 7E), suggesting that the thermal distribution of cell-mediated delivery system was more symmetrical than pristine NPs. This finding is consistent with the results of the full and quarter-width at half-maximum analysis. The number of detected counts with intensities of >50% and >25% of the total counts within the laser spot were higher in the cell-mediated system than in the pristine NR@DOX:SAs, suggesting that a more homogenous photothermal delivery was achieved in the cell-mediated system (Figure S11).
In vivo photothermal efficacy of the first and second NIR laser treatment of payload-containing macrophages, pristine nanoparticles, and PBS controls. (A) Thermal imaging and the highest temperature of different tumor-bearing mice after photothermal treatment (808 nm, 0.75 W/cm2, 3 min) at 24 h post-intratumoral injection. Inset: 3D images of NIR laser-treated mice. The (B) average temperature and (C) corresponding thermal distribution of the exposed region was further evaluated using the TAS 20.3 software program. (n = 3-5; *p < 0.05 or **p < 0.01 vs. NR@DOX:SA-treated mice). The skewness test using SPSS 13.0 (IBM) was conducted to evaluate (D) each thermal distribution and perform (E) additional quantitative analysis. On the basis of every original data in (D), the black line represented the best-fitting curve of each thermal distribution after statistical calculation using SPSS 13.0 (n = 3-5; #p < 0.05 or *p < 0.05 vs. NR@DOX:SA-treated mice).
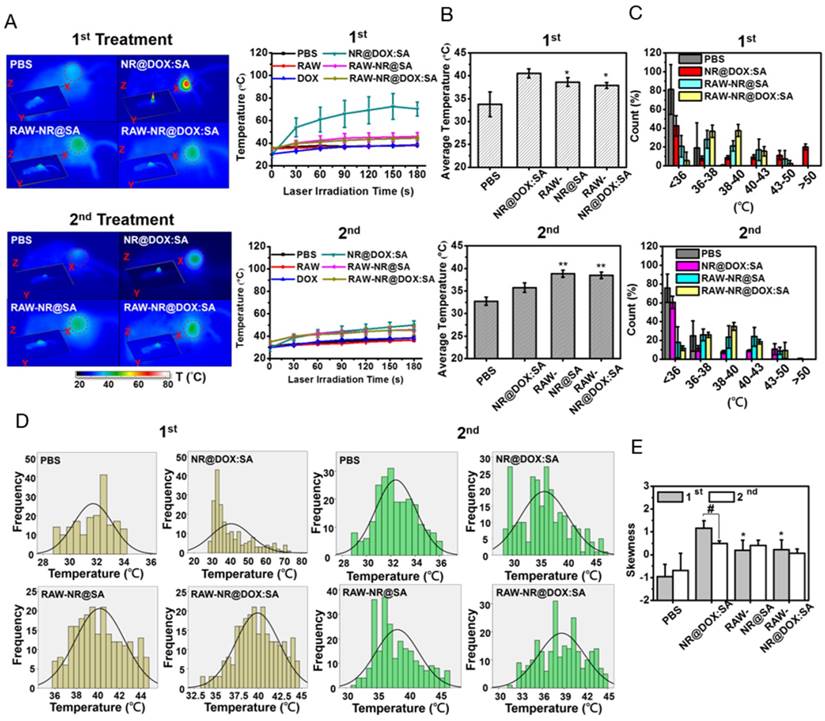
Notably, after the second NIR laser irradiation treatment, the heat generated by the pristine NPs was unexpectedly lower than that generated after the first treatment (Figure 7A). At this time in second NIR laser irradiation, the average temperature within the laser spot was approximately 36 °C, 2.5 °C lower than that of the cell-mediated delivery system, and the PBS control was still maintained at approximately 33 °C-34 °C (Figure 7B). In addition, the affected area (>38 °C) under the laser spot was decreased to approximately 30% after the second treatment, 2-fold lower than that observed after the first treatment, and the unaffected area within the exposed tumor region increased to approximately 60% after the second treatment, 1.5-fold higher than that observed after the first treatment (Figure 7C). The thermal efficacy achieved after the second treatment with pristine NR@DOX:SAs markedly decreased despite the thermal profile was closer to a symmetrical distribution (Figures 7D-7E and Figure S11). By contrast, the average temperature and corresponding thermal distribution of the cell-mediated delivery system after the second treatment were similar to those observed after the first treatment. Because of a reproducible homogenous thermal delivery, the cell-mediated system was considered more appropriate than pristine NPs for multiple treatments, particularly when combined with chemotherapy, in clinical studies.
What happened when non-homogenous photothermal drug delivery was achieved?
To determine why the resultant temperature and corresponding thermal distribution after the first and second NIR laser irradiations in pristine NR@DOX:SA-treated mice were substantially different, the biological effects induced by the photothermal treatment were investigated further. Because tumor size during the first NIR laser irradiation was controlled at 150 mm3, the tumor surface temperature detected by the IR camera was almost equivalent to the resultant temperature inside the tumor [16, 53]. In this study, all detected counts of the NIR laser-exposed regions were converted to cumulative equivalent minutes at 43 °C (CEM43). The average CEM43 of pristine NR@DOX:SAs greater than 104 was substantially larger than that of the cell-mediated system and supposedly performed more favorable tumor ablation (Figure S12A). However, the irradiated area where the CEM43 was present for more than 20 min occupied only approximately 30% of the entire exposed region and was mostly centrally localized (Figure S12B). Both an incomplete thermal ablation and collateral damage including hemorrhage, necrosis, thermal lesions [54, 55] can be highly expected. Many of these phenomena in NR@DOX:SA (+)-treated mice were visually observed after 3 d (day 4). For example, a serious burn mark was observed at 12 h post-NIR laser irradiation (inset, Figure S12C), and a damaged wound was present with slight bleeding (red arrow, Figure S12C) each day. The wound remained unhealed until at least the second NIR laser treatment. Compared with the tumor size at 12 h post-NIR treatment (inset, Figure S12C), the tumor size and margin of the damaged wound on day 4 increased continuously (Figure S12C). Moreover, two of the five mice were observed to become lame after NIR laser exposure, possibly because of irreversible bone resorption.[54, 55] By contrast, for the cell-mediated delivery system, CEM43 value was approximately 10 min, suggesting that a less pronounced but highly distributed thermal profile effectively reduced the occurrence of detrimental effects. Only some areas, possibly close to the delivery site of Au NRs [56], still induced slightly homogenous burn marks after first NIR laser irradiation (inset, Figure S12C). However, each induced burn mark appeared to heal on day 4.
Subsequently, after the treated mice were sacrificed, incomplete photothermal damage by pristine NPs was observed through hematoxylin and eosin (H&E) staining of the dissected tumor sections (Figure S12D). Microscopic images showed that the tumor core was seriously destroyed and had a large necrotic area, whereas the peripheral tumor region appeared to be unharmed. By contrast, the cell-mediated delivery system caused less pronounced but more homogenous photodamage. The damaged wound, particularly that with severe necrosis and hemorrhage, was considered to have an unfriendly microenvironment and pro-inflammatory responses that were detrimental to cancer therapy [57]. Moreover, external stimulation and induced injury have been reported to alter drug distribution, penetration and elimination [37]. Hence, the intratumoral nanoparticle distribution was further assessed by analyzing the dark-field images of the selected area in each tumor sections (Figure S12E and S12F). On the basis of the protocol described by Mooney et al. [8] and the triple standard difference methods, the recorded images were preprocessed using imaging software to flatten light fluctuations, and each count of detected scattering light over a 3-fold standard deviation (σ) of the average signal from the PBS control group was defined as the threshold. A signal beyond the threshold was considered to be the strong scattering light for detection. The received signals over the defined threshold from the cell-mediated system were distributed in the peripheral and central tumor regions, and those from NR@DOX:SA (-) were almost centrally localized (red dashed line). This finding is consistent with the results of DOX fluorescence distribution (Figure 6D), suggesting that the cell-mediated delivery system was more distributed within the tumor but pristine NR@DOX:SA was only centrally localized. On the other hand, for NR@DOX:SA-treated mice followed by NIR-irradiation (+), the tumor coverage was decreased (Figure S12G) and the average intensity of scattering light was apparently less than that of NR@DOX:SA (-) (Figure S12H). The scattering light beyond the threshold in NR@DOX:SA (+) was more localized at the tumor edges, which was away from the red dashed line. It suggests that the severe and incomplete photothermal damage in the central tumor core by the pristine NR@DOX:SA (+) induced changes in the tumor microenvironment that redistributed the tumor coverage and retention ability of the remaining pristine NR@DOX:SAs (Figure S13). This finding is consistent with the results of the statistical analysis, showing a lower temperature but a more symmetrical thermal distribution of NR@DOX:SAs after second treatment than that after the first treatment (Figure 7).
Homogenous photothermal and drug delivery by the cell-mediated system resulted in precise NIR-laser-activated drug release for improved antitumor effects
Investigation of the homogenous chemothermal and photothermal delivery of the cell-mediated system was a more effective approach to evaluate the antitumor effects; therefore, we performed immunohistochemical examinations of the frozen tumor sections on the third day after intratumoral injection. Apoptotic cancer cells were stained with a caspase-3 antibody (green) and the nuclei were counterstained with DAPI (blue). Representative images in Figure 8A showed that DOX fluorescence (red) was highly correlated with the apoptotic region, indicating that the RAW-NR@DOX:SAs were able to release DOX after NIR laser irradiation and induce apoptosis in the surrounding area. In the quantitative image analysis, both the DOX/DAPI (Figure 8B) and caspase-3/DAPI (Figure 8C) intensity ratios were significantly increased in the RAW-NR@DOX:SA (+)-treated mice compared with the nonirradiated mice (-). Furthermore, only a slight increase in the DOX/DAPI ratio was observed in the NR@DOX:SA-treated mice after NIR laser irradiation; therefore, the related toxicity was insufficient to induce desirable apoptotic death. The caspase-3/DAPI ratio did not significantly differ (p > 0.05) in the free DOX-injected and RAW-NR@DOX:SA-treated mice following NIR laser irradiation. This finding suggested that cell-mediated delivery can enhance the cytotoxicity of NR@DOX:SAs against neighboring tumor cells by combining an external trigger for drug release. Furthermore, tissue sections of the Tramp-C1 tumors pretreated with RAW-NR@DOX:SAs were stained with DAPI for nuclei identification (blue), and the CD68 marker was used to identify the macrophages (green). The DOX (red) fluorescence signals overlapped substantially with the macrophage-selective fluorescent markers before irradiation (Figure 8D). However, some DOX signals were observed within the tumor at locations distinct from the loci where the macrophages were identified, suggesting that the drug payloads had been released by the RAW-NR@DOX:SAs and had diffused into the surrounding tumor cells after NIR laser activation.
In vivo studies of antitumor effect. (A) Immunohistochemical images of tumor sections acquired from different tumor-bearing mice that received chemo- and photo-treatment on day 4. Scale bar: 100 μm. (B) The DOX/DAPI and (C) Caspase 3/DAPI fluorescence intensity ratio of each tissue image was quantified using the Image J software. (n = 5, *p < 0.05, **p < 0.01, ***p < 0.001 or n.s. > 0.05.) (D) Immunohistological studies of drug transport in tumor tissue from mice injected intratumorally with RAW-NR@DOX:SAs. DOX is shown in red, macrophages are identified by CD68 marker in green, and cell nuclei are stained with DAPI in blue. The white arrows reveal that the DOX signal spots were repositioned outside the macrophages, followed by NIR activation. Scale bar: 20 μm. (E) Resultant tumor growth curve of mice receiving various treatments. (n = 4-5, **p < 0.01, or n.s. > 0.05.) The black dashed line indicated the obvious therapeutic effect by RAW-NR@DOX:SA (+) started from day 4 after second NIR laser exposure. (F) Digital images of the tumor-bearing mice and the harvested tumors on day 11. The red-circled area of the image was further enlarged to see the detail of NR@DOX:SAs treated mice.
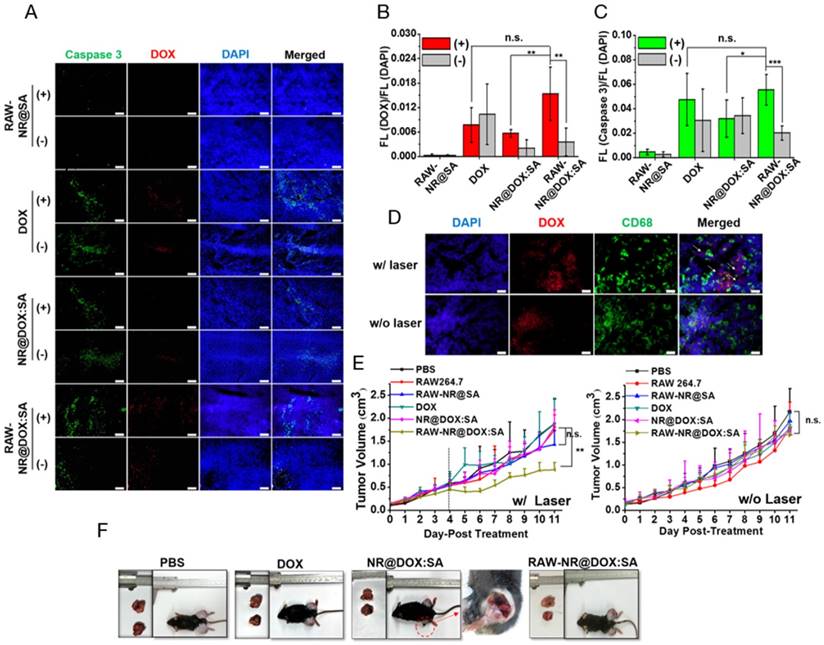
Simultaneously, the tumor growth curve for each day was plotted until day 11. In Figure 8E, photo- and chemo-combined treatment of RAW-NR@DOX:SA (+), particularly after two treatments on day 5, exhibited obvious antitumor efficacy compared with that after the first treatment of RAW-NR@SA (+) and RAW-NR@DOX:SA (-), respectively. The accumulation of long-lasting chemodrug after the first and second treatment on days 1 and 4, respectively, was believed to exert antitumor effects. By contrast, both pristine NR@DOX:SAs and free DOX exhibited limited efficacy in delaying tumor growth; the tumor sizes did not differ significantly between the pristine NR@DOX:SAs and free DOX groups and the PBS-treated control. In accordance with the aforementioned results, the tumor samples (Figure S14) harvested at day 11 from the tumor-bearing mice administered different therapeutics and the ex vivo optical images (Figure 8F) demonstrated that the mice who received combinational treatment using RAW-NR@DOX:SA (+) produced more significant tumor inhibition than the other groups. Despite the high temperature detected in the mice treated with pristine NR@DOX:SAs (Figure 7A), the accumulated energy was unequally distributed throughout the tumor (Figure 7C-7E), resulted in regional tissue injury and scarring within the irradiated area as well as rapid growth in the remaining tumor site. The twice photothermal effect produced severe biological effects resulting in an enhanced unhealable wound area (Figure 8F, red circle); this finding is similar to a previous finding of related clinical studies, suggesting severe and incomplete damages caused by thermal ablation or chemotherapy often produce adverse effects and occasionally fail to treat cancer.[38, 58] Our findings suggested that even a relatively large dose of Au NRs without homogenous drug delivery is inefficient in cancer therapy. By contrast, a less pronounced temperature increase accompanied by greater antitumor activity was observed in mice treated with RAW-NR@DOX:SAs. The free DOX was rapidly eliminated, thus reducing the desired therapeutic concentration and resulting in reduced efficacy in tumor growth suppression. This result indicated that the cell carriers were highly capable of repeatedly delivering the therapeutic payloads with adequate tumor coverage, resulting in the effective and long-lasting inhibition of tumor progression through a combinational effect.
In vivo tumor-tropic migration and specific accumulation in the hypoxic region
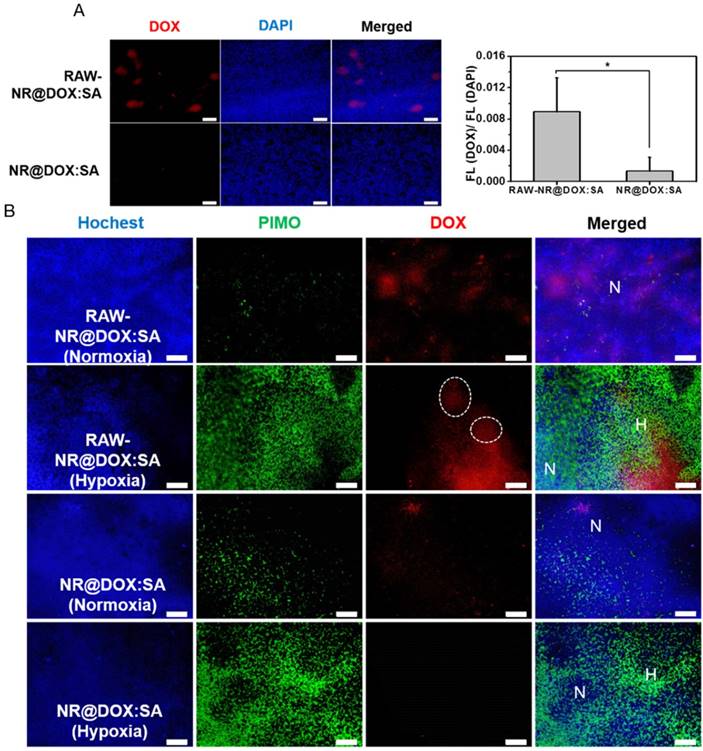
Because the drug injection site and resultant drug distribution was considerably affected by needle insertion, the evaluation of tumor-tropic migration and specific drug location could be overestimated (or underestimated) [30, 59, 60]. Therefore, the intravenous delivery of RAW-NR@DOX:SAs was evaluated to further explore the unique ability of tumor-tropic migration. When a tumor volume approached 100 mm3, nanotherapeutic agents were administered two times, followed by NIR light irradiation (808 nm, 0.75 W/cm2, 3 min) at 24 h post-injection. The RAW-NR@DOX:SA (+) group showed superior tumor growth delay (p = 0.01) compared with the other groups (p > 0.05; Figure S15A). Moreover, a substantially intense DOX fluorescence signal was detected in the xenograft tumor sections of the payload-containing macrophages compared with those in the pristine nanoparticles group (Figure 9A). To investigate the in vivo hypoxia-homing property of therapeutic macrophages, immunohistochemical examination was performed, which showed that the enhanced accumulation of DOX in both hypoxic (PIMO+) and normoxic (PIMO-) regions was promoted by cell-mediated delivery (Figure 9B). In particular, the payload macrophages were trapped and accumulated in the hypoxic regions as shown in the white circles of Figure 9B for subsequent payload delivery.[61] By contrast, the DOX signals detected in pristine NR@DOX:SAs were less pronounced in normoxic areas and were barely observed in the avascular and hypoxic areas [62]. Notably, this finding demonstrated that the tumor-tropic migration and specific accumulation of the macrophages in the hypoxic region influences the unique behaviors, resulting in enhanced drug distribution and retention ability. Despite the moderate inhibition, in the present study, tumor growth was only obtained in the RAW-NR@DOX:SA-treated mice receiving relatively low NIR irradiation (Figure 7A and Figure S15B-15C); an appreciable improvement in the treatment outcome can be achieved with higher energy input in future studies. In addition, monocytes and macrophages derived from bone marrow can be used as delivery vehicles in systematic administration to prolong circulation lifetime and improve payload accumulation in tumors under the influence of tumor-derived chemoattractants [61, 63, 64]. Furthermore, tumor growth inhibition may be more pronounced with the developed cellular-mediated therapeutics than with conventional drug delivery systems.
In vivo studies for tumortropic migration of RAW-NR@DOX:SAs by intravenous administration. (A) Fluorescence microscopic images of DOX signals were observed within the tumor tissue section harvested from mice injected intravenously with RAW-NR@DOX:SAs and NR@DOX:SAs, respectively. The fluorescence intensity ratio of DOX/DAPI from each tissue image was quantified using the Image J software. (n = 6, *p < 0.05.) Scale bar: 100 μm. (B) Immunofluorescence staining of tumor sections from therapeutic macrophage and pristine nanoparticles. Hochest (blue) was injected by intravenous administration, represented blood flow area while hypoxia area was specified by PIMO (green) marker. Normoxia region (N) referred to the area in which was passed through by blood flow (blue) without any hypoxia signal (green), and vice versa for defining hypoxia region (H). White circle indicates the therapeutic macrophages were trapped and accumulated in the hypoxic regions. Scale bar: 100 μm.
Conclusions
A specific nanotherapeutic agent, NR@DOX:SA, was successfully designed to demonstrate the underlying mechanisms of macrophage-mediated intratumoral delivery, which precisely controls the behaviors of encapsulated drug/cell carriers in combinational cancer treatment. DOX retained in the SA shell of NR@SA was retained in the macrophage carriers, which displayed superior tissue distribution and provided a more homogenous chemodrug/photothermal delivery, leading to an effective antitumor activity after NIR-activated intracellular DOX release. This enhanced drug distribution could be attributed to tumor-tropic migration and accumulation in the hypoxic regions. The more pronounced therapeutic efficacy can be increased through the use of higher-input laser power. The pristine NPs could not inhibit tumor growth because of their inefficient thermal distribution after NIR light irradiation. In addition, the generated photodamage resulted in serious unhealable wounds with various localized effects, such as hemorrhage and necrosis, and further reduced the retention ability of the NPs, which conversely could increase the obstacles for cancer therapy, particularly for multiple treatments. Free DOX with a low retention ability was rapidly eliminated for efficient antitumor effects. In particular, mounting evidence has suggested that increasing the therapeutic dose during therapy not only occasionally limits tumor growth inhibition or recurrence but also impairs quality of life in patients or even makes tumors more aggressive [35, 37, 38, 58]. Our finding suggests that enhancing therapeutic efficacy may be more practical than merely increasing the dosage. The programmable core-shell nanoplatform designed in this study not only highlights the importance of homogenous photothermal/chemodrug distribution and retention ability for achieving optimal drug delivery but also represents a promising tool for developing highly effective cell-based therapeutic agents for combinational treatment in oncology.
Acknowledgements
We appreciate financial support from the Ministry of Science and Technology (NSC 102-2113-M-007-005-MY3, 104-2627-M-007 -001, 105-2113-M-007 -021, 105-2627-M-019 -001 -) of Taiwan, ROC.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Holback H, Yeo Y. Intratumoral Drug Delivery with Nanoparticulate Carriers. Pharm. Res. 2011;28:1819-30
2. Kobayashi H, Watanabe R, Choyke PL. Improving Conventional Enhanced Permeability and Retention (EPR) Effects; What Is the Appropriate Target? Theranostics. 2014;4:81-9
3. Soo Choi H, Liu W, Misra P. et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25:1165-70
4. Tan S, Wu T, Zhang D, Zhang Z. Cell or cell membrane-based drug delivery systems. Theranostics. 2015;5:863-81
5. Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat. Rev. Drug Discovery. 2011;10:521-35
6. Su JH, Sun HP, Meng QS, Yin Q, Tang S, Zhang PC. et al. Long Circulation Red-Blood-Cell-Mimetic Nanoparticles with Peptide-Enhanced Tumor Penetration for Simultaneously Inhibiting Growth and Lung Metastasis of Breast Cancer. Adv. Funct. Mater. 2016;26:1243-52
7. Cheng Y, Morshed R, Cheng SH, Tobias A, Auffinger B, Wainwright DA. et al. Nanoparticle-Programmed Self-Destructive Neural Stem Cells for Glioblastoma Targeting and Therapy. Small. 2013;9:4123-9
8. Mooney R, Roma L, Zhao D, Van Haute D, Garcia E, Kim SU. et al. Neural Stem Cell-Mediated Intratumoral Delivery of Gold Nanorods Improves Photothermal Therapy. ACS Nano. 2014;8:12450-60
9. Gao Z, Zhang L, Hu J, Sun Y. Mesenchymal stem cells: a potential targeted-delivery vehicle for anti-cancer drug loaded nanoparticles. Nanomed-Nanotechnol. 2013;9:174-84
10. Si J, Shao S, Shen Y, Wang K. Macrophages as Active Nanocarriers for Targeted Early and Adjuvant Cancer Chemotherapy. Small. 2016;12:5108-19
11. [11] Edmundson M, Thanh NT, Song B. Nanoparticles based stem cell tracking in regenerative medicine. Theranostics. 2013;3:573-82
12. Shanmugam V, Selvakumar S, Yeh CS. Near-infrared light-responsive nanomaterials in cancer therapeutics. Chem. Soc. Rev. 2014;43:6254-87
13. Choi MR, Stanton-Maxey KJ, Stanley JK, Levin CS, Bardhan R, Akin D. et al. A Cellular Trojan Horse for Delivery of Therapeutic Nanoparticles into Tumors. Nano Lett. 2007;7:3759-65
14. Baek SK, Makkouk A, Krasieva T, Sun CH, Madsen S, Hirschberg H. Photothermal treatment of glioma; an in vitro study of macrophage-mediated delivery of gold nanoshells. J. Neurooncol. 2011;104:439-48
15. Madsen S, Baek SK, Makkouk A, Krasieva T, Hirschberg H. Macrophages as Cell-Based Delivery Systems for Nanoshells in Photothermal Therapy. Ann Biomed Eng. 2012;40:507-15
16. von Maltzahn G, Park JH, Agrawal A, Bandaru NK, Das SK, Sailor MJ. et al. Computationally guided photothermal tumor therapy using long-circulating gold nanorod antennas. Cancer. Res. 2009;69:3892-900
17. Mackey MA, Ali MRK, Austin LA, Near RD, El-Sayed MA. The Most Effective Gold Nanorod Size for Plasmonic Photothermal Therapy: Theory and In Vitro Experiments. J. Phys. Chem. B. 2014;118:1319-26
18. Li Z, Huang H, Tang S, Li Y, Yu XF, Wang H. et al. Small gold nanorods laden macrophages for enhanced tumor coverage in photothermal therapy. Biomaterials. 2016;74:144-54
19. Qin JB, Peng ZY, Li B, Ye KC, Zhang YX, Yuan FK. et al. Gold nanorods as a theranostic platform for in vitro and in vivo imaging and photothermal therapy of inflammatory macrophages. Nanoscale. 2015;7:13991-4001
20. Liu Y, Yang M, Zhang J, Zhi X, Li C, Zhang C. et al. Human Induced Pluripotent Stem Cells for Tumor Targeted Delivery of Gold Nanorods and Enhanced Photothermal Therapy. ACS Nano. 2016;10:2375-85
21. Jokerst JV, Thangaraj M, Kempen PJ, Sinclair R, Gambhir SS. Photoacoustic Imaging of Mesenchymal Stem Cells in Living Mice via Silica-Coated Gold Nanorods. ACS Nano. 2012;6:5920-30
22. Ratto F, Centi S, Avigo C, Borri C, Tatini F, Cavigli L. et al. A Robust Design for Cellular Vehicles of Gold Nanorods for Multimodal Imaging. Adv. Funct. Mater. 2016;26:7178-85
23. Chen Q, Liu Z. Albumin Carriers for Cancer Theranostics: A Conventional Platform with New Promise. Adv. Mater. 2016;28:10557-66
24. Yasun E, Li CM, Barut I, Janvier D, Qiu LP, Cui C. et al. BSA modification to reduce CTAB induced nonspecificity and cytotoxicity of aptamer-conjugated gold nanorods. Nanoscale. 2015;7:10240-8
25. Schottler S, Landfester K, Mailander V. Controlling the Stealth Effect of Nanocarriers through Understanding the Protein Corona. Angew. Chem. Int. Edit. 2016;55:8806-15
26. Kah JCY, Chen J, Zubieta A, Hamad-Schifferli K. Exploiting the Protein Corona around Gold Nanorods for Loading and Triggered Release. ACS Nano. 2012;6:6730-40
27. Cifuentes-Rius A, de Puig H, Kah JCY, Borros S, Hamad-Schifferli K. Optimizing the Properties of the Protein Corona Surrounding Nanoparticles for Tuning Payload Release. ACS Nano. 2013;7:10066-74
28. Choi JH, Hwang HJ, Shin SW, Choi JW, Um SH, Oh BK. A novel albumin nanocomplex containing both small interfering RNA and gold nanorods for synergetic anticancer therapy. Nanoscale. 2015;7:9229-37
29. Peralta DV, Heidari Z, Dash S, Tarr MA. Hybrid Paclitaxel and Gold Nanorod-Loaded Human Serum Albumin Nanoparticles for Simultaneous Chemotherapeutic and Photothermal Therapy on 4T1 Breast Cancer Cells. ACS Appl. Mater. Interfaces. 2015;7:7101-11
30. Lai P, Lechtman E, Mashouf S, Pignol JP, Reilly RM. Depot system for controlled release of gold nanoparticles with precise intratumoral placement by permanent brachytherapy seed implantation (PSI) techniques. Int. J. Pharm. 2016;515:729-39
31. Phillips WT, Bao A, Brenner AJ, Goins BA. Image-guided interventional therapy for cancer with radiotherapeutic nanoparticles. Adv. Drug Delivery Rev. 2014;76:39-59
32. Goldberg EP, Hadba AR, Almond BA, Marotta JS. Intratumoral cancer chemotherapy and immunotherapy: opportunities for nonsystemic preoperative drug delivery. J. Pharm. Pharmacol. 2002;54:159-80
33. Hohenforst-Schmidt W, Zarogoulidis P, Darwiche K, Vogl T, Goldberg EP, Huang H. et al. Intratumoral chemotherapy for lung cancer: re-challenge current targeted therapies. Drug Des. Devel. Ther. 2013;7:571-83
34. Jain S, Hirst DG, O'Sullivan JM. Gold nanoparticles as novel agents for cancer therapy. Brit. J. Radiol. 2012;85:101-113
35. Wang BK, Yu XF, Wang JH, Li ZB, Li PH, Wang H. et al. Gold-nanorods-siRNA nanoplex for improved photothermal therapy by gene silencing. Biomaterials. 2016;78:27-39
36. Abadeer NS, Murphy CJ. Recent Progress in Cancer Thermal Therapy Using Gold Nanoparticles. J. Phys. Chem. C. 2016;120:4691-716
37. Wolinsky JB, Colson YL, Grinstaff MW. Local drug delivery strategies for cancer treatment: gels, nanoparticles, polymeric films, rods, and wafers. J. Controlled Release. 2012;159:14-26
38. Rodenhuis S. The status of high-dose chemotherapy in breast cancer. Oncologist. 2000;5:369-75
39. Brincker H. Direct intratumoral chemotherapy. Crit Rev Oncol Hematol. 1993;15:91-8
40. Hayashi K, Nakamura M, Miki H, Ozaki S, Abe M, Matsumoto T. et al. Magnetically responsive smart nanoparticles for cancer treatment with a combination of magnetic hyperthermia and remote-control drug release. Theranostics. 2014;4:834-44
41. Nikoobakht B, El-Sayed MA. Preparation and growth mechanism of gold nanorods (NRs) using seed-mediated growth method. Chem Mater. 2003;15:1957-62
42. Zhang XF, Yao S, Liu C, Jiang YY. Tumor tropic delivery of doxorubicin-polymer conjugates using mesenchymal stem cells for glioma therapy. Biomaterials. 2015;39:269-81
43. Kanno SI, Yomogida S, Tomizawa A, Yamazaki H, Ukai K. et al. Combined effect of papuamine and doxorubicin in human breast cancer MCF-7 cells. Oncol Lett. 2014;8:547-50
44. von Storp B, Engel A, Boeker A, Ploeger M, Langer K. Albumin nanoparticles with predictable size by desolvation procedure. J. Microencapsul. 2012;29:138-46
45. Dreis S, Rothweller F, Michaelis A, Cinatl J, Kreuter J, Langer K. Preparation, characterisation and maintenance of drug efficacy of doxorubicin-loaded human serum albumin (HSA) nanoparticles. Int. J. Pharm. 2007;341:207-14
46. Leo E, Cameroni R, Forni F. Dynamic dialysis for the drug release evaluation from doxorubicin-gelatin nanoparticle conjugates. Int. J. Pharm. 1999;180:23-30
47. Wang FJ, Yu L, Monopoli MP, Sandin P, Mahon E, Salvati A. et al. The biomolecular corona is retained during nanoparticle uptake and protects the cells from the damage induced by cationic nanoparticles until degraded in the lysosomes. Nanomed-Nanotechnol. 2013;9:1159-68
48. See V, Free P, Cesbron Y, Nativo P, Shaheen U. et al. Cathepsin L Digestion of Nanobioconjugates upon Endocytosis. ACS Nano. 2009;3:2461-8
49. Wu Y, Ihme S, Feuring-Buske M, Kuan SL, Eisele K. et al. A core-shell albumin copolymer nanotransporter for high capacity loading and two-step release of doxorubicin with enhanced anti-leukemia activity, Adv. Healthcare Mater. 2013;2:884-94
50. Li L, Guan Y, Liu H, Hao N, Liu T, Meng X. et al. Silica Nanorattle-Doxorubicin-Anchored Mesenchymal Stem Cells for Tumor-Tropic Therapy. ACS Nano. 2011;5(9):7462-7470
51. Al-Abd AM, Hong KY, Song SC, Kuh HJ. Pharmacokinetics of doxorubicin after intratumoral injection using a thermosensitive hydrogel in tumor-bearing mice. J. Controlled Release. 2010;142:101-7
52. Jeon MJ, Ahn CH, Kim H, Chung IJ, Jung S, Kim YH. et al. The intratumoral administration of ferucarbotran conjugated with doxorubicin improved therapeutic effect by magnetic hyperthermia combined with pharmacotherapy in a hepatocellular carcinoma model. J. Exp. Clin. Cancer Res. 2014;33:57
53. Kossatz S, Ludwig R, Dähring H, Ettelt V, Rimkus G, Marciello M. et al. High Therapeutic Efficiency of Magnetic Hyperthermia in Xenograft Models Achieved with Moderate Temperature Dosages in the Tumor Area. Pharm. Res. 2014;31:3274-88
54. Dewhirst MW, Viglianti BL, Lora-Michiels M, Hanson M, Hoopes PJ. Basic principles of thermal dosimetry and thermal thresholds for tissue damage from hyperthermia. Int. J. Hyperthermia. 2003;19:267-94
55. Yarmolenko PS, Moon EJ, Landon C, Manzoor A, Hochman DW, Viglianti BL. et al. Thresholds for thermal damage to normal tissues: An update. Int. J. Hyperther. 2011;27:320-43
56. Pissuwan D, Valenzuela SM Killingsworth MC, Xu XD Cortie MB. Targeted destruction of murine macrophage cells with bioconjugated gold nanorods. J. Nanopart. Res. 2007;9:1109-24
57. Melamed JR, Edelstein RS, Day ES. Elucidating the fundamental mechanisms of cell death triggered by photothermal therapy. ACS Nano. 2015;9:6-11
58. Zagar TM, Oleson JR, Vujaskovic Z, Dewhirst MW, Craciunescu OI, Blackwell KL. et al. Hyperthermia combined with radiation therapy for superficial breast cancer and chest wall recurrence: a review of the randomised data. Int. J. Hyperthermia. 2010;26:612-7
59. Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proc. Natl. Acad. Sci. U. S. A. 1994;91:2076-80
60. Casanova F, Carney PR, Sarntinoranont M. Effect of needle insertion speed on tissue injury, stress, and backflow distribution for convection-enhanced delivery in the rat brain. PLoS One. 2014;9:e94919
61. Huang WC, Chiang WH, Cheng YH, Lin WC, Yu CF, Yen CY. et al. Tumortropic monocyte-mediated delivery of echogenic polymer bubbles and therapeutic vesicles for chemotherapy of tumor hypoxia. Biomaterials. 2015;71:71-83
62. Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224-34
63. Huang WC, Shen MY, Chen HH, Lin SC, Chiang WH, Wu PH. et al. Monocytic delivery of therapeutic oxygen bubbles for dual-modality treatment of tumor hypoxia. J. Controlled Release. 2015;220:738-50
64. Vinogradov S, Warren G, Wei X. Macrophages associated with tumors as potential targets and therapeutic intermediates. Nanomedicine (London, U. K.). 2014;9:695-707
Author contact
![]() Corresponding author: Prof. Yu-Fen Huang, Department of Biomedical Engineering and Environmental Sciences, National Tsing Hua University, Hsinchu 30013, Taiwan, ROC yufennthu.edu.tw
Corresponding author: Prof. Yu-Fen Huang, Department of Biomedical Engineering and Environmental Sciences, National Tsing Hua University, Hsinchu 30013, Taiwan, ROC yufennthu.edu.tw