Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2017; 7(8):2289-2304. doi:10.7150/thno.19439 This issue Cite
Research Paper
NPM1 Mutant Mediated PML Delocalization and Stabilization Enhances Autophagy and Cell Survival in Leukemic Cells
Qin Zou1, Shi Tan2, Zailin Yang3, Qian Zhan4, Hongjun Jin1, Jingrong Xian1, Shuaishuai Zhang1, Liyuan Yang1, Lu Wang1, Ling Zhang1 ![]()
1. Key Laboratory of Laboratory Medical Diagnostics Designated by the Ministry of Education, School of Laboratory Medicine, Chongqing Medical University, Chongqing, China;
2. Department of Clinical Laboratory, Chongqing Health Center for Women and Children, Chongqing, China;
3. Center for Hematology, Southwest Hospital, Third Military Medical University, Chongqing, China;
4. The Center for Clinical Molecular Medical detection, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China.
Received 2017-2-1; Accepted 2017-4-18; Published 2017-6-1
Abstract

Accumulating evidence has defined nucleophosmin 1 (NPM1) mutation as a driver genetic event in acute myeloid leukemia (AML), whereas the pathogenesis of NPM1-mutated AML remains to be fully elucidated. In this study, we showed that mutant NPM1 elevated autophagic activity and autophagic activation contributed to leukemic cell survival in vitro. Meanwhile, we also found high expression of promyelocytic leukemia gene (PML) and its cytoplasmic dislocation in primary NPM1-mutated AML blasts and NPM1-mA positive OCI-AML3 cells. Mechanically, mutant NPM1 interacted with PML and mediated it delocalization as well as stabilization. Notably, NPM1-mA knockdown impaired autophagic activity, while induced expression of PML reversed this effect. Finally, we confirmed that PML modulated autophagic activity via AKT signal. These findings suggest that aberrant PML expression and autophagy are beneficial to the leukemic transformation driven by NPM1 mutations. This indicates an attractive therapeutic avenue for PML targeting and/or autophagy inhibition in the treatment of NPM1-mutated AML.
Keywords: acute myeloid leukemia, nucleophosmin 1, mutation, autophagy, cell survival, promyelocytic leukemia gene, AKT.
Introduction
Acute myeloid leukemia (AML) is a clinically, cytogenetically, and molecularly heterogeneous disease. Recurrent cytogenetic abnormalities help define subgroups with different prognosis, and identify patients who might benefit from targeted therapies [1]. However, the clinical and molecular features of patients with cytogenetically normal AML (CN-AML) accounting for about 50% of all adult AML are still poorly understood [2]. High-throughout DNA sequencing of thousands of primary AML patient samples have benefited us better knowledge of molecular landscape of CN-AML [3]. Mutation in nucleophosmin 1 (NPM1) gene is one of the most frequently genetic lesions in about 30% of all AML and 50-60% of CN-AML [4]. NPM1 mutation resulted in the loss of a nucleolar localization signal (NLS) and the acquisition of a novel nuclear export signal (NES), giving rise to skewed cytoplasmic accumulation of the NPM1 mutant protein (NPM1c+) [5]. Because of distinct biological and clinical features, NPM1-mutated AML has been defined as a distinct molecular leukemia entity in the recent updated World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia [6-8]. During the past decade, intensive efforts have been done to determine the contribution of NPM1 mutation in the pathogenesis of AML. Studies in vitro have evidenced that NPM1c+ could mediate tumor suppressors such as PTEN [9] and Fbw7γ [10] depletion from the nucleus, aiding to apoptosis resistance and proliferation induction. We previously identified the regulation role of NPM1 mutation in myeloid differentiation block and invasion promotion through upregulating miRNA-10b and matrix metalloprotease (MMPs), respectively [11, 12]. Moreover, analysis derived from mouse models of NPM1-mutated AML has revealed the cooperation of NPM1 mutation with key molecular events to induce AML [13, 14]. Although NPM1 mutation has been characteristically defined as a driver genetic event in AML, the precise mechanism of action of the NPM1 mutant in AML remains to be fully elucidated, and other contributing oncogenic events are still needed to be investigated.
Autophagy, literally 'self-eating', is a highly conserved and closely regulated homeostatic process and mediates the removal of dysfunctional or damaged organelles, which are digested and recycled for cellular metabolic need in all eukaryotic cells [15, 16]. Mount of evidence has demonstrated an essential role of autophagy in the regulation of cancer progression including leukemia [17, 18]. Preclinical investigations have documented that autophagic activation induced by histone deacetylase inhibitors (HDACi) may promote a selective advantage in t (8; 21) AML cell survival [19]. Consequently, pharmacological inhibition of autophagy was showed to potentiate anticancer activity induced by chemotherapeutic drug in primary leukemia cells [20]. In addition, the genetic ablation of autophagy was found to impede megakaryopoiesis and led to severely impaired platelet production and failed hemostasis [21]. However, investigation conducted on the potential significance of autophagy in the pathogenesis of NPM1-mutated leukemia is limited.
It has become apparent that both oncogenes and tumor suppressor genes help maintain the physiological autophagic homeostasis of the cell. Dysregulation of these genes often trigger an autophagic switch leading to autophagic cell death or survival [22-24]. The tumor suppressor promyelocytic leukemia (PML) consisting of nine exons is located on chromosome 15 and was first discovered in acute promyelocytic leukemia (APL) [25]. Being the central component of discrete sub-nuclear structures named PML-nuclear bodies (PML-NBs), PML plays multiple roles in a wide variety of biological processes such as apoptosis, proliferation and cell metabolism [26-28]. In addition, PML was also defined as a member of TRIM (tripartite motif) family due to the presence of RING, B-boxes, coiled-coil (RBCC) domain, named as TRIM 19 [29]. Recently, analysis of small interfering RNA screen indicated that a large number of TRIM proteins may represent as a new class of autophagic regulators [30]. Although there is no definitive evidence that PML is involved in autophagy in cancers, Huang et al. [31] have provided compelling evidence that PML-RARα fusion protein induced constitutive autophagic activation in myeloid leukemia U937 cells. Missiroli et al. [32] have provided further evidence that PML is indeed required for autophagy regulation and cancer development. Notably, aberrant localization of PML was observed in NPM1-mutated leukemic cells. Moreover, PML degradation induced by arsenic trioxide (ATO) has been implicated a potential therapeutic relevance in NPM1-mutated AML primary samples [33, 34]. However, whether the aberrant PML expression participated in autophagic process and cell survival in NPM1-mutated AML remains unknown.
In light of these findings, we sought to evaluate the role of autophagy in NPM1-mutated leukemic cells and on the mechanistic consequences of aberrant PML expression. Here, we demonstrated that autophagic activation contributed to cell growth facilitated by NPM1 mutation type A (NPM1-mA). In addition, NPM1-mA interacted with PML and led to cytoplasmic delocalization and stabilization of PML protein. Importantly, enforced PML expression could rescue the effect of NPM1-mA knockdown on autophagy and cell proliferation. The study points to a potential therapeutic strategy for PML and/or autophagy targeting in NPM1-mutated AML therapy.
Methods
Patient Samples
The peripheral blood of 26 AML patients newly diagnosed through cytomorphology, cytogenetic and molecular genetic analysis, were obtained from Southwest Hospital of the Third Military Medical University and the First Affiliated Hospital of Chongqing Medical University. Samples were enriched for mononuclear cells by Ficoll gradient purification. The isolated mononuclear cells were used for LC3, p62 and PML relative expression analysis. Informed consent in accordance with the Declaration of Helsinki was obtained from the individuals examined, and the related study was approved by the Institutional Review Board of the Southwest Hospital of The Third Military Medical University and the First Affiliated Hospital of Chongqing Medical University. Details of the clinical characteristics of patients are provided in Table 1.
Patient characteristics
| Characteristics | Median(range) | No. of cases |
|---|---|---|
| Sex | ||
| Female | 14 | |
| Male | 12 | |
| Total | 26 | |
| Median age, y | 53.8y (26-79) y | |
| Younger than 40 y | 6 | |
| 40-60 y | 12 | |
| Older than 60 y | 8 | |
| Median WBC, ×109/L | 44 (0.3-295) | |
| Median platelets, ×109/L | 57.3 ( 3.0 - 655.0) | |
| FAB classification | ||
| M1 | 2 | |
| M2 | 4 | |
| M3 | 4 | |
| M4 | 6 | |
| M5 | 9 | |
| unclassified | 1 | |
| Karyotype | ||
| Normal | 14 | |
| t(8;21) | 2 | |
| t(15;17) | 3 | |
| inv(16) | 5 | |
| Unknown | 2 | |
| Gene mutations | ||
| NPM1 | 14 | |
| FLT3/ITD | 8 | |
| WT1 | 9 | |
| CBFβ-MYH11 | 2 |
Abbreviations: AML, acute myeloid leukemia; y, year old; WBC, white blood cell; FAB classification, French-American-British classification, a classification of acute leukemia produced by three-nation joint collaboration.
Cell culture
Human myeloid leukemia cell lines KG-1a, HL-60, THP-1, K562 and human embryonic kidney cells HEK293T were obtained from American Type Culture Collection (ATCC, USA). Human myeloid leukemia cell line OCI-AML3 (harboring NPM1 mutation type A, NPM1-mA) [35] was obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ, Germany). The myeloid leukemia cell lines were maintained in RPMI-1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS, USA) and 1% penicillin-streptomycin (Sangon biotech, China). HEK293T cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. Cultured cells were incubated in a humidity chamber (Thermo Fisher Scientific, USA) containing 5% CO2 at 37 ˚C.
Reverse transcription PCR and quantitative real-time PCR
Total RNA was isolated using TRIzol reagent (Takara, Japan) and transcribed into cDNA using PrimeScript™ RT reagent Kit (Takara, Japan). Quantitative real time-PCR (qRT-PCR) amplifications of LC3, p62, PML and NPM1-mA genes were performed using SYBR Green (KAPA Biosystems, USA) on a CFX ConnectTM real-time system (Bio-Rad, USA) with β-actin as a normalization control. Cycling conditions were 5 min at 94 °C for the initial denaturation, and amplification was performed with 40 cycles of 30 s at 94 °C, 30 s at 59 °C (for LC3), 56 °C (for p62 and NPM1-mA) or 56.4 °C (for PML), 50 s at 72 °C, and finally 10 min at 72 °C for extension. The melt curve was (65~95) °C at the rate of 0.5°C/0.05 s. Three independent experiments were performed. Details of the primer sequences used are shown in Table 2.
Primer sequences used in this study
| Genes | Sequence (5'- 3') |
|---|---|
| LC3 | F: 5'-GACCGCTGTAAGGAGGTGC-3' |
| R: 5'-CTTGACCAACTCGCTCATGTTA-3' | |
| p62 | F: 5'-GGGGACTTGGTTGCCTTTT-3' |
| R: 5'-CAGCCATCGCAGATCACATT-3' | |
| PML | F: 5'-ACATCTTCTGCTCCAACCC-3' |
| R: 5'-CGCCAAAGGCACTATCC-3' | |
| NPM1-mA | F: 5'-TGGAGGTGGTAGCAAGGTTC-3' |
| R: 5'-CTTCCTCCACTGC CAGACAGA-3' | |
| β-actin | F: 5'-TAGTTGCGTTACACCCTTTCTTG-3' |
| R: 5'-TGCTGTCACCTTCACCGTTC-3' |
Abbreviations: F stands for forward; R stands for reverse
Western blot
Following the designated treatments, cells were harvested and lysed in ice-cold RIPA lysis buffer supplemented with protease inhibitor cocktail (Roche, Switzerland). Equal amount of proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The specific primary antibodies used in this study were: rabbit monoclonal antibody LC3 І/II (Novus, USA, 1:2000), rabbit monoclonal antibody p62 (Abcam, USA, 1:2000), mouse monoclonal antibody Flag (sigma, USA, 1:1000), rabbit polyclonal mutant NPM1 antibody (Abcam, USA, 1:1000), rabbit polyclonal antibody PML (Abcam, USA, 1:1000), rabbit polyclonal antibody GFP (Abcam, USA, 1:1000), rabbit monoclonal antibody Bcl-2 and Bax (Santa, USA, 1:500), rabbit polyclonal antibody AKT and pAKT (CST, USA, 1:1000), rabbit polyclonal antibody β-actin (Proteintech, USA, 1:2000). Corresponding secondary antibodies were used, and visualization of the products was done using an ECL detection kit (Millipore, USA).
Immunocytochemistry
OCI-AML3 and THP-1 cells were washed with PBS and cytospun onto coverslips at 500 g for 5 min, fixed with 4% paraformaldehyde for 20 min and permeabilized with 1% Triton for 15 min at room temperature. Following blocking with 1% bovine serum albumin in PBS for 30 min, cells were immunostained with mouse monoclonal PG-M3 antibody (Santa, USA, 1:200) overnight at 4 °C. The primary antibody was revealed using the immunoalkaline phosphatase APAAP technique. Cells were counterstained with hematoxylin and mounted in neutral gum and analyzed using a bright field microscope.
Immunofluorescence
Following the designated treatments, cells were washed and cytospun on slides and fixed for 20 min in PBS containing 4% parafomaldehyde, permeabilized in 1% Triton X-100 for 20 min, and incubated in blocking buffer (5% FBS in PBS) for 30 min. Cells were ringed with PBS and incubated overnight at 4 °C in dilution buffer containing primary antibodies. The cells were washed three times with PBS before being incubated with an appropriate fluorochrome-conjugated secondary antibody (ZSGB-Bio, China) for 1 h at 37 °C in the dark. After nuclear counterstaining with 4, 6-diamidino-2-phenylindole (DAPI, Beyotime, China), slides were coverslipped with mounting medium and visualized using Nikon microscope. For inhibition of nuclear exportation of NPM1-mA, OCI-AML3 cells were treated with 20 nM leptomycin B (LMB; Beyotime, China) for 0 h, 2 h, 6 h, followed by immunofluorescence analysis. Quantification of cytoplasmic versus nuclear PML in leukemic cells was analyzed by Image J software. The following antibodies were used: rabbit polyclonal mutant NPM1 antibody (Abcam, USA), mouse monoclonal Flag antibody (sigma, USA), mouse monoclonal PG-M3 antibody (Santa, USA), rabbit monoclonal LC3 І/II antibody (Novus, USA), rabbit polyclonal GFP antibody (Abcam, USA). Alexa Fluor fragment of goat anti-mouse IgG and goat anti-rabbit IgG (ZSGB-Bio, China), both at 1:200 dilutions in blocking medium.
Cell transfection and infection
Plasmids encoding pEGFPC1-NPM1-mA, pEGFPC1-NPM1-wt and empty vector pEGFPC1 were kindly provided by Dr. B. Falini (Institute of Hematology, University of Perugia, Perugia, Italy). Plasmids encoding Flag-NPM1-mA, Flag-NPM1-wt and empty vector were obtained from Dr. C.J. Sherr (Genetics and Tumor Cell Biology, St, Jude Children's Research Hospital, Memphis, USA). Plasmids encoding HA-AKT, Flag-PML-IV were obtained from Dr. H.K. Lin (Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA). All transfection experiments were conducted using Lipofectamine™ 2000 (Invitrogen, USA) according to the manufacturer's instruction. After 48 h of transfection, the cells were collected for qRT-PCR, western blot or immunofluorescence analyses. shRNA targeting PML (5'-CCCGCAAGACCAACAACAT-3') and scramble lentiviral vectors were purchased from Gene Pharma (Shanghai, China), respectively. OCI-AML3 cells and THP-1 were infected with shRNA lentivirus targeting NPM1 for 48 h in the presence of 5 μg/mL polybrene (Sigma, USA), following 2 μg/mL puromycin selection for 7 days (Sigma, USA), respectively. The puromycin-resistant cells were isolated and propagated for use in the experiments.
Immunoprecipitation
The cells were harvested after 48 h of transfection and washed twice with ice-cold PBS buffer. Cells were then sonicated in IP buffer [20 mM Tris-Cl, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 % (v/v) Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM Na3VO4, and protease inhibitor cocktail (Roche), pH 7.5] at 4 °C for 3 times per 5 s by Bio-ruptor UCD-200 (Diagenode, Belgium), followed by centrifuged at 14,000 g at 4 oC for 15 min to remove the cell debris. Expressing of the indicated proteins in the lysates was checked by western blot analysis using relevant antibodies to normalize total amounts of the inputs. After normalization, the supernatants were each incubated with specific antibodies or normal IgG (as control), and equal amounts of protein A/G beads (Santa, USA) overnight at 4 oC. The protein A/G beads with the bound antibodies and interacting proteins were pelleted and washed three times with IP buffer before boiled in 2× SDS-PAGE sample. The boiled samples were then resolved in SDS-PAGE and subject to western blot analysis.
Cycloheximide assay
For cycloheximide assays, HEK293T cells were co-transfected with pEGFPC1-NPM1-mA and Flag-PML-IV. In addition, OCI-AML3 cells were infected with either shNPM1 lentivirus or vector. Next, the HEK293T and OCI-AML3 cells were treated with 100 μg/ml cycloheximide (CHX; Sigma, USA) for the indicated times, respectively. Following designated treatment, whole-cell lysates were prepared using RIPA buffer with protease inhibitors. Equal amounts of protein for each time point were analyzed by western blot.
Cell viability assay
Cell viability was determined using Cell Counting Kit-8 (Dojindo Laboratories, Japan) according to the manufacturer's instruction. In Brief, cells were seed at 2×103 cells per well in a 96-well plate for 12 h and subsequently treated with rapamycin or 3-methylademine (3-MA) reagents for indicated times. At the indicated time point, cells were treated with CCK-8 at 10 μl/well at 37°C for 3 h, and the numbers of cells per well were determined by measuring absorbance at 450 nm.
Colony formation assay
Methylcellulose clonogenic assays was carried out to determine cell colony formation ability by planting 1×103 cells per well in triplicate in 24 well-plate, and maintained in RPMI 1640 medium containing 20% FBS at 37°C in incubator. Colony numbers were scored 10 days later. The colony forming units (CFU), defined as cell clusters consisting of more than 5 cells, were counted using an inverted microscope.
The cancer genome atlas analysis
Expression levels of gene and clinical information of 200 AML patients were retrieved from The Cancer Genome Atlas (TCGA). Total of 173 samples had IlluminaGA RNASeq data for gene expression. The values (normalized RNA-Seq data) represented the expression and mutation status in the gene expression matrix and the gene mutation matrix, respectively. Median value of the measured PML expression levels based on the range of expression of PML in AML samples was used to dichotomize patients into high and low cohorts. NPM1 mutation occurs frequently in AMLs and age is one of the most important risk factors among patients with AML, therefore, 33 NPM1-mutatd cases older than 40 years of age were included in our study. Finally, comparison of the overall survival (OS) according to the levels of PML in NPM1-mutated AML cases was done by long-rank test. The OS curves were plotted according to the Kaplan-Meier methods.
Statistical analysis
All data were derived from three independent experiments. P values for comparisons of gene expression between groups were obtained using unpaired t-tests or one-way analyses of variance (ANOVA). The Kaplan-Meier estimation and the log-rank test were used to compare the survival difference. The SPSS (Version 13.0) software and GraphPad (Prism 5) was used for statistical analyses. P < 0.05 was considered statistically significant.
Results
Autophagy activation facilitated by NPM1-mA contributes to leukemic cell survival
To investigate the involvement of autophagy in NPM1-mutated leukemic cell growth, we firstly assessed the levels of autophagy marker in the NPM1-mutated cell lines and primary blasts. The results showed that mutant NPM1-expressing OCI-AML3 cell line had higher LC3 I/II levels and lower p62 levels, as compared to the wild-type NPM1-expressing THP-1 and KG-1a cell lines (Figure 1A-B). Similar results were obtained by Immunofluorescence analysis, as indicated by the accumulated LC3 puncta in OCI-AML3 cells (Figure 1C). In addition, higher LC3 I/II and lower p62 mRNA levels were also observed in primary NPM1-mutated AML blasts, as compared to primary NPM1-unmutated AML blasts (Figure 1D).
The levels of autophagy marker in AML cell lines and primary blasts. (A, B) qRT-PCR and western blot showing the expression of LC3 and p62 mRNA and protein in KG-1a, THP-1 and OCI-AML3 cell lines. β-actin served as the loading controls. Data are represented as mean ± s.d. of three independent experiments. * P<0.05. (C) Representative microscopy images of LC3 puncta in the AML cell lines. LC3 was stained with anti-LC3 I/II (green), nucleus (blue) were visualized by DAPI (×400). (D) Levels of LC3 and p62 in 14 primary NPM1-mutated AML cases assessed by qRT-PCR, and compared with 12 primary NPM1-unmutated AML cases.
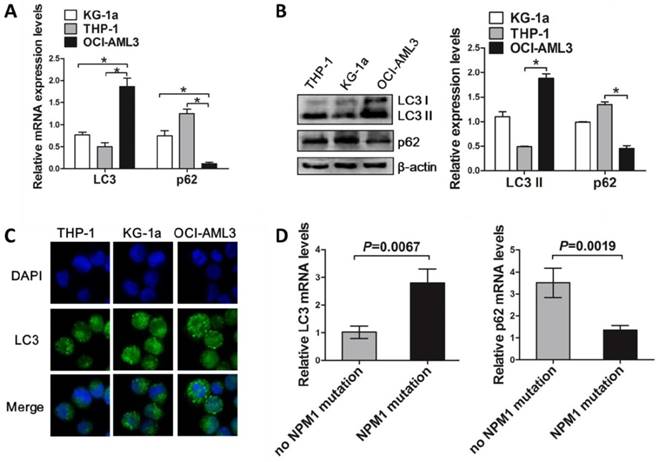
Next, we assessed the autophagic activity upon NPM1-mA expression. THP-1 cells derived from human monocytic leukemia were transiently transfected with Flag-NPM1-mA, Flag-NPM1-wt, and empty vector, and designated as vector group, NPM1-wt group and NPM1-mA group, respectively. The data showed that NPM1-mA overexpression increased endogenous LC3-II and decreased p62 levels (Figure 2A). We also found that overexpression of NPM1-mA resulted in increased accumulation of LC3 puncta (Figure 2B). To validate the effect of NPM1-mA on autophagy flux, 3-methyladenine (3-MA), an autophagy inhibitor, was added in NPM1-mA-enforced THP-1 cells. Consistent with previous observation revealing the capacity of 3-MA to prevent autophagy in leukemic cells [31], 3-MA treatment suppressed LC3-II accumulation and p62 downregulation induced by NPM1-mA overexpression (Figure 2C). Furthermore, we observed that enforced NPM1-mA expression promoted cell proliferation in vitro, whereas exposure to 3-MA abrogated the enhancement of NPM1-mA-induced autophagic activation and growth advantage (Figure 2D). Similar results were obtained from another wild-type NPM1-expressing KG-1a cell line (Figure 2E-H).
Autophagy activation facilitated by NPM1-mA enhances leukemic cell survival. (A, E, I) Western blot of LC3 I/II and p62 levels in NPM1-mA-enforced THP-1 and KG-1a cells as well as NPM1-mA-silenced OCI-AML3 cells. β-actin served as the loading controls. Data are represented as mean ± s.d. of three independent experiments. (B, F, J) Representative microscopy images of LC3 puncta in THP-1, KG-1a and OCI-AML3 cells. wild-type NPM1 and NPM1-mA was stained with anti-Flag (red), LC3 stained with anti-LC3 I/II (green), nucleus (blue) were visualized by DAPI (×400). (C, G, K) Western blot of LC3 I/II and p62 protein levels in THP-1 and KG-1a treated with 3-MA (2 mM), and OCI-AML3 cells treated with rapamycin (5 μM) for 6 h, respectively. Data are represented as mean ± s.d. of three independent experiments. (D, H, L) Evaluation of cell proliferation in THP-1 and KG-1a cells treated with 3-MA (2 mM), and in the NPM1-mA-silenced OCI-AML3 cells treated with rapamycin (5 μM) for indicated times by CCK-8 assay. Three independent experiments were performed. * P<0.05, as comparison to vector and NPM1-wt group, respectively. # P<0.05, as comparison to 3-MA or rapamycin treatment group.
In complementary loss-of-function studies, we established the NPM1-mA-silenced OCI-AML3 cells, which has been previously characterized and used for this type of analysis [11]. The results showed that depletion of NPM1-mA decreased LC3-II levels and increased p62 levels, along with the reduced accumulation of LC3 puncta numbers in OCI-AML3 cells (Figure 2I-J). Additionally, treatment with autophagy activator rapamycin reversed the changes in autophagic activity and cell proliferation caused by NPM1-mA knockdown (Figure 2K-L). Collectively, these results demonstrated that autophagic activity was enhanced upon NPM1-mA expression and autophagic activation provided a survival benefit to leukemic cells in vitro.
PML is aberrantly expressed in NPM1-mutated leukemic cells
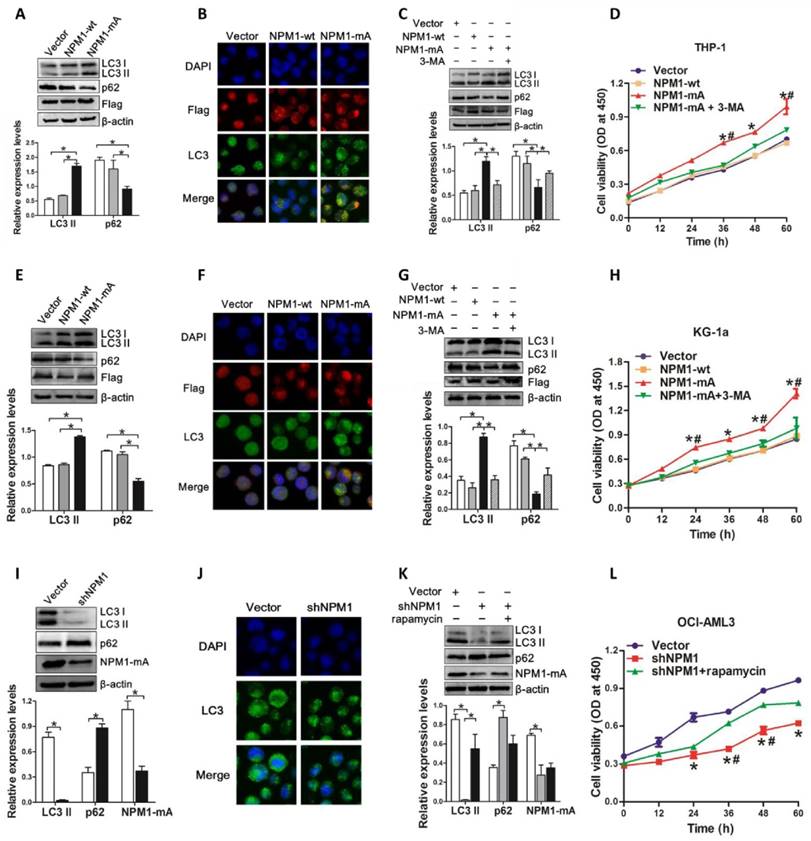
Recent studies have implicated the critical role of PML in the regulation of autophagy in cancer cells [32]. We hypothesized that aberrant PML might participate in autophagy and cell survival in NPM1-mutated AMLs. To test this notion, we first evaluated PML expression and intracellular distribution in myeloid leukemia cell lines. Most PML isoforms were detected at various levels in five myeloid leukemia cell lines, and relative high expression levels of PML were observed in OCI-AML3 cells harboring NPM1-mA (Figure 3A-B). Consistent with above data, we also found that PML expression were higher in NPM1-mutated AML samples compared with that in NPM1-unmutated AMLs (Figure 3C). In addition, analysis from immunocytochemistry staining showed that, unlike the mainly restrained to the nuclear fractions in THP-1 cells, PML exhibited nucleoplasm and/or cytoplasmic expression patterns in OCI-AML3 cells by the presence of the red precipitate particles (Figure 3D). Similar results were obtained by immunofluorescence analysis (Figure 3E). Importantly, we detected the endogenous co-localization of PML and mutant NPM1 protein in OCI-AML3 cells (Figure 3F). These results confirmed the aberrant expression of PML in OCI-AML3 cells and indicated the potential relationship between PML and mutant NPM1 protein.
Aberrant expression and intracellular distribution of PML in NPM1-mutated leukemic cells. (A-B) qRT-PCR and western blot showing the levels of PML mRNA and protein in five myeloid leukemia cell lines. Three independent experiments were performed. * P<0.05. (C) Expression levels of PML in primary NPM1-mutated AML samples (n=14) compared with NPM1-unmutated AMLs (n=12). (D-E) Representative results of cytoplasm localization of PML (red) detected by (D) immunocytochemistry staining (APAAP, ×400) and (E) immunofluorescence analysis (×400) in OCI-AML3 versus THP-1 cells. PML was stained with anti-PG-M3 (red). Quantification of cytoplasmic versus nuclear PML in OCI-AML3 and THP-1 cells was analyzed by Image J software. (F) The localization of endogenous mutant NPM1 (green) and PML (red) in OCI-AML3 cells analyzed by immunofluorescence (×600).
PML delocalization and stabilization is mediated by mutant NPM1
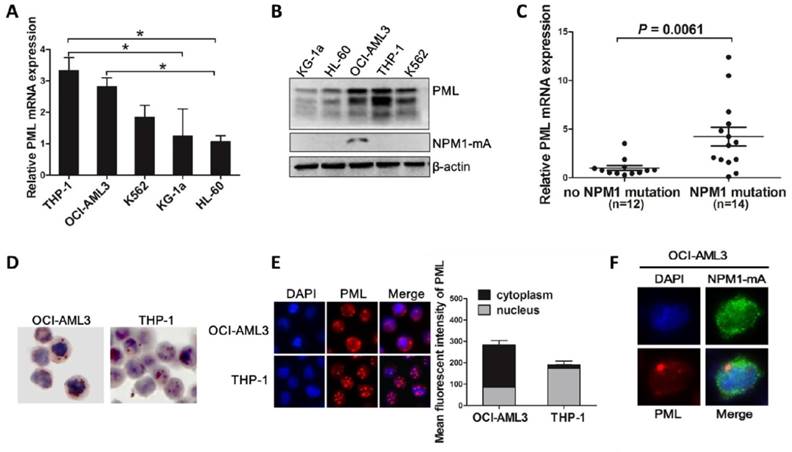
Based on the above data and the critical role of NPM1 in proper nucleolar localization and stability of some nuclear proteins [36, 37], we proposed that NPM1-mA may be involve in regulation of PML delocalization and stabilization. Firstly, we examined the effect on PML localization upon NPM1-mA expression. THP-1 cells were transfected with pEGFPC1-NPM1-mA plasmids, followed by immunofluorescence analysis of PML localization. The results showed that PML was delocalized to the cytoplasm of the transfected cells in NPM1-mA group while restrained to the nuclear in the two control groups (Figure 4A). Since normal nucleo-cytoplasmic traffic of NPM1 occurs in a CRM1-dependent manner, we then tested the effects of the specific CRM1 inhibitor leptomycin (LMB) treatment on NPM1 mutant as well as PML localization. Our data showed that treatment with LMB relocated cytoplasmic NPM1 mutant to the nucleus in OCI-AML3 cells, accompanied by restoration of PML nuclear localization (Figure 4B). Then, we determined whether NPM1 mutant protein physically interacts with PML. Co-immunoprecipitation (Co-IP) analysis revealed that immunoprecipitation with mutant NPM1 antibody pulled down endogenous PML along with NPM1-mA in OCI-AML3 cells (Figure 4C). Similarly, immunoprecipitation with PML antibody pulled down endogenous NPM1-mA along with PML (Figure 4D). In support of this result, HEK293T cells were transfected with pEGFPC1-NPM1-mA and Flag-PML-IV (the best studied PML isoform), and the interaction between exogenous of NPM1-mA and PML was confirmed (Figure 4E-F). These data indicated that mutant NPM1 interacted with PML, giving rise to cytoplasmic delocalization of PML protein in OCI-AML3 cells.
NPM1-mA interacts with PML and leads to cytoplasmic delocalization of PML in OCI-AML3 cells. (A) Immunofluorescence analysis (×400) of GFP (green) and PML (red) localization in pEGFPC1 group, pEGFPC1-NPM1-wt group and pEGFPC1-NPM1-mA group, respectively. Cytoplasmic versus nuclear GFP and PML in the transfected THP-1 cells were quantified and data were shown as mean ± s.d of three independent experiments. (B) Immunofluorescence analysis (×600) of endogenous NPM1-mA (green) and PML (red) localization in OCI-AML3 cells treated with 20 nM leptomycin B (LMB) for 0 h, 2 h and 6 h. Cytoplasmic versus nuclear NPM1-mA and PML in the transfected THP-1 cells were quantified and data were shown as mean ± s.d of three independent experiments. (C-D) Total lysates from OCI-AML3 cells were immunoprecipitated with (C) anti- mutant NPM1 or control rabbit IgG antibodies and (D) anti-PML or control mouse IgG antibodies, followed by immunoblotting for PML and mutant NPM1, respectively. (E-F) HEK293T cells were transiently transfected with Flag-PML-IV and pEGFPC1-NPM1-mA, cell lysates were immunoprecipitated with (E) anti-GFP antibody and (F) anti-Flag antibody, followed by immunoblotting for anti-Flag and anti-GFP, respectively.
Next, we investigated whether NPM1-mA plays a role in the turnover of PML protein. pEGFPC1-NPM1-mA was transfected into THP-1 cells and PML protein levels were monitored. Our results revealed that enforced NPM1-mA expression (Figure 5A) led to upregulation of PML protein (Figure 5B). In contrast, shRNA-mediated NPM1-mA downregulation decreased PML protein levels in OCI-AML3 cells (Figure 5C), whereas the levels of PML remained constant in NPM1-silenced THP-1 cells (Figure 5D). Next, we undertook to identify the effect of NPM1-mA on half-life of PML protein. HEK293T cells were co-transfected with pEGFPC1-NPM1-mA and Flag-PML-IV, followed by cycloheximide (CHX, 100 μg /mL) to block de novo protein synthesis. Our data showed that NPM1-mA overexpression in HEK293T cells alleviated the degradation of exogenous PML protein caused by CHX treatment in a time-dependent manner (Figure 5E). In contrast, NPM1-mA knockdown in OCI-AML3 cells sped up the degradation of endogenous PML protein (Figure 5F). Next, we sought to determine the potential mechanism of which mutant NPM1 regulated PML stability. Consistent with previous report that PML is subject to proteasome-dependent proteolysis [38], our data showed that treatment with CHX resulted in PML protein levels decreasing in a time-dependent manner, whereas addition of proteasome inhibitor MG132 (10 μM) reversed the changes in PML protein levels caused by CHX treatment (Figure 5G). Further experiments demonstrated that the addition of MG132 could alleviate the downregulation of PML mediated by NPM1-mA knockdown in OCI-AML3 cells (Figure 5H). These data supported that mutant NPM1 mediated PML stabilization through inhibiting proteolysis. Additionally, we also determined the changes in PML mRNA levels upon NPM1-mA expression (Figure 5I), indicating other potential mechanisms underlying aberrant PML expression in NPM1-mutated AML cells.
NPM1-mA stabilizes PML in OCI-AML3 cells. (A-B) qRT-PCR and western blot analysis of NPM1-mA mRNA and PML protein levels in THP-1 cells following NPM1-mA overexpression. ***P<0.001. (C-D) OCI-AML3 and THP-1 cells were infected with shRNA lentivirus targeting NPM1, respectively. qRT-PCR analysis of NPM1-mA and/or wild-type NPM1 mRNA levels, western blot analysis of PML protein levels. β-actin served as the loading controls. (E-F) Western blot analysis of PML protein levels in (E) HEK293T cells following NPM1-mA overexpression and (F) OCI-AML3 cells following shRNA-mediated NPM1-mA downregulation in the presence of cycloheximide (CHX, 100 μg/mL) for the indicated times. β-actin served as the loading controls. (G) OCI-AML3 cells were treated with CHX for indicated times, followed by MG132 (10 μM) treatment, the levels of PML protein were monitored by western blot. (H) Western blot analysis of changes in endogenous PML levels in NPM1-mA-silenced OCI-AML3 cells in the presence of MG132 for 4 h. β-actin served as the loading controls. (I) Effect of NPM1-mA on PML mRNA levels by gain-and loss-of function experiments. Three independent experiments were performed. * P<0.05, ** P<0.01.
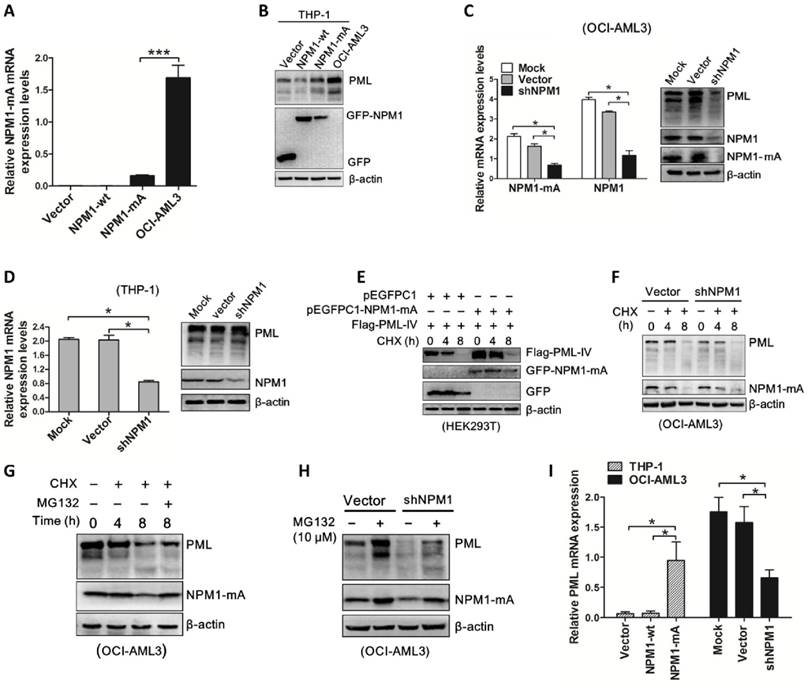
PML knockdown inhibits cell growth through suppressing autophagy in OCI-AML3 cells
To gain evidence that the effect of aberrant PML expression mediated by NPM1 mutant is biologically relevant, OCI-AML3 cells were stably infected with shRNA lentivirus targeting PML and cell growth was observed. As expected, PML mRNA and protein levels were downregulated caused by shRNA mediated PML depletion in OCI-AML3 (Figure 6A-B). Importantly, results from CCK-8 analysis revealed that loss of PML resulted in cell proliferation inhibition (Figure 6C). In line with the data, colony formation assay showed decreased number of cell clusters morphologically and lower proportion of colony forming units in the PML-silenced OCI-AML3 cells as compared to scramble group (Figure 6D). Additionally, we measured the changes in apoptosis-associated protein Bcl-2 and Bax levels and found that silenced PML decreased Bcl-2 levels and increased Bax protein levels (Figure 6E). In support of these results, we accessed to the RNA-Seq data and clinical information of 33 NPM1-mutated AMLs from TCGA database to analyze the possible correlation between PML expression and prognosis. The results revealed that NPM1-mutated AML patients expressing high PML levels had a shorter survival compared with those expressing low PML expression (Figure 6F). Collectively, these results revealed that knockdown of PML could suppress cell growth in OCI-AML3 cells.
PML knockdown suppresses cell growth in OCI-AML3 cells. (A-B) qRT-PCR and western blot analysis of PML mRNA and protein levels in OCI-AML3 stably infected with shRNA lentivirus targeting PML. Three independent experiments were performed, ** P<0.01. (C) CCK-8 analyzed the effect of PML knockdown on cell proliferation in OCI-AML3 cells. Three independent experiments were performed, ** P<0.01. (D) Representative results of colony formation units in OCI-AML3 cells infected with shRNA targeting PML and the percentage of colony formation units observed under a light microscope (×100). Data are represented as mean ± s.d of three independent experiments. (E) Western blot of Bcl-2 and Bax protein in OCI-AML3 cells infected with shRNA lentivirus targeting PML and scramble. β-actin served as the loading controls. (F) Overall survival according to the levels of PML in NPM1-mutated leukemia patients with the log-rank test applied for comparison.
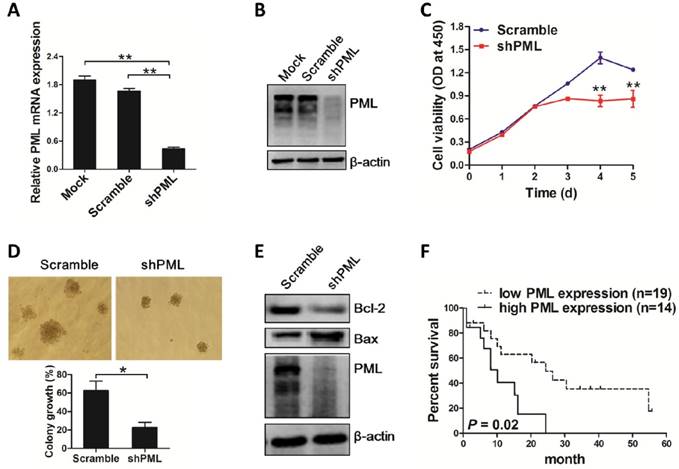
Next, we evaluated the effect of PML knockdown on autophagic activity in OCI-AML3 cells. Results from western blot analysis revealed that PML depletion decreased the levels of endogenous LC3-II and increased p62 levels (Figure 7A). Consistent with these observations, we found that loss of PML resulted in reduced LC3 puncta distribution (Figure 7B). In addition, rapamycin treatment could reverse the changes in LC3-II conversion and p62 levels mediated by PML depletion (Figure 7C). Furthermore, autophagic activation by rapamycin rescued the inhibiting effect of PML depletion-mediated cell proliferation (Figure 7D). Finally, we sought to explore the potential mechanism by which PML modulated autophagy. As a key regulator of cell proliferation and survival, the phosphoinositide 3-kinase (PI3-kinase)-AKT signaling pathway has been implicated an important role in the regulation of autophagy [39]. Thus, we assessed whether AKT was involved in autophagy regulation mediated by PML. Results from western blot showed that the levels of total AKT and phosphorylated AKT were decreased in PML-silenced OCI-AML3 cells (Figure 7E). Notably, enforced expression of AKT could alleviate the impairment of PML depletion-mediated autophagy in OCI-AML3 cells, as indicated by the increased LC3-II and decreased p62 levels (Figure 7F). These results indicated that PML was involved in autophagy regulation at least partially due to AKT signal in OCI-AML3 cells.
PML knockdown impairs autophagic activity of OCI-AML3 cells via AKT. (A) Western blot of LC3 I/II and p62 levels in OCI-AML3 stably infected with shRNA lentivirus targeting PML and scramble. β-actin served as the loading controls. Data are represented as mean ± s.d of three independent experiments, * P<0.05. (B) Representative microscopy images of LC3 puncta in OCI-AML3 cells stably expressing shPML. LC3 was stained with anti-LC3 (green), nucleus (blue) were visualized by DAPI (×400). (C) OCI-AML3 cells were stably infected with shRNA lentivirus targeting PML, followed by rapamycin (5 μM) treatment for 6 h, and western blot determined LC3 I/II and p62 levels. Data are represented as mean ± s.d of three independent experiments, * P<0.05. (D) CCK-8 analysis of cell proliferation activity in PML-depleted OCI-AML cells followed by rapamycin induction for indicated times. Three independent experiments were performed. * P<0.05, as comparison to scramble group; # P<0.05, as comparison to rapamycin treatment group. (E) Western blot of AKT and pAKT levels in OCI-AML3 cells stably expressing shPML. β-actin served as the loading controls. Data are represented as mean ± s.d of three independent experiments, * P<0.05. (F) The PML-depleted OCI-AML3 cells were transfected with HA-AKT, western blot analyzed the levels of LC3-II, p62, AKT and pAKT. β-actin served as the loading controls. Data are represented as mean ± s.d of three independent experiments, * P<0.05.
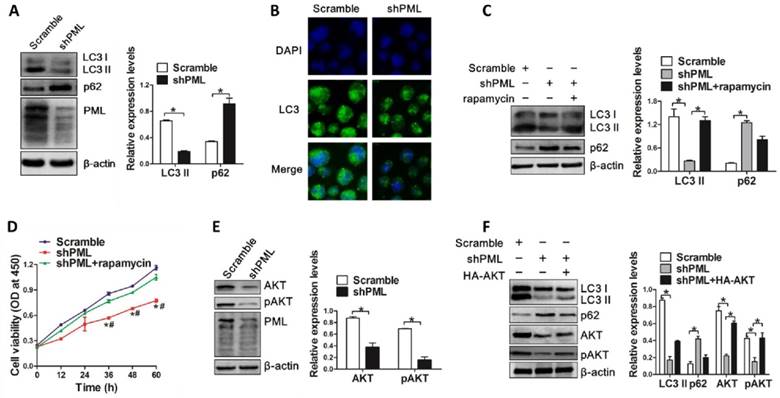
PML expression is critical for NPM1-mA-mediated autophagy and cell survival
On the basis of above results, we further evaluated the pivotal role of PML in NPM1-mA mediated autophagy and cell growth. The NPM1-mA-silenced OCI-AML3 cells were transiently transfected with Flag-PML-IV plasmid. We found that introduction of exogenous PML resulted in LC3-II accumulation and p62 downregulation (Figure 8A). In addition, we also observed the increased accumulation of LC3 puncta in PML-transfected cells as compared to vector group (Figure 8B). Next, we examined the effect of PML overexpression on cell growth in NPM1-mA-silenced OCI-AML3 cells. Results from CCK-8 analysis confirmed that NPM1-mA knockdown resulted in cell growth inhibition, whereas overexpression of PML bypassed the effect of NPM1-mA depletion on cell proliferation (Figure 8C). Additionally, we measured the changes in apoptosis-associated protein and found that PML overexpression increased Bcl-2 levels and decreased Bax levels in NPM1-mA-silenced OCI-AML3 cells (Figure 8D). Collectively, our data indicated that PML was involved in NPM1-mA-mediated autophagy and cell proliferation in OCI-AML3 cells.
PML is critical for NPM1-mA-mediated autophagy and cell proliferation. (A) Western blot of LC3 I/II and p62 levels in NPM1-mA-silenced OCI-AML3 cells following PML overexpression. β-actin served as the loading controls. Data are represented as mean ± s.d of three independent experiments, * P<0.05. (B) Representative microscopy images of LC3 puncta in the NPM1-mA-silenced OCI-AML3 cells followed by PML overexpression, PML was stained with anti-Flag (red), LC3 was stained with anti-LC3 (green), nucleus (blue) were visualized by DAPI staining (×400). (C) Changes in cell proliferation activity in the NPM1-mA-silenced OCI-AML3 cells followed by PML overexpression, as assessed by CCK-8. * P<0.05, as comparison to vector group; # P<0.05, as comparison to Flag-PML-IV treatment group. (D) Western blot of Bcl-2 and Bax protein levels in NPM1-mA-silenced OCI-AML3 cells with PML overexpression. β-actin served as the loading controls. Data are represented as mean ± s.d of three independent experiments, * P<0.05.
Discussion
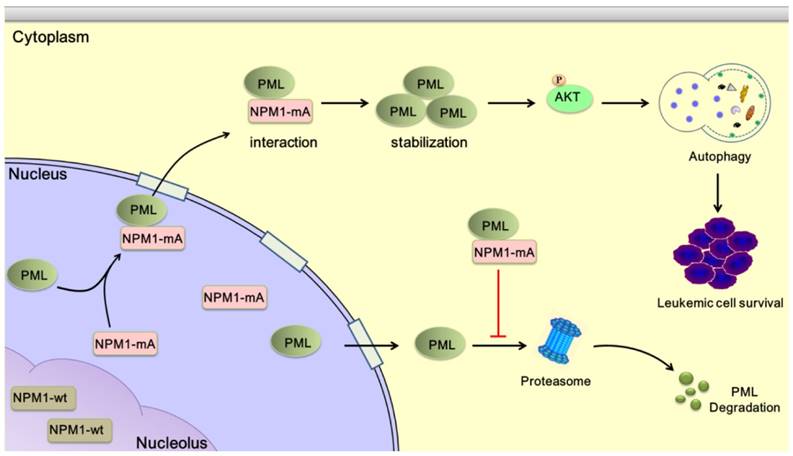
The NPM1 mutation is one of the most frequent genetic alterations thus far identified in AML. Despite progress in the clinical and biological characterization of NPM1-mutated AML, the role played by the NPM1 mutations in AML development has not been fully elucidated. Herein, our data showed that NPM1 mutant mediated tumor suppressor PML delocalization and stabilization, further enhanced autophagic activity and cell survival in AML (Figure 9).
Schematic diagram describing the function significance of NPM1-mA in leukemic cells. NPM1-mA mediates PML delocalization to the cytoplasm and enhances PML stability through inhibiting proteasomal degradation, further promotes autophagy via AKT signal, and consequently contributes to leukemic cell survival.
In recent years, autophagy being suggested as an important mechanism in leukemia pathogenesis has aroused much attention [40]. Notably, increased autophagy-associated transcripts levels were reported in NPM1-mutated AML during analysis of the potential target of autophagy pathway in human cancers with recurrent molecular alteration [41]. In this study, we determined the autophagic activity in leukemia cell lines as well as primary blasts and found that enforced NPM1-mA expression could promote autophagy in leukemia cells. It has been reported that NPM1 was essential for autophagy when the nucleolus was disrupted by inhibiting RNA polymerase I (Pol I) transcription [42]. Indeed, NPM1 is found mainly in the nucleolus. Consequent to the mutations, the NPM1 leukemic mutants export from nucleus to cytoplasm, leading wild-type NPM1 to cytoplasm dislocation [43]. These findings indicated that NPM1 mutant may be involved in autophagy in leukemia. Additionally, our data revealed that NPM1 mutation promoted leukemic cell growth in vitro. Evidence from transgenic mouse model showing the critical role of NPM1 mutant in myeloproliferation induction further confirmed our notion [44]. In addition, our data showed that autophagy inhibitor 3-MA treatment could abrogate the enhancement of NPM1-mA-mediated autophagic activation on cell survival. In line with our data, disruption of autophagy by 3-MA was shown a synergism antineoplastic effect with HDAC inhibitors in chronic myeloid leukemia Ba/F3 cells [45]. These findings and our data were consistent with the conception that activated autophagy enhance cell survival in the established tumors [40].
In addition to the well-documented role of autophagy-related gene (ATG) in autophagy, the function of tumor suppressor genes such as p53 linking autophagy in tumor development has been proposed in recent years [46]. PML, a pleiotropic tumor suppressor controlling a variety of biological processes, has been recently defined a regulation role in autophagy [32]. Notably, aberrant PML expression was reported in NPM1-mutated AMLs [33]. In the present study, we confirmed the aberrant localization of PML protein in mutant NPM1-expressing OCI-AML3 cells. It has been acknowledged that alternative splicing of C-terminal exon 6 encoding the nuclear localization signal can give rise to cytoplasmic localization of PML protein [47]. More recent research by Buczek et al. [48] has evidenced that the cytoplasmic localization of PML may also be mediated by its nuclear export in a chromosomal maintenance 1 (CRM1)-dependent manner. Here, we provided the first evidence that the cytoplasmic localization of PML could be mediated by interacting with mutant NPM1. Indeed, NPM1 mutant protein has been previously reported to delocalize some nuclear protein such as ARF to cytoplasm [49]. The exact domain required for interaction between mutant NPM1 and PML needs to be further determined.
In addition to the aberrant localization of PML, we also observed higher expression of PML in NPM1-mutated AML blasts and cultured cells. Furthermore, loss- and gain-of function assays revealed that PML was subject to similar-mutant NPM1-dependent regulation. Mechanically, we verified that NPM1-mA could stabilize PML through inhibiting proteasome-mediated degradation. Aberrant PML post-translational modifications are the most characterized mechanism accounting for PML loss in cancer. Recent studies have demonstrated the pivotal roles of ubiquitination in PML stability mediated by an E3 ligase UHRF1 or deubiquitinase USP11 in cancers [50, 51]. Considering that NPM1 mutant regulated PETN stabilization through blocking HAUSP-mediated deubiquitination in AML [9], we aim to identify whether the interaction of mutant NPM1 with PML may protect PML protein from ubiquitination-mediated degradation in the future study. In addition, we also observed the change in PML mRNA levels upon NPM1-mA expression. Indeed, the transcriptional up-regulation of PML was previously reported to be accompanied by elevated protein levels [28, 52]. Certainly, other potential multiple mechanisms underlying aberrant PML expression in NPM1-mutated AML cells are worthy to be further investigated.
The activity of PML has been intensively investigated in the context of suppression in tumorigenesis. Here, we uncovered an unexpected function of PML in NPM1-mutated leukemic cells. Preliminarily in vitro experiments showed that PML knockdown led to cell proliferation suppression and colony formation ability impairment, as well as anti-apoptotic Bcl-2 downregulation and pro-apoptotic Bax upregulation. Further analysis of TCGA AML dataset revealed a negative correlation between PML expression and overall survival in NPM1-mutated AMLs, which is consistent with the observation that high PML expression correlated to poor prognosis in chronic myeloid leukemia [53]. In fact, evidence of the function of PML beyond tumor suppression in certain cellular settings is quickly accumulating [28, 54]. More recent study has showed PML upregulation in breast cancer cells and pharmacological inhibition of PML by arsenic trioxide reduced the tumor formation capacity in vivo [55]. Interestingly, a growing body of clinical evidence has also implicated that arsenic trioxide-induced PML degradation has a potential therapeutic role in NPM1-mutated AMLs [33, 34].
Recent studies have evidenced that aberrant PML promoted breast cancer cell survival and hematopoietic stem cell maintenance through regulation of peroxisome proliferator-activated receptor (PPAR) signaling and fatty acid oxidation metabolism [28, 56]. Here, we showed that the inhibiting effect of PML knockdown on cell proliferation might be associated with impaired autophagic activity, which is consistent with recent observation that autophagy is a survival mechanism of AML precursors during dual mTORC2/mTORC1 targeting [57]. It is well established that a variety of kinases was involved in autophagy-signaling network. AKT, also known as protein kinase B, has profound effects on autophagy via the activation of kinase target of rapamycin (TOR) protein, which antagonizes autophagy [58]. Unexpectedly, our data showed that PML knockdown downregulated AKT levels. Moreover, ectopic expression of AKT could rescue the autophagy inhibition mediated by PML depletion in NPM1-mutated leukemic cells. These data are in agreement with the observation recently reported by Khan et al. [59] that sucrose induced autophagy in chondrocytes dependent on the activation of AKT/mTOR/P70S6K signaling pathways. In addition, a recent paper has revealed that pharmacological inhibition of AKT reduced autophagic activity and enhanced radiosensitivity in prostate cancer cells [60]. Thus, defining the precise roles of AKT in autophagy depends on the specific cellular context. Finally, we performed a rescue assay and further highlighted the critical role of PML in NPM1-mA-mediated autophagy and cell proliferation.
In the present study, OCI-AML3 cell line that is accessible and preferable mutant NPM1-expressing cell line was selected for research. Our observation warrant further research on the effects of NPM1 mutations on autophagy including clinical studies and experiments with animal models. Further studies are needed to elucidate the role of PML in cell proliferation and autophagic activity as well as pro- versus anti-apoptotic cell markers using another mutant NPM1-expressing IMS-M2 cell line and MEF PML-/- versus MEF PML+/+ in the context of NPM1 mutation. In addition, the use of primary blasts from wild type or mutant NPM1- expressing AML patients will be tried in the continuous study to better translate the biology of the disease.
In summary, our data suggest for the first time that autophagy is beneficial to leukemogenesis driven by NPM1 mutation. Additionally, a mechanistic link between NPM1-mA expression and PML delocalization and stabilization illustrates an anticipated role of PML in NPM1-mutated AML. Collectively, these results conclusively indicate that pharmacological inhibitors of PML and/or autophagy are potential therapeutics for NPM1-mutated AML therapy.
Abbreviations
AML, acute myeloid leukemia; APL, acute promyelocytic leukemia; ATO, arsenic trioxide; ATG, autophagy-related gene; CN-AML, cytogenetically normal AML; CHX, cycloheximide; CFU, colony forming units; CRM1, chromosomal maintenance 1; DAPI, 4,6-diamidino-2-phenylindole; HDACi, histone deacetylase inhibitors; LMB, leptomycin; MMPs, matrix metalloprotease; 3-MA, 3-methyladenine; NPM1, nucleophosmin; NLS, nucleolar localization signal; NES, nuclear export signal; NPM1c+, cytoplasmic mutant NPM1; NPM1-mA, NPM1 mutation type A; PML, promyelocytic leukemia; PML-NBs, PML-nuclear bodies; PI3-kinase, phosphoinositide 3-kinase; Pol I, polymerase I; PPAR, peroxisome proliferator-activated receptor; RBCC, RING, B-boxes, coiled-coil; TRIM, tripartite motif; TCGA, The Cancer Genome Atlas; WHO, World Health Organization.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 81271913) and Graduate Fellowship in research innovation from the Chongqing Municipal Education Commission (grant no. CYS15134, CYS16140).
Availability of data and materials
The RNA-seq datasets and clinical information of NPM1-mutated AMLs for overall survival analysis in the current study were retrieved from TCGA database (https://genome-cancer.ucsc.edu and https://tcga-data.nci.nih.gov/ tcga/).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Falini B, Martelli MP. Impact of genomics in the clinical management of patients with cytogenetically normal acute myeloid leukemia. Best Pract Res Clin Haematol. 2015;28:90-7
2. Cagnetta A, Adamia S, Acharya C, Patrone F, Miglino M, Nencioni A. et al. Role of genotype-based approach in the clinical management of adult acute myeloid leukemia with normal cytogenetics. Leuk Res. 2014;38:649-59
3. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND. et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374:2209-21
4. Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L. et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254-66
5. Falini B, Bolli N, Shan J, Martelli MP, Liso A, Pucciarini A. et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood. 2006;107:4514-23
6. Martelli MP, Sportoletti P, Tiacci E, Martelli MF, Falini B. Mutational landscape of AML with normal cytogenetics: biological and clinical implications. Blood Rev. 2013;27:13-22
7. Sportoletti P, Varasano E, Rossi R, Mupo A, Tiacci E, Vassiliou G. et al. Mouse models of NPM1-mutated acute myeloid leukemia: biological and clinical implications. Leukemia. 2015;29:269-78
8. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391-405
9. Noguera NI, Song MS, Divona M, Catalano G, Calvo KL, Garcia F. et al. Nucleophosmin/B26 regulates PTEN through interaction with HAUSP in acute myeloid leukemia. Leukemia. 2013;27:1037-43
10. Bonetti P, Davoli T, Sironi C, Amati B, Pelicci PG, Colombo E. Nucleophosmin and its AML-associated mutant regulate c-Myc turnover through Fbw7 gamma. J Cell Biol. 2008;182:19-26
11. Zou Q, Tan S, Yang Z, Wang J, Xian J, Zhang S. et al. The human nucleophosmin 1 mutation A inhibits myeloid differentiation of leukemia cells by modulating miR-10b. Oncotarget. 2016;7:71477-90
12. Xian J, Shao H, Chen X, Zhang S, Quan J, Zou Q. et al. Nucleophosmin Mutants Promote Adhesion, Migration and Invasion of Human Leukemia THP-1 Cells through MMPs Up-regulation via Ras/ERK MAPK Signaling. Int J Biol Sci. 2016;12:144-55
13. Vassiliou GS, Cooper JL, Rad R, Li J, Rice S, Uren A. et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat Genet. 2011;43:470-5
14. Sportoletti P, Varasano E, Rossi R, Bereshchenko O, Cecchini D, Gionfriddo I. et al. The human NPM1 mutation A perturbs megakaryopoiesis in a conditional mouse model. Blood. 2013;121:3447-58
15. White E, Mehnert JM, Chan CS. Autophagy, Metabolism, and Cancer. Clin Cancer Res. 2015;21:5037-46
16. Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814-22
17. Duffy A, Le J, Sausville E, Emadi A. Autophagy modulation: a target for cancer treatment development. Cancer Chemother Pharmacol. 2015;75:439-47
18. Watson AS, Mortensen M, Simon AK. Autophagy in the pathogenesis of myelodysplastic syndrome and acute myeloid leukemia. Cell Cycle. 2011;10:1719-25
19. Torgersen ML, Engedal N, Boe SO, Hokland P, Simonsen A. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells. Blood. 2013;122:2467-76
20. Zhu S, Cao L, Yu Y, Yang L, Yang M, Liu K. et al. Inhibiting autophagy potentiates the anticancer activity of IFN1@/IFNα in chronic myeloid leukemia cells. Autophagy. 2013;9:317-27
21. Cao Y, Cai J, Zhang S, Yuan N, Li X, Fang Y. et al. Loss of autophagy leads to failure in megakaryopoiesis, megakaryocyte differentiation, and thrombopoiesis in mice. Exp Hematol. 2015;43:488-94
22. Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P. et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276:35243-6
23. Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R. et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013;494:323-7
24. White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308-16
25. Kakizuka A, Miller WH Jr, Umesono K, Warrell RP Jr, Frankel SR, Murty VV. et al. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell. 1991;66:663-74
26. Bernardi R, Pandolfi PP. Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene. 2003;22:9048-57
27. Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8:1006-16
28. Carracedo A, Weiss D, Leliaert AK, Bhasin M, de Boer VC, Laurent G. et al. A metabolic prosurvival role for PML in breast cancer. J Clin Invest. 2012;122:3088-100
29. Jensen K, Shiels C, Freemont PS. PML protein isoforms and the RBCC/TRIM motif. Oncogene. 2001;20:7223-33
30. Mandell MA, Jain A, Arko-Mensah J, Chauhan S, Kimura T, Dinkins C. et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell. 2014;30:394-409
31. Huang Y, Hou JK, Chen TT, Zhao XY, Yan ZW, Zhang J. et al. PML-RARalpha enhances constitutive autophagic activity through inhibiting the Akt/mTOR pathway. Autophagy. 2011;7:1132-44
32. Missiroli S, Bonora M, Patergnani S, Poletti F, Perrone M, Gafa R. et al. PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development. Cell Rep. 2016;16:2415-27
33. Martelli MP, Gionfriddo I, Mezzasoma F, Milano F, Pierangeli S, Mulas F. et al. Arsenic trioxide and all-trans retinoic acid target NPM1 mutant oncoprotein levels and induce apoptosis in NPM1-mutated AML cells. Blood. 2015;125:3455-65
34. El Hajj H, Dassouki Z, Berthier C, Raffoux E, Ades L, Legrand O. et al. Retinoic acid and arsenic trioxide trigger degradation of mutated NPM1, resulting in apoptosis of AML cells. Blood. 2015;125:3447-54
35. Quentmeier H, Martelli MP, Dirks WG, Bolli N, Liso A, Macleod RA. et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia. 2005;19:1760-7
36. Colombo E, Bonetti P, Lazzerini Denchi E, Martinelli P, Zamponi R, Marine JC. et al. Nucleophosmin is required for DNA integrity and p19Arf protein stability. Mol Cell Biol. 2005;25:8874-86
37. Korgaonkar C, Hagen J, Tompkins V, Frazier AA, Allamargot C, Quelle FW. et al. Nucleophosmin (B23) targets ARF to nucleoli and inhibits its function. Mol Cell Biol. 2005;25:1258-71
38. Gurrieri C, Capodieci P, Bernardi R, Scaglioni PP, Nafa K, Rush LJ. et al. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst. 2004;96:269-79
39. Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G. et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338:956-9
40. Sehgal AR, Konig H, Johnson DE, Tang D, Amaravadi RK, Boyiadzis M. et al. You eat what you are: autophagy inhibition as a therapeutic strategy in leukemia. Leukemia. 2015;29:517-25
41. Lebovitz CB, Robertson AG, Goya R, Jones SJ, Morin RD, Marra MA. et al. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy. 2015;11:1668-87
42. Katagiri N, Kuroda T, Kishimoto H, Hayashi Y, Kumazawa T, Kimura K. The nucleolar protein nucleophosmin is essential for autophagy induced by inhibiting Pol I transcription. Sci Rep. 2015;5:8903
43. Falini B, Martelli MP. NPM1-mutated AML: targeting by disassembling. Blood. 2011;118:2936-8
44. Cheng K, Sportoletti P, Ito K, Clohessy JG, Teruya-Feldstein J, Kutok JL. et al. The cytoplasmic NPM mutant induces myeloproliferation in a transgenic mouse model. Blood. 2010;115:3341-5
45. Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L. et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110:313-22
46. Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A. et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296-300
47. Nisole S, Maroui MA, Mascle XH, Aubry M, Chelbi-Alix MK. Differential Roles of PML Isoforms. Front Oncol. 2013;3:125
48. Buczek ME, Miles AK, Green W, Johnson C, Boocock DJ, Pockley AG. et al. Cytoplasmic PML promotes TGF-beta-associated epithelial-mesenchymal transition and invasion in prostate cancer. Oncogene. 2016;35:3465-75
49. Colombo E, Martinelli P, Zamponi R, Shing DC, Bonetti P, Luzi L. et al. Delocalization and destabilization of the Arf tumor suppressor by the leukemia-associated NPM mutant. Cancer Res. 2006;66:3044-50
50. Guan D, Factor D, Liu Y, Wang Z, Kao HY. The epigenetic regulator UHRF1 promotes ubiquitination-mediated degradation of the tumor-suppressor protein promyelocytic leukemia protein. Oncogene. 2015;34:5206
51. Wu HC, Lin YC, Liu CH, Chung HC, Wang YT, Lin YW. et al. USP11 regulates PML stability to control Notch-induced malignancy in brain tumours. Nat Commun. 2014;5:3214
52. Carracedo A, Rousseau D, Douris N, Fernandez-Ruiz S, Martin-Martin N, Weiss D. et al. The promyelocytic leukemia protein is upregulated in conditions of obesity and liver steatosis. Int J Biol Sci. 2015;11:629-32
53. Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y. et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453:1072-8
54. Iwanami A, Gini B, Zanca C, Matsutani T, Assuncao A, Nael A. et al. PML mediates glioblastoma resistance to mammalian target of rapamycin (mTOR)-targeted therapies. Proc Natl Acad Sci U S A. 2013;110:4339-44
55. Martin-Martin N, Piva M, Urosevic J, Aldaz P, Sutherland JD, Fernandez-Ruiz S. et al. Stratification and therapeutic potential of PML in metastatic breast cancer. Nat Commun. 2016;7:12595
56. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE. et al. A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18:1350-8
57. Altman JK, Szilard A, Goussetis DJ, Sassano A, Colamonici M, Gounaris E. et al. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin Cancer Res. 2014;20:2400-9
58. F OF, Rusten TE, Stenmark H. Phosphoinositide 3-kinases as accelerators and brakes of autophagy. FEBS J. 2013;280:6322-37
59. Khan NM, Ansari MY, Haqqi TM. Sucrose, But Not Glucose, Blocks IL1-beta-Induced Inflammatory Response in Human Chondrocytes by Inducing Autophagy via AKT/mTOR Pathway. J Cell Biochem. 2017;118:629-39
60. Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ. et al. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014;5:e1437
Author contact
![]() Corresponding author: Ling Zhang, School of Laboratory Medicine, Chongqing Medical University, No.1, Yixueyuan Road, Chongqing, 400016, China Tel: +86 023-68485240, Fax: +86 023-68485239 Email: lingzhangedu.cn
Corresponding author: Ling Zhang, School of Laboratory Medicine, Chongqing Medical University, No.1, Yixueyuan Road, Chongqing, 400016, China Tel: +86 023-68485240, Fax: +86 023-68485239 Email: lingzhangedu.cn