Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(10):2673-2689. doi:10.7150/thno.18915 This issue Cite
Research Paper
Microvesicles as Potential Biomarkers for the Identification of Senescence in Human Mesenchymal Stem Cells
Qian Lei1#, Teng Liu2#, Fei Gao1, Hui Xie1, Li Sun1, Aiqi Zhao1, Wenxiang Ren1, Hao Guo1, Liming Zhang3, Hongxiang Wang4, Zhichao Chen1 ![]() , An-Yuan Guo2
, An-Yuan Guo2 ![]() , Qiubai Li1
, Qiubai Li1 ![]()
1. Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China;
2. Department of Bioinformatics and Systems Biology, Key Laboratory of Molecular Biophysics of the Ministry of Education, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, China;
3. Department of Hematology, JingZhou Central Hospital, Tongji Medical College, Huazhong University of Science and Technology, Jingzhou, China;
4. Department of Hematology, Wuhan Central Hospital, Wuhan, China.
# These authors contributed equally to the study.
Received 2016-12-25; Accepted 2017-4-12; Published 2017-7-6
Abstract

Senescence in human mesenchymal stem cells (MSCs) not only contributes to organism aging and the development of a variety of diseases but also severely impairs their therapeutic properties as a promising cell therapy. Studies searching for efficient biomarkers that represent cellular senescence have attracted much attention; however, no single marker currently provides an accurate cell-free representation of cellular senescence. Here, we studied characteristics of MSC-derived microvesicles (MSC-MVs) that may reflect the senescence in their parental MSCs. We found that senescent late passage (LP) MSCs secreted higher levels of MSC-MVs with smaller size than did early passage (EP) MSCs, and the level of CD105+ MSC-MVs decreased with senescence in the parental MSCs. Also, a substantially weaker ability to promote osteogenesis in MSCs was observed in LP than EP MSC-MVs. Comparative analysis of RNA sequencing showed the same trend of decreasing number of highly-expressed miRNAs with increasing number of passages in both MSCs and MSC-MVs. Most of the highly-expressed genes in LP MSCs and the corresponding MSC-MVs were involved in the regulation of senescence-related diseases, such as Alzheimer's disease. Furthermore, based on the miRNA profiling, transcription factors (TF) and genes regulatory networks of MSC senescence, and the datasets from GEO database, we confirmed that expression of miR-146a-5p in MSC-MVs resembled the senescent state of their parental MSCs. Our findings provide evidence that MSC-MVs are a key factor in the senescence-associated secretory phenotype of MSCs and demonstrate that their integrated characteristics can dynamically reflect the senescence state of MSCs representing a potential biomarker for monitoring MSC senescence.
Keywords: mesenchymal stem cells, senescence, microvesicles, biomarker.
Introduction
Since their discovery by Friedenstein in 1970 [1], mesenchymal stem cells (MSCs) have been found to broadly distribute throughout the body and reside in bone marrow, adipose tissues, heart, gut, lung, liver, placenta, umbilical cord, amniotic fluid, and periodontal ligament [2]. These cells have the potential to differentiate into lineages of mesenchymal tissues such as bone, fat, and cartilage cells [3, 4]; they also have endodermic [5] and neuroectodermic differentiation potential [6] and play an integral role in physiological processes, such as hematopoiesis [7] and immunomodulation [8]. More recently, MSCs have become a promising tool for cell-based therapy in tissue engineering and regenerative medicine.
There is considerable evidence that MSC senescence is considered as a contributing factor to aging and aging-related diseases [9, 10] and replicative senescence impairs the regenerative potential of MSCs [11]. To better understand and monitor cell senescence in MSCs, it is necessary to have a reliable biomarker for identification of these cells. Unique phenotypic alterations of senescent MSCs have been reported including enlarged morphology, arrested proliferative capability, increased β-galactosidase activity, telomere shortening, accumulation of DNA damage, alteration of chromatin organization, reduced expression of surface antigen markers, up-regulation of cell cycle inhibitors (P16INK4A and P21WAF1), and senescence-associated secretory phenotype (SASP) [11, 12]. Since surface and external factors can be detected without intracellular delivery of a probe and without harming the cells, they can serve as ideal biomarkers to identify senescent cells. Senescent MSCs release a specific secretome, including matrix metalloproteinases (MMP2, TIMP2) [13], cytokines (IL-6) [14], insulin like growth factors binding proteins (IGFBP4, IGFBP7) [15], and monocyte chemoattractant protein-1 (MCP-1) [16]. The role of these factors has been investigated in the identification of MSC senescence.
As a key component of the cell secretome, microvesicles (MVs) are shed from cell surface by their parental cells into the extracellular environment [17]. Recent reports indicate that these small vesicles can mirror the molecular and functional characteristics of their parental cells and participate in important biological processes [18], such as the surface-membrane trafficking and the horizontal transfer of proteins and RNAs among neighboring cells [19, 20]. A growing body of evidences has shown that MVs shed by MSCs (MSC-MVs) express MSC-related markers [21, 22], such as CD29, CD44, CD73, CD90, and CD105, which act as key effectors of MSCs. Many biological functions have been attributed to MSC-MVs, such as tissue repair, hematopoietic support, immunomodulatory regulation, and inhibition of tumor growth [22-24]. Recently, it has been reported that old rat MSC-MVs have unique miRNAs and significantly inhibited TGF-β1-mediated epithelial-mesenchymal transition [21]; however, no information is available on whether MSC-MVs could represent characteristics of their parental cells in senescence.
In the present study, we investigated the changes in MSC-MVs when their parental MSCs experienced senescence, including MSC-MV size distribution, concentration, surface antigens, osteogenesis-related functions and miRNA content, to characterize these senescent MSC-MVs and evaluate their ability to resemble their parental senescent MSCs.
Results
Characterization and validation of senescent MSCs
An in vitro replicative senescent cell model was established using long-term cultured human umbilical cord MSCs. After passage 13 (P13), MSCs gradually became larger with flat and irregular shape which is the typical morphology of senescent cells (Figure 1A). Surface immunophenotypic markers of MSCs were detected at passage 5, 14, and 22 by Flow Cytometry (FCM). All MSCs samples were positive for CD29, CD44, CD73, CD90, and CD105 (Figure 1B), and negative for CD34 and CD45 (Figure S1). There were more β-galactosidase (β-gal)-stained cells in cultured MSCs after passage 14 (Figure 1C). FCM cell cycle analysis also showed that with increasing passage numbers, the fraction of cells in G1/G0 increased (P5, 67.50 ± 0.4890 %; P13, 70.39 ± 0.1338 %; P22, 82.24 ± 0.1155 %, P < 0.001), whereas that in G2/S/M phase decreased (P5, 32.50 ± 0.4890 %; P13, 29.61 ± 0.1338 %; P22, 17.76 ± 0.1155 %, P < 0.001) (Figure 1D). Furthermore, mRNA levels of cell senescent molecular markers, p16 and p21, in cultured MSCs were significantly increased from P3 to P18 (Figure 1E). The secreted levels of IL-6, IGFBP4, IGFBP7 and MCP-1, the SASP markers of MSCs, were also remarkably increased from P5 to P20 (Figure 1F). These data demonstrated that late passage (LP) MSCs (P13 or later) displayed a senescent phenotype, compared to the earlier passages of MSCs.
Characterization and validation of senescent MSCs. (A) The morphology of MSCs observed under an inverted microscope at passage 4, 13, 17, and 22 (P4, 13, 17, and 22). (×100; arrows indicate senescent cells). (B) Immunophenotypic analysis of surface markers CD29, CD44, CD73, CD90 and CD105 in MSCs at P5, 14, and 22 by FCM. (C) β-gal staining (blue) of MSCs observed under an inverted microscope at P5, 14, 17, and 22. (×100; arrows indicate senescent cells). (D) Cell cycle characteristics of different passage MSCs (P5, 13, and 22) by FCM. (E) RT-PCR demonstrating mRNA expression levels of p16 and p21, n = 4. (F) ELISA analysis of IL-6, IGFBP4, IGFBP7, and MCP-1 in different passage MSCs (P5, 15, and 20). n = 5. (G-H) Oil Red O staining (G) and Alizarinl Red S staining of different passage MSCs (P5, 18) observed under an inverted microscope at 2 weeks. (×200). Each experiment was confirmed in four different donors, and the representative data are shown. Abbreviations: AIM: adipogenic induction medium; CM: complete medium; AIM: adipogenic induction medium; OIM: osteogenic induction medium.
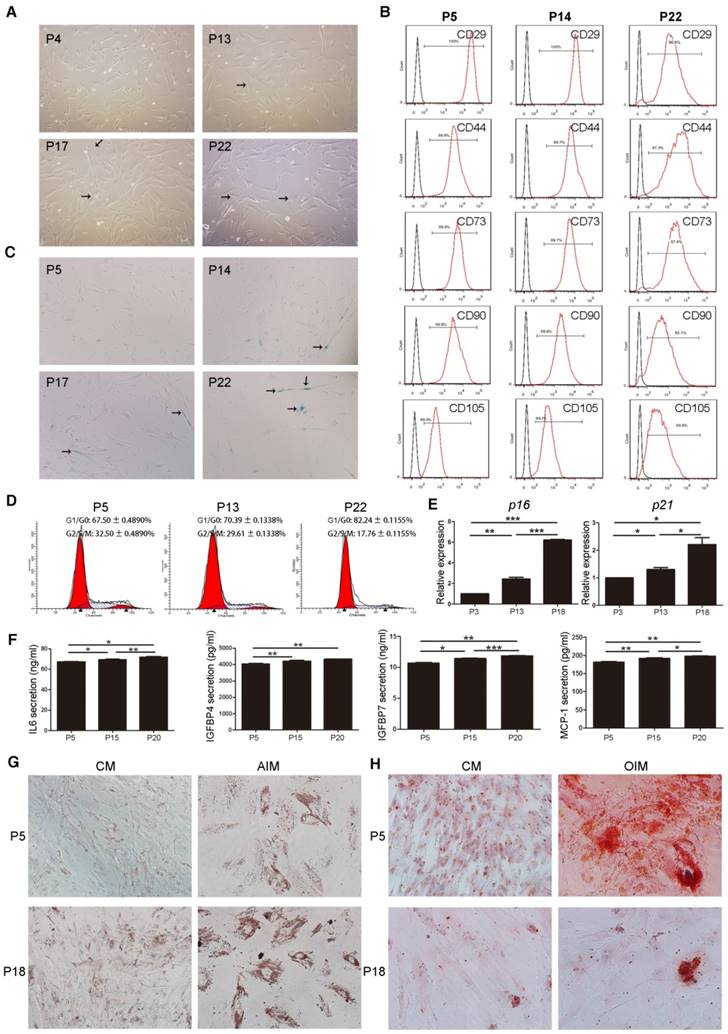
To test the adipogenic differentiation potential, MSCs were cultured in complete medium (CM) or adipogenic induction medium (AIM) followed by Oil red O staining at 2 weeks. Lipid droplet formation was much more effective in MSCs at P18 than P5, indicating the increased adipogenic differentiation ability in the late passage senescent MSCs (Figure 1G, Figure S2). Likewise, MSCs were cultured in CM or osteogenic induction medium (OIM) followed by Alizarin red staining at 2 weeks to test the osteogenesis differentiation potential. Calcium salt formation was much weaker in MSCs at P18 than P5, revealing that late passage senescent MSCs exhibited decreased differentiation potential in osteogenesis (Figure 1H, Figure S3).
Characterization and validation of MSC-MVs
MSC-MVs were isolated from MSCs supernatants by successive differential centrifugation [22]. Consistent with previous studies, MSC-MVs displayed a spheroidal shape surrounded by a double-layer membrane [25] (Figure 2A). CFSE-labeled MSC-MVs (most of them were less than 1,000 nm in diameter) could be observed under confocal microscopy (Figure 2B). The tunable resistive pulse sensing (TRPS) [26] analysis confirmed the heterogeneity of the MSC-MV samples, with a size distribution profile ranging from 200 to 1000nm in diameter (Figure 2C). Western blotting demonstrated that both MSCs and MSC-MVs expressed general EV markers including cytosolic proteins (TSG101, Alix) and cytoskeletal protein (β-actin) (Figure 2D); the expression level of Alix was lower in MSC-MVs than in MSCs (Figure 2D).
Characterization and validation of MSC-MVs. (A) Representative micrograph of MSC-MVs observed by transmission electron microscopy (TEM). Scale bar, 0.5 μm. (B) CFSE-labelled MSC-MVs observed by confocal microscopy. Scale bar, 2 μm. (C) TRPS analysis of the size distribution of MSC-MV samples. (D) Western blot analysis of proteins isolated from MSC-MVs or MSCs. (E) Representative flow cytometry plots of MSC-MV samples using 1 μm standard beads. (F) Integrity of MSC-MVs samples detected by FCM. (G) FCM analysis of the surface markers of MSC-MVs.
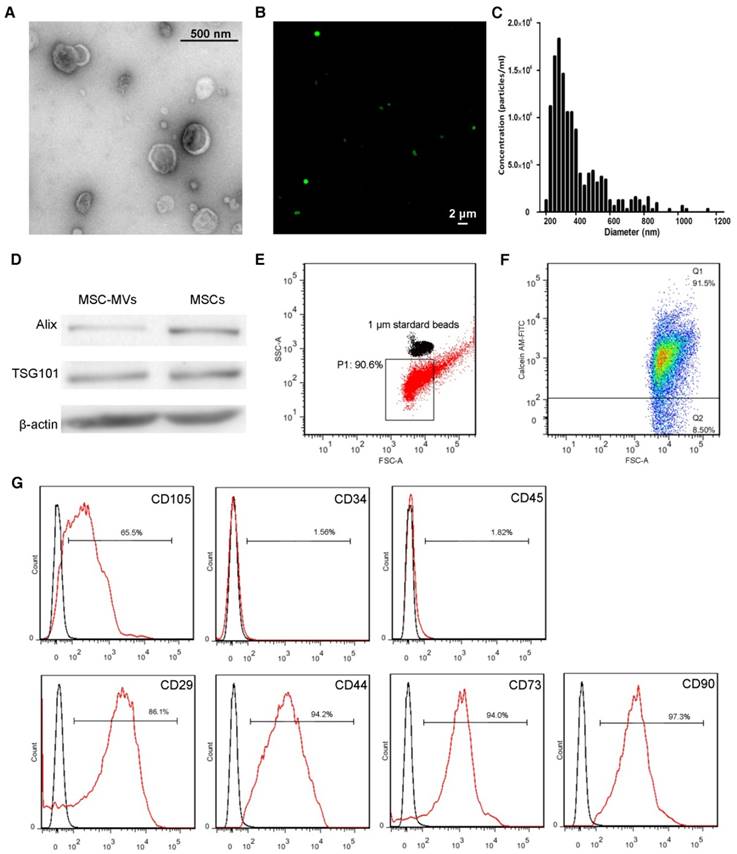
The sizes of MSC-MVs were judged by 1μm standard beads (Figure 2E) and their integrity was determined by staining with calcein-AM, which only stained intact MSC-MVs but not the debris [27]. The flow cytometric data showed the integrity of MSC-MV samples was 91.5% by calcein-AM staining (Figure 2F) and We further detected the immunophenotype of the intact MSC-MVs. Consistent with previous studies, MSC-MVs were positive for CD29, CD44, CD73, CD90 and CD105, and negative for CD34 and CD45, similar to their parental cells (Figure 2G, 1B). Interestingly, the expression level of CD105 was lower on the surface of MSC-MVs than on MSCs (Figure 2G, 1B).
Differences in concentration, size, and immunophenotype between MSC-MVs derived from early and late passage MSCs
We isolated EP (P3 to P7) and LP (P13 to P22) MSC-MVs from equal volumes (17 ml) of supernatant of MSCs (3 × 106) that had been cultured for 48 hours. TEM showed no obvious differences in spherical structures between EP MSC-MVs (P3-P7) and LP MSC-MVs (P13-P22) (Figure 3A). TRPS analysis showed that LP MSCs released more MVs than EP MSCs. The concentration (particles/ml) was 4-fold higher in LP MSC-MVs than in EP MSC-MVs (Figure 3B, 3C). Size analysis demonstrated that both EP and LP MSC-MVs displayed similar size distribution profiles with diameters ranging from 200 to 1000 nm (Figure 3B), but the average size of EP MSC-MVs (397.3 ± 4.410 nm) was significantly larger than that of LP MSC-MVs (358.7 ± 4.485 nm) (P = 0.0036) (Figure 3D). Based on the percentage makeup of distribution, we found that there were more particles larger than 500 nm in EP MSC-MVs than in LP MSC-MVs (Figure 3E).
Differences in concentration, size, and immunophenotype between MSC-MVs derived from EP and LP MSCs. (A) TEM images of EP and LP MSC-MVs. Scale bar, 500nm. n = 3. (B) Size distribution of EP and LP MSC-MVs. n = 3. (C) Concentration of EP and LP MSC-MVs. n = 3. (D) Average size of EP and LP MSC-MVs. n = 3. (E) Size composition of EP and LP MSC-MVs. (F) FCM analysis of the expression of surface markers on EP and LP MSC-MVs. Each experiment was confirmed in five different donors and the representative data are shown. Abbreviations: EP: early passage; LP: late passage.
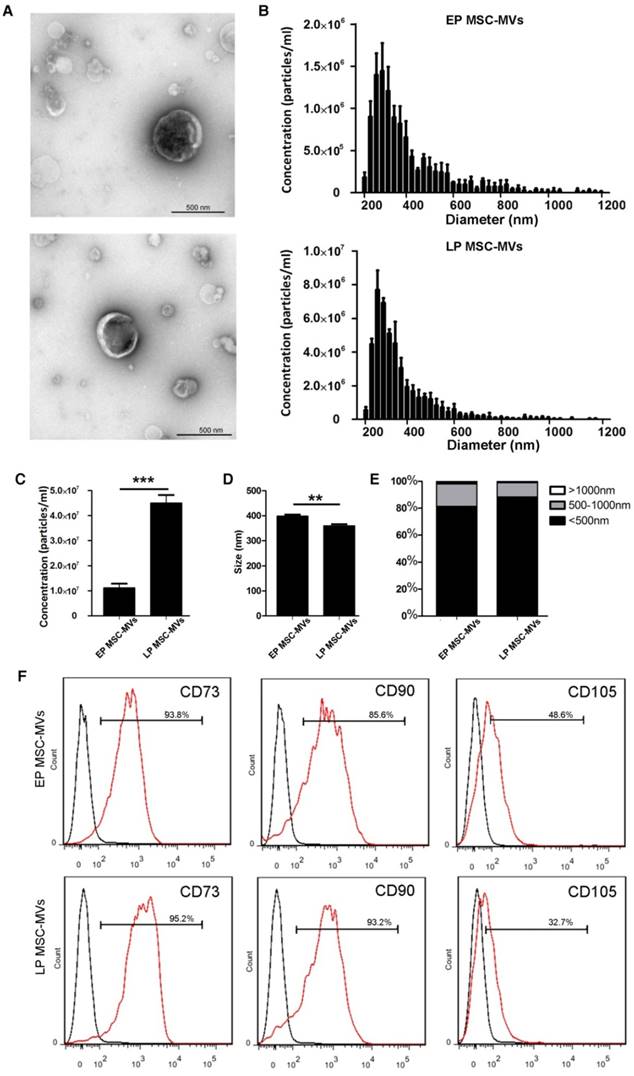
To examine the possible changes in immunophenotypic markers of MSC-MVs derived from EP to LP MSCs, we analyzed the expression levels of CD73, CD90, CD105 by FCM analysis. There was no significant difference in the levels of CD73 (EP vs LP, 95.30 ± 0.7550 % vs 94.40 ± 0.4041 %, P = 0.3526) and CD90 (EP vs LP, 93.83 ± 4.118 % vs 93.93 ± 0.4667 %, P = 0.9819) between EP and LP MSC-MVs, but the level of CD105 (EP vs LP, 46.37 ± 1.146 % vs 34.43 ± 0.9135 %, P = 0.0012) on the surface of LP MSC-MVs was significantly lower than that of EP MSC-MVs (Figure 3F). The data were validated in five different donor samples. These results indicated that the changes in concentration, size, and immunophenotype of MSC-MVs can reflect and resemble their parental MSC senescence.
Decreased pro-osteogenesis function in LP MSC-MVs versus EP MSC-MVs
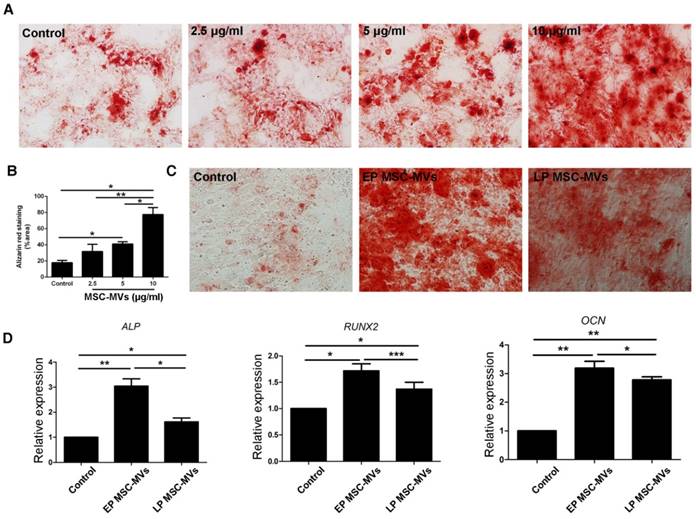
It had been reported that exosomes, another type of EVs derived from bone marrow MSCs (BMSCs), can promote osteogenic differentiation of BMSCs [28]. To investigate if MSC-MVs can exert a similar osteogenic effect, BMSCs at a density of 3×104/ml in a six-well plate were treated with 2.5, 5, and 10 μg/ml EP MSC-MVs (P3-P7) once a day for 14 days and assayed for osteogenic differentiation. Alizarin Red staining analysis showed more calcium deposits in BMSCs treated with MSC-MVs than in the control group, and the level of calcium deposits was higher in the 10 μg/ml group than in both 2.5 and 5 μg/ml groups (Figure 4A, 4B). Next we treated BMSCs with EP or LP MSC-MVs at 10 μg/ml to compare their pro-osteogenesis abilities. Similar to the decreased capacity of osteogenesis in senescent MSCs, LP MSC-MVs showed significantly lower pro-osteogenesis effect in BMSCs than EP MSC-MVs (Figure 4C). Accordingly, the levels of osteogenic genes, including ALP, RUNX2, and OCN, were lower in LP MSC-MVs than in EP MSC-MVs (Figure 4D). Taken together, these results indicate that MSC-MVs promote BMSC osteogenic differentiation, and the decreased pro-osteogenesis effect in LP MSC-MVs resemble that of decreased osteogenesis in LP MSCs.
Decreased pro-osteogenesis function in LP MSC-MVs than EP MSC-MVs. (A) Alizarin Red staining of the dose-dependent assay of EP MSC-MVs (2.5, 5 and 10 μg/ml) on osteogenic differentiation of BMSCs. (B) Percentage of alizarin red-positive area over total area. n = 3. (C) Alizarin Red staining of the mineralization of BMSCs, EP, and LP MSC-MVs (10μg/ml, once a day for 14 days). n = 3. (D) Real-time PCR showing the expression of ALP, RUNX2, and OCN in BMSCs. n = 4. Dilution of MSC-MVs using LG-DMEM medium, LG-DMEM medium-treated cells serving as a control. Each experiment was confirmed in four different donors and the representative data are shown. Abbreviations: EP: early passage; LP: late passage.
Gene and miRNA expression profiles in MSCs and MSC-MVs during senescence
We examined the gene expression changes in MSC senescence by performing RNA-Seq and miRNA-Seq in both EP and LP MSCs and their corresponding MSC-MVs. The sequencing reads and mapping information are shown in Table S1-S2. We applied RPM (reads per million reads) and RPKM (reads per kilobase per million reads) to quantify the miRNA and gene expressions (see methods). Approximately 1000 miRNAs and 20000 genes were detected in the four samples (Figure 5A). In each sample, a relatively high expression of 22% miRNA (RPM ≥ 50) and 3.3%-12.4% genes (RPKM ≥ 50) was detected. We also observed that the number of highly expressed miRNAs decreased in senescent MSCs and even more so in MSC-MVs (Figure 5B). Furthermore, we compared the differentially expressed miRNAs (DEMs) and genes (DEGs) between EP and LP (Figure 5C, 5D). Interestingly, most of the DEMs were down-regulated in both MSCs and MSC-MVs during the senescence, and the number of down-regulated miRNAs in MSC-MVs was much higher than that in MSCs. This suggested that it is possible to find miRNAs in MSC-MVs that would represent the MSCs senescence.
Gene and miRNA expression profiles and differential expression in MSCs and MSC-MVs during senescence. (A) miRNA and gene expression distribution of different passage MSCs and MSC-MVs. (B) Venn graphs of highly expressed miRNAs and genes in MSCs and MSC-MVs. Yellow parts have more highly expressed miRNAs or genes and the blue section has less. (C) DEMs and DEGs between EP to LP MSCs and MSC-MVs, respectively. (D) The expression cluster of DEMs in MSCs and MSC-MVs. (E) Top 10 pathways of KEGG enrichment of DEGs in MSCs and MSC-MVs. Abbreviations: DEGs: differentially expressed genes; DEMs: differentially expressed miRNAs; EP: early passage; LP: late passage.
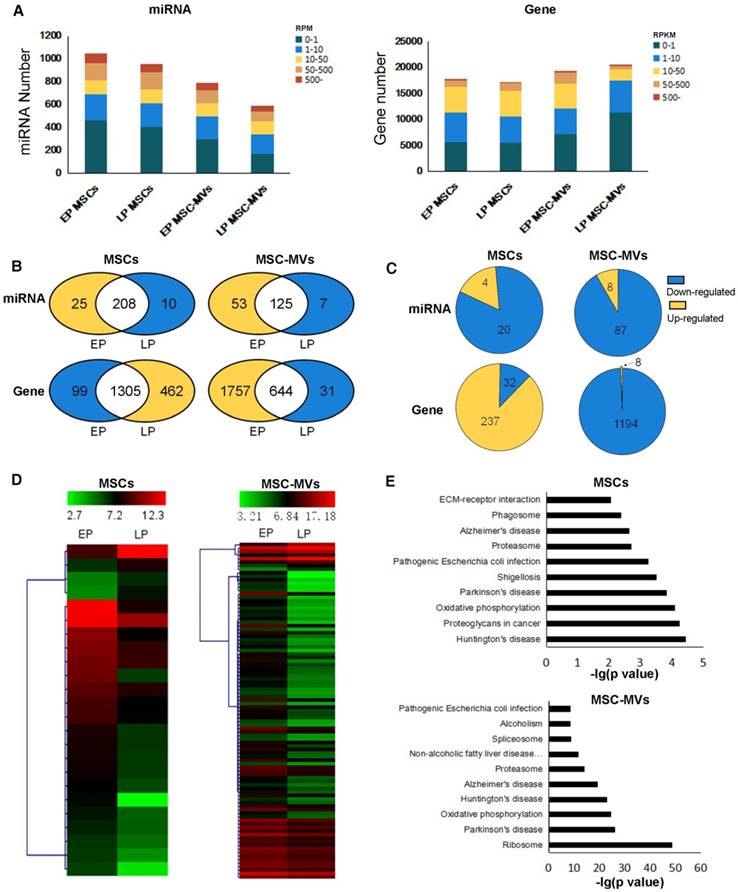
For genes, the number of highly expressed genes was increased in MSCs but decreased in MSC-MVs during senescence (Figure 5B). Similarly, most of DEGs were up-regulated in MSCs but down-regulated in MSC-MVs (Figure 5C). The top 10 KEGG pathways enriched by DEGs in MSCs or MSC-MVs are shown in Figure 5E, and their intersection pathways contained proteasome, oxidative phosphorylation, Alzheimer's disease, Huntington's disease, Parkinson's disease and Pathogenic Escherichia coli infection. Especially, the oxidative phosphorylation pathways and senescence-related diseases (such as Alzheimer's disease, Parkinson's disease and Huntington's disease) were enriched. These pathways were up-regulated in MSCs, but down-regulated in MSC-MVs.
miR-146a-5p expression change in MSC-MVs resembled that in MSCs during senescence
In an attempt to identify key regulators and potential markers of senescence, we investigated the gene expression regulation by miRNAs and transcription factors (TF) of DEGs in the MSC senescence. We constructed the miRNA-TF co-regulatory network [29] with 269 DEGs, 24 DEMs and 25 differentially expressed TFs between EP and LP MSCs by identifying and merging the TF-miRNA-Gene feed forward loops (FFLs) (Figure 6A). About 15% DEGs, 50% DEMs and 44% TFs were in the network. Since many papers reported that miRNAs are the key molecules in MVs [30, 31] and the trend of miRNA expressions is similar in MSCs and MSC-MVs, our analysis focused on miRNAs. The top 50% miRNAs ordered by node degrees in the network were miR-301a-3p, miR-301b, miR-335-5p, miR-148a-3p, miR-146a-5p, and miR-204-5p (Figure 6B), which were also the core hubs in DEMs network (Figure S4) and had the same expression alteration trends in both MSCs and MSC-MVs from EP and LP (Figure 6C). We subsequently chose these miRNAs for further validation.
Changes of miR-146a-5p expression in MSC-MVs resembling that in MSCs during senescence. (A) miRNA and TF co-regulatory network. (B) miRNA degree of distribution in the network. (C) Clusters of the hub miRNAs. (D) Relative expression of miR-146a-5p, miR-335-3p, miR-148a-5p, miR-301a-3p, and miR-301b (GSE68932). (E) Expression value of miR-146a-5p and miR-335-3p in samples of different ages of dataset GSE67630. (F) Expression of miR-146a-5p in MSCs and MSC-MVs analyzed by RT-PCR. The data were confirmed in three different donors. Abbreviations: EP: early passage; LP: late passage.
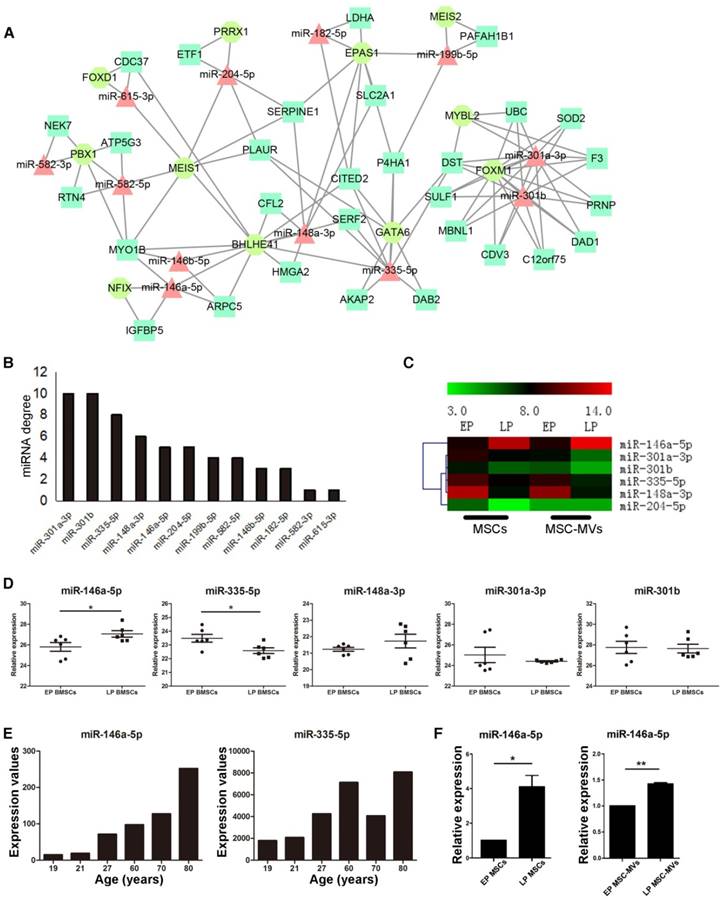
First, we used two datasets from NCBI GEO database to validate the top six miRNAs. The dataset GSE68932 was from 6 EP (P3-P4) and 6 LP (P6-P7) human BMSC samples. miR-204-5p was not detected but the expression of other 5 miRNAs was compared between EP and LP samples (Figure 6D). Consistent with our data, miR-146a-5p was up-regulated (P < 0.05), and miR-335-5p was down-regulated (P < 0.05) (Figure 6D). To examine the expression of miR-146a-5p and miR-335-5p in physiological aging, we used another dataset, GSE67630, which contains 6 human BMSC samples from different ages (19, 21, 27, 60, 70 and 80 years old). Compared to our findings, miR-146a-5p continually increased during physiological aging but miR-335-5p had the opposite trend (Figure 6E). We next validated the expression of miR-146a-5p, miR-148a-3p, miR-204-5p, miR-301a-3p, miR-301b, and miR-335-5p in different passages of MSCs and MSC-MVs by RT-PCR (Figure 6F, S5) and confirmed the up-regulation of miR-146a-5p in both MSCs and MSC-MVs during senescence (Figure 6F).
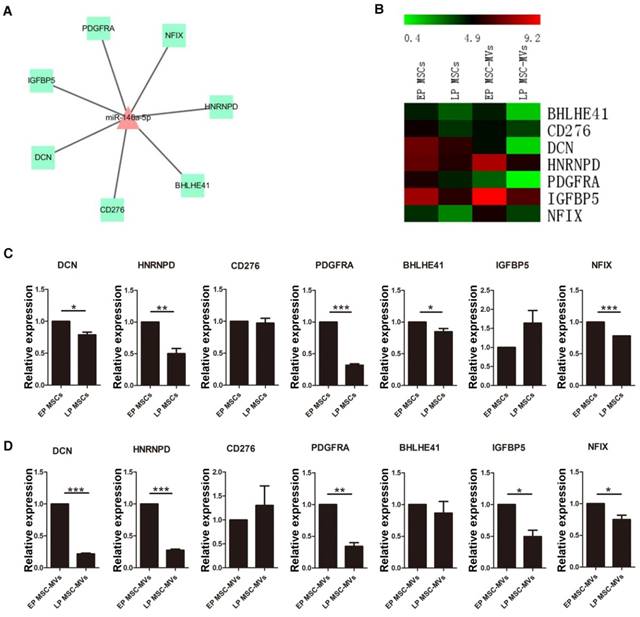
To further identify the potential of miR-146a-5p in MSC-MVs to resemble the senescence state of their parental MSCs, the expression of seven target genes of miR-146a-5p with a negative expression trend (Figure 7A, 7B) was validated in various passages MSCs and MSC-MVs by RT-PCR (Figure 7C, 7D). Among these target genes, DCN, HNRNPD, PDGFRA, BHLHE41 and NFIX were down-regulated in both MSCs and MSC-MVs during senescence. However, the expression of CD276 and IGFBP5 was different from the RNA sequencing data. Thus, the expression of miR-146a-5p was up-regulated and most of its target genes were down-regulated in both MSCs and MSC-MVs during senescence, suggesting that the expression of miR-146a-5p in MSC-MVs can serve as a potential senescent marker of the parental MSCs.
Alteration in the target genes of miR-146a-5p in MSC-MVs resembling that in MSCs during senescence. Network (A) and expression profile (B) of the target genes of miR-146a-5p. Expression of the target genes of miR-146a-5p in MSCs (C) and MSC-MVs (D) analyzed by RT-PCR. Each experiment was confirmed in three different donors. Abbreviations: EP: early passage; LP: late passage.
CD105 and miR-146a-5p expression changes in MSC-MVs resemble those in MSCs during senescence under serum-reduced culture condition
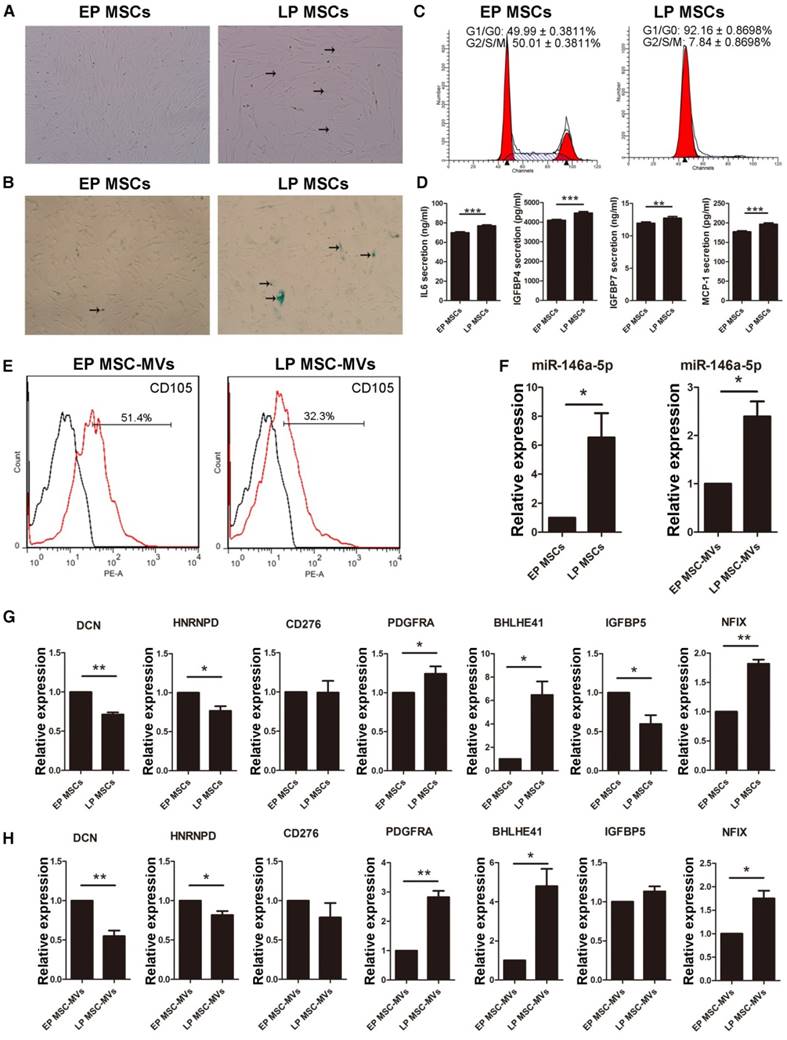
Since the serum used in the culture medium can partially be the source of the MVs, and/or affect the secreted MVs, we cultured MSCs in DMEM/F-12 containing reduced 5% FBS to validate the main findings. LP MSCs appeared large with flat shape (Figure 8A) and the expression of β-gal was increased in the culture medium with reduced FBS (Figure 8B). There was an increase and decrease in LP MSCs in G0/G1 (P < 0.001) and G2/S/M phases (P < 0.001), respectively (Figure 8C). We also examined the levels of the SASP markers and detected a significant increase in their levels from EP MSCs to LP MSCs (Figure 8D). We then examined the expression level of CD105 in MSC-MVs derived from EP and LP MSCs. Similar to the results observed in the regular 10% serum-supplemented medium (Figure 3F), the level of CD105 on LP MSC-MVs was significantly lower than that of EP MSC-MVs (52.07 ± 0.3383 % vs 32.27 ± 0.4910 %, P < 0.001) (Figure 8E). We also analyzed the expression levels of miR-301a-3p, miR-301b, miR-335-5p, miR-148a-3p, miR-146a-5p, and miR-204-5p, and detected a significant up-regulation of miR-146a-5p in both MSCs and MSC-MVs during senescence (Figure 8F), other miRNAs were down-regulated not in MSC-MVs but MSCs (Figure S6). Additionally, we examined the expression of the target genes of miR-146a-5p by RT-PCR and found that the levels of DCN, HNRNPD, and CD276 were down-regulated while PDGFRA, BHLHE41 and NFIX were up-regulated in both MSCs and MSC-MVs during senescence (Figure 8G, 8H). These results suggested that reducing the serum concentration in the culture medium did not alter the capacity of CD105 or miR-146a-5p to resemble that of their parental cells in senescence.
Changes of CD105 and miR-146a-5p expression in MSC-MVs resembling MSC senescence state under serum-reduced culture condition. (A) The morphology of MSCs observed under an inverted microscope. (×100; arrows indicate senescent cells). (B) β-gal staining (blue; arrows: senescent cells) of MSCs observed under an inverted microscope. (×100). (C) Cell cycle analysis of MSCs (P4 and P19) by FCM. (D) ELISA analysis of IL-6, IGFBP4, IGFBP7, and MCP-1 in EP and LP MSCs (P4 and P19). n = 5. (E) Representative FCM analysis of the expression of CD105 on EP and LP MSC-MVs. (F-H) Expression of miR-146a-5p in MSCs and MSC-MVs (F), and the target genes of miR-146a-5p in MSCs (G) and MSC-MVs (H), analyzed by RT-PCR. Each experiment was confirmed in three different donors, and the representative data are shown. Abbreviations: EP: early passage; LP: late passages.
Discussion
Currently, there is an aggravating trend of aging population world-wide, and the incidence of age-related diseases, such as Alzheimer's and cancer is rising [32]. One of the variety of aging factors is cellular senescence, which is an underlying determinant for aging of various organs, albeit at different rates [33]. Recent studies have shown that regular elimination of senescent cells slows time-dependent functional decline and prolongs lifespan in mice [34]. No similar studies are available in humans because of the lack of reliable biomarkers to identify and eliminate senescent cells [12]. Many studies have focused on EVs as new components of SASP and have been extensively reviewed by Urbanelli, et al. [35]. Our study has identified MVs, a component of cell secretome, as a novel source manifesting senescence in their parental cells in terms of concentration, size distribution, phenotypic markers, specific functions, and miRNA content. It is believed that secreted factors associated with SASP have an excellent potential as biomarkers because they can be readily detected and monitored. Our findings provide evidence that MVs have considerable potential as secreted biomarkers to accurately identify their senescent parental cells.
MSCs have been used as a promising tool for cell-based therapy in tissue engineering and regenerative medicine [36, 37]. For this, in vitro expansion of primary MSCs is necessary. However, the expansion potential of MSCs is limited as the in vitro aging leads to loss of multipotency and replicative senescence [11]. Thus, a robust cell-free biomarker to identify senescence in the cultured MSCs without harming the cells is necessary, especially for the cells obtained from aged donors. Numerous studies have demonstrated that the number and function of MSCs decline with aging associated with increased senescent cells in MSCs pool [38, 39]. Senescence of MSCs is believed to be the underlying cause in osteoporosis [40], systemic lupus erythematosus [41], and Hutchinson Gilford progeria syndrome [38]. Therefore, identification and even elimination of senescent MSCs in human body are attractive to both basic research and clinical practice. To date, MSC senescence in vivo remains poorly understood and there are no available markers to monitor it [12]. In the present study, we demonstrated that MSC-MVs actually resembled their parental MSC senescence in combinative signatures (Figure 9). These findings give support to MSC-MVs as a key factor in the senescence-associated secretory phenotype of MSCs and demonstrate that their integrated characteristics can dynamically resemble the MSC senescence state, representing a potential biomarker to precisely identify MSC senescence in vitro. With the development of modern techniques, such as high resolution FCM [42], and the development of researches in seeking tissue-specific MSC markers, we believe that MSC-MVs could be developed as a potentially practical and cell-free marker to identify and real-time monitor MSC senescence in vivo. However, there is a quite long way ahead.
Schematic diagram of combinative signatures of MSC-MVs to identify MSC senescence. Abbreviations: EP: early passage; LP: late passage.
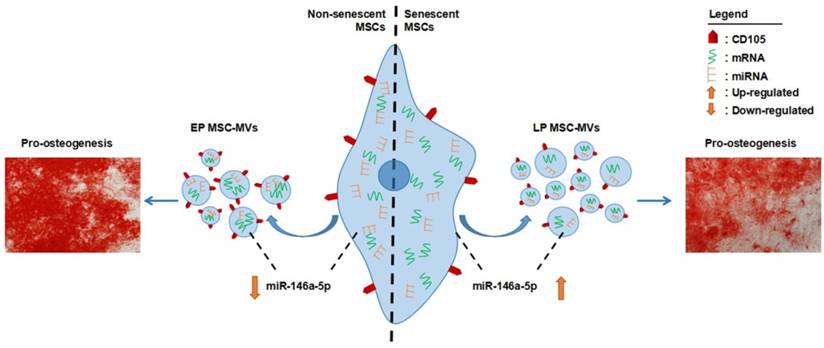
Many studies reported the level of plasma MVs associated with aging and age-associated diseases. The levels of platelet MVs in plasma of elderly people were increased [35] and the levels of myeloid MVs in cerebrospinal fluid are positively correlated with neurodegenerative disease [43]. Furthermore, senescent tumor cells [44] and endothelial cells [45] release higher levels of MVs. Consistent with these observations, our study also showed that senescent LP MSCs released higher levels of MSC-MVs than EP MSCs. MV size, another characteristic of MSC-MVs, has been shown to be larger in anti-retroviral therapy-naive patients than healthy control subjects and correlated with the progression of HIV [46]. Our TRPS analysis of MSC-MVs demonstrated that senescent LP MSC-MVs were clearly smaller than EP MSC-MVs, although they had similar size distributions. In the future, it is important to extend these observations to MVs released from other senescent cells. We further demonstrated that MSC-MVs resembled their parental MSCs in terms of the decreased expression of CD105, which is defined as one of the essential surface molecules [47] and can be easily detected. It is also important to understand the mechanisms underlying the decrease of CD105 on MSC-MVs during MSC senescence.
Although the functions of MSC-MVs in disease progression and tissue repair have been well-studied [24], very little information is available on the pro-osteogenic function of MSC-MVs in MSCs [48]. The present study uncovered the pro-osteogenic function of MSC-MVs and showed that EP MSC-MVs were more efficient than LP senescent MSC-MVs. This observation was in line with the decreased osteogenesis differentiation in senescent MSCs implying that MSC-MVs have the potential to be not only a biomarker but a functional senescence-related factor. It would be interesting to study other functions, such as hematopoiesis support and immune depression of MSC-MVs when the parental MSCs experience senescence.
It has been reported that miRNAs are key regulators and reliable markers of cellular senescence [35, 49]. A growing body of studies as well as our previous studies have uncovered the key functions of MV-packed miRNAs [30, 50]. In this study, to determine the potential of miRNAs encapsulated in MVs to serve as a MSC senescence marker, we sequenced mRNAs and miRNAs from different passages of MSCs and MSC-MVs. The data demonstrated, for the first time, that most of the DEMs were consistently down-regulated in both MSCs and MVs. Most of the DEGs, however, were up-regulated from EP to LP in MSCs, while down-regulated in their MVs. Previous studies reported up-regulation of most of the DEGs [51, 52] but down-regulation of most of the DEMs [53] in cells undergoing senescence, which is consistent with our study. However, there are no studies focusing on the gene and miRNA expression changes of MSC-MVs during senescence of their parental MSCs. A significant finding from our study was the down-regulation of most of the DEMs in both MSCs and MSC-MVs during senescence with a sharper decrease in MSC-MVs than in MSCs. Thus, changes in the expression of miRNAs can be more readily detected in MSC-MVs than in MSCs, miRNAs in MSC-MVs may be the sensitive molecular markers indicative of the parental MSC senescence.
Previous studies have demonstrated that miR-146a-5p contributes to the senescence of hepatocytes [54], macrophages [55], and endothelial cells [56]. Similarly, miR-146a-5p can be as a marker of senescence-related pro-inflammatory status in the vasculature [57]. It has also been shown to be an NF-kB-responsive and well-established SASP-modulating miRNA involved in the control of cellular senescence [58]. Another recent study reported that miR-146a-5p exists in exosomes and can serve as one of the diagnostic markers for esophageal cancer [59]. We used a multi-pronged approach and identified miR-146a-5p as MVs-encapsulated miRNA, which was present as a hub miRNA in our regulatory network [29, 30, 60-62]. First, we showed that the expression trend of miR-146a-5p was similar in MSC-MVs and MSCs during senecence. Second, we used a published dataset from EP and LP BMSC samples to validate it. Third, we also used another dataset from BMSC samples in different ages representing the physiological aging to validate it. And, finally, we used RT-PCR to confirm the up-regulation of miR-146a-5p and down-regulation of most of its target genes in MSC-MVs and MSCs during senescence. Thus, we characterized MVs-encapsulated miR-146a-5p as a potential senescence-associated marker to identify senescent MSCs. The functional role of miR-146a-5p in SASP-modulation of MSCs requires further investigations.
In conclusion, our findings have identified MSC-MVs as a key factor in the senescence-associated secretory phenotype of MSCs. The multidimensional characterization of MSC-MVs strongly suggest that their integrated characteristics can dynamically resemble the MSC senescence status representing a potential biomarker for identifying and monitoring MSC senescence.
Materials and Methods
Isolation of MSCs and culture conditions
Human umbilical cords (UCs; n = 5) were obtained from consenting patients delivering full-term (38-40 weeks) infants by cesarean section. The study was approved by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology and followed the principles in the Declaration of Helsinki. UCs were rinsed with phosphate buffered saline (PBS; Hyclone, Logan, UT, USA) to remove the blood cells. Subsequently, samples were cut into 1cm segments, and UC veins, arteries, and amnion were removed. UCs were then minced into 1-3 mm3 pieces. The minced pieces were placed in a 55 cm2 culture dish and incubated in DMEM/F-12 (Hyclone) supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA) and 100 U/ml of penicillin/streptomycin (Gibco). After 2 weeks, the UC tissue was removed and the adherent cells were passaged with 0.25% trypsin (Gibco) for 1min at 37ºC. The obtained cells were centrifuged at 250g for 5 min, and subcultured at a density of 4,000 cells/cm2. To reduce the effect of serum on the secreted content of MSCs, P4 and P19 MSCs were cultured in DMEM/F-12 (Hyclone) containing reduced serum (5% FBS, Gibco) of the same lot (LOT: 1652792).
Human bone mesenchyme stem cells (BMSCs) were commercially obtained from Cyagen Biosciences Inc. (Guangzhou, China). BMSCs were cultured in α-MEM (Hyclone) supplemented with 10% FBS (Gibco) and 100 U/ml of penicillin/streptomycin (Gibco). BMSCs (P4) were used in the osteogenic differentiation assay. All cells were maintained at 37°C in a humidified atmosphere with 5% CO2.
Isolation of MSC-MVs
MSC-MVs were isolated from the culture supernatants of MSCs by successive differential centrifugation, as previously described [22]. Briefly, MSCs were cultured in complete medium. Medium was collected every 48h and centrifuged at 750g for 15 min and 1,500g for 20 min to remove cells and debris. The supernatant was further centrifuged at 16,000g for 60 min at 4ºC to pellet MSC-MVs. Then the MSC-MVs pellets were washed with PBS, and centrifuged at 16,000g for 60 min at 4ºC. MSC-MV samples were kept at 4ºC and used within 24 hours.
Transmission electron microscopy
The isolated MSC-MVs were fixed in 100 μl of 3% glutaraldehyde for 2 hours. A drop (10 μl) of MSC-MVs was transferred to a formvar-carbon-coated grid. After rinsing, the grids were stained with 1% uranyl acetate solution for 5 min and examined with transmission electron microscopy (Hitachi HT7700, Tokyo, Japan) at 80 kV.
Laser confocal fluorescence microscopy
The isolated MSC-MVs were labeled with carboxyfluorescein succinimidyl ester (CFSE; Invitrogen, Carlsbad, CA, USA) for 20 minutes at 37 ºC. After washing in PBS for three times, MSC-MVs were observed under a laser confocal fluorescence microscope (Olympus, Tokyo, Japan).
Tunable resistive pulse sensing
The size distribution and concentration of MSC-MVs were measured with TRPS (qNano, Izon Science Ltd, Christchurch, New Zealand) using a NP400 nanopore at a 44.8 mm stretch. The concentration of MSC-MVs was standardized using calibration with CPC800 carboxylated polystyrene beads (size 660 nm, concentration of 1.2 ×1011 particles/ml).
Flow cytometry analysis
MSCs were collected for immunophenotypic analysis at P5, 14, and 22. Phycoerythrin (PE)-labeled anti-CD29, CD44, CD73, CD90, CD105, CD34, CD45 and IgG isotype control (all from BD Biosciences, San Diego, CA, USA) were incubated with MSCs for 30 minutes at 4 ºC. After washing in PBS for three times, MSCs were analyzed by flow cytometry (BD Biosciences).
To determine the immunophenotypic profile of MSC-MVs, 1 μm of standard microbeads (Sigma Aldrich, St. Louis, MO) were added to the MSC-MV samples. All samples were stained with Calcein-AM (Sigma), which was used to define the MSC-MVs with intact membrane structure. Subsequently, phycoerythrin (PE)-labeled anti-CD29, CD44, CD73, CD90, CD105, CD34, CD45 and IgG isotype control (BD Biosciences) were incubated with MSC-MV samples for 30 min at 4ºC. After washing in PBS for three times, MSC-MV samples were analyzed by FCM.
For cell cycle analysis, MSCs were trypsinized, centrifuged and washed for three times in PBS. Cells were then resuspended and fixed in 70% ethanol at 4ºC overnight. Fixed cells were washed at 500g for 5 min and stained with 10 μg/ml PI, 100 μg/ml DNase-free RNase A, 0.1% Triton X-100 and kept in the dark for 30 min. Cell suspensions were filtered and measured by FCM.
Osteogenic differentiation assay
For osteogenic differentiation, MSCs (3×104/ml) were plated into a 6-well plate and treated with or without MSC-MVs (10 μg/ml; Dilution of MSC-MVs using LG-DMEM medium) once a day for 14 days. MSC-MVs were quantified according to the total protein content using a BCA protein assay kit (Beyotime, Shanghai, China). The cells were cultured in osteogenic induction medium (OIM; Cyagen) that was changed every 3 days. After 14 days, the cells were fixed with 10% formalin and stained with Alizarin Red (Sigma) for analysis. The positive area was measured with Image J software.
Adipogenic differentiation assay
For adipogenic differentiation, MSCs (3×104/ml) were plated into a 6-well plate and cultured in adipogenic induction medium (AIM; Cyagen). The medium was changed every 3 days. After 14 days, adipogenic potential of MSCs was analyzed using oil red O (Sigma) staining. The positive area was measured with Image J software.
Senescence-associated β-galactosidase staining
The activity of senescence-associated β-galactosidase (β-gal) was measured in different passages of MSCs using SA-β-Gal staining kit (Beyotime). Briefly, MSCs (5×104/ml) were grown in a 12-well plate and fixed in fixative solution for 15 min. Subsequently, cells were washed with PBS and stained with SA-β-Gal staining solution at 37ºC for 12 hours. The positive cells presented a blue color; images were obtained with an inverted microscope (Olympus).
Enzyme-linked immunosorbent assay
MSCs were cultivated for another 24 hours with fresh serum-free medium. Supernatants were collected and clarified by centrifugation at 1000 g for 20 min. IL-6, IGFBP4, IGFBP7 and MCP-1 were detected by ELISA (Abcam, Cambridge, UK) according to the manufacturer's instructions. Results were obtained by measuring absorbance at 450 nm.
Real-time PCR
Total RNA was isolated from MSCs using Trizol reagent (Invitrogen) according to the manufacturer's instructions. RNA quality and purity were examined using Nucleic Acid/Protein Analyzer (Beckman Coulter, Brea, CA, USA); the A260/A280 ratio values were in the range of 1.8-2.0. Approximately, 1μg RNA was reverse transcribed using the FastQuant RT Kit (TIANGEN, Beijing, China). The real-time reverse transcriptase polymerase chain reaction (RT-PCR) was performed with the SYBR green PCR master mix (TIANGEN) on an Applied Biosystems 7500 Real-Time PCR System. The specific primer sequences are displayed in Supplementary Material (Table S3). β-actin was used as an invariant housekeeping gene. For miRNA expression, normalization was done using U6 as the reference [63]. Each reaction was performed in triplicate. Relative expression levels of RNA were calculated using the ΔΔCt method.
Western blot analysis
MSCs or MSC-MVs were lysed in ice-cold RIPA buffer (Beyotime) for 30 min at 4ºC. Lysates (25 μg of protein) were analyzed by 10% sodium-dodecyl sulfate-polyacrylamide gel electrophoresis (Sigma) and transferred to nitrocellulose membranes (Merck Millipore, Billerica, MA, USA), which were hybridized with anti-Alix (1:1000; Cell Signaling Technology, Beverly, MA, USA), anti-TSG101 (1:1000, Abcam), or anti-ACTIN (1:10000, sigma). Membranes were further incubated with HRP-coupled secondary antibodies (1:10000, Abcam).
mRNA and small RNA sequencing and analysis
To reduce variability, MSCs were obtained by serial passaging from the same donor (38 weeks, male). Total RNAs including small RNAs were isolated from MSCs and MVs in EP (MSCs: P5; MSC-MVs: P3-P6) and LP (MSCs and MSC-MVs: P13-14) status. mRNA-seq and small RNA-seq were performed with HiSeq2500 at WuXi AppTec (Shanghai, China). The low-quality reads were removed by in-house scripts. Clean reads were mapped to human reference genome hg19 and Refseq mRNAs or miRBase pre-miRNAs (Table S1, S2). We normalized the gene and miRNA expression level by RPKM (reads per kilobase per million mapped reads) and RPM (reads per million mapped reads), respectively, which are the common quantification methods used in mRNA-seq and small RNA-seq.
Identification of differentially expressed genes and miRNAs in cells and MSC-MVs
The cutoff for DEGs was the RPKM ≥ 100 in either stage and fold change ≥ 1.5 or ≤ 0.6666. The cutoff for DEMs was the RPM ≥ 50 and |log2(fold change)| ≥ 1. The differentially expressed miRNAs were hierarchically clustered using an average linkage algorithm and a Euclidean distance for the distance measure. We utilized MeV to visualize the clustered data and applied the DAVID: Functional Annotation Tools to do the KEGG enrichment analysis.
MSC senescence FFL network construction
Since the expression of TFs may be lower than other protein coding genes, we applied the cutoff RPKM ≥ 10 and |log2 (fold change)| ≥ 1 to obtain differentially expressed TFs between EP and LP MSCs. Combined with differentially expressed TFs, miRNAs and genes, we applied the method described in our previous review [29] to construct the miRNA and TF co-regulatory network. Targets of miR-146-a-5p were predicated by our miRNA target datasets combined with multiple methods and databases. A negative expression trend between miR-146a-5p and its target was required in miR-146a-5p senescence network. We utilized Cytoscape to show the network.
Data availability
The mRNA-seq and small RNA-seq data are available at BIGD Genome Sequence Archive (GSA) with the accession number PRJCA000325.
Statistical analysis
Results were presented as mean ± standard deviation. Two tailed Student's t-test was conducted using Graph-Pad Prism 5 Software, P values < 0.05 were deemed statistically significant (*P < 0.05; **P < 0.01; ***P < 0.001). All results are representative of data generated from three independent experiments.
Supplementary Material
Supplementary figures and tables.
Abbreviations
MSCs: mesenchymal stem cells; MSC-MVs: MSCs-derived microvesicles; BMSCs: bone marrow mesenchymal stem cells; EP: early passage; LP: late passage; FCM: Flow Cytometry; CM: complete medium; AIM: adipogenic induction medium; OIM: osteogenic induction medium; TRPS: tunable resistive pulse sensing; DEMs: differentially expressed miRNAs; DEGs: differentially expressed genes; TF: transcription factor; UCs: umbilical cords.
Acknowledgements
This study was supported in whole or part by grants from the National Natural Science Foundation of China (NSFC; 81272624 to Q. Li, 31471247 to A.Y. Guo, and 81470330 and 81170497 to Z. Chen).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Friedenstein AJ, Chailakhjan RK, Lalykina KS. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet. 1970;3:393-403
2. Karantalis V, Hare JM. Use of mesenchymal stem cells for therapy of cardiac disease. Circ Res. 2015;116:1413-30
3. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD. et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143-7
4. Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR. et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418:41-9
5. Krause DS. Bone marrow-derived cells and stem cells in lung repair. Proc Am Thorac Soc. 2008;5:323-7
6. Kopen GC, Prockop DJ, Phinney DG. Marrow stromal cells migrate throughout forebrain and cerebellum, and they differentiate into astrocytes after injection into neonatal mouse brains. Proc Natl Acad Sci U S A. 1999;96:10711-6
7. Sa da Bandeira D, Casamitjana J, Crisan M. Pericytes, integral components of adult hematopoietic stem cell niches. Pharmacol Ther. 2016 [Epub ahead of print]
8. Ansboro S, Roelofs AJ, De Bari C. Mesenchymal stem cells for the management of rheumatoid arthritis: immune modulation, repair or both? Curr Opin Rheumatol. 2016 [Epub ahead of print]
9. Liu H, Xia X, Li B. Mesenchymal stem cell aging: Mechanisms and influences on skeletal and non-skeletal tissues. Exp Biol Med (Maywood). 2015;240:1099-106
10. Reitinger S, Schimke M, Klepsch S, de Sneeuw S, Yani SL, Gassner R. et al. Systemic impact molds mesenchymal stromal/stem cell aging. Transfus Apher Sci. 2015;52:285-9
11. Wagner W, Ho AD, Zenke M. Different facets of aging in human mesenchymal stem cells. Tissue Eng Part B Rev. 2010;16:445-53
12. Matjusaitis M, Chin G, Sarnoski EA, Stolzing A. Biomarkers to identify and isolate senescent cells. Ageing Res Rev. 2016;29:1-12
13. Ozcan S, Alessio N, Acar MB, Mert E, Omerli F, Peluso G. et al. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging (Albany NY). 2016;8:1316-29
14. O'Hagan-Wong K, Nadeau S, Carrier-Leclerc A, Apablaza F, Hamdy R, Shum-Tim D. et al. Increased IL-6 secretion by aged human mesenchymal stromal cells disrupts hematopoietic stem and progenitor cells' homeostasis. Oncotarget. 2016;7:13285-96
15. Severino V, Alessio N, Farina A, Sandomenico A, Cipollaro M, Peluso G. et al. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis. 2013;4:e911
16. Jin HJ, Lee HJ, Heo J, Lim J, Kim M, Kim MK. et al. Senescence-Associated MCP-1 Secretion Is Dependent on a Decline in BMI1 in Human Mesenchymal Stromal Cells. Antioxid Redox Signal. 2016;24:471-85
17. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373-83
18. Maas SL, Breakefield XO, Weaver AM. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol. 2016 [Epub ahead of print]
19. Lu JF, Pokharel D, Padula MP, Bebawy M. A novel method to detect translation of membrane proteins following microvesicle intercellular transfer of nucleic acids. J Biochem. 2016;160:281-9
20. Cossetti C, Iraci N, Mercer TR, Leonardi T, Alpi E, Drago D. et al. Extracellular vesicles from neural stem cells transfer IFN-gamma via Ifngr1 to activate Stat1 signaling in target cells. Mol Cell. 2014;56:193-204
21. Wang Y, Fu B, Sun X, Li D, Huang Q, Zhao W. et al. Differentially expressed microRNAs in bone marrow mesenchymal stem cell-derived microvesicles in young and older rats and their effect on tumor growth factor-beta1-mediated epithelial-mesenchymal transition in HK2 cells. Stem Cell Res Ther. 2015;6:185
22. Xie H, Sun L, Zhang L, Liu T, Chen L, Zhao A. et al. Mesenchymal Stem Cell-Derived Microvesicles Support Ex Vivo Expansion of Cord Blood-Derived CD34(+) Cells. Stem Cells Int. 2016;2016:6493241
23. Xie H, Wang Z, Zhang L, Lei Q, Zhao A, Wang H. et al. Development of an angiogenesis-promoting microvesicle-alginate-polycaprolactone composite graft for bone tissue engineering applications. PeerJ. 2016;4:e2040
24. Akyurekli C, Le Y, Richardson RB, Fergusson D, Tay J, Allan DS. A systematic review of preclinical studies on the therapeutic potential of mesenchymal stromal cell-derived microvesicles. Stem Cell Rev. 2015;11:150-60
25. Phinney DG, Di Giuseppe M, Njah J, Sala E, Shiva S, St Croix CM. et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat Commun. 2015;6:8472
26. Lobb RJ, Becker M, Wen SW, Wong CS, Wiegmans AP, Leimgruber A. et al. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J Extracell Vesicles. 2015;4:27031
27. Sun L, Wang HX, Zhu XJ, Wu PH, Chen WQ, Zou P. et al. Serum deprivation elevates the levels of microvesicles with different size distributions and selectively enriched proteins in human myeloma cells in vitro. Acta Pharmacol Sin. 2014;35:381-93
28. Narayanan R, Huang CC, Ravindran S. Hijacking the Cellular Mail: Exosome Mediated Differentiation of Mesenchymal Stem Cells. Stem Cells Int. 2016;2016:3808674
29. Zhang HM, Kuang S, Xiong X, Gao T, Liu C, Guo AY. Transcription factor and microRNA co-regulatory loops: important regulatory motifs in biological processes and diseases. Brief Bioinform. 2015;16:45-58
30. Zhang HM, Li Q, Zhu X, Liu W, Hu H, Liu T. et al. miR-146b-5p within BCR-ABL1-Positive Microvesicles Promotes Leukemic Transformation of Hematopoietic Cells. Cancer Res. 2016;76:2901-11
31. Kosaka N, Yoshioka Y, Fujita Y, Ochiya T. Versatile roles of extracellular vesicles in cancer. J Clin Invest. 2016;126:1163-72
32. de Cabo R, Carmona-Gutierrez D, Bernier M, Hall MN, Madeo F. The search for antiaging interventions: from elixirs to fasting regimens. Cell. 2014;157:1515-26
33. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513-22
34. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J. et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184-9
35. Urbanelli L, Buratta S, Sagini K, Tancini B, Emiliani C. Extracellular Vesicles as New Players in Cellular Senescence. Int J Mol Sci. 2016;17:E1408
36. Galvez-Martin P, Sabata R, Verges J, Zugaza JL, Ruiz A, Clares B. Mesenchymal Stem Cells as Therapeutics Agents: Quality and Environmental Regulatory Aspects. Stem Cells Int. 2016;2016:9783408
37. Kadam P, Van Saen D, Goossens E. Can mesenchymal stem cells improve spermatogonial stem cell transplantation efficiency? Andrology. 2017;5:2-9
38. Kubben N, Zhang W, Wang L, Voss TC, Yang J, Qu J. et al. Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell. 2016;165:1361-74
39. Pan H, Guan D, Liu X, Li J, Wang L, Wu J. et al. SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res. 2016;26:190-205
40. Roman F, Urra C, Porras O, Pino AM, Rosen CJ, Rodriguez JP. Real-Time H2 O2 Measurements in Bone Marrow Mesenchymal Stem Cells (MSCs) Show Increased Antioxidant Capacity in Cells From Osteoporotic Women. J Cell Biochem. 2016 [Epub ahead of print]
41. Gu Z, Tan W, Ji J, Feng G, Meng Y, Da Z. et al. Rapamycin reverses the senescent phenotype and improves immunoregulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling pathway. Aging (Albany NY). 2016;8:1102-14
42. Montoro-Garcia S, Shantsila E, Tapp LD, Lopez-Cuenca A, Romero AI, Hernandez-Romero D. et al. Small-size circulating microparticles in acute coronary syndromes: relevance to fibrinolytic status, reparative markers and outcomes. Atherosclerosis. 2013;227:313-22
43. Joshi P, Turola E, Ruiz A, Bergami A, Libera DD, Benussi L. et al. Microglia convert aggregated amyloid-beta into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014;21:582-93
44. Lehmann BD, Paine MS, Brooks AM, McCubrey JA, Renegar RH, Wang R. et al. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008;68:7864-71
45. Burger D, Kwart DG, Montezano AC, Read NC, Kennedy CR, Thompson CS. et al. Microparticles induce cell cycle arrest through redox-sensitive processes in endothelial cells: implications in vascular senescence. J Am Heart Assoc. 2012;1:e001842
46. Hubert A, Subra C, Jenabian MA, Tremblay Labrecque PF, Tremblay C, Laffont B. et al. Elevated Abundance, Size, and MicroRNA Content of Plasma Extracellular Vesicles in Viremic HIV-1+ Patients: Correlations With Known Markers of Disease Progression. J Acquir Immune Defic Syndr. 2015;70:219-27
47. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D. et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315-7
48. Bobis-Wozowicz S, Kmiotek K, Sekula M, Kedracka-Krok S, Kamycka E, Adamiak M. et al. Human Induced Pluripotent Stem Cell-Derived Microvesicles Transmit RNAs and Proteins to Recipient Mature Heart Cells Modulating Cell Fate and Behavior. Stem Cells. 2015;33:2748-61
49. Hammond SM, Sharpless NE. HMGA2, microRNAs, and stem cell aging. Cell. 2008;135:1013-6
50. Zhu X, You Y, Li Q, Zeng C, Fu F, Guo A. et al. BCR-ABL1-positive microvesicles transform normal hematopoietic transplants through genomic instability: implications for donor cell leukemia. Leukemia. 2014;28:1666-75
51. Wagner W, Horn P, Castoldi M, Diehlmann A, Bork S, Saffrich R. et al. Replicative senescence of mesenchymal stem cells: a continuous and organized process. PLoS One. 2008;3:e2213
52. Wagner W, Bork S, Horn P, Krunic D, Walenda T, Diehlmann A. et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS One. 2009;4:e5846
53. Mori MA, Raghavan P, Thomou T, Boucher J, Robida-Stubbs S, Macotela Y. et al. Role of microRNA processing in adipose tissue in stress defense and longevity. Cell Metab. 2012;16:336-47
54. Smith EJ, Shay KP, Thomas NO, Butler JA, Finlay LF, Hagen TM. Age-related loss of hepatic Nrf2 protein homeostasis: Potential role for heightened expression of miR-146a. Free Radic Biol Med. 2015;89:1184-91
55. Jiang M, Xiang Y, Wang D, Gao J, Liu D, Liu Y. et al. Dysregulated expression of miR-146a contributes to age-related dysfunction of macrophages. Aging Cell. 2012;11:29-40
56. Prattichizzo F, Giuliani A, Recchioni R, Bonafe M, Marcheselli F, De Carolis S. et al. Anti-TNF-alpha treatment modulates SASP and SASP-related microRNAs in endothelial cells and in circulating angiogenic cells. Oncotarget. 2016;7:11945-58
57. Olivieri F, Lazzarini R, Recchioni R, Marcheselli F, Rippo MR, Di Nuzzo S. et al. MiR-146a as marker of senescence-associated pro-inflammatory status in cells involved in vascular remodelling. Age (Dordr). 2013;35:1157-72
58. Olivieri F, Albertini MC, Orciani M, Ceka A, Cricca M, Procopio AD. et al. DNA damage response (DDR) and senescence: shuttled inflamma-miRNAs on the stage of inflamm-aging. Oncotarget. 2015;6:35509-21
59. Warnecke-Eberz U, Chon SH, Holscher AH, Drebber U, Bollschweiler E. Exosomal onco-miRs from serum of patients with adenocarcinoma of the esophagus: comparison of miRNA profiles of exosomes and matching tumor. Tumour Biol. 2015;36:4643-53
60. Ye H, Liu X, Lv M, Wu Y, Kuang S, Gong J. et al. MicroRNA and transcription factor co-regulatory network analysis reveals miR-19 inhibits CYLD in T-cell acute lymphoblastic leukemia. Nucleic Acids Res. 2012;40:5201-14
61. Lin Y, Sibanda VL, Zhang HM, Hu H, Liu H, Guo AY. MiRNA and TF co-regulatory network analysis for the pathology and recurrence of myocardial infarction. Sci Rep. 2015;5:9653
62. Lin Y, Zhang Q, Zhang HM, Liu W, Liu CJ, Li Q. et al. Transcription factor and miRNA co-regulatory network reveals shared and specific regulators in the development of B cell and T cell. Sci Rep. 2015;5:15215
63. Occhipinti G, Giulietti M, Principato G, Piva F. The choice of endogenous controls in exosomal microRNA assessments from biofluids. Tumour Biol. 2016;37:11657-65
Author contact
![]() Corresponding authors: Qiubai Li, MD Ph.D and Zhichao Chen, MD Ph.D, Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1277 Jiefang Avenue, Wuhan 430022, China; Tel and Fax: +86-27-8572-6387. An-Yuan Guo, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, 1037 Luoyu Road, Wuhan 430074, China. Emails: qiubailiedu.cn (Q.L), chenzhichaoedu.cn (Z.C) and guoayedu.cn (A.Y.G)
Corresponding authors: Qiubai Li, MD Ph.D and Zhichao Chen, MD Ph.D, Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1277 Jiefang Avenue, Wuhan 430022, China; Tel and Fax: +86-27-8572-6387. An-Yuan Guo, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, 1037 Luoyu Road, Wuhan 430074, China. Emails: qiubailiedu.cn (Q.L), chenzhichaoedu.cn (Z.C) and guoayedu.cn (A.Y.G)