Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(12):3021-3033. doi:10.7150/thno.18992 This issue Cite
Research Paper
Glutamic Pyruvate Transaminase GPT2 Promotes Tumorigenesis of Breast Cancer Cells by Activating Sonic Hedgehog Signaling
Yuan Cao1*, Shu-Hai Lin1*, Yongbin Wang1, Y. Eugene Chin2, Lan Kang3, Jun Mi1 ![]()
1. Department of Biochemistry and Molecular Cell Biology; Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, Shanghai Jiao Tong University School of Medicine;
2. Shanghai Institute for Biological Science, Chinese Academy of Sciences;
3. Institute of Cancer Stem Cell, Dalian Medical University.
* These authors contributed equally to this work.
Received 2016-12-30; Accepted 2017-6-2; Published 2017-7-22
Abstract

Increased glutamine metabolism is a hallmark of cancer. Mitochondrial glutamic pyruvate transaminase (GPT2) catalyzes the reversible transamination between alanine and α-ketoglutarate (α-KG), also known as 2-oxoglutarate, to generate pyruvate and glutamate during cellular glutamine catabolism. However, the precise role of GPT2 in tumorigenesis remains elusive. Here, we report that in breast cancer tissue samples and breast cancer cell lines, GPT2 expression level was markedly elevated and correlated with the pathological grades of breast cancers. GPT2 overexpression increased the subpopulation of breast cancer stem cells in vitro and promoted tumorigenesis in mice. GPT2 reduced α-KG level in cells leading to the inhibition of proline hydroxylase 2 (PHD2) activity involved in the regulation of HIF1α stability. Accumulation of HIF1α, resulting from GPT2-α-KG-PHD2 axial, constitutively activates sonic hedgehog (Shh) signaling pathway. Overall, GPT2 promotes tumorigenesis and stemness of breast cancer cells by activating the Shh signaling, suggesting that GTP2 is a potential target for breast cancer therapy.
Introduction
Increased glutamine metabolism is a hallmark of cancer. Proliferating tumor cells utilize glutamine's carbon to maintain pools of TCA cycle intermediates for energy production and its nitrogen to produce nonessential amino acids, hexosamine, nucleotides, and other molecules (1). For example, renal cell carcinomas are glutamine addicted (2). Moreover, the oncogenic function of PIK3CA mutations is dependent on glutamine metabolism in colorectal cancer cells (3). Intracellular glutamate in cancer cells is primarily a product of glutamine metabolism with a proportion of glutamate destined for secretion (4-6). Glutaminase generates glutamate from glutamine and its activity was reported to correlate with tumor growth rate (7-9). In addition, glutamate is an intracellular signaling molecule in many tissues (10). Amplified secretion of glutamate, as well as other aspects of dysregulated glutamatergic signaling, has been shown to correlate with a malignant phenotype (11-13).
The Hedgehog (Hh), Wnt and Notch signaling pathways are considered three major signaling events regulating stemness of cancer cells (14-16). Hh signaling is initiated by the binding of Shh ligand to the Patched-1 receptor (PTCH1) relieving repression of the transducer protein Smoothened (SMO) which then triggers the activation of the GLI family of transcription factors. The genes activated by GLI include PTCH1 and GLI itself (17). On the other hand, Suppressor of Fused Homolog (SUFU) suppresses Hh signaling by regulating the localization of the transcription factor GLI (18).
The glutamic pyruvate transaminase (GPT) is an alanine transaminase that catalyzes the reversible transamination between alanine and α-ketoglutarate to generate pyruvate and glutamate. Alanine transaminases play important roles in gluconeogenesis and amino acid metabolism in many tissues including skeletal muscle, kidney, and liver (19). GPT1 locates in cytosol which is a biomarker used clinically in liver diseases. The GPT2 protein is more abundant than GPT1, especially in muscle and fat, suggesting a novel role of GPT2 in the metabolism and homeostasis of glucose, amino acids, and fatty acids (20). Under metabolic stress, GPT2 expression in hepatocyte cell lines is upregulated by the activating transcription factor 4. Proliferating breast tumor cells express high levels of GPT2 and the viability of pancreatic cancer cells was decreased when the activity of GPT2 was inhibited (21, 22). GPT2 expression was also upregulated in ductal breast carcinomas (23). Moreover, GPT2 and α-ketoglutarate - the tricarboxylic acid cycle intermediate obtained from the catalysis of glutamine by glutaminase, is essential for xenograft tumour growth of colorectal cancers with PIK3CA mutations (3). These observations indicate that GPT2 is important for tumor growth. However, its role in tumorigenesis remains elusive.
Here, we report that GPT2 overexpression reduced the intracellular α-ketoglutarate (α-KG) and increased the stability of HIF-1α, which, in turn, increased the stemness of breast cancer cells through activating sonic hedgehog (Shh) signaling. Our observations suggest that GPT2 is a potential target for breast cancer therapy.
Results
GPT2 protein levels correlate with the pathological stages of breast cancer
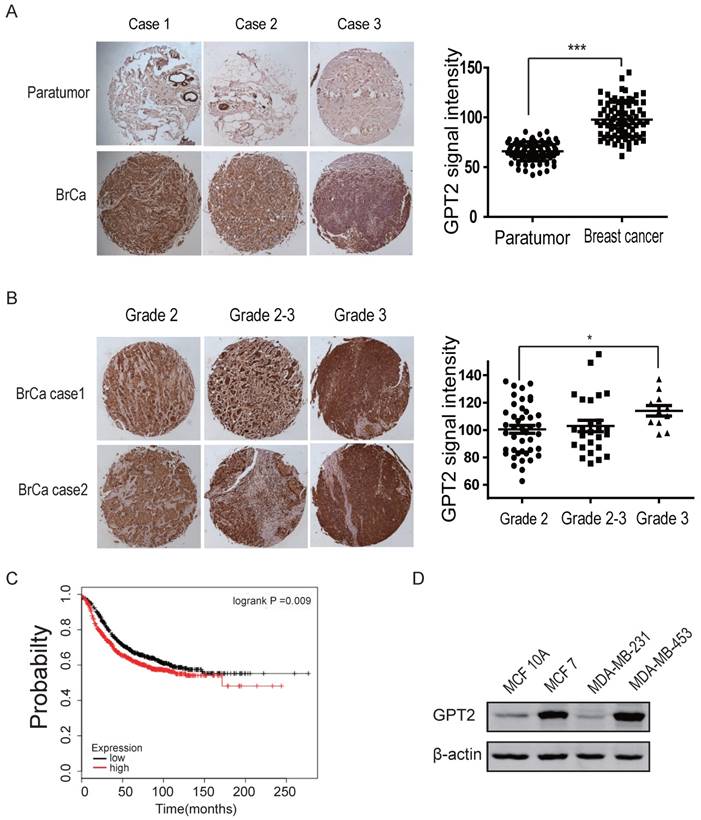
Because of the importance of GPT2 in glutamate metabolism, we performed immunohistochemistry staining to examine GPT2 expression in breast cancers from 82 cancer patients. GPT2 expression in breast cancers was increased as compared to para-carcinoma tissues (Figure 1A). Furthermore, the protein level of GPT2 was also analyzed in breast cancers of different pathological grades. As shown in Figure 1B, higher grade samples had higher levels of GPT2 expression. The Kaplan-Meier survival analysis (n = 1764) (kmplot.com) showed breast cancer patients with higher GPT2 mRNA level had a worse prognosis than those with lower GPT2 mRNA level (p < 0.01, Figure 1C) (24). Elevated GPT2 protein levels were also detected in most of breast cancer cell lines including MCF7 and MDA-MB-453 (Figure 1D). These findings suggest GPT2 protein levels correlate with progression of breast cancer.
GPT2 expression levels correlate with the pathological stages of breast cancer patients A. GPT2 immunohistochemistry analysis of breast cancer or paratumor tissues. The data represent the levels of GPT2 protein in breast carcinoma and normal breast tissue, which were quantified by densitometry. The difference between two groups was analyzed by T-test. ***: P < 0.001. B. GPT2 immunohistochemistry analysis of breast cancers of different pathologic grades. The graph represents relative GPT2 protein levels, quantified by densitometry. *: P < 0.05. C. Kaplan-Meier analysis of breast cancer patients based on GPT2 mRNA levels with low level (882 cases) vs high level (882 cases). P-values were calculated by log-rank test. D. Analysis of GPT2 expression in normal (MCF10A) and breast cancer (MDA-MB-231 and MCF7) cell lines. The Western blot shown here is a representative of three independent experiments.
GPT2 is tumorigenic and enhances breast cancer cell stemness
To determine the tumorigenic property of GPT2, the cell viability and cell growth assays were first analyzed. We overexpressed GPT2 in breast cancer MDA-MB-231 cells with low level of endogenous GPT2 or knocked-down GPT2 in breast cancer MCF7 cells with high level of endogenous GPT2 protein (Figure S1A). The cell counting data shows that overexpression of GPT2 promoted cell growth while knockdown of GPT2 inhibited cell growth (Figure S1B); however, overexpression of GPT2 had no significant effects on cell viability (Figure S1C). Therefore, the colony formation assay and soft agar assays were further performed. As shown in Figure 2A, the overexpression of GPT2 significantly increased while the knockdown of GPT2 decreased colony numbers of MDA-MB-231 and MCF7 cells, respectively. Consistent with this observation, the soft agar assay also showed that the overexpression of GPT2 increased colony numbers of MDA-MB-231 cells while GPT2 knockdown decreased the colony numbers of MCF7 cells (Figure 2B & Figure S1D). Moreover, reducing glutamine concentration in the culture medium also decreased colony formation of MDA-MB-231 cells (Figure 2C). These observations indicated that GPT2 promotes tumorigenesis of breast cancer cells.
GPT2-induced breast cancer cell stemness and tumorigenesis A. & B. GPT2 promotes colony formation of breast cancer cells. A. Colony formation assay (n = 3), ***: P < 0.001; **: P < 0.01. B. Soft agar assay (n = 3). ***: P < 0.001 C. The effects of glutamine on colony formation. The basic medium was without glutamine. D. Flow cytometry analysis of CD44 and CD24 expression in GPT2 overexpressing MDA-MB-231 and GPT2 knockdown MCF7 cells. The values represent percentage of CD44+CD24- cells. **: P < 0.01;*: P < 0.05. E. GPT2 and ALDH expression was analyzed in GPT2 overexpressing MDA-MB-231 and GPT2 knockdown MCF7 cells by immunofluorescence staining. Red and green represent GPT2 and ALDH, respectively (20 X). F. Representative Western blots show the expression of ALDH, SOX2, OCT4 and NANOG in GPT2 overexpressing MDA-MB-231 and GPT2 knockdown MCF7 cells. G. GPT2 and ALDH expression was analyzed in clinical breast tumors and paratumor tissues by immunofluorescence staining (n= 6 cases). Red and green represent GPT2 and ALDH, respectively (10 X). The values on the histogram represent cell numbers per view. The big white bars represent GPT2 positive cell numbers while the small shading bars inside indicate ALDH positive cell numbers. GPT2 overexpression cells were MDA-MB-231 while GPT2 knockdown cells were MCF7. Data in (A-G) represent three independent experiments. Error bars: standard deviation. ***: P < 0.001; **: P < 0.01; *: P < 0.05.
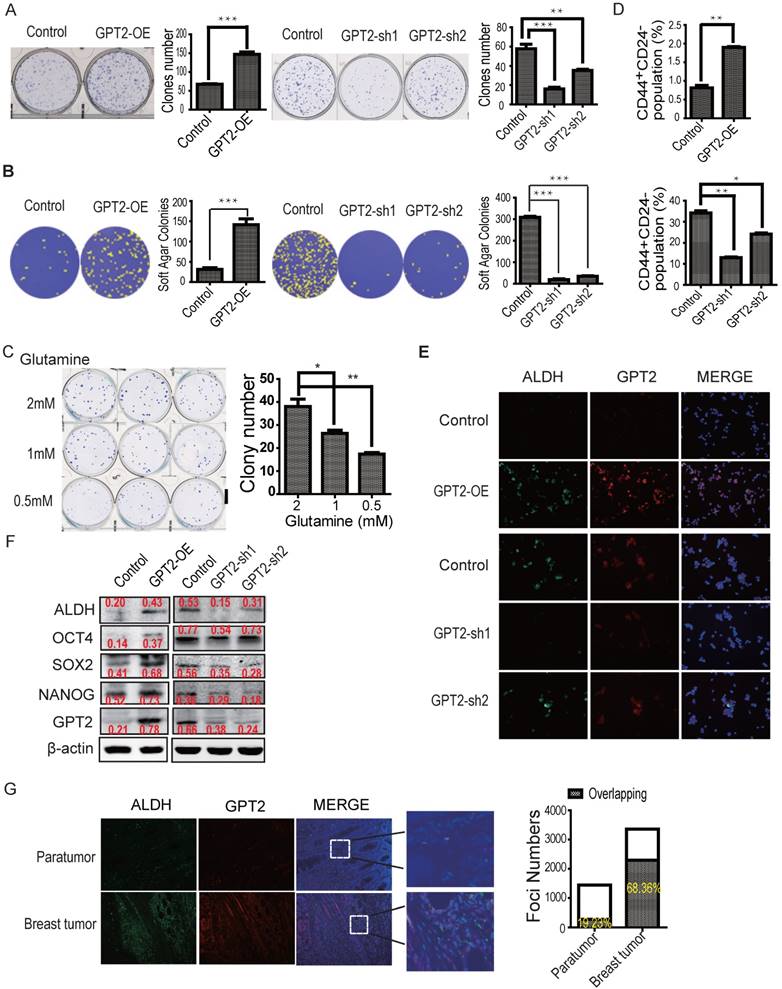
The subpopulation of cancer stem cells (CSCs) have been defined by their tumor-initiating properties in multiple types of cancer (25, 26). The CD44+/CD24 has been widely used as a marker for breast cancer stem cells (BCSCs) (27-30). Therefore, we analyzed the percentage of CD44+CD24- breast cancer cells by flow cytometry,-in MDA-MB-231 cells overexpressing GPT2 or MCF7 cells depleted of GPT2. As shown in Figure 2D, the overexpression of GPT2 increased the percentage of CD44+CD24- cells from 0.88% to 1.83% while the knockdown of GPT2 decreased the percentage of CD44+CD24- cells from 33.11% to 13.24% or 23.61%.
Since the aldehyde dehydrogenase (ALDH) is another recognized biomarker for BCSCs (31), the expression of ALDH1 was analyzed by immunofluorescence staining of MDA-MB-231 and MCF7 cells. As shown in Figure 2E, the overexpression of GPT2 increased the percentage of ALDH1 positive cells in the MDA-MB-231 cell line (from 33.3% to 84.1%), while the knockdown of GPT2 decreased the percentage of ALDH1 positive cells in the MCF7 cell line (from 71.3% to average 37.6%) (Figure S1E). To further determine whether GPT2 promotes the stemness of breast cancer cells, the expression of stem cell markers including ALDH1, SOX2, OCT4 and NANOG were determined. As shown in Figure 2F, the critical markers for the stem cell were upregulated in MDA-MB-231 cells overexpressing GPT2. In contrast, the knock down of GPT2 decreased the expression of these markers in MCF cells.
The overlap of GPT2 and ALDH expression was also determined by immunofluorescence staining in clinical breast cancer samples. As shown in Figure 2G, 68.36% of GPT2 positive cells were also ALDH positive in breast cancer specimens but only 19.23% of GPT2 positive cells were ALDH positive in paratumor tissues. These observations suggest GPT2 promotes the stemness of breast cancer cells.
GPT2 activates the sonic hedgehog signaling by increasing HIF-1α protein level
To explore the mechanism by which GPT2 induced breast cancer cell stemness, we examined the critical molecules in the Wnt, Hedgehog, and Notch pathways, which play a critical role in the self renewal and differentiation of cancer stem-like cells (14, 16, 32). As displayed in Figure 3A, GPT2 overexpression activated the Shh signaling as reflected by the upregulated expression of Shh, PTCH1, PTCH2 and Gli1. The GPT2 knockdown, on the other hand, downregulated the expression of these activators of hedgehog signaling. GPT2 overexpression and knockdown efficiency were verified by both Western blot and q-PCR (Figure 3A & S2A). However, GPT2 did not activate Wnt or Notch signalingas indicated by non-alteration in the expression of their critical effector β-catenin or RBPJ. β-catenin and RBPJ are transcriptional regulators important in the Wnt or Notch signaling pathway, respectively. The reason for the upregulation of Notch1 and phospho-GSK3β in MDA-MB-231 cells overexpressing GPT2 was not clear (Figure S2B & S2C) (33-35).
GPT2 activates the sonic hedgehog signaling by increasing HIF-1α stability A. GPT2 activates hedgehog signaling pathway. Representative Western blots are shown as indicated. B. Representative Western blots show Shh, Gli1 and ALDH protein levels in GPT2 overexpressing MDA-MB-231 cells treated with or without Cyclopamine (a SMO inhibitor). ALDH is a marker for breast cancer stem cells. The final concentration of cyclopamine was 10 μM. C. Representative Western blots show the protein and hydroxylation levels of HIF1α in MDA-MB-231 and MCF7 cells treated with or without CoCl2, a HIF1α stabilizer, the final concentration of CoCl2 was 1mM. D. Representative Western blots show the protein and hydroxylation levels of HIF1α in GPT2 overexpressing MDA-MB-231 and GPT2 knockdown MCF7 cells. E. HIF1α mRNA level analyzed by realtime PCR in GPT2 overexpressing MDA-MB-231 and GPT2 knockdown MCF7 cells. #, P > 0.05. F. Representative Western blots show Shh and Gli1 protein levels in GPT2-overexpressing MDA-MB-231 cells treated with or without YC-1 (a HIF1α inhibitor). The final concentration of YC-1 was 5 μM. All Western blots and Q-PCR data are representative of three independent experiments.
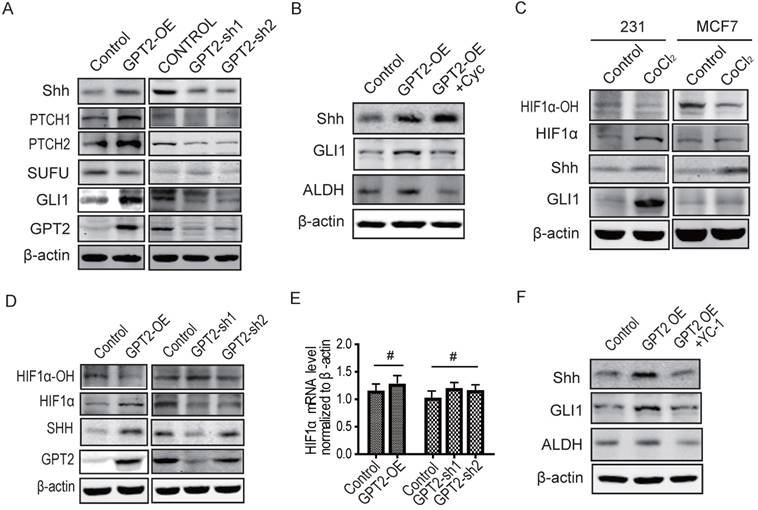
To further confirm the effect of GPT2 on the hedgehog signaling, GPT2 overexpressing MDA-MB-231 cells were treated with hedgehog signaling inhibitor cyclopamine (36). Figure 3B shows that cyclopamine decreased the protein level of Gli1, but did not affect Shh, the upstream regulator of Gli1, which was upreguated by GPT2. These results indicated that GPT2 might increase the stemness of breast cancer cells through activating hedgehog signaling.
Recent data indicated that hypoxia-induced factor HIF1α is an important factor in the regulation of cancer cell stemness (17, 37, 38). We also examined the protein levels of Gli1 and Shh in breast cancer cells treated with HIF1α stabilizer CoCl2 to verify the role of HIF1α in the regulation of breast cancer cell stemness. Figure 3C indicates that CoCl2 increased Gli1 expression in both MDA-MB-231 cells and MCF7 cells, suggesting that HIF1α regulates hedgehog signaling in breast cancer cells. The final concentration of CoCl2 was determined in MDA-MB-231 cells by dose response assay (Figure S2D).
Meanwhile, CoCl2 also increased GPT2 expression in MDA-MB-231 cells (Figure S2E). To verify whether GPT2 activated hedgehog signaling through HIF1α, the protein levels of HIF1α were first determined in the MDA-MB-231 and MCF7 cells. As shown in Figure 3D, overexpression of GPT2 reduced HIF1α hydroxylation and increased HIF1α protein level in MDA-MB-231 while the knockdown of GPT2 reduced HIF1α protein level in MCF7 cells, but not the mRNA levels (Figure 3E), suggesting GPT2 stabilizes HIF1α protein. Also, HIF1α inhibitor YC-1 (39) abolished the GPT2-induced increase in Shh and Gli1 expression (Figure 3F), and reduced GPT2 expression too (Figure S2E), suggesting HIF1α mediates GPT2-activated hedgehog signaling and regulates GPT2 expression.
GPT2 overexpression inhibits proline hydrolyase activity by decreasing cellular α-ketoglutarate
GPT2 is a pyridoxal enzyme that catalyzes the reversible transamination between alanine and α-ketoglutarate to generate pyruvate and glutamate, and the effective cellular α-KG (the ratio of α-KG to fumarate and succinate) regulates dioxygenase activity (40). The HIF1α proline hydroxylase (PHD) is a member of dioxygenase family. We, therefore, speculated that GPT2 reduced PHD activity by downregulating the effective α-KG in breast cancer cells. To determine whether GPT2 decreases HIF1α hydroxylation through suppressing PHD activity, the cellular concentration of α-KG, fumarate and succinate were first analyzed in GPT2 overexpressing MDA-MB-231 cells and GPT2 knockdown MCF7 cells. Figure 4A shows that the cellular content of α-KG was decreased in MDA-MB-231 cells expressing GPT2; in line with this, we observed that α-KG content increased in MCF7 cells depleted of GPT2. Both fumarate and succinate had no significant changes in breast cancer cells with different GPT2 levels. Moreover, the cellular alanine, α-KG, glutamate and pyruvate, the four substrates involved in the conversion were also analyzed. Figure 4B shows that the cellular content of α-KG and pyruvate were much lower than alanine and glutamate. In contrast to α-KG, the pyruvate increased in GPT2 overexpressing MDA-MB-231 cells and decreased in GPT2 knockdown cells compared to control cells. However, there were no significant changes in the content of alanine and glutamate.
GPT2 overexpression inhibits proline hydrolyase activity by decreasing cellular effective α-ketoglutarate A. Cellular α-KG, fumarate and succinate content examined by GC-MS in GPT2-overexpressing MDA-MB-231 and GPT2 knockdown MCF7 cells. *: P < 0.05. B. The cellular content of α-KG, alanine, pyruvate and glutamate detected by GC-MS. C. Mass isotopologue analysis of α-KG and glutamate in MDA-MB-231cells cultured with [U-13C5]-glutamine and unlabelled glucose (n = 3 cultures from a representative experiment). D. Mass isotopologue analysis of α-KG and glutamate in MDA-MB-231cells cultured with [U-13C6]-glucose and unlabelled glutamine (n = 3 cultures from a representative experiment). E. The ODD luciferase activity measured in 293 cells. The final concentration of CoCl2 was 1mM (***: P < 0.001). F. The ODD luciferase activity measured in 293 cells treated with α-KG and/or succinate. The final concentration of α-KG and succinate were 100μM and 200μM, respectively. The values were normalized to control (*: P < 0.05). G. α-KG increased the hydroxylation and reduced the stability of HIF1α in MDA-MB-231 cells with or without GPT2 overexpression. The final concentrations of α-KG and succinate were 100μM and 200μM, respectively. H. PHD2, but not PHD1 nor PHD3 increased the expression of Shh. Data in (A-H) represent three independent experiments. Error bars, s.d. ***: P < 0.001; **: P < 0.01; *: P < 0.05.
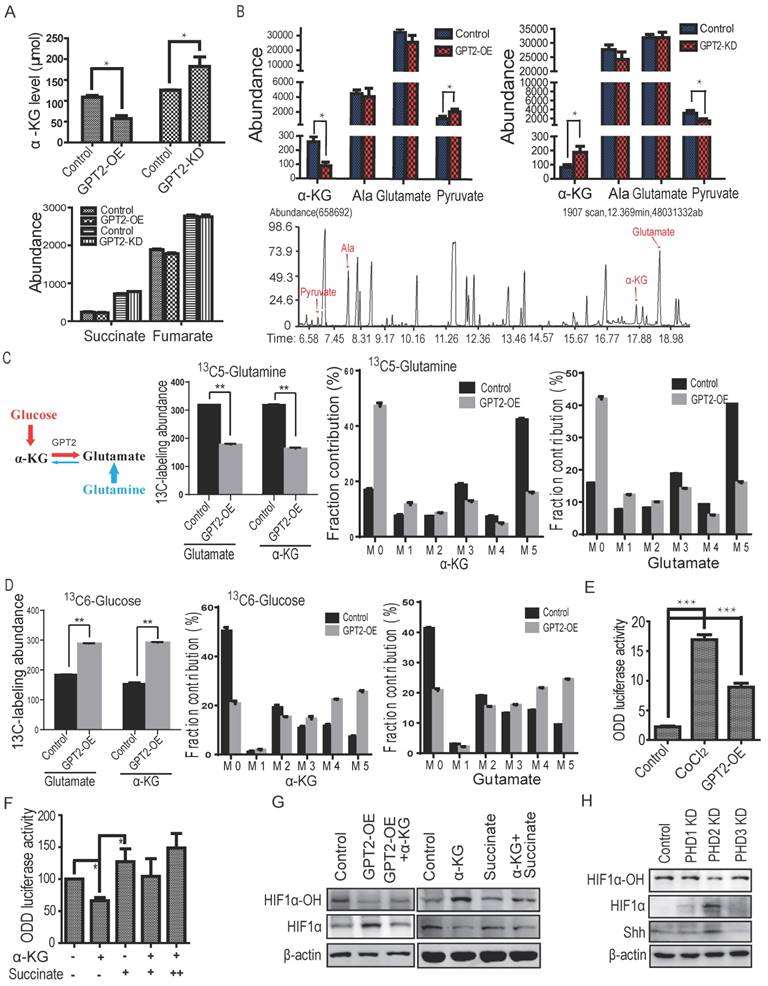
To further verify GPT2-catalyzed conversion of α-KG to glutamate, we used stable isotope-labeled nutrients ([U-13C6]-glucose and [U-13C5]-glutamine) as the tracers in cell culture, respectively. The mass isotopomer distribution (MID) describes the fractional contribution of each isotopologue normalized to the sum of all possible isotopologues. The metabolites α-KG and glutamate with 5 carbon atoms can have 0 to 5 of its carbon atoms labeled with 13C, resulting in isotopologues that increase in mass (M) from M0 (all carbons unlabeled i.e. 12C) to M5 (5 carbons labeled i.e. 13C). As shown in Figure 4C & 4D, GPT2 overexpression increased 13C labeling of α-KG and glutamate derived from glucose carbons compared to control group, but decreased 13C labeling of α-KG and glutamate derived from glutamine carbons. In contrast, GPT2 knockdown reversed this process (Figure S3A & 3B), supporting the notion that GPT2 promotes the conversion of α-KG to glutamate resulting in lower concentration of α-KG.
To examine whether GPT2 regulates PHD activity, the ODD (oxygen-dependent degradation domain) luciferase assay was performed on 293 cells expressing GPT2. Overexpression of GPT2 as well as CoCl2 treatment increased ODD luciferase activity (Figure 4E), suggesting that overexpression of GPT2 suppresses PHD activity. To determine whether GPT2 regulates HIF1α stability through α-KG, the ODD luciferase assay was performed on 293 cells treated with α-KG, succinate or a combination of α-KG and succinate. As shown in Figure 3F, α-KG decreased the ODD luciferase activity but succinate restored the ODD activity in a dose-dependent manner. Moreover, α-KG restored the HIF1α hydroxylation decreased by GPT2 overexpression and reduced the protein level of HIF1α and stem cell markers in MDA-MB-231 cells. In contrast, succinate decreased HIF1α hydroxylation levels and restored HIF1α protein level as well as stem cell markers (Figure 4G & S3C).
We also knocked down three PHD isoforms (PHD1, PHD2, or PHD3) individually in MDA-MB-231 cells to verify which isoform was responsible for HIF1α hydroxylation (Figure S3D). We found that PHD2 knockdown, but not PHD1 or PHD3 knockdown, reduced HIF1α hydroxylation (Figure 4H), suggesting PHD2 is the primary enzyme responsible for HIF1α hydroxylation and that GPT2 increases HIF1α hydroxylation through regulating PHD activity in breast cancer cells.
GPT2 promotes mammary tumorigenesis
To further determine whether GPT2 promotes breast cancer cell tumorigenesis, 1 x 106 MDA-MB-231 cells overexpressing GPT2 or MCF7 cells depleted of GPT2 were injected into the armpits of nude mice as shown in the schematic drawing (Figure 4A & 4B). In the MDA-MB-231 group with GPT2 overexpression, 4 out of 8 injected mice grew tumors in 3 weeks after injection while only 1 out of 8 injected mice with control MDA-MB-231 cells had tumor (Figure 5A).
GPT2 promotes mammary tumorigenesis A. GPT2 overexpression promotes tumor formation of breast cancer cells. Mice injected with GPT2 overexpressing MDA-MB-231 cells at the indicated cell numbers. The tumors in the blue circles on the left are controls; the tumors in the red circles on the right are overexpression group. B. Depletion of GPT2 reduces tumor formation of breast cancer cells. Mice injected with GPT2 knockdown MCF7 cells at the indicated cell numbers. The tumors in the blue circles on the left are control. C. The expression of GPT2 and ALDH1 was detected by immunofluorescence staining. The data show representative images from three independent experiments. The values on the histogram represent foci numbers per view, 6 views for each group were analyzed. D. Detection of α-KG in xenografted tumors by α-KG assay kit. The data shown is presented as the means ± SD of three independent experiments. E. Expression analysis of GPT2 and HIF1α by Western blot and immunofluorescence staining in cells and in xenografted tumors. The data show representative images from three independent experiments.
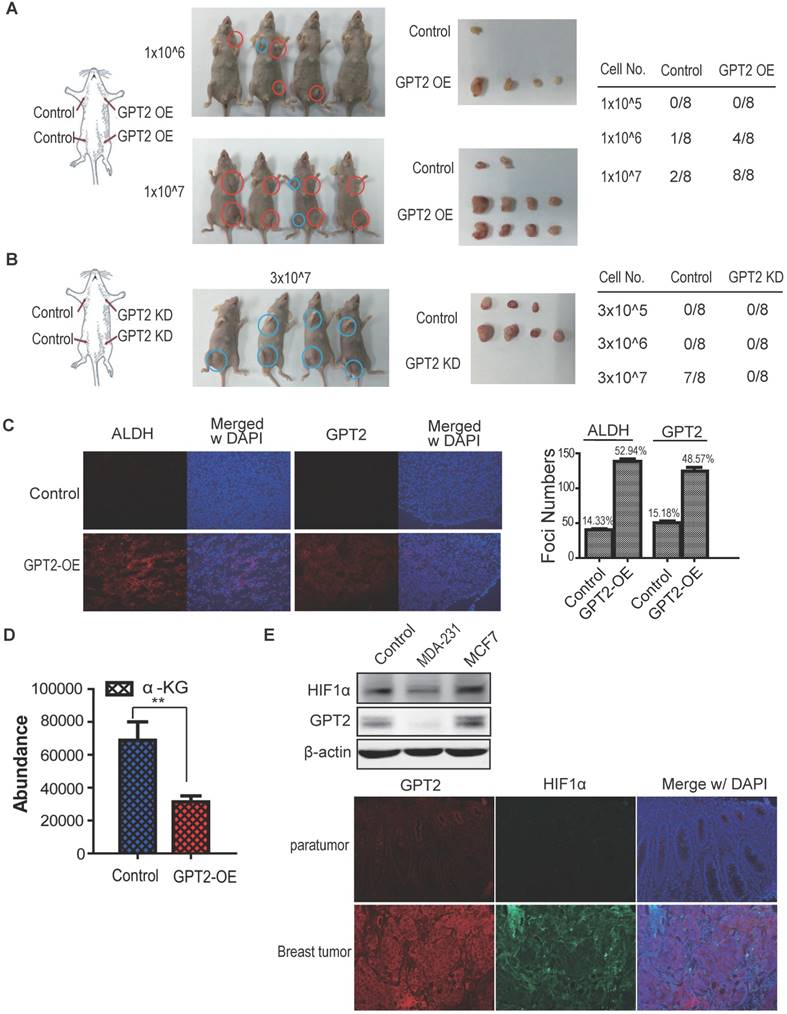
When the cell number was increased to 1 x 107 for each injection, all mice with GPT2 expressing MDA-MB-231 cells had tumors (8 out of 8) in 3 weeks after injection, whereas only two tumors were observed in the control group (2 out of 8). In contrast, GPT2 knockdown dramatically suppressed tumor formation. When 3 x 107 of MCF7 cells were injected, GPT2 depleted group did not grow any tumors (0 out of 8) while 7out 8 mice had tumors in the control group (Figure 5B).
To verify whether GPT2 promotes stemness of breast cancer cells in vivo, the immunofluorescence staining of ALDH1, a biomarker of breast cancer stem-like cell, was performed on xenografted tumors samples. As is evident from Figure 5C, the percentage of ALDH1 positive tumor cells was increased (from 14.33% to 52.94%) in tumors overexpressing GPT2 compared to the control group. The α-KG content in xenografted tumors was also evaluated by the α-KG assay kit. Figure 5D showed that the content of α-KG decreased in MDA-MB-231 xenografted tumors and increased in MCF7 xenografted tumors. Furthermore, we detected the expression of HIF1α and GPT2 in breast cancer cell lines and xenograft tumor specimens, our immunofluorescence data show that to some extent HIF1α protein levels are correlated with GPT2 expression (Figure 5E). These observations suggest GPT2 promotes tumorigenesis of breast cancer cells through HIF1α.
Discussion
Previous studies demonstrated that glutamine, as well as glucose metabolism is a hallmark of cancer. Glutamine catabolism is increased in highly proliferating cells for biosynthesis and energy production (41-43). Moreover, the binding of glutamate to its receptors activates SRC family kinases and downstream signaling stimulating cancer cell proliferation, apoptosis resistance, migration and invasion in different cancer cell lines (44). Glutamine metabolism is directly regulated by oncogenes and tumor suppressors (45-48). Among these genes, Myc was shown to be a critical regulator of glutamine metabolism and glutamine addiction in cancer cells (49-51).
Glutamate pyruvate transaminase (GPT) catalyzes α-ketoglutarate to glutamate or reversibly converts α-ketoglutarate into glutamate. GPT1 is located in the cytoplasma whereas GPT2 is a mitochondrial isoform. Both isoforms possess similar enzymatic activity for alanine and pyruvate but with different Km and Kcat values (52). GPT2 has been reported to increase the content of α-ketoglutarate during the transformation of cancer-associated fibroblasts (CAF) (53). However, we observed that GPT2 overexpression reduced the content of α-ketoglutarate by converting it to glutamate in breast cancer MDA-MB-231 cells suggesting that GPT2 plays different roles in CAFs and cancer cells. Highly proliferating cancer cells require more “building blocks” for anabolism that can be acquired by catalyzing α-ketoglutarate from TCA cycle into glutamate; in contrast, the CAFs with decreased proliferation (54), might not need glutamate required for anabolic processes. Interestingly, metabolomics analysis with stable isotope-labeling revealed overexpressing GPT2 increases 13C mass isotopologue distributions (MIDs) in α-KG and glutamate derived from glucose but decreases 13C MIDs in α-KG and glutamate derived from glutamine, supporting the notation that overexpressing GPT2 converses α-KG to glutamate in breast cancer cells.
Cancer stem-like cells have aberrantly regulated hedgehog (Hh) pathway as well as abnormalities in Wnt, Notch and PTEN pathways inducing tumorigenesis (55, 56). Several studies have elucidated the molecular networks in these signaling pathways. Hh proteins bind Patched-1 (PTCH) receptor derepressing the membrane protein Smoothened (SMO) and activated SMO then triggers Gli and its subsequent downstream target SUFU (57). Previous studies have reported the existence of an activated Shh pathway in lung cancer and other cancer models resulting in tumorigenesis (58). Small cell lung cancer (SCLC) cells were dependent on Hh signaling for their malignant behavior and underwent autocrine regulation (59). Although the mechanism of Hh signaling is not completely understood, aberrant Hh signaling is believed to result in tumor growth, proliferation, aggression, and metastasis.
In the present study, we have shown that GTP2 overexpression results in HIF1α protein stabilization, which plays an important role in Hedgehog signaling. HIF1α stability is regulated by ubiquitin-dependent proteasome degradation. HIF1α ubiquitination is triggered by hydroxylation at P402 and/or P564. PHD2 was found to directly regulate HIF1α hydroxylation (60-62) and itself is activated by oxygen and α-KG but suppressed by fumarate and succinate, two inhibitory metabolic structural analogs of α-KG. Our results showed that in breast cancer cells GPT2 upregulation decreased α-KG content, resulting in PHD2 inhibition and HIF1α stabilization.
In brief, our data have demonstrated that GPT2 promotes tumorigenesis of breast cancer cells by converting α-ketoglutarate, which downregulates Shh signaling, into glutamate. Our results suggest GTP2 is a potential target for breast cancer therapy and local delivery of molecules inhibiting GPT2 could avoid its possible side effects to muscle and liver.
Materials and Methods
Cells and reagents
MDA-MB-231 cells and MCF7 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) with 10% fetal bovine serum (FBS). SYBR Green PCR master mix was purchased from ABI. Monoclonal antibodies against GPT2 were purchased from Proteintech. The antibody against ALDH were obtained from BD (Cat # :611195). Antibodies against CD24 PE (Cat #:12-0247), CD44 APC (Cat #:17-0441) were acquired from eBioscience. Antibodies against Shh (Cat # :2207), HIF1α (Cat # :3716), HIF1α hydroxylation (Cat # :3434), SUFU (Cat # :2520), PTCH1 (Cat # :2468), PTCH2 (Cat # :2470), Gli1 (Cat # :3538), Notch Receptor Interaction Antibody Sampler Kit (Cat # :8658), Notch Isoform Antibody Sampler Kit (Cat # :3640), β-cantenin (Cat # :8480), p-GSK3β(Cat # :5558s), GSK3β(Cat # :9315L), OCT4 (Cat # :2750), SOX2 (Cat # :2748S), NANOG (Cat # :4903) were purchased from Cell Signaling Technology. The antibody against β-actin came from Santa Cruz Biotechnology (Cat # :47778). The α-Ketoglutarate Colorimetric/Fluorometric Assay Kit were obstained from BioVision Technologies. The human breast tumor tissue microarrays were purchased from Shanghai Outdo Biotech Co., Ltd. Dimethyl succinate (Cat # V900547) and Dimethyl 2-oxoglutarate (Cat # 349631) were obtained from Sigma-Aldrich.
Western blotting
The cells were harvested and lysed on ice for 15 min in lysis buffer containing 150 mM NaCl, 0.1% SDS, 1% NaMoO4, 1% NP40, 50 mM Tris-HCl (pH 7.5), and 0.02% NaN3 supplemented with 1 mM phenylmethanesulfonyl fluoride (PMSF). Protein concentration was detected by using the BCA assay kit (Ding Guo Biotechnology, Shanghai, China). Subsequently, the proteins were separated on polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were separately incubated with various antibodies.
Clinical samples
Tissue collection was approved by the Shanghai Jiao Tong University Medical Ethical Committee. Fresh and paraffin-embedded breast cancer blocks or peripheral normal breast tissue were collected after informed consent from patients undergoing surgery.
Colony formation assay
MDA-MB-231 cells overexpressing GPT2 or MCF7 cells depleted of GPT2 were seeded into 100 mm plastic dish plates at a cell density of 1000 cells/well and were allowed to grow for 10-14 days until clones were visible. PBS-washed cells were fixed with 4% paraformaldehyde and stained with 1% crystal violet. The stained clones were counted.
Soft agar colony formation assay
The anchorage-independent growth of MDA-MB-231 cells overexpressing GPT2 or MCF7 cells depleted of GPT2 was monitored by the soft agar colony formation assay. In brief, cells (500/well) were re-suspended in 1.5 ml mixture of 1.2% low-melt agarose and 2× DMEM with 10% fetal bovine serum (v:v = 1:1). Subsequently, cells were loaded in 6-well plates in triplicate on top of the solidified bottom agar comprising a mixture of equal-volumes of 0.7 % low-melt agarose and DMEM with 10% fetal bovine serum. The cells were incubated at 37°C, 5% CO2 for 2 weeks. The colonies composed of more than 50 cells were counted.
Luciferase assay
For the ODD luciferase assay, the ODD domain of HIF-1a (GenBank accession number U59496) was fused to the 5 ' end of the firefly luciferase reporter gene (63). As a control, a luciferase expression vector was used, in which the luciferase gene was driven by the CMV promoter. GPT2 overexpressing cells and control cells were transfected with1 μg ODD-firefly luciferase reporter gene. The renilla plasmid pRL-TK (10ng) was used as an internal control. Cells were harvested 48 hr after transfection and analyzed with the Dual-Luciferase Reporter Assay kit. Firefly luciferase values were normalized to renilla luciferase values from the same cells. The assays were repeated at least three times in independent experiments.
Flow cytometry analysis
MDA-MB-231 cells overexpressing GPT2 or MCF7 cells depleted of GPT2 were stained with anti-human CD24-PE (eBioscience) and anti-human/mouse CD44-APC (eBioscience) for 30 min. At least 1 x 106 cells were analyzed by a FACS Aria II(BD). Cells were gated based on their forward and side scatter properties.
Quantitative real-time PCR
Total cellular RNA was extracted using the guanidinium isothiocyanate and phenol-chloroform method. For mRNA quantification, 1μg total RNA was reverse transcribed into cDNA with MMLV reverse transcriptase according to the manufacturer's instructions. Quantitative real-time PCR reactions were performed in triplicate in a 96-well plate using 1 μl cDNA. The β-actin gene was used as an internal control for normalization. The primers for q-PCR were: GPT2 primers, sense: 5'-GGAGCTAGTGACGGCATTTCTACGA-3', and antisense: 5'-CCCAGGGTTGATTATGCAGAGCA-3'; HIF1α primers, sense: 5'-AAACAGAGCAGGAAAAGGAGG-3', and antisense: 5'-TCAAAGCGACAGATAACACG-3'; β-actin primers, sense: 5'-GCGGGAAATCGTGCGTGACATT-3', and antisense: 5'- GATGGAGTTGAAGGTAGTTTCG-3'.
Lentiviral shRNA knockdown and gene overexpression
Two short-hairpin RNA (shRNA) fragments specifically targeting human GPT2 were synthesized (Sangon Biotech, Shanghai,China) and inserted into the Mirzip lentiviral shRNA mir vector (Open Biosystems, Lafayette, CO). These lentiviral plasmids were co-transfected into 293T cells with pVSVg, pRSV-Rev, and pMDL to generate the lentiviruses using Lipofectamine 2000 (Invitrogen). Viral stocks were collected from the transduced 293T cells 72 hr post-transfection, and were used to infect MCF7 cells with 10 μg/ml polybrene. The viruses containing non-sense shRNA were used as control. The full length GPT2 cDNA fragment was cloned into the pHRSIN lentiviral vector. These lentiviral plasmids were co-transfected into 293T cells with psPAX2 and pMD2G to generate the lentiviruses using Lipofectamine 2000 (Invitrogen). Viral stocks were collected from the transduced 293T cells 72 hr post-transfection, and were used to infect MDA-MB-231 with 10 μg/ml polybrene.
Immune staining
Immunofluorescence staining was performed on cultured cells and tissue sections. Cells or sections were first fixed for 20 min in PBS containing 4% paraformaldehyde. After three washes, the fixed cells or tissue sections were permeabilized by 0.4% Triton X-100 for 10 min. Cells or tissue sections were then incubated with primary antibodies at 4°C overnight, followed by the appropriate Alexa Fluor 488- or 594-conjuated secondary antibodies at 37°C for 1 h. Subsequently, the cells and tissue sections were visualized by fluorescence microscopy.
For immunohistochemistry, breast tumors and normal breast tissues were fixed in 4% (v/v) phosphate-buffered paraformaldehyde overnight. The sections were deparaffinized with xylene, followed by rehydration through successive washes of ethanol/water (from 100% ethanol successively to 70% ethanol). Slides were then incubated in methanol with 3% H2O2 at room temperature. After incubation in 10 mM sodium citrate buffer at 96 °C for 1 hr, the sections were blocked with 10% goat serum for 1 hr, then incubated with primary antibodies at 4°C overnight. After incubation with horse radish peroxidase-conjugated anti-rabbit polyclonal antibody, sections were visualized with a DAB substrate. Images were captured by microscopy and analyzed by densitometry method through Image J software.
Cellular a-KG detection
Cells (2 x 106) were rapidly homogenized with 100 μl of ice cold α-KG assay buffer. Add 1- 50 μl samples were added to duplicate wells of a 96-well plate and the volume was brought to 50 μl with assay buffer. α-KG was detected using Ketoglutarate Colorimetric/Fluorometric Assay Kits which were purchased from BioVision.
Sample Preparation and GC-MS for metabolomics analysis
The cell samples were collected to the same cell number in 1 mL of 50% aqueous methanol (1.2*106/mL). Then, the same volume of chloroform was added and the mixture was processed by 7 cycles of 2 min ultra-sonication and 2 min interval in ice-bath. The cell lysates were kept at 4 °C for 1h prior to centrifugation at 14,000 g and 4 °C for 15 min. A total 600 μL of supernatant was mixed with 10 μL of internal standards (0.05 mg/mL of 13C3-15N-L-alanine, 13C5-15N-L-valine, 13C6-L-leucine and 13C6-15N L-isoleucine), and the mixture was evaporated to dryness, reconstituted in 50 μL of 20 mg/mL methoxyamine hydrochloride pyridine, and incubated at 37 °C for 90 min. Following the supplementation of another 50 μL of BSTFA (with 1% TMCS), the sample was derivatized at 70 °C for 60 min prior to GC-MS metabolomics analysis. Quality control (QC) sample pooled from representative plasma samples in each group were prepared and analyzed with the same procedure as that for the experiment samples.
Metabolomics instrumental analysis was performed on an Agilent 7890A gas chromatography system coupled to an Agilent 5975C inert MSD system (Agilent Technologies Inc., CA, USA). A HP-5ms fused-silica capillary column (30 m × 0.25 mm × 0.25 μm; Agilent J&W Scientific, Folsom, CA) was utilized to separate the derivatives. Helium (>99.999%) was used as a carrier gas at a constant flow rate of 1 mL/min through the column. Injection volume was 1 μL in splitless mode, and the solvent delay time was 6 min. The initial oven temperature was held at 70 °C for 2 min, ramped to 160 °C at a rate of 6 °C/min, to 240 °C at a rate of 10 °C/min, to 300 °C at a rate of 20 °C/min, and finally held at 300 °C for 6 min. The temperatures of injector, transfer line, and electron impact ion source were set to 250 °C, 250 °C, and 230 °C, respectively. The impact energy was 70 eV, and data was collected in a full scan mode (m/z 50-600).
Sample Preparation and GC-MS for metabolic flux analysis
The cell samples were collected to the same cell number in 1 mL of 50% aqueous methanol (1.2*106/mL). Then, the same volume of chloroform was added and the mixture was processed by 7 cycles of 2 min ultra-sonication and 2 min interval in ice-bath. The cell lysates were kept at 4 °C for 1 h prior to centrifugation at 14, 000 g and 4 °C for 15 min. A total 600 μL of supernatant was evaporated to dryness under nitrogen stream, reconstituted in 30 μL of 20 mg/mL methoxyamine hydrochloride in anhydrous pyridine, and incubated at 37 °C for 90 min. Then, a tert-butyldimethylsilyl (TBDMS) derivatization was initiated by adding 30 μL of MTBSTFA (with 1% TBDMCS, Regis Technologies) and incubating at 55 °C for 60 min.
Sample analysis was performed on an Agilent 7890A gas chromatography system coupled to an Agilent 5975C inert MSD system (Agilent Technologies Inc., CA, USA). A HP-5MS fused-silica capillary column (30 m × 0.25 mm × 0.25μm; Agilent J&W Scientific, Folsom, CA) was utilized to separate the derivatives. Helium (>99.999%) was used as a carrier gas at a constant flow rate of 1 mL/min through the column. Injection volume was 1 μL by splitless mode, and the solvent delay time was 5.5 min. The initial oven temperature was held at 100 °C for 2 min, ramped to 180 °C at a rate of 10 °C/min, to 260 °C at a rate of 5 °C/min, to 320 °C at a rate of 10 °C/min and finally held for 8 min. The temperatures of injector, transfer line, and electron impact ion source were set to 250 °C, 250 °C, and 230 °C, respectively. The electron energy was 70 eV, and data was collected in a full scan mode (m/z 50-600). Steady state metabolic flux was calculated by 13C mass isotopologue distributions (MIDs) for α-KG and glutamate with home-made package in R language, which applies an elementary metabolite unit framework to efficiently simulate MIDs and deducts natural isotope abundance.
Breast cancer xenograft model
Six-week-old BALB/c nude mice were obtained from the Shanghai animal laboratory. The animals were handled according to the protocol approved by the Shanghai Jiao Tong University Institutional Animal Care and Use Committee. Tumor xenografts were generated by subcutaneous injections with varying numbers of GPT2-OE and GPT2-sh1 cells (the total cell numbers in each injection were 1 × 105, 1 × 106, 1 × 106 and 3 × 105, 3 × 106, 3 × 107 ) into the armpits and groin bilaterally. After 5 weeks, the mice were sacrificed and the tumors were excised.
Statistical analysis
All data are presented as the means ± SD, and are representative of at least three independent experiments. The differences between groups were assessed by Student's t test; all reported differences are p < 0.05 unless otherwise stated.
Acknowledgements
We would like to thank Shanghai ProfLeader Biotech Co., Ltd for GC-MS analysis and thank Dr. Xuefeng Wu for useful discussion. This study was supported by grants from the National Program on Key Basic Research Project (973 Program) (2012CB910102), the Shanghai Committee of Science and Technology (11DZ2260200), and the National Natural Science Foundation of China (81372194) (81572300) to Dr. Mi, and the National Natural Science Foundation of China (81672911) to Dr. Lin.
Author contribution statement
Y. C., S. L., Y.W. and L.K. performed most of the experiments; Y. Q. provided reagents and revised the paper; J. M. designed the project and wrote the article; all authors reviewed the manuscript.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Willard SS, Koochekpour S. Glutamate signaling in benign and malignant disorders: current status, future perspectives, and therapeutic implications. International journal of biological sciences. 2013;9:728-42
2. Shroff EH, Eberlin LS, Dang VM. et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:6539-44
3. Hao Y, Samuels Y, Li Q. et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nature communications. 2016;7:11971
4. Carrascosa JM, Martinez P, Nunez de Castro I. Nitrogen movement between host and tumor in mice inoculated with Ehrlich ascitic tumor cells. Cancer research. 1984;44:3831-5
5. Collins CL, Wasa M, Souba WW. et al. Determinants of glutamine dependence and utilization by normal and tumor-derived breast cell lines. Journal of cellular physiology. 1998;176:166-78
6. Seidlitz EP, Sharma MK, Saikali Z. et al. Cancer cell lines release glutamate into the extracellular environment. Clinical & experimental metastasis. 2009;26:781-7
7. Daye D, Wellen KE. Metabolic reprogramming in cancer: unraveling the role of glutamine in tumorigenesis. Seminars in cell & developmental biology. 2012;23:362-9
8. Knox WE, Horowitz ML, Friedell GH. The proportionality of glutaminase content to growth rate and morphology of rat neoplasms. Cancer research. 1969;29:669-80
9. Linder-Horowitz M, Knox WE, Morris HP. Glutaminase activities and growth rates of rat hepatomas. Cancer research. 1969;29:1195-9
10. Fazzari J, Lin H, Murphy C. et al. Inhibitors of glutamate release from breast cancer cells; new targets for cancer-induced bone-pain. Scientific reports. 2015;5:8380
11. Namkoong J, Shin SS, Lee HJ. et al. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer research. 2007;67:2298-305
12. Speyer CL, Smith JS, Banda M. et al. Metabotropic glutamate receptor-1: a potential therapeutic target for the treatment of breast cancer. Breast cancer research and treatment. 2012;132:565-73
13. Takano T, Lin JH, Arcuino G. et al. Glutamate release promotes growth of malignant gliomas. Nature medicine. 2001;7:1010-5
14. McGovern M, Voutev R, Maciejowski J. et al. A "latent niche" mechanism for tumor initiation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11617-22
15. Peacock CD, Wang Q, Gesell GS. et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:4048-53
16. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843-50
17. Spivak-Kroizman TR, Hostetter G, Posner R. et al. Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer. Cancer research. 2013;73:3235-47
18. Barnfield PC, Zhang X, Thanabalasingham V. et al. Negative regulation of Gli1 and Gli2 activator function by Suppressor of fused through multiple mechanisms. Differentiation; research in biological diversity. 2005;73:397-405
19. Qian K, Zhong S, Xie K. et al. Hepatic ALT isoenzymes are elevated in gluconeogenic conditions including diabetes and suppressed by insulin at the protein level. Diabetes/metabolism research and reviews. 2015;31:562-71
20. Yang RZ, Blaileanu G, Hansen BC. et al. cDNA cloning, genomic structure, chromosomal mapping, and functional expression of a novel human alanine aminotransferase. Genomics. 2002;79:445-50
21. Coloff JL, Murphy JP, Braun CR. et al. Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell metabolism. 2016;23:867-80
22. Itkonen HM, Gorad SS, Duveau DY. et al. Inhibition of O-GlcNAc transferase activity reprograms prostate cancer cell metabolism. Oncotarget. 2016;7:12464-76
23. Richardson AL, Wang ZC, De Nicolo A. et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9:121-32
24. Gyorffy B, Lanczky A, Eklund AC. et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123:725-31
25. Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Current opinion in biotechnology. 2007;18:460-6
26. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nature reviews Cancer. 2008;8:755-68
27. Perrone G, Gaeta LM, Zagami M. et al. In situ identification of CD44+/CD24- cancer cells in primary human breast carcinomas. PloS one. 2012;7:e43110
28. Ponti D, Costa A, Zaffaroni N. et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer research. 2005;65:5506-11
29. Wang LB, He YQ, Wu LG. et al. [Isolation and characterization of human breast tumor stem cells]. Xi bao yu fen zi mian yi xue za zhi = Chinese journal of cellular and molecular immunology. 2012;28:1261-4
30. Wright MH, Calcagno AM, Salcido CD. et al. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast cancer research: BCR. 2008;10:R10
31. Ginestier C, Hur MH, Charafe-Jauffret E. et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555-67
32. Peacock CD, Wang Q, Gesell GS. et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A. 2007;104:4048-53
33. Takebe N, Harris PJ, Warren RQ. et al. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8:97-106
34. Grigoryan T, Wend P, Klaus A. et al. Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes & development. 2008;22:2308-41
35. Dumont E, Fuchs KP, Bommer G. et al. Neoplastic transformation by Notch is independent of transcriptional activation by RBP-J signalling. Oncogene. 2000;19:556-61
36. Taipale J, Chen JK, Cooper MK. et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005-9
37. Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465-72
38. Mohyeldin A, Garzon-Muvdi T, Quinones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell. 2010;7:150-61
39. Wan J, Wu W, Zhang R. Local recurrence of small cell lung cancer following radiofrequency ablation is induced by HIF-1alpha expression in the transition zone. Oncology reports. 2016;35:1297-308
40. Zhang D, Wang Y, Shi Z. et al. Metabolic reprogramming of cancer-associated fibroblasts by IDH3alpha downregulation. Cell Rep. 2015;10:1335-48
41. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029-33
42. Daye D, Wellen KE. Metabolic reprogramming in cancer: unraveling the role of glutamine in tumorigenesis. Seminars in cell & developmental biology. 2012;23:362-9
43. DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313-24
44. Hu H, Takano N, Xiang L. et al. Hypoxia-inducible factors enhance glutamate signaling in cancer cells. Oncotarget. 2014;5:8853-68
45. Hu W, Zhang C, Wu R. et al. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010;107:7455-60
46. Suzuki S, Tanaka T, Poyurovsky MV. et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A. 2010;107:7461-6
47. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
48. Wang JB, Erickson JW, Fuji R. et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18:207-19
49. Wise DR, DeBerardinis RJ, Mancuso A. et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782-7
50. Yuneva M, Zamboni N, Oefner P. et al. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. 2007;178:93-105
51. Gao P, Tchernyshyov I, Chang TC. et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762-5
52. Glinghammar B, Rafter I, Lindstrom AK. et al. Detection of the mitochondrial and catalytically active alanine aminotransferase in human tissues and plasma. Int J Mol Med. 2009;23:621-31
53. Zhang D, Wang Y, Shi Z. et al. Metabolic reprogramming of cancer-associated fibroblast by IDH3a. Cell Rep. 2015;10:14
54. Wu J, Fu R, Huang X. et al. Cell proliferation downregulated by TGF-beta2-triggered G1/S checkpoint in clinical CAFs. Cell Cycle. 20160
55. Reya T, Morrison SJ, Clarke MF. et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11
56. Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895-902
57. Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349-54
58. Yuan Z, Goetz JA, Singh S. et al. Frequent requirement of hedgehog signaling in non-small cell lung carcinoma. Oncogene. 2007;26:1046-55
59. Watkins DN, Berman DM, Burkholder SG. et al. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422:313-7
60. Lee KA, Lynd JD, O'Reilly S. et al. The biphasic role of the hypoxia-inducible factor prolyl-4-hydroxylase, PHD2, in modulating tumor-forming potential. Mol Cancer Res. 2008;6:829-42
61. Tennant DA, Frezza C, MacKenzie ED. et al. Reactivating HIF prolyl hydroxylases under hypoxia results in metabolic catastrophe and cell death. Oncogene. 2009;28:4009-21
62. Minervini G, Masiero A, Moro S. et al. In silico investigation of PHD-3 specific HIF1-alpha proline 567 hydroxylation: A new player in the VHL/HIF-1alpha interaction pathway?. FEBS Lett. 2013
63. Dewhirst MW, Cao Y, Li CY. et al. Exploring the role of HIF-1 in early angiogenesis and response to radiotherapy. Radiother Oncol. 2007;83:249-55
Author contact
![]() Corresponding author: Jun Mi: jmeiedu.cn
Corresponding author: Jun Mi: jmeiedu.cn