Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
CTC Biology
Current enrichment methodologies...
Selection of appropriate...
Future frontiers for CTC...
Conclusions
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(10):2606-2619. doi:10.7150/thno.18588 This issue Cite
Review
Circulating Tumor Cells: Moving Biological Insights into Detection
Lichan Chen, Ann M Bode, Zigang Dong ![]()
The Hormel Institute, University of Minnesota, 801 16th Ave NE, Austin, Minnesota 55912
Received 2016-12-1; Accepted 2017-4-19; Published 2017-6-25
Abstract

Circulating tumor cells (CTCs) have shown promising potential as liquid biopsies that facilitate early detection, prognosis, therapeutic target selection and monitoring treatment response. CTCs in most cancer patients are low in abundance and heterogeneous in morphological and phenotypic profiles, which complicate their enrichment and subsequent characterization. Several methodologies for CTC enrichment and characterization have been developed over the past few years. However, integrating recent advances in CTC biology into these methodologies and the selection of appropriate enrichment and characterization methods for specific applications are needed to improve the reliability of CTC biopsies. In this review, we summarize recent advances in the studies of CTC biology, including the mechanisms of their generation and their potential forms of existence in blood, as well as the current CTC enrichment technologies. We then critically examine the selection of methods for appropriately enriching CTCs for further investigation of their clinical applications.
Introduction
In tumor development, cancer cells acquire sequential genetic and epigenetic alterations, each of which confers some form of increased adaption, resulting in heterogeneous tumor cell populations [1]. Some of these heterogeneous populations can leave the primary tumor and relocate to a distant site where they adapt to the new environment and evolve to novel clones to form metastases [1-7]. This spatiotemporal dynamic heterogeneity in tumor cell population, caused by adaption-related evolution, is one of the major reasons for the development of resistance to chemotherapy or radiotherapy. Thus obtaining real-time disease information is highly desirable for cancer treatment. Although traditional imaging and tissue biopsies remain as the standard in cancer diagnosis and in monitoring treatment response, these approaches cannot overcome the constraints caused by the spatiotemporal dynamic heterogeneity of cancer cell populations and do not allow visualization of minimal residual disease. For this reason, an alternative diagnostic method, liquid biopsy, which may overcome these constraints, has gained increased attention in cancer research in the last few years. Liquid biopsy is a technique for sampling and analyzing critical biomarkers in nonsolid biological tissue, primarily blood.
Circulating tumor cells (CTCs) are derived from tumors as an early step in blood-borne metastasis, which is transient in blood with a half-life of 1 to 2.4 hours [8-10]. Studies from both animal models and cancer patients demonstrate that CTCs have a promising potential as a liquid biopsy that can facilitate early detection, prognosis, therapeutic target selection, and monitoring therapeutic response. CTCs in most cancer patients are low in abundance-usually lower than 10 cells per milliliter [11-15]. However, in some rare outliers, CTCs might be as high as hundreds or even thousands per milliliter of blood. Therefore, a CTC biopsy generally begins with an enrichment step to raise the concentration of CTCs by several log units before they can be characterized. A number of methodologies based on biological and physical differences between CTCs and blood cells have been developed for CTC enrichment over the past few years. However, huge divergences in the characteristics of CTCs are harvested from different enrichment methods due to the similarities between CTCs and blood cells, as well as innate CTC heterogeneity [16-18]. Improving the reliability of CTC biopsy, a deep understanding of phenotype variation of tumor cells on the molecular level during their release from the primary tumor and while traveling in the circulation, even during disease development and therapeutic processes, is highly desirable for improving CTC enrichment and characterization technologies. In this review, we summarize recent advances from studies focusing on the biology of CTCs, including the mechanisms of their generation and their potential forms of existence in blood, as well as current CTC enrichment technologies. We then examine the selection of methods for appropriately enriching CTCs for further study of their clinical applications.
CTC Biology
During their metastasis to a distant site mainly through circulating blood, cancer cells often execute a cascade of intravasation-translocation-extravasation-colonization. This process includes their release from the primary tumor, entrance into the bloodstream, infiltration from the blood vessels to distant tissues where the cells as latent seeds of metastases need to survive and adapt to the supportive niches, and eventually develop into overt metastasis [1]. Cancer cells tightly adhere to their neighboring cells in part through their tight junctions, desmosomes and hemi-desmosomes and, at the same time, are surrounded by a complex stroma that is composed of an extracellular matrix (ECM) and neo-angiogenic blood vessels with or without a basal membrane (BM) [1, 19]. Initially, the property of adherence and the stroma serve as physical constraints to prevent distant metastasis. Hence, carcinoma cells have to overcome these barriers by enhancing motility in the stroma and invading into the blood through EMT or non-EMT mediated active or passive entry. The living CTCs in circulation might eventually arrest at the primary tumor, other metastatic lesions or at new distant sites. The arrested CTCs might then extravasate, possibly supported by the mesenchymal-to-epithelial transition (MET), and undergo dormancy or engraftment and colonization. The current findings on the mechanisms explaining how CTCs are generated as well as their forms of existence in blood are summarized in Fig. 1.
Mechanisms of circulating tumor cell (CTC) generation and the potential CTC subpopulations existing in the blood circulation. Two main mechanisms are involved in cancer cells leaving the tumor and moving to the blood. 1) CTCs undergo epithelial-mesenchymal transition (EMT) or 2) non-EMT-mediated invasion. In EMT-mediated invasion, cancer cells actively adopt cellular programs to break down the basement membrane (BM), migrate through the extracellular matrix (ECM), and enter the blood. In contrast, in non-EMT-mediated invasion, cancer cells enter the blood either through centrosome amplification-triggered invasion or through passive infiltration initiated by external forces. In both mechanisms, CTCs can leave tumors as single cells or in clusters. Once reaching the blood, CTCs might be present in one or more of the following subpopulations: (a-d) single cells with a different EMT phenotype that might further bind to platelets and/or macrophages; or (e-i) in clusters with a different EMT phenotype that may be further cloaked by platelets, macrophages, and/or reactivated stromal cells.
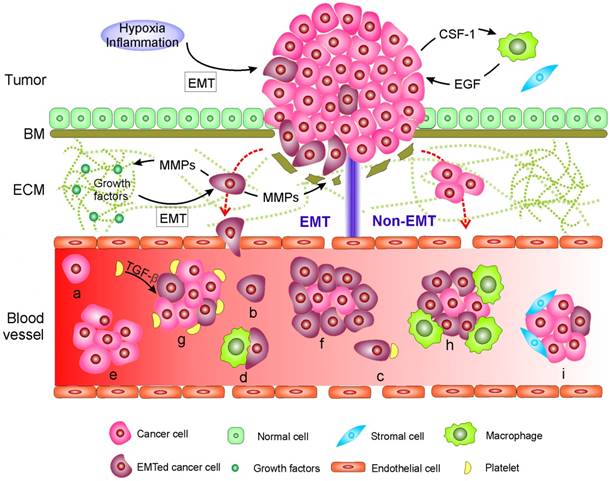
EMT and non-EMT-mediated release of CTCs into the blood
Two main biological mechanisms are involved in the release of CTCs from the tumor into the circulating blood. The first occurs through the epithelial-mesenchymal transition (EMT) and the second involves non-EMT-mediated invasion. EMT is a dedifferentiation process in which epithelial cells depolarize, partially or completely lose intracellular contacts by ending the expression of epithelial markers, and gain mesenchymal traits that confer stem-like properties and favor migration [20, 21]. Not surprisingly, metastasis usually involves EMT [20-23]. During carcinogenesis, epithelial cells gradually lose their apicobasal polarity and growth control [24]. Concurrently, a cancer-associated stromal niche is gradually formed [1, 19]. Activation of EMT in cancer cells often requires interactions between these cells and reactivated stromal cells in their surroundings, which is also designated as an inflammation environment [25]. The cancer cells recruit stromal cells [25, 26], respond to various EMT-inducing signals released from the stromal cells, such as various cytokines, and activate certain transcription factors (e.g., SNAIL, TWIST, and ZEB), microRNAs [27], epigenetic machinery [28], and post-translational regulation [29], leading to EMT [1, 21, 26, 29]. Activation of an EMT program can also be induced by hypoxia [30]. To promote further migration through the BM and ECM, invasive cancer cells remodel their surrounding stroma by up-regulating various ECM-degrading proteases, such as matrix-metalloproteinases (MMPs) and cathepsins, to excavate passageways [31]. Note that during the degradation of ECM components, MMPs also catalyze the release of certain cell-surface and matrix-bound growth factors and cytokines from ECM, all of which further facilitate the growth and survival of cancer cells as well as constructing a supporting tumor microenvironment [31]. The synergistic effect of EMT-induced cytoskeletal rearrangements within cancer cells, MMP-mediated passageway excavation, and adhesive interactions between cancer cells and ECM all drive the invasion and migration of cancer cells through the stroma. Invasive cancer cells enter the blood by their synergistic effects of passive infiltration and active transendothelial migration. Passive infiltration benefits from the intrinsic leaky and torturous vasculature in the tumor environment, which is generated to facilitate the supply of nutrients and oxygen needed for tumor growth [32]. Molecular programs might be further adopted by cancer cells to regulate vasculature permeability and promote transendothelial migration [33-37].
Invasive cancer cells can migrate through the stroma and blood vessels as single cells or in clusters, which are designated microemboli [38-44]. Single cells often exhibit EMT-associated changes, whereas clusters appear to maintain cell-cell connections and harbor a mesenchymal edge [41]. Clusters can either exclusively contain cancer cells or consist of cancer cells and reactive stromal cells (e.g., cancer-associated fibroblasts) [45-47], in which distinct cell clones cooperate to promote mutual survival and metastatic ability [40, 43].
Invasion and metastasis can also occur independently of EMT. This idea is based on recent studies with transgenic mouse models suggesting that EMT might be dispensable for initiation of metastasis [48, 49]. These transgenic mouse models harbor deletion of transcription factors that regulate EMT (e.g., SNAIL or TWIST) and suggest that suppression of EMT in the primary tumor does not alter the emergence of systemic metastasis [49]. Furthermore, Godinho, et al. [50] reported that centrosome amplification can trigger cancer cell invasion by disrupting cell-cell adhesion by increasing Arp2/3-dependent actin polymerization. In addition, passive infiltration initiated by external forces, such as tumor growth and mechanical forces (e.g., surgery), might cause the formation of “accidental CTCs” in circulating blood [51-53]. The disseminating cancer cells originating from non-EMT-mediated invasion might also consist of single cells or clusters and retain their epithelial phenotype [48],[53].
Challenges of maintaining CTC survival in the vasculature
Once cancer cells reach the circulating blood, most CTCs will undergo apoptosis due to a lack of a correct cellular/ECM attachment or other hazards present in the blood environment, including components of the innate immune system, shear force or oxidative stress [54-57]. Only camouflaged cancer cells tend to adapt to the circulatory environment and evade the immune defenses to complete the rigorous metastatic process [58-60]. To resist death, CTCs essentially reshape the integrin expression profile and activate cellular signaling such as the Akt signal transduction pathway through context-dependent mechanisms [61]. To escape immune surveillance, cancer cells might up-regulate various surface proteins, such as CD47 [14, 62-64], PD-L1 [65], and vascular cell adhesion molecule 1 (VCAM-1) [66], which bind to macrophages to evade phagocytosis. To avoid oxidative stress, cancer cells show an increased dependence on the reduced nicotinamide adenine dinucleotide phosphate (NADPH)-generating enzyme in the folate pathway [67]. Recent studies reported that cancer cells expressing tissue factor proteins on their surfaces tend to attract platelets [68, 69]. The association with platelets not only stimulates reversible metabolic changes in cancer cells that enable them to withstand oxidative stress [67, 70], but also links tumor cells with CD11b+ macrophages, establishing microclots to protect CTCs in the circulating blood [71].
Existence of heterogeneous forms of CTCs
CTCs in the blood of patients with cancer usually exist as apoptotic or viable cells and might contain one or more of the following subpopulations: 1) single cells with a different EMT phenotype that may further bind to platelets and/or macrophages; or 2) clusters with different EMT phenotypes that may be further disguised by platelets, macrophages, and/or reactive stromal cells (Fig. 1 and Fig. 2A).
Physical heterogeneity of CTCs. (A) CTCs exist as intact cells, clusters, or apoptotic cells. (B) Quantitative analysis of the effective diameter (maximum feret diameter) for individual CTCs and WBCs isolated in three cases, e.g., two different melanoma patients (M1 and M2) and a breast cancer patient (B3). (C) Relationship between circulating tumor cell (CTC) nuclear size and metastatic status. A-C were reprinted with permission from ref [15] [74] [78], respectively.
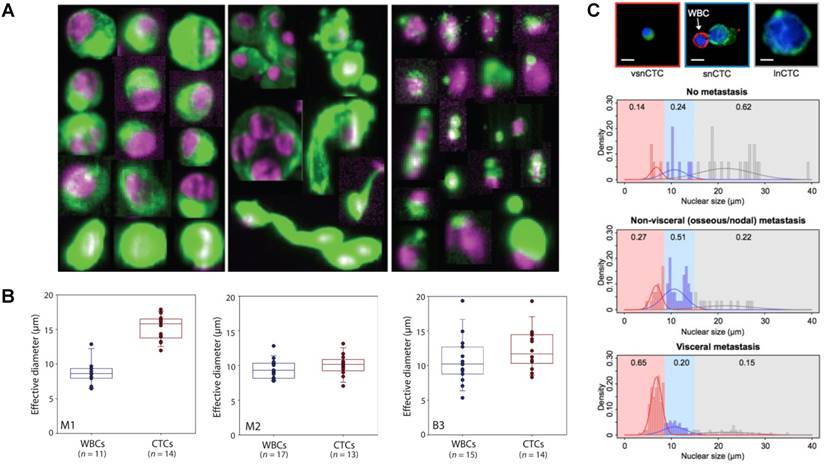
Numerous analyses on the characteristics of CTCs have demonstrated the broad physical heterogeneity of CTCs [15, 72-75]. For example, CTCs derived from different types of tissue, or even from the same tissue, harbor large variations in CTC size [74, 76]. Most CTCs show large overall size and a high nuclear to cytoplasmic ratio compared to surrounding white blood cells (WBCs); while some CTCs show considerable size similarity with surrounding leukocytes (Fig. 2B) [74, 77]. The nuclear to cytoplasmic ratios between single CTCs and CTC clusters are similar, but cells contained in CTC clusters have less area and length, on average, than single CTCs [72, 77]. The nuclear size of CTCs has been associated recently with disease stages. Chen et al. [78] demonstrated that three distinct CTC subpopulations with large, small, and very small nuclear (vsn) sizes were detected in the blood of patients with prostate cancer (Fig. 2C). Small nuclear CTCs and vsnCTCs were identified in patients with metastatic disease, whereas vsnCTCs counts alone were found to be elevated in patients with visceral metastases. Furthermore, comparative studies showed that the total net charge of cancer cells is significantly larger than that of blood cells [79-84].
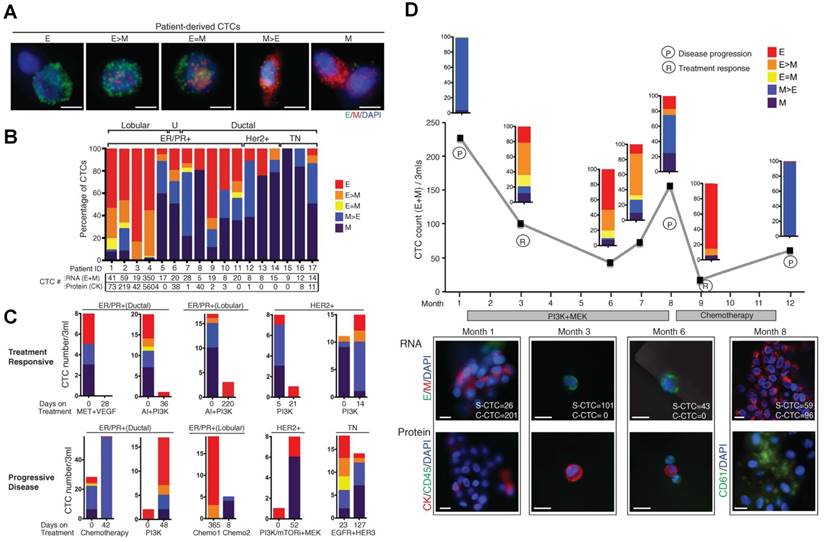
At a more practical level, the composition of CTC subpopulations and the biological features of each subpopulation vary by cancer type, by disease stage, as well as course of therapy. In general, increased numbers of CTCs are detected as the disease progresses in individual patients [15, 85]. CTCs from a given patient may present as intact single cells with round or odd shapes, intact clusters, apoptotic cells, and/or cell fragments [15, 74, 85]. Among the patients exhibiting CTCs, the majority have only single CTCs that are detectable and a minority of patients, particularly those with advanced disease, simultaneously harbor single CTCs and clusters of CTCs that are detectable [40, 45, 75, 77]. Yu, et al. [58] recently demonstrated that circulating breast cancer cells exhibit dynamic phenotypical composition, which varies by histological subtype and over the course of therapy (Fig. 3). In the blood of a particular patient with breast cancer, CTCs can present multiple phenotypes ranging from exclusively epithelial (E) to intermediate (E > M, E = M, E < M), and exclusively mesenchymal (M). Within one subtype, the EMT features in CTCs show individuality as well as generality. For example, the CTCs from a patient with ER+/PR+ breast cancer were predominantly epithelial; whereas those from patients with HER2+ and triple negative (TN; ER-/PR-/HER2-) breast cancer were predominantly mesenchymal. Reversible shifts between the fate of CTCs accompanied each cycle of response to therapy and disease progression. Declining numbers of M+ CTCs and CTC clusters and a switch to predominantly E+ CTCs and single migratory cells were observed in a post-treatment sample that accompanied the response to therapy; whereas an increasing number of M+ CTCs and CTC clusters were observed in the post-treatment sample once the disease relapsed. Miyamoto, et al. [86] also showed that circulating prostate cancer cells exhibit dynamic phenotypical composition over the course of therapy. CTCs in a patient with castration-sensitive prostate cancer showed a transformation from the “androgen receptor (AR)-on” (PSA+/PSMA-) to the “AR-off” (PSA-/PSMA+) phenotype when treated with leuprolide to induce therapeutic androgen deprivation.
Circulating breast cancer cells exhibit dynamic phenotypical composition, which varies by histological subtype (A and B) and over the course of therapy (C and D). Figures were reprinted with permission from ref [58].
Current enrichment methodologies and their limitations
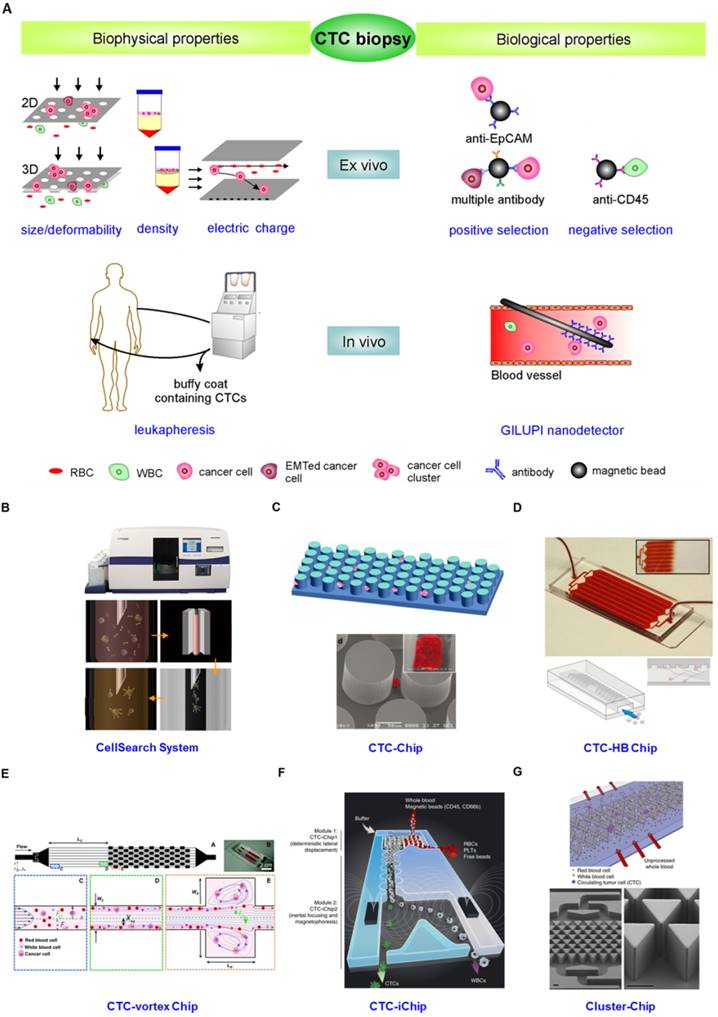
From the description above, the idea that the innate heterogeneity of CTC could complicate their enrichment is not surprising. Various enrichment methodologies based on physical and biological differences between epithelial-derived CTCs and blood cells have been exploited (Fig. 4). The physical property-dependent enrichment methodologies include size/deformability-based filtration, density-gradient centrifugation, and electrical property-based di-electrophoresis (DEP) separation (Fig.4A). A biological property-dependent enrichment methodology can be either a positive selection procedure based on targeting surface markers exclusively expressed on CTCs or a negative selection method based on depletion of blood cells (Fig. 4A). The advantages and limitations of these methodologies are summarized in Table 1.
(A) CTC enrichment methodologies can be summarized into physical and biological property-dependent methodologies. Physical property-dependent methodologies include size-, density-, and electrical charge-based strategies. Among the biological property-dependent methodologies, the EpCAM antibody is commonly used for positive selection. Multiple antibodies for positive selection as well as CD-45-mediated negative depletion have been introduced to eliminate loss of CTC subpopulations. Leukapheresis and GLUPI nanodetection enable the enrichment of CTCs in vivo. Devices developed to improve the enrichment efficiency include (B) the CellSearch system; (C) the CTC-Chip, the first microfluidic chip integrated into CTC enrichment; (D) the CTC-HB chip, in which a herringbone design enables passive mixing, increasing interactions between CTCs and an antibody-coated channel surface; (E) the CTC-vortex Chip, which combines the use of micro-scale vortices and inertial focusing; (F) the CTC-iChip, capable of either positive or negative selection of CTCs; (G) the Cluster-Chip, capable of isolating CTC clusters through specialized bifurcating traps. B-G were reprinted with permission from ref [155], [12], [108], [91], [74], [75], respectively.
Advantages and limitations of current enrichment methodologies.
| Method | Advantage | Limitation | Current improvement | |
|---|---|---|---|---|
| Physical properties | Size/ deformability based filtration | Easy to use High throughput High CTC cluster capture efficacy | Loss of CTCs with size equal to and smaller than pore size of filter Low purity CTC damage due to hemodynamic stress | Introduce CTC size-amplification strategy to reduce the loss of CTC with small size Employ 3D filter to improve cell viability Use microfluidic devices to reduce shear forces and to exclusively enrich clusters |
| Density-gradient centrifugate | Easy to use High throughput | Loss of CTCs with high density Low purity | Depletion of leukocytes by bi-specific antibodies to improve purity Integrate filter into centrifuge tube to reduce the loss of cluster and CTC with large size Use microfluidic devices designed with inertial focusing and vortex function | |
| Electric property based dielectro-phoresis | High CTC viability | Loss of CTCs with electrical property similar to leukocytes Low purity | ||
| Biological properties | Positive selection | High purity | Loss of CTC subpopulations including EMTed CTCs, clusters, and CTCs cloaked by blood cells Tumor-specific assays may be required since CTC phenotype varies by cancer type and over disease progression and treatment course | Search new markers, e.g. actin bundling protein plastin 3 Utilize combination of antibodies Use microfluidic devices integrated with micromixer to increase capture efficacy |
| Negative selection | Potential to enrich all CTC subpopulations | Low purity | Introduce microfluidic devices to improve purity |
Physical property-dependent enrichment methodologies
Epithelial-derived CTCs were initially believed to be larger and stiffer compared to leukocytes (7-12 μm in diameter). Thus various filtration-based devices have been developed to isolate CTCs over past decades [87-91]. In this method, blood is filtered through pores that are usually 8 μm in diameter so as to trap molecules larger than the maximum pore sizes. The success of filtration is determined by factors that include blood flow rate, pore size uniformity, and membrane rigidity. High flow rates can cause CTCs, especially EMT-associated CTCs with high deformability, to 'squeeze' through pores and cause the membrane to warp, whereas slow flow rates lead to excess accumulation of leukocytes, clotting of blood and prolonged processing time [92]. Obviously, filtration would lose CTCs the same size or smaller than the pore diameter while capturing leukocytes and other molecules that are larger. Nonetheless, because of its advantage of ease-of-use, high-throughput, and good recovery efficacy of CTC clusters, filtration technologies are continuing to be improved. In particular, improvements include using three dimensional microfilters to minimize hemodynamic stress on cells, thus sustaining cell viability [89] and adopting a CTC size-amplification strategy to reduce the loss of small-sized CTCs [93].
Density-gradient centrifugation enriches CTCs in the mononucleocyte fraction in a buoyant density taking advantage of their similarity [94]. Although it is easy-to-use and is high-throughput, density-gradient centrifugation inevitably loses CTCs, showing a maximum recovery rate of ~70%. To increase the purity of enriched CTCs, bi-specific antibodies against antigens on leukocytes and erythrocytes have been used to form large multicellular rosettes of leukocytes and erythrocytes, which are easily removed from the mononucleocyte fraction by centrifugation [95]. To reduce the loss of CTCs with high density, a combined density- and size-based separation (e.g., OncoQuick technology), has been developed by inserting a porous barrier into a centrifuge tube [94].
By using dissimilarities in morphological and electrical properties of different cell types, di-electrophoresis (DEP) cell separation technology has been used for enriching CTCs with high viability [79-82]. DEP field flow fractionation combines electric force fields with hydrodynamic and sedimentation forces to pull tumor cells away from blood cells. The success of the process is determined by factors that include electric field frequency, electric voltage, blood flow rates and buffer formulations. Although the viability of cells enriched by DEP is greater than 97%, the average cell recovery of DEP is still only about 70% [82].
Biological property-dependent enrichment methodologies
Beyond physical difference-dependent enrichment technologies, the most popular CTC enrichment technologies involve antibody-mediated isolation. Antibodies targeting epithelial cell surface markers (e.g., epithelial cell adhesion molecule EpCAM), which are present on carcinoma cells, but absent on the mesenchymal leukocyte surfaces, are frequently used in the positive selection of carcinoma cells from the blood [96]. However, positive selection exclusively involving the EpCAM antibody suffers from loss of EMT-associated CTCs, “poorly differentiated, and stem-cell-like” cells and clusters, which might represent the most aggressive CTC subpopulations [16-18, 97]. To resolve this problem, many research groups are currently focusing on a search for new CTC surface markers. For example, the actin bundling protein, plastin 3, has been identified as a novel marker that is not down-regulated by CTCs during their EMT and is not expressed in blood cells [98]. Another improvement for successful capture of heterogeneous CTC subpopulations is the use of a combination of antibodies [58, 99]. A cocktail of antibodies against mesenchymal or stem cell antigens (e.g., c-MET, N-cadherin, CD318, and mesenchymal stem cell antigen), against tissue-specific markers (e.g., PSA for prostate cancer [86], and mammaglobin for breast cancer [100]), and against various tumor-specific markers (e.g., HER2, MUC1, EGFR, folate-binding protein receptor, TROP-2, CA9, and CD147) has been created to enrich CTCs [101-103]. However, the use of a cocktail of antibodies might increase the risk of contamination by blood cells that also express at least one of these markers, and might be useless when CTCs are hidden by platelets and macrophages or when surface markers of CTCs change as the disease progresses or over the course of therapy. Importantly, both the quality of antibodies and the position of the epitope that an antibody recognizes also can influence the efficacy of positive selection.
Obviously, positive selection requires an assumption about the unknown nature of CTCs in a given blood sample. This bias could be avoided by negative selection in which CTCs are enriched by depletion of leukocytes from the blood using antibodies against antigens exclusively expressed on leukocytes (e.g., anti-CD45) [74, 104]. Notably, to improve purity, cascade-negative selection might be required to deplete normal cells.
Improvement of enrichment efficiency by the development of new devices
Besides the development of new assays, great progress has been made in developing new devices to further improve enrichment efficiency. Whereas the CellSearch system represents a breakthrough in CTC enrichment technology both in principle and in clinical application (Fig. 4B) [105], the low yield of CTC capture is a major concern for its clinical value. The low enrichment efficiency is caused by several factors, including the exclusive use of the EpCAM antibody, which induces the loss of EMT-associated CTCs and CTC clusters, the ineffective collection of magnetic bead-labeled CTCs through a sticky blood sample, and the multistep batch purification process. To increase the collection efficiency of magnetic bead-labeled CTCs from sticky blood, Tasalaz, et al. [106] developed a device that sweeps magnetic rods through the whole area of the capture wells. Integrating microfluidics into CTC enrichment technology created another milestone to improve the enriched yield and purity of CTCs. Microfluidics is a powerful technological system that is capable of performing separation and detection of small quantities of samples with high sensitivity in a short time frame [107]. Nagrath, et al. [12] pioneered a silicon-based micropost CTC-Chip for enriching CTCs from blood (Fig. 4C). Thousands of microposts fitted with the EpCAM antibody increased encounters between flowing cells and antibodies, thereby improving the enrichment efficiency of CTCs. However, the device is likely to disrupt CTC clusters due to the tight distances between microposts that presumably prevent the passage of large cellular clusters. Following the CTC-Chip, plenty of microfluidic devices integrated with a nanomixer and/or nanomaterials have been developed for improving enrichment efficiency as well as enabling on-chip characterization [75, 91, 108-111]. For example, the Toner group [108] released a transparent herringbone CTC-Chip that made use of passive mixing to increase the interactions between CTCs and the antibody-coated channel surface (Fig. 4D). The use of transparent materials allowed on-chip characterization of the enriched CTCs. Microfluidics capable of high-throughput processing under low shear conditions have also been integrated into physical difference-dependent enrichment technologies. For instance, Sollier, et al. [91] developed a microfluidic chip combining micro-scale vortices and inertial focusing for the extraction of CTCs from blood with a purity of 57-94% (Fig. 4E). Hou, et al. [111] constructed a spiral microchannel with inherent centrifugal forces for continuous, size-based separation of CTCs from blood. Microfluidics incorporated with negative selection enable enrichment of CTCs from virtually all types of cancer. The Toner group [74] further developed a CTC-iChip capable of enrichment of CTCs with either positive or negative selection (Fig. 4F). The chip first achieved size-based separation of the nucleated cells including CTCs and WBCs from RBCs, platelets and plasma. The nucleated cells were then arrayed in a near-single line due to inertial focusing as they traveled through specially configured curved channels. Inertial-focused tagged leukocytes were magnetically deflected as they traveled through microfluidic channels equipped with magnets. The untagged CTCs were delivered at an average purity of 1% in solution, which can be subjected to subsequent molecular characterization. Circulating CTC clusters, ranging from large tumor cell clusters, blood clots carrying tumor cells to clumps of tumor cells mixed with reactive stromal cells [47, 112, 113] have been observed to establish metastasis more efficiently than single cells [40]. Thus the Toner group [75] developed a Cluster-Chip to examine the true prevalence and significance of CTC clusters (Fig. 4G). The CTC clusters were isolated through specialized bifurcating traps under low-shear conditions that preserve cluster integrity and CTC clusters in 30-40% of patients with metastatic breast, prostate, or melanoma were identified.
These CTC enrichment technologies described above are conducted to enrich CTCs from blood specimens drawn from patients with cancer. The volume of blood needed is usually less than 10 mL. However, CTC heterogeneity necessitates analysis of all CTC subpopulations in sufficient numbers. CTCs enriched from blood samples with limited volume cannot represent the whole CTC population in patients, which decreases the reliability of CTC biopsies [14, 114]. Two approaches that are capable of in vivo enrichment of CTCs with statistically meaningful numbers have been developed (Fig. 4A). One approach involves the collection of CTCs by putting a GLUPI nanodetector in a peripheral arm vein. Up to 1.5 L of blood in 30 min is supposed to pass the nanodetector coated with the EpCAM antibody [115]. Clinical studies of patients with breast and lung cancer demonstrated that the nanodetector can enrich a larger number of CTCs compared to the CellSearch assay [115]. The other approach is to use leukapheresis, which is a standard clinical method frequently conducted to isolate mononuclear cells from several liters of blood for various applications. Leukapheresis can recover a large number of CTCs from non-metastatic cancer patients (i.e., a median of 7,500 CTCs per patient) [13]. Although leukapheresis shows a promising potential to enrich CTCs with statistically meaningful numbers, it is much more invasive than drawing a blood sample, which is a critical issue for clinical use. In addition to enrichment, ex vivo cultures of primary CTCs allow subpopulations of CTCs to be more fully characterized [14, 116], and enable drug sensitivity to be monitored [117].
Selection of appropriate enrichment and characterization methods for specific applications
Although many CTC enrichment technologies based on new markers and devices have been developed, each methodology has its own advantages and limitations. The physical difference- dependent enrichment technologies share the same limitation originating from physical similarities between CTCs and leukocytes, whereas antibody-mediated positive selection might suffer from the heterogeneous CTC phenotype, which varies by cancer type or subtype, disease progression, and over the course of therapy. Therefore, none of the current enrichment technologies satisfy all the CTC applications. Choosing an enrichment strategy based upon the aim of further analysis would be optimal. According to CTC biology as well as current enrichment technologies, we now examine the selection of appropriate enrichment and characterization methods for specific applications (Table 2).
Selection of appropriate enrichment and characterization methodologies for specific CTC applications. The challenges in each application as well as their appropriate enrichment and characterization strategies are listed.
| Concept | Focus | Challenges | Enrichment methodology | Characterization | |
|---|---|---|---|---|---|
| Early detection | Screen patients to detect cancer early | Positive or negative | Sensitivity and Reliability | Negative selection integrated with microfluidics | Identification by immunostaining, molecular characterization including RNA, DNA and protein analysis |
| As prognostic maker | Assess how a disease will behave | Enumeration of all CTC subpopulations in blood sample with a given volume | Enrich all cell subpopulations | Negative selection integrated with microfluidics | Identification and enumeration by immunostaining |
| As predictive marker | Predict whether a drug or other therapies will be effective, or monitor the effectiveness of treatment | Selection of therapeutic target; Sensitive or resistant response to a specific therapy; CDX model for drug screening | Enrich all CTC subpopulations in living state with statistical significance number; CTCs may exhibit dynamic phenotypical profiles over disease progression and therapy course | Negative selection integrated with microfluidics; Methods combing density- and size- based separation; Leukapheresis | Genomic characterizations in assemble or single-cell level by RT-PCR, in situ hybridization (ISH), comparative genomic hybridization (CGH), mutational assays, and next-generation sequencing (NGS); Susceptibilities to various drug regimens |
Early detection of cancer and characterization of tumor origin and metastatic sites
Studies with animal models and clinical subjects demonstrated that CTCs exist in the blood and migrate to distant organs long before a tumor is diagnosed [23, 26], indicating the potential of CTCs as a diagnostic parameter of early disease [11-13, 26]. In addition, abundant evidence shows that cancer cells in the primary tumor are able to metastasize to different organs and CTCs reaching new specific organs may adapt to the acquisition of an “organ-mimetic” phenotype [118-120]. Thus CTCs circulating from the primary tumor or metastases would present specific signatures capable of identifying their origin as well as their disseminating destination [120, 121]. The reliability of CTCs in early cancer detection and in characterization of tumor origin and metastatic sites depends on the sensitivity of method adopted and the blood volume sampled. Due to the ultralow concentration of CTCs in blood from patients with early disease, enrichment of CTCs from a large blood volume is required to achieve reliable detection. Thus in vivo enrichment methods, including the GLUPI nanodetector coated with a combination of antibodies and leukapheresis, would be ideal choices. However, the invasiveness of in vivo platforms would be a barrier for the screening of patients for early detection of cancer. CTCs in the enriched product can be identified by either immunostaining or RT-PCR [122, 123]. For identification by immunostaining, a CTC is defined as a cell that is positive for EpCAM and CK8, 18 and 19, negative for CD45, with an intracellular nucleus and at least 4 μm × 4 μm in size [15]. Notably, down-regulation of EpCAM and CK of CTCs due to EMT might result in false negative results during molecular identification. Furthermore, CK+ and CD45- cells can also occur in patients with benign diseases of the colon [108, 124]. By using RT-PCR, before labeling a sample as positive for cancer, a validated cutoff value is required to overcome the problem of false-positives. Identification of tumor origins can be achieved by characterization of CTCs with different markers. For example, CK7 and PSA/PSMA have been used to identify CTCs for lung cancer and prostate cancer, respectively [125]. A recent study showed that CTCs in patients with breast cancer brain metastases have a specific protein expression signature of HER2+-EGFR+-HPSE+-NOTCH1+-EPCAM- [116].
Enumeration of CTCs as prognostic indicators
One well-validated clinical application of CTCs is the assessment of their prognostic value at pretreatment baseline in patients with known metastatic cancers including breast [126], prostate [127, 128], melanoma [129], and colon [130-132]. The main challenge of using CTCs as a prognostic marker is enumeration of all CTC subpopulations in a blood sample with a given volume. The main methodology adopted in current clinical trials is the CellSearch assay [95]. However, as indicated above, exclusive EpCAM antibody-mediated positive selection usually results in false negative results due to loss of EMT-associated CTCs and CTC clusters. To enrich all CTC subpopulations in a given blood sample, negative selection integrated with microfluidics (e.g., CTC-iChip) would be ideal. Furthermore, direct detection of CTCs in blood samples by microscopy with automated scanning technology is an ideal alternative in which the CTCs are mounted directly on a slide that is labeled with flurophore-conjugated antibodies [133-136].
CTCs as predictive markers
Treatment of cancer, especially metastatic disease, is usually complicated by the systemic nature of the disease, the heterogeneity of metastases, the multitude of genes and pathways involved in different organs, as well as drug resistance [58]. Studies on CTC characteristics showed that the genomic profiles of CTCs are largely comparable to primary tumors and metastases, suggesting that CTCs can provide a snapshot of the molecular landscape of a patient's overall tumor characteristics at the time of blood draw [137, 138]. This can potentially provide a predictive marker for guiding tailored choices of precise treatment for specific patients at distinct times [139-141]. For example, Heitzer, et al. found that in CTCs from colorectal cancer patients, mutations in known-driver genes, such as apc, pic3ca, and kras, could be detected in the matched primary tumor, metastases and CTCs, suggesting the promising potential of CTCs as a predictive marker in colorectal cancer [142]. Furthermore, detection of CTCs can reveal tumor cells that present at a subclonal level in the primary tumor and metastases, which might be ignored in a tissue biopsy. For example, ALK-rearranged CTCs were detected in ALK-negative patients with NSCLC [143], which will be useful in guiding therapy because ALK-rearrangement-positive NSCLC patients do not benefit from EGFR tyrosine-kinase-inhibitor (TKI) therapy and need to be treated with an ALK inhibitor [144]. In addition, establishing CTC-derived xenograft (CDX) models to test their susceptibilities toward various clinically relevant drug regimens could provide promising potential in personalized cancer therapy because CTCs represent real-time disease information [14, 117, 145, 146]. Enrichment of CTCs, especially those with high metastatic and therapy-resistance potential, such as CTCs with high plastic EMT-associated phenotype and various CTC clusters [14, 28, 47, 112, 113], in a living state with a statistically significant number is of utmost importance to establish the CDX model.
The challenge of these applications is characterizing all CTC subpopulations in numbers sufficient for statistical significance. As indicated earlier in the CTC biology section, CTCs might be cloaked by platelets, macrophages and/or reactive stromal cells and could exhibit dynamic molecular and phenotypical composition as disease progresses and over the course of therapy. Thus to enrich the numbers of all CTC subpopulations to reach statistical significance, negative selection integrated with microfluidics (e.g., CTC-iChip) and a method combining density and size-based separation (e.g., OncoQuick technology) would be ideal methods for blood samples with a high concentration of CTCs (e.g., blood samples from patients with SCLC in advanced stages) [40], while leukapheresis might be appropriate for patients harboring ultralow concentrations of CTCs in their blood. Besides enrichment methodologies, selecting the appropriate characterizations is also crucial to gain meaningful results from analysis. Genomic characterizations by various genotyping techniques are usually involved in the identification of genomic profiles of CTCs, including point mutations, rearrangements, small insertions/deletions and gene amplification. The genotyping techniques include RT-PCR, in situ hybridization (ISH), comparative genomic hybridization (CGH), mutational assays, next-generation sequencing (NGS), and single-cell analysis [147].
Notably, when using partially purified CTC populations, digital subtraction of background leukocyte levels is essential for deriving CTC-based expression profiles [9, 148]. Single-cell analysis of CTCs could provide important advantages in eliminating the effect of background leukocytes. Among the genotyping techniques developed, single-cell analysis of CTCs not only provides an important advantage in eliminating the effect of background leukocytes, but also offers great promise for more comprehensive genomic and transcriptomic coverage, shedding light on the intra- and inter-patient genetic heterogeneity as well as changes after treatment. Initial single-cell analysis of CTCs mainly focused on DNA-single cell sequencing (SCS) to profile copy number and assess DNA mutations in CTCs for comparison to matched primary and metastatic tumors [138, 142, 149, 150], and also centered on RNA-SCS to study the transcriptional programs of CTCs [40, 151, 152]. Genomic analyses showed that although the copy number profile of CTCs was highly similar to that of primary and metastatic tumors, point mutations displayed extensive variability. For example, Ni et al. [150] performed exome sequencing of single CTCs from eleven lung adenocarcinoma cancer patients. Only 59% of the point mutations found in the primary and metastatic tumors were detected in CTCs. Recently, DNA-SCS and RNA-SCS of CTCs have been used to investigate genome evolution and transcriptomes in response to therapy [153, 154]. Particularly, the interpretation of genetic information might not be sufficient to elucidate the activated pathways that are ultimately the target of therapeutic intervention, and thus epigenetic-, proteomic- and metabolomics- analyses of CTCs are desirable, again ideally down to the single cell level. To establish the CDX model, the enriched product can either be directly injected into mice or injected into mice after isolation of specific CTC subpopulations by fluorescence activated cell sorting (FACS).
Future frontiers for CTC detection
Progress towards sensitive and rapid CTC detection has been impressive; nevertheless, significant opportunities to address more fully the goals of this field still exist. As CTC biology indicates, CTCs might be cloaked by platelets, macrophages and/or reactive stromal cells and could exhibit dynamic phenotypical composition as the disease progresses and over the course of therapy; however, physical properties such as size remain relatively stable during disease progression. Therefore, compared to positive selection, physical property-dependent enrichment methodologies and negative selection might be more reliable and adaptable for in vitro enrichment of CTCs from the blood of patients with most types of cancer. However, further advances in these methods are needed to obtain a population of highly pure CTCs, which would facilitate the accuracy of subsequent molecular characterization. In addition, automatic platforms integrating CTC enrichment with subsequent molecular characterization are highly desirable to enable the generation of accurate results even in the absence of a highly trained operator. Well-designed microfluidic devices that integrate enrichment strategies involving cascade physical property-dependent and antibody-based selections with subsequent molecular characterization might provide a fully automated platform. Furthermore, to improve the capability of CTCs to serve as a predictive marker, technologies for high-throughput single cell analysis will be in high demand because of CTC heterogeneity.
Conclusions
CTCs derived from tumors released into the blood are low in abundance and show high heterogeneity in their morphological and phenotypic profiles. Recent studies further demonstrate that CTCs exhibit dynamic biological profiles, which vary by cancer type, as disease progresses and over the course of treatment, which further complicates their enrichment and characterization. This review presented the most recent advances in CTC biology, summarized current CTC enrichment methodologies and examined the selection of appropriate enrichment and characterization methods for specific clinical applications. The hope is that the present framework will stimulate further improvements in the detection and characterization of CTCs and assist in translating the CTC biopsy from the laboratory to clinical practice.
Acknowledgements
This work was supported by The Hormel Foundation and National Institutes of Health grants CA166011, CA187027 and CA196639.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chaffer CL, Weinberg RA. A Perspective on Cancer Cell Metastasis. Science. 2011;331:1559-64
2. Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127:679-95
3. Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW. et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999-1005
4. Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJ, Witt H. et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature. 2012;482:529-33
5. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B. et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114-7
6. Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A. et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809-13
7. Kuukasjärvi T, Karhu R, Tanner M, Kähkönen M, Schäffer A, Nupponen N. et al. Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 1997;57:1597-604
8. Alix-Panabieres C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer. 2014;14:623-31
9. Haber DA, Velculescu VE. Blood-Based Analyses of Cancer: Circulating Tumor Cells and Circulating Tumor DNA. Cancer Disc. 2014;4:650-61
10. Plaks V, Koopman CD, Werb Z. Circulating Tumor Cells. Science. 2013;341:1186-1188
11. Stott SL, Lee RJ, Nagrath S, Yu M, Miyamoto DT, Ulkus L. et al. Isolation and Characterization of Circulating Tumor Cells from Patients with Localized and Metastatic Prostate Cancer. Sci Transl Med. 2010;2:25ra3
12. Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L. et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450:1235-9
13. Fischer JC, Niederacher D, Topp SA, Honisch E, Schumacher S, Schmitz N. et al. Diagnostic leukapheresis enables reliable detection of circulating tumor cells of nonmetastatic cancer patients. Proc Natl Acad Sci USA. 2013;110:16580-5
14. Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V. et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotech. 2013;31:539-44
15. Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C. et al. Tumor Cells Circulate in the Peripheral Blood of All Major Carcinomas but not in Healthy Subjects or Patients With Nonmalignant Diseases. Clin Cancer Res. 2004;10:6897-904
16. Farace F, Massard C, Vimond N, Drusch F, Jacques N, Billiot F. et al. A direct comparison of CellSearch and ISET for circulating tumour-cell detection in patients with metastatic carcinomas. Br J Cancer. 2011;105:847-53
17. Krebs MG, Hou J-M, Sloane R, Lancashire L, Priest L, Nonaka D. et al. Analysis of Circulating Tumor Cells in Patients with Non-small Cell Lung Cancer Using Epithelial Marker-Dependent and -Independent Approaches. J Thorac Oncol. 2012;7:306-15
18. Hofman V, Ilie MI, Long E, Selva E, Bonnetaud C, Molina T. et al. Detection of circulating tumor cells as a prognostic factor in patients undergoing radical surgery for non-small-cell lung carcinoma: comparison of the efficacy of the CellSearch Assay™ and the isolation by size of epithelial tumor cell method. Intl J Cancer. 2011;129:1651-60
19. Wan L, Pantel K, Kang Y. Tumor metastasis: moving new biological insights into the clinic. Nat Med. 2013;19:1450-64
20. Nieto MA. Epithelial Plasticity: A Common Theme in Embryonic and Cancer Cells. Science. 2013:342
21. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell. 2009;139:871-90
22. Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY. et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell. 2008;133:704-15
23. Rhim Andrew D, Mirek Emily T, Aiello Nicole M, Maitra A, Bailey Jennifer M, McAllister F. et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell. 2012;148:349-61
24. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546-58
25. López-Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med. 2009;1:303-14
26. Kang Y, Pantel K. Tumor Cell Dissemination: Emerging Biological Insights from Animal Models and Cancer Patients. Cancer Cell. 2013;23:573-81
27. Pencheva N, Tavazoie SF. Control of metastatic progression by microRNA regulatory networks. Nat Cell Biol. 2013;15:546-54
28. Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19:1438-49
29. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97-110
30. Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN. et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA. 2012;109:2784-9
31. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52-67
32. Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17:1359-70
33. Sonoshita M, Aoki M, Fuwa H, Aoki K, Hosogi H, Sakai Y. et al. Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer Cell. 2011;19:125-37
34. Reymond N, Im JH, Garg R, Vega FM, d'Agua BB, Riou P. et al. Cdc42 promotes transendothelial migration of cancer cells through β1 integrin. J Cell Biol. 2012;199:653-68
35. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275-92
36. Padua D, Zhang XH-F, Wang Q, Nadal C, Gerald WL, Gomis RR. et al. TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133:66-77
37. Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C. et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007;446:765-70
38. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA. et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110-22
39. Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, Papaemmanuil E. et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353-7
40. Aceto N, Bardia A, Miyamoto David T, Donaldson Maria C, Wittner Ben S, Spencer Joel A. et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell. 2014;158:1110-22
41. Friedl P, Locker J, Sahai E, Segall JE. Classifying collective cancer cell invasion. Nat Cell Biol. 2012;14:777-83
42. Cheung KJ, Ewald AJ. A collective route to metastasis: Seeding by tumor cell clusters. Science. 2016;352:167-9
43. Tabassum DP, Polyak K. Tumorigenesis: it takes a village. Nat Rev Cancer. 2015;15:473-83
44. Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445-57
45. Hou J-M, Krebs MG, Lancashire L, Sloane R, Backen A, Swain RK. et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J Clinic Oncol. 2012;30:525-32
46. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9:285-93
47. Duda DG, Duyverman AMMJ, Kohno M, Snuderl M, Steller EJA, Fukumura D. et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci USA. 2010;107:21677-82
48. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472-6
49. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525-30
50. Godinho SA, Picone R, Burute M, Dagher R, Su Y, Leung CT. et al. Oncogene-like induction of cellular invasion from centrosome amplification. Nature. 2014;510:167
51. Camara O, Kavallaris A, Nöschel H, Rengsberger M, Jörke C, Pachmann K. Seeding of epithelial cells into circulation during surgery for breast cancer: the fate of malignant and benign mobilized cells. World J Surg Oncol. 2006;4:1
52. Förnvik D, Andersson I, Dustler M, Ehrnström R, Rydén L, Tingberg A. et al. No evidence for shedding of circulating tumor cells to the peripheral venous blood as a result of mammographic breast compression. Breast Cancer Res Treat. 2013;141:187-95
53. Sawabata N, Funaki S, Hyakutake T, Shintani Y, Fujiwara A, Okumura M. Perioperative circulating tumor cells in surgical patients with non-small cell lung cancer: does surgical manipulation dislodge cancer cells thus allowing them to pass into the peripheral blood?. Surg Today. 2016:1-8
54. Mitchell MJ, King MR. Computational and experimental models of cancer cell response to fluid shear stress. Front Oncol. 2013;3:3389
55. Chambers AF, Groom AC, MacDonald IC. Metastasis: Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563-72
56. Rossi E, Basso U, Celadin R, Zilio F, Pucciarelli S, Aieta M. et al. M30 Neoepitope Expression in Epithelial Cancer: Quantification of Apoptosis in Circulating Tumor Cells by CellSearch Analysis. Clin Cancer Res. 2010;16:5233-43
57. Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A. et al. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature. 2016;531:513-7
58. Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT. et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science. 2013;339:580-4
59. Labelle M, Begum S, Hynes Richard O. Direct Signaling between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-Like Transition and Promotes Metastasis. Cancer Cell. 2011;20:576-90
60. Wels J, Kaplan RN, Rafii S, Lyden D. Migratory neighbors and distant invaders: tumor-associated niche cells. Genes Dev. 2008;22:559-74
61. Douma S, van Laar T, Zevenhoven J, Meuwissen R, van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430:1034-9
62. Chao MP, Weissman IL, Majeti R. The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24:225-32
63. Weiskopf K, Ring AM, Ho CCM, Volkmer J-P, Levin AM, Volkmer AK. et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341:88-91
64. Steinert G, Schölch S, Niemietz T, Iwata N, García SA, Behrens B. et al. Immune escape and survival mechanisms in circulating tumor cells of colorectal cancer. Cancer Res. 2014;74:1694-704
65. Mazel M, Jacot W, Pantel K, Bartkowiak K, Topart D, Cayrefourcq L. et al. Frequent expression of PD-L1 on circulating breast cancer cells. Molecular Oncol. 2015;9:1773-82
66. Chen Q, Zhang XH-F, Massagué J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell. 2011;20:538-49
67. Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z. et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015
68. Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood. 2004;104:397-401
69. Nieswandt B, Hafner M, Echtenacher B, Männel DN. Lysis of Tumor Cells by Natural Killer Cells in Mice Is Impeded by Platelets. Cancer Res. 1999;59:1295-300
70. Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C. et al. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med. 2015;7:308re8-re8
71. Gil-Bernabé AM, Ferjančič Š, Tlalka M, Zhao L, Allen PD, Im JH. et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood. 2012;119:3164-75
72. Daniel CL, Edward HC, Madelyn SL, Thomas JM, Maria Loressa U, Melissa T. et al. Cytometric comparisons between circulating tumor cells from prostate cancer patients and the prostate-tumor-derived LNCaP cell line. Phys Biol. 2012;9:016002
73. Phillips KG, Kuhn P, McCarty OJ. Physical Biology in Cancer. 2. The physical biology of circulating tumor cells. Am J Physiol-Cell Ph. 2014;306:C80-C8
74. Ozkumur E, Shah AM, Ciciliano JC, Emmink BL, Miyamoto DT, Brachtel E. et al. Inertial Focusing for Tumor Antigen-Dependent and -Independent Sorting of Rare Circulating Tumor Cells. Sci Transl Med. 2013;5:179ra47
75. Sarioglu AF, Aceto N, Kojic N, Donaldson MC, Zeinali M, Hamza B. et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat Meth. 2015;12:685-691
76. Harouaka RA, Nisic M, Zheng S-Y. Circulating tumor cell enrichment based on physical properties. J Lab Auto. 2013;18:455-68
77. Edward HC, Marco W, Madelyn L, Craig Y, Dena M, Daniel L. et al. Characterization of circulating tumor cell aggregates identified in patients with epithelial tumors. Phys Biol. 2012;9:016001
78. Chen J-F, Ho H, Lichterman J, Lu Y-T, Zhang Y, Garcia MA. et al. Subclassification of prostate cancer circulating tumor cells by nuclear size reveals very small nuclear circulating tumor cells in patients with visceral metastases. Cancer. 2015;121:3240-51
79. Shim S, Stemke-Hale K, Tsimberidou AM, Noshari J, Anderson TE, Gascoyne PR. Antibody-independent isolation of circulating tumor cells by continuous-flow dielectrophoresis. Biomicrofluidics. 2013;7:011807
80. Gascoyne PR, Noshari J, Anderson TJ, Becker FF. Isolation of rare cells from cell mixtures by dielectrophoresis. Electrophoresis. 2009;30:1388-98
81. Wang X-B, Yang J, Huang Y, Vykoukal J, Becker FF, Gascoyne PRC. Cell Separation by Dielectrophoretic Field-flow-fractionation. Anal Chem. 2000;72:832-9
82. Gupta V, Jafferji I, Garza M, Melnikova VO, Hasegawa DK, Pethig R. et al. ApoStream™, a new dielectrophoretic device for antibody independent isolation and recovery of viable cancer cells from blood. Biomicrofluidics. 2012;6:024133
83. Voldman J. ELECTRICAL FORCES FOR MICROSCALE CELL MANIPULATION. Annu Rev Biomed Eng. 2006;8:425-54
84. Gawad S, Schild L, Renaud P. Micromachined impedance spectroscopy flow cytometer for cell analysis and particle sizing. Lab Chip. 2001;1:76-82
85. Jorge N, Marco W, Madelyn SL, Dena M, Lyudmila B, Anand K. et al. High-definition imaging of circulating tumor cells and associated cellular events in non-small cell lung cancer patients: a longitudinal analysis. Phys Biol. 2012;9:016004
86. Miyamoto DT, Lee RJ, Stott SL, Ting DT, Wittner BS, Ulman M. et al. Androgen Receptor Signaling in Circulating Tumor Cells as a Marker of Hormonally Responsive Prostate Cancer. Cancer Disc. 2012;2:995-1003
87. Lin HK, Zheng S, Williams AJ, Balic M, Groshen S, Scher HI. et al. Portable Filter-Based Microdevice for Detection and Characterization of Circulating Tumor Cells. Clin Cancer Res. 2010;16:5011-8
88. Vona G, Sabile A, Louha M, Sitruk V, Romana S, Schütze K. et al. Isolation by size of epithelial tumor cells: a new method for the immunomorphological and molecular characterization of circulating tumor cells. Amer J Pathol. 2000;156:57-63
89. Zheng S, Lin HK, Lu B, Williams A, Datar R, Cote RJ. et al. 3D microfilter device for viable circulating tumor cell (CTC) enrichment from blood. Biomed Microdevices. 2011;13:203-13
90. Desitter I, Guerrouahen BS, Benali-Furet N, Wechsler J, Jaenne PA, Kuang Y. et al. A new device for rapid isolation by size and characterization of rare circulating tumor cells. Anticancer Res. 2011;31:427-41
91. Sollier E, Go DE, Che J, Gossett DR, O'Byrne S, Weaver WM. et al. Size-selective collection of circulating tumor cells using Vortex technology. Lab Chip. 2014;14:63-77
92. Attard G, de Bono JS. Utilizing circulating tumor cells: challenges and pitfalls. Current Opin Gene Develop. 2011;21:50-8
93. Lee HJ, Oh JH, Oh JM, Park J-M, Lee J-G, Kim MS. et al. Efficient Isolation and Accurate In Situ Analysis of Circulating Tumor Cells Using Detachable Beads and a High-Pore-Density Filter. Angew Chem Int Ed. 2013;52:8337-40
94. Gertler R, Rosenberg R, Fuehrer K, Dahm M, Nekarda H, Siewert JR. Detection of circulating tumor cells in blood using an optimized density gradient centrifugation. Molecular Staging Cancer: Springer. 2003:149-55
95. Ramirez J-M, Fehm T, Orsini M, Cayrefourcq L, Maudelonde T, Pantel K. et al. Prognostic Relevance of Viable Circulating Tumor Cells Detected by EPISPOT in Metastatic Breast Cancer Patients. Clin Chem. 2014;60:214-21
96. Went PT, Lugli A, Meier S, Bundi M, Mirlacher M, Sauter G. et al. Frequent EpCam protein expression in human carcinomas. Human Pathol. 2004;35:122-8
97. Bednarz-Knoll N, Alix-Panabières C, Pantel K. Plasticity of disseminating cancer cells in patients with epithelial malignancies. Cancer Metastasis Rev. 2012;31:673-87
98. Yokobori T, Iinuma H, Shimamura T, Imoto S, Sugimachi K, Ishii H. et al. Plastin3 is a novel marker for circulating tumor cells undergoing the epithelial-mesenchymal transition and is associated with colorectal cancer prognosis. Cancer Res. 2013;73:2059-69
99. Alix-Panabières C, Pantel K. Circulating Tumor Cells: Liquid Biopsy of Cancer. Clin Chem. 2013;59:110-8
100. Markou A, Strati A, Malamos N, Georgoulias V, Lianidou ES. Molecular characterization of circulating tumor cells in breast cancer by a liquid bead array hybridization assay. Clin Chem. 2011;57:421-30
101. Pecot CV, Bischoff FZ, Mayer JA, Wong KL, Pham T, Bottsford-Miller J. et al. A Novel Platform for Detection of CK+ and CK- CTCs. Cancer Disc. 2011;1:580-6
102. Ignatiadis M, Rothe F, Chaboteaux C, Durbecq V, Rouas G, Criscitiello C. et al. HER2-positive circulating tumor cells in breast cancer. PloS one. 2011;6:e15624
103. Liu S, Tian Z, Zhang L, Hou S, Hu S, Wu J. et al. Combined cell surface carbonic anhydrase 9 and CD147 antigens enable high-efficiency capture of circulating tumor cells in clear cell renal cell carcinoma patients. Oncotarget. 2016;7:59877-59891
104. Yang L, Lang JC, Balasubramanian P, Jatana KR, Schuller D, Agrawal A. et al. Optimization of an enrichment process for circulating tumor cells from the blood of head and neck cancer patients through depletion of normal cells. Biotech Bioeng. 2009;102:521-34
105. Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C. et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clinic Cancer Res. 2004;10:6897-904
106. Talasaz AH, Powell AA, Huber DE, Berbee JG, Roh K-H, Yu W. et al. Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc Natl Acad Sci USA. 2009;106:3970-5
107. Whitesides GM. The origins and the future of microfluidics. Nature. 2006;442:368-73
108. Stott SL, Hsu C-H, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA. et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci USA. 2010;107:18392-7
109. Yoon HJ, Kozminsky M, Nagrath S. Emerging Role of Nanomaterials in Circulating Tumor Cell Isolation and Analysis. ACS Nano. 2014;8:1995-2017
110. Yoon HJ, Kim TH, Zhang Z, Azizi E, Pham TM, Paoletti C. et al. Sensitive capture of circulating tumour cells by functionalized graphene oxide nanosheets. Nat Nano. 2013;8:735-41
111. Hou HW, Warkiani ME, Khoo BL, Li ZR, Soo RA, Tan DS-W. et al. Isolation and retrieval of circulating tumor cells using centrifugal forces. Sci Rep. 2013;3:1259
112. Molnar B, Ladanyi A, Tanko L Sréter L, Tulassay Z. Circulating tumor cell clusters in the peripheral blood of colorectal cancer patients. Clin Cancer Res. 2001;7:4080-5
113. Brandt B, Junker R, Griwatz C, Heidl S, Brinkmann O, Semjonow A. et al. Isolation of prostate-derived single cells and cell clusters from human peripheral blood. Cancer Res. 1996;56:4556-61
114. Lalmahomed ZS, Kraan J, Gratama JW, Mostert B, Sleijfer S, Verhoef C. Circulating tumor cells and sample size: the more, the better. J Clinic Oncol. 2010;28:e288-e9
115. Saucedo-Zeni N, Mewes S, Niestroj R, Gasiorowski L, Murawa D, Nowaczyk P. et al. A novel method for the in vivo isolation of circulating tumor cells from peripheral blood of cancer patients using a functionalized and structured medical wire. Intel J Oncol. 2012;41:1241-50
116. Zhang L, Ridgway LD, Wetzel MD, Ngo J, Yin W, Kumar D. et al. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci Transl Med. 2013;5:180ra48-ra48
117. Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC. et al. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345:216-20
118. Fidler IJ. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat Rev Cancer. 2003;3:453-8
119. Smid M, Wang Y, Zhang Y, Sieuwerts AM, Yu J, Klijn JG. et al. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008;68:3108-14
120. Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274-84
121. Bos PD, Zhang XHF, Nadal C, Shu W, Gomis RR, Nguyen DX. et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005-9
122. Yu M, Stott S, Toner M, Maheswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J Cell Biol. 2011;192:373-82
123. Pantel K, Alix-Panabières C. Circulating tumour cells in cancer patients: challenges and perspectives. Trends Mole Med. 2010;16:398-406
124. Pantel K, Denève E, Nocca D, Coffy A, Vendrell J-P, Maudelonde T. et al. Circulating epithelial cells in patients with benign colon diseases. Clin Chem. 2012;58:936-40
125. Lu S-H, Tsai W-S, Chang Y-H, Chou T-Y, Pang S-T, Lin P-H. et al. Identifying cancer origin using circulating tumor cells. Cancer Biol Therapy. 2016;17:430-8
126. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC. et al. Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer. New Engl J Med. 2004;351:781-91
127. de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H. et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14:6302-9
128. Danila DC, Heller G, Gignac GA, Gonzalez-Espinoza R, Anand A, Tanaka E. et al. Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin Cancer Res. 2007;13:7053-8
129. Mocellin S, Hoon D, Ambrosi A, Nitti D, Rossi CR. The prognostic value of circulating tumor cells in patients with melanoma: a systematic review and meta-analysis. Clin Cancer Res. 2006;12:4605-13
130. Cohen SJ, Alpaugh RK, Gross S, O'Hara SM, Smirnov DA, Terstappen LW. et al. Isolation and characterization of circulating tumor cells in patients with metastatic colorectal cancer. Clinical Colorect Cancer. 2006;6:125-32
131. Cohen S, Punt C, Iannotti N, Saidman B, Sabbath K, Gabrail N. et al. Prognostic significance of circulating tumor cells in patients with metastatic colorectal cancer. Annal Oncol. 2009;20:1223-9
132. Cohen SJ, Punt CJ, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY. et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clinic Oncol. 2008;26:3213-21
133. Krivacic RT, Ladanyi A, Curry DN, Hsieh HB, Kuhn P, Bergsrud DE. et al. A Rare-Cell Detector for Cancer. P Natl Acad Sci USA. 2004;101:10501-4
134. Werner SL, Graf RP, Landers M, Valenta DT, Schroeder M, Greene SB. et al. Analytical validation and capabilities of the Epic CTC platform: enrichment-free circulating tumour cell detection and characterization. J Circ Biomark. 2015;4:3
135. Ntouroupi TG, Ashraf SQ, McGregor SB, Turney BW, Seppo A, Kim Y. et al. Detection of circulating tumour cells in peripheral blood with an automated scanning fluorescence microscope. Br J Cancer. 2008;99:789-95
136. Deng G, Herrler M, Burgess D, Manna E, Krag D, Burke JF. Enrichment with anti-cytokeratin alone or combined with anti-EpCAM antibodies significantly increases the sensitivity for circulating tumor cell detection in metastatic breast cancer patients. Breast Cancer Res. 2008;10:1-11
137. Onstenk W, de Klaver W, de Wit R, Lolkema M, Foekens J, Sleijfer S. The use of circulating tumor cells in guiding treatment decisions for patients with metastatic castration-resistant prostate cancer. Cancer Treat Rev. 2016;46:42-50
138. Lohr JG, Adalsteinsson VA, Cibulskis K, Choudhury AD, Rosenberg M, Cruz-Gordillo P. et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotech. 2014;32:479-84
139. Bidard F-C, Mathiot C, Degeorges A, Etienne-Grimaldi M-C, Delva R, Pivot X. et al. Clinical value of circulating endothelial cells and circulating tumor cells in metastatic breast cancer patients treated first line with bevacizumab and chemotherapy. Annal Oncol. 2010;21:1765-71
140. Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV. et al. Detection of Mutations in EGFR in Circulating Lung-Cancer Cells. New Engl J Med. 2008;359:366-77
141. Riethdorf S, Müller V, Zhang L, Rau T, Loibl S, Komor M. et al. Detection and HER2 expression of circulating tumor cells: prospective monitoring in breast cancer patients treated in the neoadjuvant GeparQuattro trial. Clin Cancer Res. 2010;16:2634-45
142. Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM. et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013;73:2965-75
143. Pailler E, Adam J, Barthélémy A, Oulhen M, Auger N, Valent A. et al. Detection of circulating tumor cells harboring a unique ALK rearrangement in ALK-positive non-small-cell lung cancer. J Clinic Oncol. 2013;31:2273-81
144. Kwak EL, Bang Y-J, Camidge DR, Shaw AT, Solomon B, Maki RG. et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. New Engl J Med. 2010;363:1693-703
145. Morrow CJ, Trapani F, Metcalf RL, Bertolini G, Hodgkinson CL, Khandelwal G. et al. Tumourigenic Non-Small Cell Lung Cancer Mesenchymal Circulating Tumour Cells-A Clinical Case Study. Annal Oncol. 2016 mdw122
146. Hodgkinson CL, Morrow CJ, Li Y, Metcalf RL, Rothwell DG, Trapani F. et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med. 2014;20:897-903
147. Cote RJ, Datar RH. Circulating Tumor Cells. Springer. 2016
148. Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S. et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012;487:510-3
149. Jiang R, Lu Y-T, Ho H, Li B, Chen J-F, Lin M. et al. A comparison of isolated circulating tumor cells and tissue biopsies using whole-genome sequencing in prostate cancer. Oncotarget. 2015;6:44781-93
150. Ni X, Zhuo M, Su Z, Duan J, Gao Y, Wang Z. et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. P Natl Acad Sci USA. 2013;110:21083-8
151. Ramskold D, Luo S, Wang Y-C, Li R, Deng Q, Faridani OR. et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotech. 2012;30:777-82
152. Ting David T, Wittner Ben S, Ligorio M, Vincent Jordan N, Shah Ajay M, Miyamoto David T. et al. Single-Cell RNA Sequencing Identifies Extracellular Matrix Gene Expression by Pancreatic Circulating Tumor Cells. Cell Rep. 2014;8:1905-18
153. Suzuki A, Matsushima K, Makinoshima H, Sugano S, Kohno T, Tsuchihara K. et al. Single-cell analysis of lung adenocarcinoma cell lines reveals diverse expression patterns of individual cells invoked by a molecular target drug treatment. Genome Biol. 2015;16:66
154. Dago AE, Stepansky A, Carlsson A, Luttgen M, Kendall J, Baslan T. et al. Rapid phenotypic and genomic change in response to therapeutic pressure in prostate cancer inferred by high content analysis of single circulating tumor cells. PloS one. 2014;9:e101777
155. Park Y, Kitahara T, Urita T, Yoshida Y, Kato R. Expected clinical applications of circulating tumor cells in breast cancer. World J Clin Oncol. 2011;2:303-10
Author contact
![]() Corresponding author: Email: zgdongumn.edu Telephone: 507-437-9615 Fax: 507-437-9606
Corresponding author: Email: zgdongumn.edu Telephone: 507-437-9615 Fax: 507-437-9606