Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods and Materials
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(8):2186-2203. doi:10.7150/thno.18407 This issue Cite
Research Paper
Interleukin-32α Inhibits Endothelial Inflammation, Vascular Smooth Muscle Cell Activation, and Atherosclerosis by Upregulating Timp3 and Reck through suppressing microRNA-205 Biogenesis
Dong Ju Son1*, Yu Yeon Jung2*, Young Sik Seo3, Heonyong Park3, Dong Hun Lee1, Sanghyeon Kim4, Yoon-Seok Roh1, Sang Bae Han1, Do Young Yoon5 ![]() , Jin Tae Hong1
, Jin Tae Hong1 ![]()
1. College of Pharmacy & Medical Research Center, Chungbuk National University, Cheongju, Chungbuk 28160, Korea;
2. Department of Dental Hygiene, Gwangyang Health Sciences University, Gwnagyang, Jeonnam 57764, Korea;
3. Department of Molecular Biology, Dankook University, Yongin, Gyeonggi 16890, Korea;
4. Stanley Brain Research Laboratory, Stanley Medical Research Institute, 9800 Medical Center Drive, Rockville, MD 20850, USA;
5. Department of Bioscience and Biotechnology, Konkuk University, Seoul 05029, Korea.
*These authors contributed equally to this work.
Received 2016-11-18; Accepted 2017-2-13; Published 2017-6-1
Abstract

Interleukin-32 (IL-32) is a multifaceted cytokine that promotes inflammation and regulates vascular endothelial cell behavior. Although some IL-32 isoforms have been reported to contribute to vascular inflammation and atherosclerosis, the functional role of IL-32α in vascular inflammation and atherogenesis has not been studied.
Methods: IL-32α function was assessed in cells with transient IL-32α overexpression or treated with recombinant human IL-32α by western blotting and mRNA expression analysis. Vascular smooth muscle cell (VSMC) proliferation and migration was examined by BrdU incorporation and wound healing assays, respectively. In addition, the participation of IL-32α on vascular inflammation, arterial wall thickening, and atherosclerosis in vivo was monitored in human IL-32α transgenic (hIL-32α-Tg) mice with or without ApoE knockout (ApoE-/-/hIL-32α-Tg).
Results: Our analyses showed that IL-32α suppresses genes involved in the inflammatory and immune responses and cell proliferation, and by limiting matrix metalloproteinase (MMP) function. In vivo, administration of hIL-32α inhibited vascular inflammation and atherosclerosis in hIL-32α-Tg and ApoE-/-/hIL-32α-Tg mice. Subsequent microarray and in silico analysis also revealed a marked decreased in inflammatory gene expression in hIL-32α-Tg mice. Collectively, our studies demonstrated that IL-32α upregulates the atheroprotective genes Timp3 and Reck by downregulating microRNA-205 through regulation of the Rprd2-Dgcr8/Ddx5-Dicer1 biogenesis pathway.
Conclusion: Our findings provide the first direct evidence that IL-32α is an anti-inflammatory and anti-atherogenic cytokine that may be useful as a diagnostic and therapeutic protein in atherosclerosis.
Keywords: Interleukin 32, Timp3, Reck, Vascular inflammation, Atherosclerosis.
Introduction
Atherosclerosis is a chronic inflammatory disease of the arterial system that leads to myocardial infarction, ischemic stroke, and peripheral arterial disease [1, 2]. The vascular endothelium is composed of a monolayer of endothelial cells (ECs) lining the lumen of the blood vessels and has emerged as a key regulator of vascular homeostasis and functions, including cell adhesion and vascular inflammation, that are involved in the initial events of atherogenesis [3-5]. Pro-inflammatory stimuli and perturbed flow trigger signal transduction, gene regulation, endothelial cytokine secretion, and the upregulation of leukocyte adhesion molecules in ECs, ultimately resulting in endothelial dysfunction and vascular inflammation [6, 7]. These inflammatory responses in the vascular endothelium and recruitment of leukocytes into the subendothelial space stimulate the phenotypic change of the neighboring vascular smooth muscle cells (VSMCs) to a pro-inflammatory state, and these cells also play important roles in atherosclerosis [1, 8].
Cytokines are involved in all stages of atherosclerosis and have a profound influence on disease pathogenesis [9-11]. A detailed analysis of local vascular inflammation and cytokine expression in atherosclerotic plaques revealed that a balance between pro-inflammatory cytokines—such as interleukin (IL)-1, IL-2, IL-12, interferon (IFN)-γ, and tumor necrosis factor (TNF)-α—and anti-inflammatory cytokines—such as IL-4, IL-5, IL-10, and IL-13—is crucial for lesion development [12]. IL-32 has also been implicated as a critical regulator of EC function and vascular inflammation [13-16]; therefore, it is likely to participate in the development of atherosclerosis. IL-32 is reportedly involved in the pathogenesis of chronic diseases including rheumatoid arthritis, inflammatory bowel disease, Alzheimer's disease, cancer, and diabetes [17-23]. Notably, IL-32 is expressed as several distinct isoforms generated through alternative splicing [24, 25], suggesting the individual isoforms may play different roles and be expressed by different cell types. However, the functional specificity of each isoform regarding the regulation of EC inflammation and atherogenesis has not been fully studied. Three major isoforms (IL-32α, IL-32β, and IL-32γ) have been intensively studied thus far. IL-32β is the most inducible isoform in inflamed ECs [13], and highly expressed in the human atherosclerotic arterial wall [16], and propagates vascular inflammation [26]. IL-32γ has also been reported as a pro-inflammatory factor that contributes to atherogenesis and plaque instability by increasing the production of chemokine (C-C motif) ligand 2 (CCL2), soluble vascular cell adhesion molecule-1 (VCAM-1) and matrix metalloproteinases (MMPs) [16]. A previous evaluation implicated IL-32β and IL-32γ in vascular inflammation and atherosclerosis [16, 26]; however, while IL-32α is the most prominently expressed isoform in unstimulated human ECs [13], no evidence exists for a functional role of IL-32α in atherosclerosis. An earlier study by our group showed that IL-32α overexpression enhances IL-6 production in THP-1 human monocytic leukemia cells upon treatment with phorbol 12-myristate 13-acetate (PMA), but IL-6 upregulation was only weakly induced by IL-32α overexpression itself in the absence of PMA [27]. We have also shown that IL-1β, TNFα, and IL-8 were not induced by IL-32α overexpression in THP-1 cells [27], suggesting that IL-32α may have distinct biological roles under different physiological conditions. Thus, the current study focused on investigating the functional roles of IL-32α in vascular inflammation and atherosclerosis, which have not previously been studied.
In the present study, we examined the roles of IL-32α in vascular inflammation and atherogenesis and its underlying mechanisms. Notably, human IL-32α (hIL-32α) expression inhibited EC inflammation and VSMC proliferation and migration in vitro. Moreover, hIL-32α potently decreased arterial wall thickening and atherosclerotic plaque development in hIL-32α-expressing transgenic (hIL-32α-Tg) mice and apolipoprotein knockout (ApoE-/-)/hIL-32α-Tg mice by upregulating tissue inhibitor of metalloproteinase 3 (Timp3) and reversion-inducing cysteine-rich protein with Kazal motifs (Reck) by suppressing miR-205 processing.
Methods and Materials
The methods and materials are available the Methods section in the Supplementary Materials.
Results
Human IL-32α inhibits endothelial inflammation in vitro
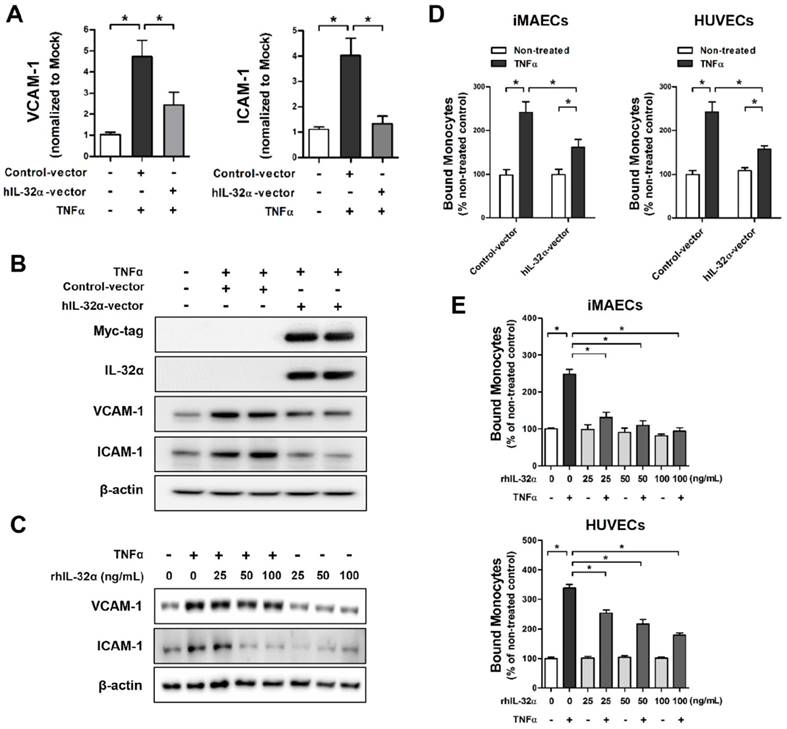
To evaluate the biological functions of IL-32α on vascular inflammation and atherosclerosis, we initially tested the effect of IL-32α on endothelial inflammatory gene expression in vitro. Since IL-32 may regulate diverse intracellular functions through interactions among its different isoforms [25], we used immortalized mouse arterial endothelial cells (iMAECs) that lack endogenous IL-32 expression, to rule out the possibility of interaction effects among endogenous IL-32 isoforms. Interestingly, iMAECs transiently transfected with an hIL-32α expression vector showed a significant reduction in TNFα-induced expression of the EC inflammation markers VCAM-1 and intracellular adhesion molecule-1 (ICAM-1) at the mRNA and protein level as compared to vector-only controls (Figs. 1A, B). Moreover, we showed that treatment with recombinant hIL-32α protein (rhIL-32α) also effectively decreased TNFα-induced VCAM-1 and ICAM-1 expression in iMAECs (Fig. 1C). Furthermore, in vitro monocyte-EC adhesion assays using THP-1 monocytes, iMAECs, and human umbilical vein endothelial cells (HUVECs) showed significantly decreased monocyte adhesion to TNFα-stimulated ECs in both hIL-32α-expressing and rhIL-32α-treated ECs compared with their controls (Figs. 1D, E). In contrast, the monocyte-EC adhesion in unstimulated cells was not affected by hIL-32α overexpression or rhIL-32α treatment (Figs. 1D, E). Taken together, these findings suggested that IL-32α might inhibit EC inflammation through regulating inflammatory gene expression.
IL-32α inhibits endothelial inflammation in vitro. (A,B) Immortalized mouse arterial endothelial cells (iMAECs) were cultured in 24-well culture plates for 24 h, then transfected with human IL-32α-expressing pcDNA3.1+-6 × Myc vector (hIL-32α-vector) or control vector. After 24 h, cells were treated with recombinant tumor necrosis factor α protein (TNFα) (10 ng/mL; 24 h), and (A) VCAM-1 and ICAM-1 mRNA expression was determined by qPCR (data shown as mean ± SEM; n = 5, *p < 0.05 by Student's t-test) and (B) Myc-tag, IL-32α, VCAM-1, and ICAM-1 proteins were analyzed by western blotting. (C) iMAECs were treated with recombinant human IL-32α protein (rhIL-32α) for 24 h, and then treated with TNFα for 24 h. Cells were lysed and analyzed by western blotting with antibodies against VCAM-1 and ICAM-1, using β-actin as a loading control. For monocyte adhesion assays, iMAECs and human umbilical vein endothelial cells (HUVECs) were (D) transfected with hIL-32α-vector or (E) treated with rhIL-32α for 24 h, then treated with TNFα (10 ng/mL; 24 h), and THP-1 monocyte adhesion to endothelial cells was determined (data shown as mean ± SEM; n = 6, *p < 0.05 by Student's t-test).
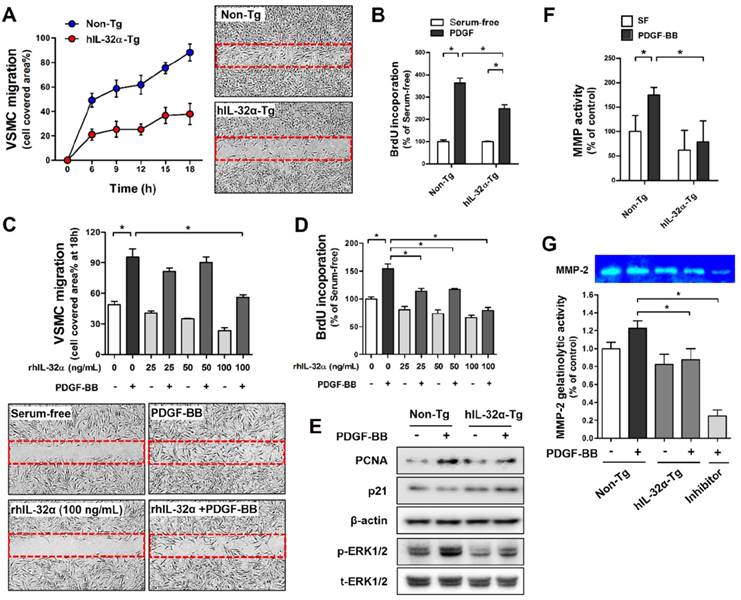
Human IL-32α inhibits PDGF-BB-induced VSMC migration and proliferation in vitro
We next investigated the regulatory role of IL-32α on VSMC migration and proliferation using primary mouse VSMCs from the thoracic aorta of hIL-32α-Tg and non-Tg littermate control mice. VSMC migration and proliferation was assessed by wound healing assay and bromodeoxyuridine (BrdU)-incorporation assay, respectively. Since platelet-derived growth factor (PDGF)-BB is a primary stimulus for the abnormal VSMC migration and proliferation and a critical contributor to vascular diseases [28]. Notably, hIL-32α-Tg VSMCs exhibited a marked decrease in cell migration and proliferation in response to PDGF-BB as compared to those from non-Tg mice (Figs. 2A, B). We further confirmed that rhIL-32α treatment also effectively inhibited PDGF-BB-induced cell migration and proliferation in non-Tg mice VSMCs (Figs. 2C, D). We next sought to determine whether IL-32α could direct regulate genes associated with migration and proliferation. The results showed a notable decrease in proliferating cell nuclear antigen (PCNA) expression and ERK phosphorylation (p-ERK1/2)in Tg cells, whereas p21 expression was increased (Fig. 2E). In addition, we showed that hIL-32α-Tg VSMCs also exhibited a marked reduction in MMP gelatinase activity, especially for MMP-2, which is important for VSMC migration (Figs. 2F, G) [29]. Taken together, these results demonstrated that IL-32α had an inhibitory effect on PDGF-BB-induced VSMC migration and proliferation, which likely occurred by regulating the expression of factors involved cell proliferation and migration and reducing MMP activity.
IL-32α inhibits vascular smooth muscle cell (VSMC) migration and proliferation. VSMCs were isolated from thoracic aortas of hIL-32α-Tg and non-Tg mice. (A) VSMCs were cultured in an ibidi μ-Dish with culture inserts for 24 h. Cells were then treated with PDGF-BB (25 ng/mL; 24 h) in serum-free media, and cell migration was monitored for 18 h by taking pictures every 3 h. The area of migrated cells was quantified (n = 3). Images shown are representative microscopy images at 18 h. (B) VSMCs were cultured in 96-well plates for 24 h, serum-starved for 12 h, then treated with PDGF-BB (25 ng/mL; 24 h), and cell proliferation was determined by BrdU incorporation assay (n = 5 each). (C-D) VSMCs from non-Tg mice were treated with rhIL-32α (25-100 ng/mL) for 24 h in serum-free medium, then cells were treated with PDGF-BB (25 ng/mL; 24 h). (C) cell migration (n = 3) and (D) cell proliferation (n = 5) were determined as described above (data shown as mean ± SEM, *p < 0.05 by Student's t-test). Images shown in C are representative microscopy images for each group. (E) VSMCs were lysed and analyzed by western blotting with antibodies against PCNA, p21, phospho-ERK1/2, and total-ERK1/2, using β-actin as a loading control. (F) For in vitro MMP activity assays, cells were cultured in 96-well plates for 24 h, then treated with PDGF-BB (25 ng/mL; 24 h). MMP activity was determined by cell-based enzyme-linked immunosorbent assay (ELISA) using DQ™-gelatin (data shown as mean ± SEM; n = 8, *p < 0.05 as determined by Student's t-test). (G) To evaluate specific MMPs activity, cells were cultured in 6-well plates for 24 h, then treated with PDGF-BB (25 ng/mL; 24 h), then gelatinolytic activity (MMP-2; 72 kDa) was determined in culture medium using gelatin zymography followed by Coomassie blue staining. GM6001 (40 μM), a broad-spectrum MMP inhibitor, was used as negative control. Bar diagrams presented fold change of MMP-2 considering the serum-free control density as 1 (data shown as mean ± SEM, n = 3, *p < 0.05 as determined by Student's t-test).
Human IL-32α expression inhibits vascular adhesion protein expression and arterial wall thickening in mice
Next, we examined whether our in vitro findings were relevant in vivo using hIL-32α-Tg mice. First, we verified arterial hIL-32α expression in Tg mice with reverse transcription-polymerase chain reaction (RT-PCR) and immunohistochemistry. As expected, IL-32α mRNA and protein were clearly expressed in hIL-32α-Tg mice arteries, particularly in the endothelial layer, but not in those from non-Tg littermate controls (Fig. 3A; Supplementary Fig. 1). Interestingly, we also found that basal VCAM-1 and ICAM-1 expression was lower in the aortas of hIL-32α-Tg mice as compared to non-Tg counterparts (Figs. 3B-E).
Vascular adhesion protein expression is suppressed in the artery endothelium of transgenic hIL-32α (hIL-32α-Tg) mice. (A-C) Thoracic aorta sections of hIL-32α-Tg and littermate non-Tg control mice were immunohistochemically stained for (A) IL-32α, (B) VCAM-1, and (C) ICAM-1. Images shown are representative microscopy images (n = 3 each). The red rectangle indicates the magnified area shown in the lower panel. Nuclei (blue) and protein expression (brown) are shown. Scale bar, 100 μm. Expression of (D) VCAM-1 and (E) ICAM-1 (red) in the endothelium was determined by immunofluorescence double-stained with a specific endothelium marker CD31 (cyan). Auto-fluorescence (yellow) shows internal elastic lamina (IEL). Nuclei were stained with DAPI (blue). The red rectangle indicates the magnified area shown in the lower panel. (F) VCAM-1 and ICAM-1 mRNA expression was determined by qPCR using endothelial-enriched RNA obtained from the left carotid artery (LCA) and right carotid artery (RCA) following partial carotid ligation in hIL-32α-Tg or non-Tg mice at 2 days post-ligation (data shown as mean ± SEM; n = 8, *p < 0.05 as determined by Student's t-test). L, lumen.
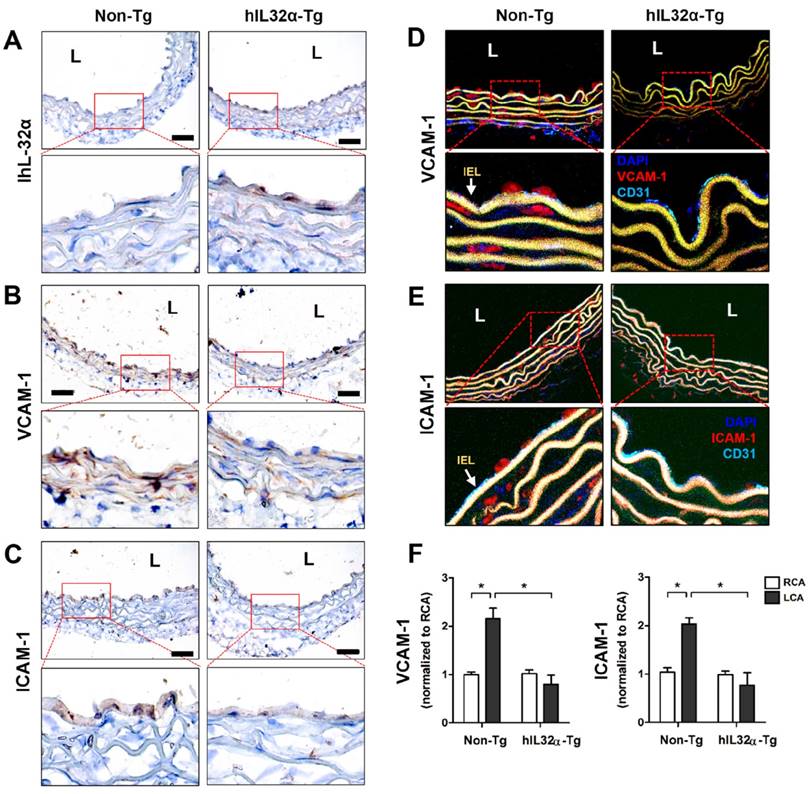
We next investigated whether IL-32α could induce VCAM-1 and ICAM-1 gene expression changes in arteries during vascular inflammation and atherosclerosis. For this, we employed the partial carotid ligation model, a mouse model of acute vascular inflammation and atherosclerosis [30, 31]. For this, endothelial-enriched RNA was collected from the partially-ligated left common carotid artery (LCA) and untouched right common carotid artery (contralateral control, RCA) of hIL-32α-Tg and non-Tg mice at 48 h post-ligation as previously described [30]. As expected, increased endothelial VCAM-1 and ICAM-1 expression was observed in partially ligated LCAs compared to that in the untouched RCA in non-Tg mice as previously shown [30, 31]. In comparison, hIL-32α-Tg mice showed reduced VCAM-1 and ICAM-1 expression in LCA as compared to those in non-Tg mice (Fig. 3F).
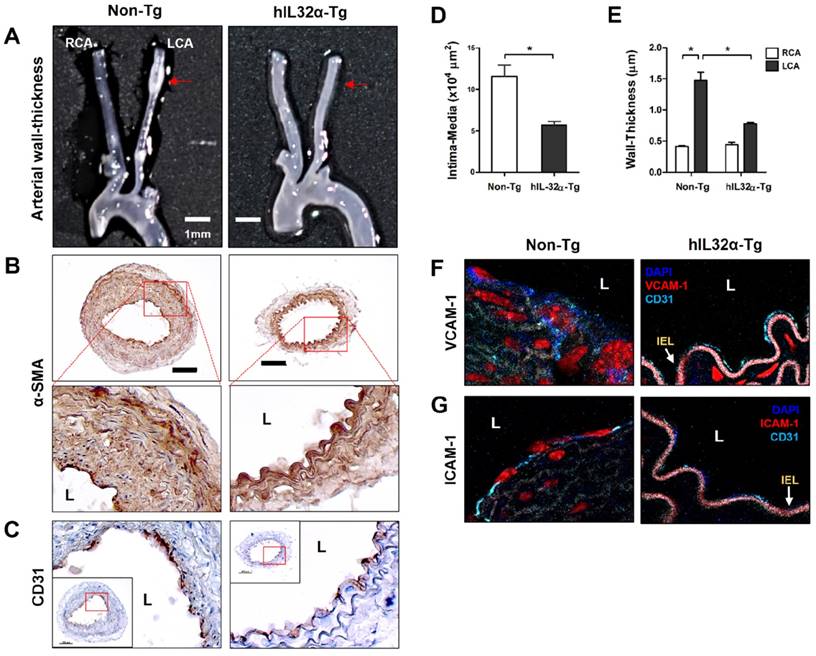
To assess the impact obesity on arterial wall thickening, we compared carotid artery wall thickness using the same ligation model in mice fed a high-fat diet. Significantly, partial LCA ligation induced arterial wall thickening—characterized by increased VSMCs in the intima-media and artery wall—in non-Tg mice fed a high-fat diet for 4 weeks, whereas hIL-32α-Tg mice showed significantly smaller lesion areas and arterial wall thicknesses (Figs. 4A-E). We further found that VCAM-1 and ICAM-1 expression in endothelium was dramatically decreased in hIL-32α-Tg-mice with LCA compared to non-Tg counterparts (Figs. 4F, G). These results clearly demonstrated that IL-32α overexpression inhibits vascular adhesion protein expression and arterial wall thickening.
Arterial wall thickening is diminished in hIL-32α-Tg mice. (A-C) For the assessment of arterial wall thickening, hIL-32α-Tg and non-Tg littermate control mice (n = 6 each) were partially ligated and fed a high-fat diet for 4 weeks. (A) Aortic trees including the carotid arteries were dissected and examined by bright-field imaging. Scale bar, 1 mm. (B-C) Frozen sections prepared from the lesion area of LCA, denoted by red arrows in A, were stained for (B) alpha-smooth muscle actin (α-SMA), a vascular smooth muscle cell marker, and (C) CD31, an endothelium marker. The red rectangle indicates the magnified area shown in the lower panel. Scale bar, 100 μm. The thicknesses of the (D) intima media and (E) wall were quantified (data shown as mean ± SEM; n = 6, *p < 0.05 as determined by Student's t-test). Expression of (D) VCAM-1 and (E) ICAM-1 (red) in the intimal layer was determined by immunofluorescence double-stained with a specific endothelium marker CD31 (cyan). Auto-fluorescence (pink) shows internal elastic lamina (IEL). Nuclei were stained with DAPI (blue). L, lumen.
Identification of genes regulated by human IL-32α in the mouse arterial endothelium
Since IL-32α appeared to an effective regulator of vascular inflammation and arterial wall thickening, we decided to perform a DNA microarray study using endothelial-enriched RNA obtained from the carotid arteries of hIL-32α-Tg and non-Tg mice 48 h post-partial ligation. As shown in the heat maps (Fig. 5A), the results showed remarkably low variation within each group, demonstrating the reproducibility of the data. Notably, we found 2195 genes (1876 upregulated and 261 downregulated) significantly altered by ≥2-fold in hIL-32α-Tg mice when assuming a 10% false discovery rate (Fig. 5C), while 4251 genes (3557 upregulated and 517 downregulated) were changed in the non-Tg LCA endothelium (Fig. 5B). Moreover, 487 genes (431 upregulated and 56 downregulated) were significantly changed (> 2-fold) in the untouched RCA endothelium of hIL-32α-Tg mice compared with non-Tg counterparts (Fig. 5D). In contrast, 33 genes were significantly upregulated, whereas 336 genes were significantly downregulated in the partially ligated LCA endothelium of hIL-32α-Tg mice compared to that in the non-Tg mice LCA endothelium (Fig. 5E). These findings suggest that IL-32α modulates the expression of endothelial genes under both intact and atherogenic conditions. Since the purpose of this microarray was to identify IL-32α regulatory genes involved in its inhibitory role on endothelial inflammation and atherosclerosis, we focused on the 369 genes whose expression significantly differed between LCA endothelium samples from hIL-32α-Tg and non-Tg mice. The detailed list of the significantly changed genes is shown in Supplementary Table 1.
Global gene expression profiles in the carotid artery endothelium of hIL-32α-Tg mice. Total RNAs were obtained from the intima of mouse LCA and RCA at 48 h after ligation. Affymetrix GeneChip® Mouse Gene 2.0 ST arrays containing 770,317 mouse gene probes were used. (A) Hierarchical clustering analysis results indicating the genes whose expression significantly differed between hIL-32α-Tg mice LCA endothelium and non-Tg mice LCA endothelium are shown as heat maps. Each column represents a single sample pooled from 3 different LCAs, and each row represents a single gene probe. (B-E) Scatter plots show normalized intensities of each probe under 4 comparison conditions: (B) non-Tg LCA vs. non-Tg RCA, (C) hIL-32α-Tg LCA vs. hIL-32α-Tg RCA, (D) hIL-32α-Tg RCA vs. non-Tg RCA, and (E) hIL-32α-Tg LCA vs. non-Tg LCA after ligation. Genes are shown that were up-regulated (red) or down-regulated (blue) (≥2-fold difference) at a false discovery rate of ≤10% in LCA compared with RCA. Microarray results for selected (F) pro-atherogenic and (G) anti-atherogenic genes are shown as the fold-increase or fold-decrease of gene expression in hIL-32α-Tg LCA compared with that in non-Tg LCA on a log2 scale (mean ± SEM). All genes shown in the graphs were significantly different between groups (n = 3, p < 0.05 as determined by Student's t-test).
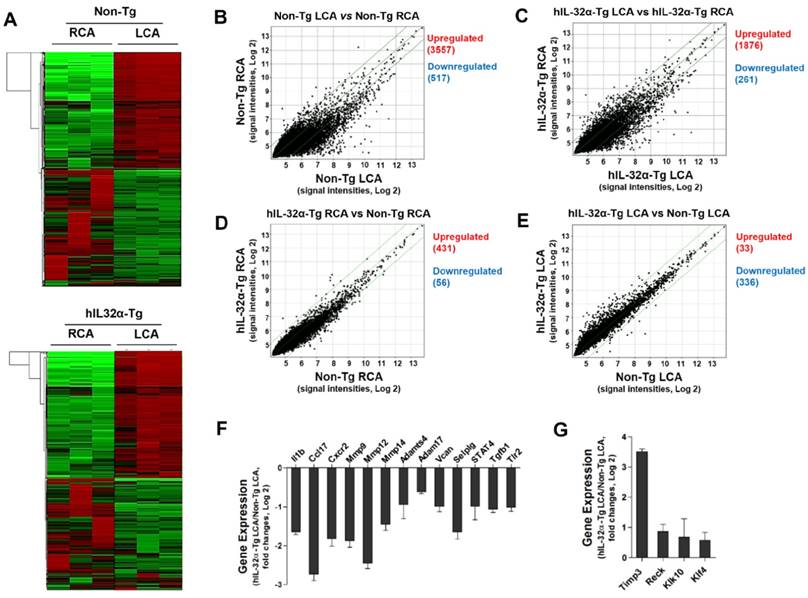
To understand the functional importance of these significantly changed genes at the systems level, we performed co-expression network analysis on the microarray data. Of the 23 co-expression modules, the IL-32α_M14 module was significantly associated with hIL-32α expression in the mouse model of endothelial inflammation and atherosclerosis (Supplementary Table 2). In particular, analysis of the hIL-32α-Tg mice LCA endothelium showed a significant downregulation of genes involved in the immune and inflammatory responses, cell migration, and apoptosis (Supplementary Fig. 2; Supplementary Table 3), suggesting that hIL-32α overexpression may directly inhibit immune and inflammatory gene expression in this context.
We then compared the network associated with IL-32α to that from a previous cell culture study to further explore potential physiological mechanisms. For this, we generated a co-expression network from publicly available microarray data on TNFα-stimulated HUVECs without and with exposure to anti-inflammatory oligomeric procyanidins (OPCs) [32]. Of the 25 co-expression modules, two were significantly associated with exposure to the oligomers in TNF-α treated HUVECs (Supplementary Table 4). Specifically, the OPC_M18 module—consisting of genes related to apoptosis, cell migration, and cell cycle—was negatively associated with OPCs treatment (Supplementary Table 5), while the opposite was found with the OPC_M25 module that includes genes related to viral immunity, angiogenesis, and immune response (Supplementary Table 6). A substantial number of nodes (genes) were shared between the IL-32α_M14 module and the 2 modules associated with OPC treatment (Supplementary Fig. 3), suggesting while IL-32α may directly inhibit the expression of genes involved in the immune and inflammatory responses, OPCs may also regulate these genes indirectly.
To further explore the functional importance of IL-32α-regulated genes, we performed functional annotation analysis on the list of the significantly changed genes. Notably, KEGG pathway analysis showed a marked enrichment in genes involved in disease processes mediated through changes in signal transduction, cell adhesion, cell proliferation, cell differentiation, cell cycle, immune response, inflammatory response, cell behavior, and apoptosis (Supplementary Table 7), such as autoimmune diseases, cancer, diabetes, and cardiovascular diseases. These include inflammation (Il1b, Esm1, Prg4, Ccl17, Ccl22, Ccl3, Il1r2, Il1f9, Cxcr2, Cxcr4, Il18rap, Ccr1, and Il17ra), adhesion molecules (Pgm5, Itgax, and Cd80), cell response (Hba-a2), and extracellular matrix (ECM) degradation (Timp3, Reck, Mmp9, Mmp12, Adamts4, and Vcan). Genes related to inflammation, cell cycle, and cell proliferation that showed significant changes in expression are listed in Supplementary Table 8. Moreover, we found that several pro-atherogenic genes were downregulated (Fig. 5F), whereas anti-atherogenic genes were upregulated in the hIL-32α-Tg LCA endothelium (Fig. 5G). These results suggested that hIL-32α expression modulates genes that regulate inflammatory and immune responses, as well as those involved in cell proliferation and migration in the mouse arterial endothelium.
Human IL-32α inhibits endothelial inflammation and VSMC activation by upregulating Timp3 and Reck
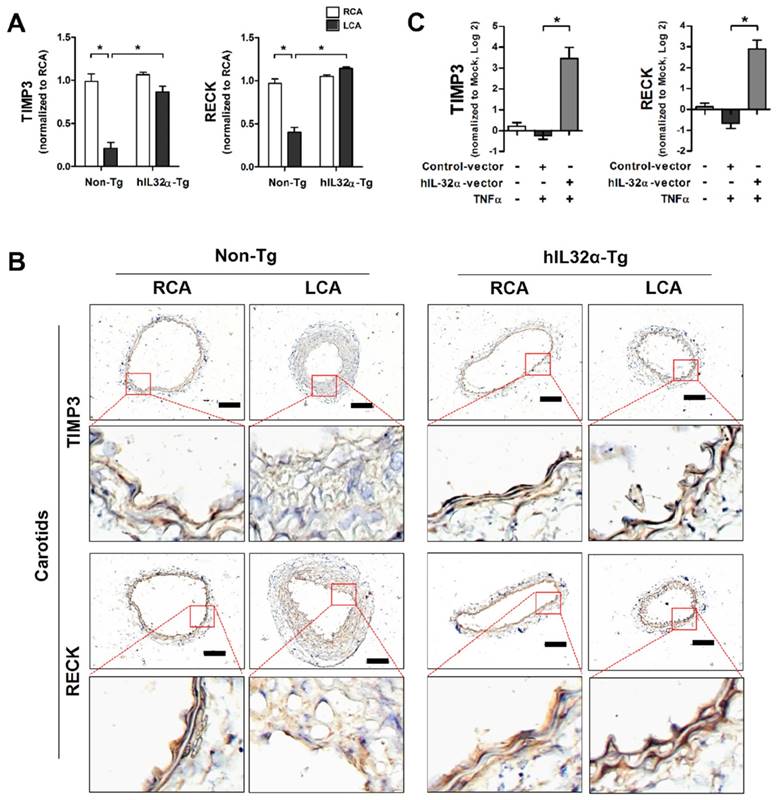
Timp3 and Reck are endogenous vascular protective genes reported to have functional importance in vascular disease development and progression [33, 34]. As such, we decided to focus on these genes to determine the functional significance of IL-32α in vascular dysfunction and disease. Importantly, quantitative polymerase chain reaction (qPCR) and immunohistochemistry analysis showed that Timp3 and Reck mRNA and protein expression were significantly decreased in partially-ligated LCA compared with contralateral RCA in non-Tg control mice (Figs. 6A, B), consistent with previous findings [34, 35], but upregulated in the LCA and aorta tissue from hIL-32α-Tg-mice LCA (Figs. 6A, B; Supplementary Fig. 4A). This result was further confirmed in hIL-32α-overexpressing and rhIL-32α-treated ECs in vitro (Fig. 6C; Supplementary Fig. 4B). In addition, endothelial constitutive nitric oxide synthase (eNOS; Nos3) mRNA expression was decreased in the non-Tg LCA endothelium by partial ligation, but upregulated in hIL-32α-Tg-mice (Supplementary Fig. 5A). Moreover, IL-1β and MMP9 were markedly downregulated in hIL-32α-Tg-mice LCA endothelium compared with that of controls (Supplementary Figs. 5B, C). These results demonstrated that hIL-32α expression rescues the loss of endogenous vascular protective genes (Timp3, Reck, and eNOS) and suppresses pro-atherogenic genes (IL-1β and MMP9) in inflamed vascular arteries.
IL-32α upregulates Timp3 and Reck expression. (A) Timp3 and Reck mRNA expression was determined by qPCR using endothelial-enriched RNA obtained from the LCA and RCA following partial carotid ligation in hIL-32α-Tg or non-Tg mice at 2 days post-ligation (n = 8 each), or (C) RNA obtained from the human IL-32α -expressing pcDNA3.1+-6×Myc vector or control vector (hIL-32α-vector) transfected iMAECs (n = 4) (data shown as mean ± SEM, *p < 0.05 as determined by Student's t-test). (B) Frozen sections of carotid arteries (partially ligated LCA and untouched RCA, 2 days post-ligation) from hIL-32α-Tg and littermate non-Tg control mice were stained with antibodies against Timp3 and Reck. Images shown are representative microscopy images (n = 4 each). The red rectangle indicates the magnified area shown in the lower panel. Nuclei (blue) and protein expression (brown) are shown. Scale bar, 100 μm.
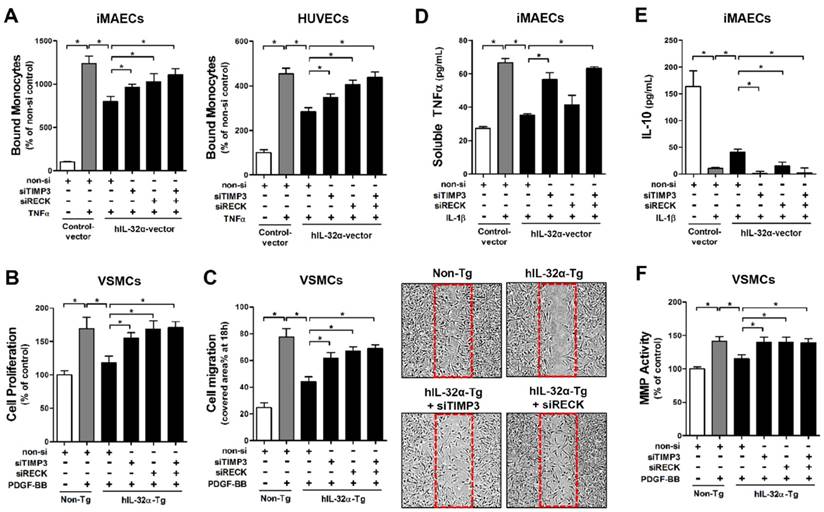
Since we confirmed that IL-32α positively regulates Timp3 and Reck expression, we next tested whether IL-32α could inhibit EC inflammation and VSMC proliferation and migration. For this, we performed monocyte-EC adhesion assays and VSMC proliferation and migration assays with cells transfected with siRNA specific to Timp3 or Reck. Notably, we found that the inhibitory effect of hIL-32α on monocyte-EC binding was attenuated by Timp3 or Reck knockdown in iMAECs and HUVECs (Fig. 7A), as well as VSMC proliferation and migration in hIL-32α-Tg mice VSMCs (Figs. 7B, C). Because Timp3 and Reck are endogenous MMP and a disintegrin and metalloprotease (ADAM) inhibitors, we next examined their importance in IL-32α-mediated MMP and ADAM inhibition. The results showed that IL-1β-induced soluble-TNFα release into the conditioned medium—an indicator of ADAM17 (also known as TACE) activity—was reduced in hIL-32α-expressing iMAECs, and was attenuated by the knockdown of Timp3, but not Reck (Fig. 7D). Interestingly, the decrease in IL-10 production mediated by IL-1β was rescued in hIL-32α-expressing iMAECs, which was attenuated by knockdown of either Timp3 or Reck (Fig. 7E). In addition, IL-32α-dependent MMP inhibition in VSMCs was also blunted in Timp3 or Reck knockdown cells (Fig. 7F). Together, these results indicated that IL-32α upregulates the Timp3 and Reck expression, which in turn inhibits EC inflammation and VSMC proliferation and migration.
IL-32α inhibits EC inflammation and VSMC proliferation through Timp3 and Reck. (A) iMAECs and HUVECs were transfected with Timp3 or Reck siRNA (siTimp3 or siReck, 150 nM) in the presence of human IL-32α-expressing pcDNA3.1+-6×Myc vector (hIL-32α-vector). Cells were then treated with TNFα (10 ng/mL; 24 h), and monocyte-EC adherence was determined (n = 6 each). (B-C) VSMCs isolated from the thoracic aortas of hIL-32α-Tg and non-Tg mice were transfected with siTimp3 or siReck (150 nM), then VSMC (B) proliferation and (C) migration were determined by wound healing (n = 3) and BrdU incorporation assays (n = 5), respectively, as described in Methods. Images shown in C lower panels are representative microscopy images of each group. (D-E) iMAECs were transfected with siTimp3 or siReck (150 nM), then stimulated with IL-1β (10 ng/mL), and (D) soluble TNFα and (E) IL-10 in conditioned media was determined by ELISA (n = 3). (F) VSMCs isolated from the thoracic aortas of hIL-32α-Tg and non-Tg mice were transfected with siTimp3 or siReck (150 nM), then MMP activity was determined by cell-based ELISA using DQ™-gelatin (n = 8). All data shown as mean ± SEM, *p < 0.05 as determined by Student's t-test.
IL-32α inhibits miR-205 expression by regulating microRNA biogenesis
Previous studies demonstrate that miR-205 blocks Timp3 and Reck expression through direct binding to their 3ʹ untranslated regions (3′UTR), causing EC inflammation, vascular dysfunction, atherosclerosis, and abdominal aortic aneurysm (AAA) [33-35]. Moreover, miR-205 interacts with IL-32 in prostate cancer cells to regulate cell cycle and viability [36]. Consistent with previous studies [33-35], our dual-luciferase reporter assay confirmed that miR-205 directly binds to the 3′UTR of Timp3 and Reck and inhibits their expression in iMAECs (Supplementary Fig. 6). However, it is unknown whether the regulatory role of IL-32α on Timp3 and Reck expression has any association with the expression and function of miR-205. To address this knowledge gap, we focused on miR-205 to investigate the possibility that IL-32α might increase Timp3 and Reck expression by downregulating miR-205 even if other miRNAs may be involved in this phenomenon. Thus, we first examined whether IL-32α could regulate miR-205 expression in ECs. Consistent with a previous study [34], miR-205 expression (both precursor and mature forms) was upregulated in partially-ligated LCA endothelium compared with RCA in non-Tg mice, and markedly decreased in hIL-32α-Tg LCA compared with that in non-Tg LCA (Fig. 8A). We further confirmed that IL-32α overexpression significantly suppressed TNFα-induced miR-205 expression in both iMAECs and HUVECs (Fig. 8B). These results demonstrated that IL-32α effectively inhibits miR-205 expression in inflamed ECs in vitro and in vivo. To examine the biological relevance of IL-32α-dependent miR-205 downregulation, we examined whether IL-32α was sufficient to rescue Timp3 and Reck expression in cells treated with a miR-205 mimic. Consistent with previous studies [33, 34], treatment with the miR-205 mimic (mature form) significantly decreased Timp3 and Reck expression in iMAECs (Supplementary Fig. 7A), and was unable to be rescued by hIL-32α expression (Supplementary Fig. 7A). In addition, miR-205-induced monocyte-EC adhesion and VSMC proliferation and migration were not altered by hIL-32α expression (Supplementary Figs. 7B, C), suggesting that IL-32α suppresses miR-205 expression but may not otherwise influence its biological functions.
IL-32α inhibits microRNA-205 expression by regulating microRNA biogenesis. (A) Expression of precursor and mature forms of miR-205 was determined by qPCR using endothelial-enriched RNA obtained from the LCA and RCA following partial carotid ligation in hIL-32α-Tg or non-Tg mice at 2 days post-ligation (n = 5 each). (B) iMAECs and HUVECs were cultured for 24 h, then transfected with human IL-32α-expressing pcDNA3.1+-6×Myc vector (hIL-32α-vector) or control vector. After 24 h, cells were treated with TNFα (10 ng/mL; 24 h), and the expression of mature miR-205 was determined by qPCR (n = 4). (C) Dgcr8, Drosha, and Dicer1 mRNA expression in mouse LCA (2 days post-ligation) was determined by qPCR (n = 5). (D) iMAECs were transfected with Dgcr8, Drosha, or Dicer1 siRNA (150 nM) for 24 h. The cells were then treated with TNFα and the expression of mature miR-205 was determined by qPCR (n = 5 each). (E) Rprd2 and Ddx5 expression in mouse RCA and LCA (2 days post-ligation) was determined by qPCR (n = 5). (F) iMAECs were transfected with hIL-32α or control vector. After 24 h, cells were treated with TNFα (10 ng/mL; 24 h), and Rprd2 and Ddx5 mRNA expression was determined by qPCR (n = 5 each). (G-H) iMAECs were transfected with Rprd2 siRNA (200 nM) for 24 h, and the expression of precursor and mature forms of (G) miR-205, and (H) Timp3 and Reck mRNA was determined by qPCR (n = 6 each). All data shown as mean ± SEM, *p < 0.05 as determined by Student's t-test.
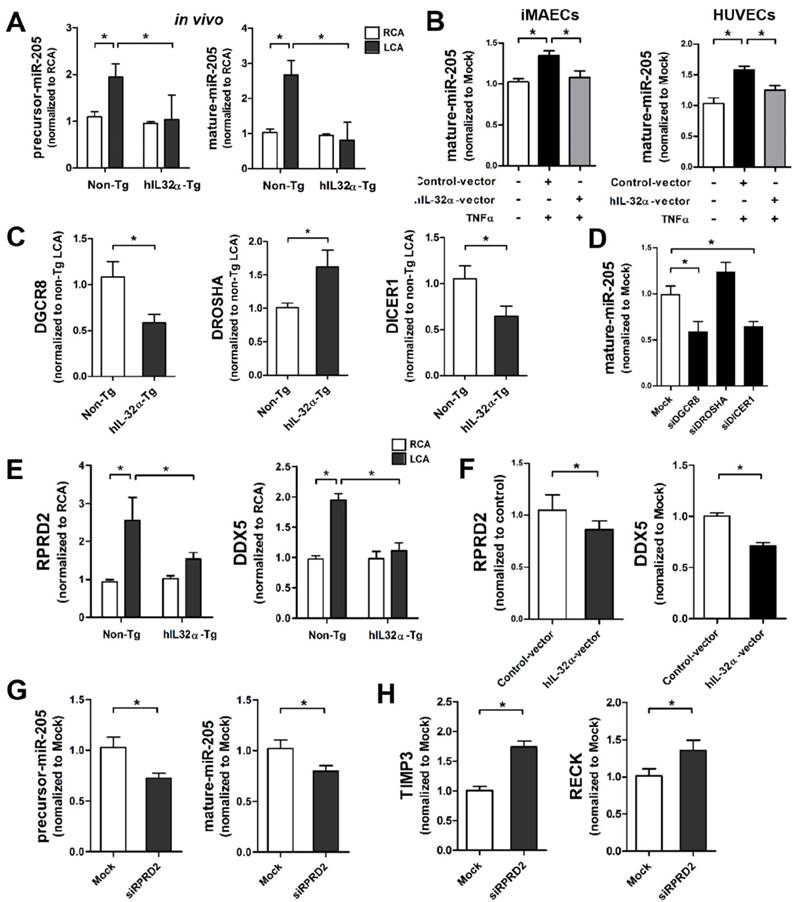
Since IL-32α was unable to affect the biological functions of miR-205, we speculated that it might inhibit miR-205 expression by regulating miRNA biogenesis. Thus, we first investigated whether IL-32α affected the expression of Dgcr8, Drosha, and Dicer1, which are important components of the canonical miRNA biogenesis pathway [37, 38]. Interestingly, Dgcr8 and Dicer1 expression was significantly decreased in hIL-32α-Tg mice LCA endothelium compared with that of non-Tg mice LCA, whereas Drosha was increased (Fig. 8C). These results suggest that IL-32α may inhibit miR-205 biogenesis by regulating Dgcr8 and Dicer1 independent of Drosha. To confirm this theory, we knocked down Dgcr8 and Dicer1 using specific siRNAs, and then analyzed miR-205 expression in ECs. Notably, Dgcr8 and Dicer1 knockdown significantly decreased mature miR-205 expression in iMAECs, whereas the Drosha knockdown cells showed an insignificant increase in miR-205 expression (Fig. 8D). These results indicated that miR-205 is processed via a Dgcr8- and Dicer1-dependent mechanism, but independent of Drosha. Additionally, we found that Dgcr8 and Dicer1 knockdown inhibited monocyte-EC binding and VSMC proliferation and migration (Supplementary Figs. 8A, B), suggesting that Dgcr8 and Dicer1 have important roles in EC inflammation and VSMC activation.
To further investigate the regulatory role of IL-32α on Dgcr8 and Dicer1 expression, we performed a Gene Ontology (GO) analysis using GeneMania (http:www.genemania.org) to generate interaction networks between IL-32α and Dgcr8 and Dicer1. This strategy uncovered tight networking of the 3 input genes (IL-32, Dgcr8, and Dicer1) with 20 associated genes including Tarbp2 and Ago2, which are well-known miRNA processing cofactors (Supplementary Fig. 9). Of these 20 genes, we selected Rprd2, Khsrp (also known as KSRP), and Ddx5 as IL-32α-regulated components that may have important roles in the regulation of Dgcr8- and Dicer1-dependent miR-205 biogenesis based on their known roles in RNA synthesis and miRNA processing [39-42]. The qPCR results showed increased Rprd2 and Ddx5 mRNA expression in partially-ligated LCA as compared to unligated RCA in non-Tg control mice, while that of Khsrp was not significantly changed (Fig. 8E; Supplementary Fig. 10A). Interestingly, we found that expression of Rprd2 and Ddx5, but not Khsrp, was markedly decreased in hIL-32α-Tg-mice LCA compared to non-Tg mice LCA (Fig. 8E; Supplementary Fig. 10A). This result was further confirmed in hIL-32α expression vector-transfected ECs in vitro (Fig 8F, Supplementary Fig. 10B), suggesting that IL-32α may inhibit Dgcr8- and Dicer1-dependent miR-205 biogenesis by regulating Rprd2 and Ddx5 expression.
Although RPRD family genes, including Rprd2, are involved in transcription by interacting with RNA polymerase II [41, 43], which transcribes miRNAs, their roles in miRNA biogenesis have not been reported yet. Therefore, we examined the involvement of Rprd2 in miR-205 biogenesis and found that its knockdown resulted in the decreased the expression of both precursor and mature miR-205 in ECs (Fig. 8G), increased Timp3 and Reck expression (Fig. 8H), and the inhibitory effects of IL-32α on monocyte-EC binding and VSMC proliferation and migration (Supplementary Figs. 11A, B). These results suggested that Rprd2 regulates miR-205 biogenesis and its biological functions.
Additionally, we observed that expression of exoribonuclease XRN1—a recently identified non-canonical pathway miRNA processor [34]—was decreased in LCA endothelium by partial ligation and rescued in IL-32α-Tg mice (Supplementary Fig. 12A). This observation suggested that IL-32α may regulate the expression of other miRNAs by influencing Xrn1-dependent non-canonical miRNA biogenesis. Consistent with the notion, we found decreased expression of miR-712—an atypical miRNA derived from 45S pre-ribosomal RNA (RN45S) via a XRN1-dependent non-canonical processing—in hIL-32α-Tg mice LCA endothelium as compared to non-Tg mice (Supplementary Fig. 12B). Interestingly, miR-712 was previously identified as a murine homologue of miR-205 and shown to induce vascular inflammation, atherosclerosis, and AAA by targeting Timp3 and Reck [33, 34]. This raises the possibility that IL-32α could inhibit vascular inflammation, vascular wall thickening, and atherosclerosis by inhibiting pro-atherogenic miRNA biogenesis through Rprd2-Dgcr8/Ddx5-Dicer1-dependent canonical and XRN1-dependent non-canonical pathway regulation. To the best of our knowledge, this is the first report demonstrating that IL-32α can regulate miRNA expression through both the canonical and non-canonical miRNA biogenesis pathways.
Human IL-32α inhibits atherosclerosis development in ApoE-/- mice
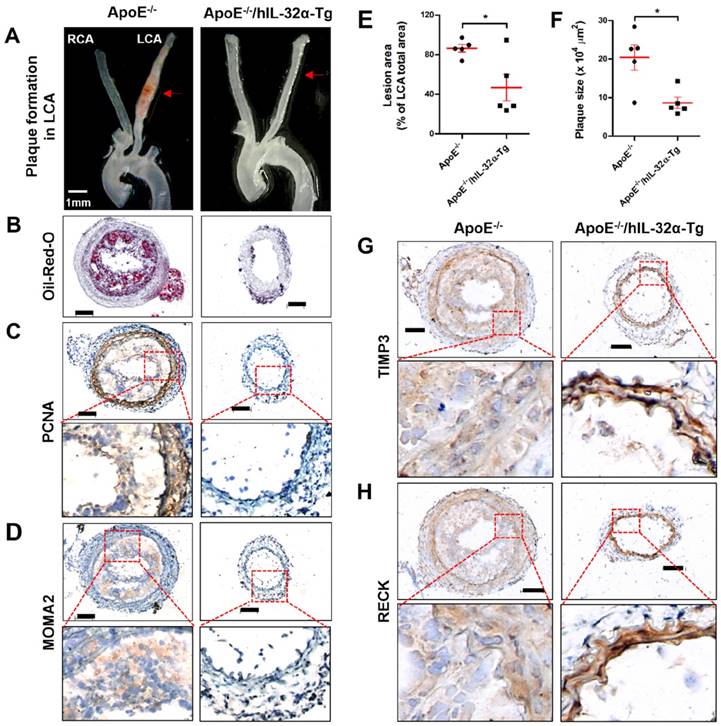
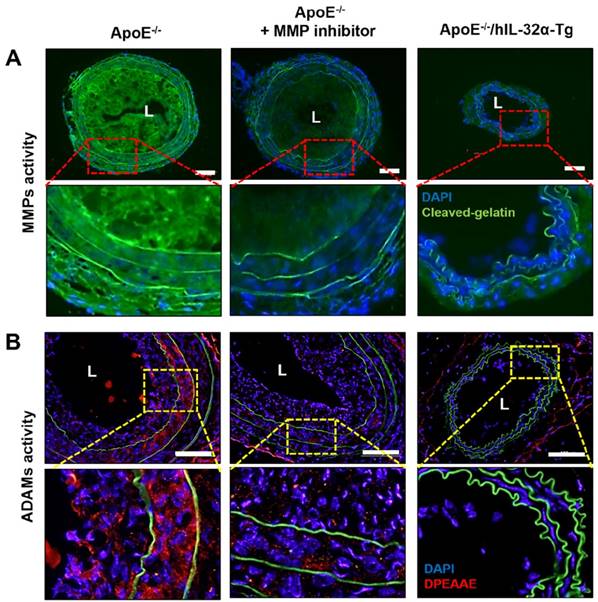
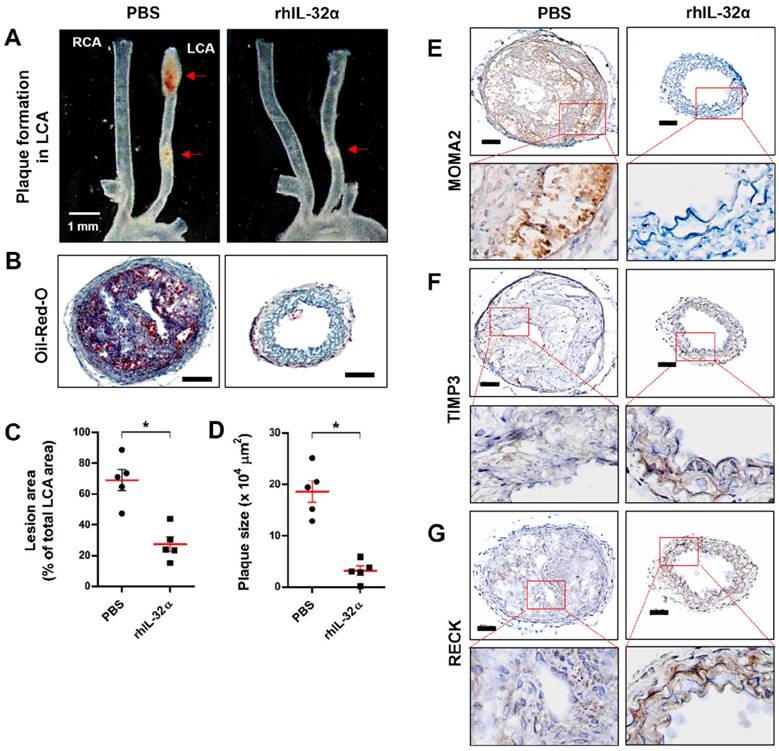
Finally, we tested the role of IL-32α in atherosclerotic plaque development in the carotid arteries of ApoE-/-/hIL-32α-Tg mice and ApoE-/-/non-Tg littermates using an acute partial ligation model of atherosclerosis [30, 34]. As expected, partial ligation of the LCA rapidly induced atherosclerosis in ApoE-/-/non-Tg littermate control mice given a high-fat diet by 2 weeks post-ligation, whereas the contralateral RCA remained lesion-free (Figs. 9A, B). In comparison, ApoE-/-/hIL-32α-Tg mice showed significantly less lesion development (Figs. 9A, B, E, F). Moreover, PCNA and MOMA2 immunostaining revealed increased atherosclerosis and leukocyte infiltration in the LCA of ApoE-/-/non-Tg mice, respectively, but staining for both markers was dramatically lower in ApoE-/-/hIL-32α-Tg mice (Figs. 9C, D). We further confirmed that Timp3 and Reck expression was higher in ApoE-/-/hIL-32α-Tg mice than in ApoE-/-/non-Tg littermate controls (Figs. 9G, H; Supplementary Fig. 13), consistent with our observations in hIL-32α-Tg mice. Furthermore, increased MMP and ADAM activity was observed in the LCA of ApoE-/-/non-Tg controls, but strongly blunted in ApoE-/-/hIL-32α-Tg mice (Fig. 10A, B). In addition, we found that the administration of rhIL-32α protein (1 μg/mouse) potently inhibited atherosclerotic lesion development (Figs 11. A-D) and leukocyte infiltration (Fig. 11E), and increased Timp3 (Fig. 11F) and Reck (Fig. 11G) expression in the LCA of ApoE-/-/non-Tg mice. Collectively, these results provide strong evidence for a direct hierarchical link connecting IL-32α with Timp3 and Reck and their effects on MMP activity, EC inflammation, arterial wall thickening, and atherogenesis.
hIL-32α inhibits atherosclerosis in ApoE-/-/hIL-32α-Tg mice. ApoE-/-/hIL-32α-Tg and littermate ApoE-/-/non-Tg control mice were partially ligated and fed a high-fat diet for 2 weeks. (A) Aortic trees including the carotid arteries were dissected and examined by bright-field imaging, and the lesion area was quantified in E (data shown as mean ± SEM; n = 5, *p < 0.05 as determined by Student's t-test). Scale bar, 1 mm. (B) Frozen sections prepared from the lesion area of LCA, denoted by red arrows in A, were stained with oil red O and plaque size was quantified in F (data shown as mean ± SEM; n = 5, *p < 0.05 as determined by Student's t-test). Scale bar, 100 μm. (C-D, G-H) LCA frozen sections were used for immunostaining with antibodies against (C) PCNA, (D) MOMA2, (G) Timp3, and (H) Reck. Images shown are representative microscopy images (n = 5 each). The red rectangle indicates the magnified area shown in the lower panel. Nuclei (blue) and protein expression (brown) are shown. Scale bar, 100 μm.
MMP and ADAM activity is dampened in the atherosclerotic plaques of ApoE-/- /hIL-32α-Tg mice. (A-B) ApoE-/-/hIL-32α-Tg and littermate ApoE-/-/non-Tg control mice were partially ligated and fed a high-fat diet for 2 weeks. Frozen sections prepared from these mice. (A) MMP activity in LCA was determined by in situ zymography using DQ™-gelatin (green). The red rectangle indicates the magnified area shown in the lower panel. DAPI (blue); autofluorescent elastic lamina (green line). (J) ADAM activity in LCA was determined by immunofluorescence staining with an antibody specific to the versican fragment peptide DPEAA (red). The yellow rectangle indicates the magnified area shown in the lower panel. DAPI (blue); autofluorescent elastic lamina (green line). Sections from ApoE-/-/non-Tg mice were incubated with the MMP inhibitor GM6001 (50 μM) as a negative control. Scale bar, 100 μm.
Administration of recombinant human IL-32α protein inhibits atherosclerosis in ApoE-/- mice. (A-C) ApoE-/- mice were pretreated twice with rhIL-32α (1 μg/mouse, administered by intraperitoneal injection) or PBS 1 and 2 days before partial ligation. Mice were then fed a high-fat diet and rhIL-32α treatments were administered every 2 days for 2 weeks. Aortic trees including the carotid arteries were dissected and examined by bright-field imaging, and the lesion area was quantified in C (data shown as mean ± SEM; n = 5, *p < 0.05 as determined by Student's t-test). Scale bar, 1 mm. (B) Frozen sections prepared from the lesion area of LCA, denoted by red arrows in A, were stained with oil red O and plaque size was quantified in D (data shown as mean ± SEM; n = 5, *p < 0.05 as determined by Student's t-test). Scale bar, 100 μm. (E-G) LCA frozen sections were used for immunostaining with antibodies against (E) MOMA2, (F) Timp3, and (G) Reck. Images shown are representative microscopy images (n = 5 each). The red rectangle indicates the magnified area shown in the lower panel. Nuclei (blue) and protein expression (brown) are shown. Scale bar, 100 μm.
Discussion
Although the association of IL-32 with vascular inflammation and atherosclerosis has been suggested previously [13, 15, 16], compelling evidence for its functional role and the underlying mechanism has been scarce, largely because of a lack of adequate animal models and methods to test these functions in vivo. In the current study, we employed a partial carotid ligation mouse model that rapidly induces gene expression changes, endothelial inflammation, and atherosclerosis [30, 31, 44]. We also used a powerful method to extract endothelial RNA from the mouse carotid intima [30], which enabled us to perform genome-wide studies to identify IL-32α-regulated genes in hIL-32α-Tg mice. Additionally, we generated ApoE-/-/hIL-32α-Tg-mice to provide direct evidence for the inhibitory role of IL-32α on atherosclerosis development in vivo.
While investigating the role of IL-32α in vascular inflammation and atherosclerosis, IL-32α was initially identified as an anti-inflammatory isoform based on its effects on inflammatory marker expression and leukocyte adhesion in TNFα-stimulated ECs in vitro. We further found that PDGF-BB-induced cell proliferation and migration were decreased in primary-cultured aortic VSMCs from hIL-32α-Tg mice. Subsequent in vivo functional studies showed that hIL-32α expression potently inhibits vascular inflammatory gene expression and arterial wall thickening in the mouse partial carotid ligation model. Furthermore, the results from ApoE-/-/hIL-32α-Tg mice and rhIL-32α-administrated ApoE-/- mice provided clear evidence that IL-32α potently blocks atherosclerosis by inhibiting cell proliferation and monocyte infiltration. These results suggested that IL-32α is an anti-inflammatory and anti-atherogenic cytokine that inhibits vascular inflammation and atherosclerosis.
To investigate the anti-inflammatory and anti-atherogenic mechanisms of IL-32α, we performed in vivo endothelial DNA microarray experiments to identify IL-32α-regulated genes using the partial carotid ligation mouse model and the endothelial RNA extraction method. We used endothelial-enriched RNAs rather than whole aortic wall samples containing RNA from multiple cell types including ECs, VSMCs, and immune cells to reduce the variation among different samples, which could largely dictate the outcomes of the array results depending on the proportions of cell types in each tissue sample. From this, we identified 369 genes (33 up-regulated and 336 down-regulated) that changed significantly at 48 h after partial ligation in the hIL-32α-Tg mice LCA endothelium compared with the control non-Tg LCA. Furthermore, in silico analyses demonstrated that hIL-32α potently suppresses co-expression gene networks associated with immune and inflammatory response, cell migration, and apoptosis in the inflamed endothelium. In addition, comparative network analysis between our microarray experiment and previously published results from TNFα-treated HUVECs suggested that IL-32α directly regulates the expression of genes related to immune and inflammatory responses. This is the first genome-wide DNA microarray study revealing the gene expression changes in response to IL-32α expression in vivo.
One of the interesting findings of this study was the observation that pro-atherogenic genes were downregulated, whereas anti-atherogenic genes were upregulated in the hIL-32α-Tg mice LCA endothelium. In particular, Timp3 and Reck are vascular protective genes that inhibit EC inflammation, endothelial permeability, and ECM remodeling by inhibiting metalloproteinases—such as MMP2/9, ADAM10, ADAMTS4, and ADAM17—that are well-established disease mediators in atherosclerosis and AAA [33, 34, 45-48]. Timp3 has been identified in atherosclerotic plaque macrophages between in the necrotic/lipid-rich core and the protective fibrous cap [49], and consequently plays a protective role against plaque rupture and AAA by suppressing local proteolysis [50]. VSMC migration from the medial layer to the intima along with ECM remodeling are results of the dynamic balance of matrix synthesis and degradation and associated with MMP activity and their respective endogenous inhibitors including Timp3 and Reck [51]. In vascular remodeling, MMPs not only synthesize ECM, but also induce ECM degradation and remodeling, as well as VSMC proliferation and migration [52]. However, the interaction between the MMPs and Timp3/Reck are not well described and the subject of active investigation. Furthermore, a correlation between IL-32α treatment and Timp3 and Reck expression has not previously been reported. In the present study, we found that Timp3 and Reck expression increased in hIL-32α-expressing mice arteries and in response to rhIL-32α treatment. In further experiments, we found that the inhibitory effects of IL-32α on ADAM17 and MMPs were blunted by the knockdown of Timp3 and Reck, which blocked the inhibitory role of IL-32α on EC inflammation and VSMC proliferation and migration. These results suggest that IL-32α inhibits EC inflammation, VSMC activation, and atherosclerosis by upregulating of Timp3 and Reck expression.
Over the last decade, cytokines have been found to modulate the functions of ECs, VSMCs, and macrophages by interacting with miRNAs to influence vascular inflammation and atherosclerosis [53-57]. IL-32 also has been reported to interact with miRNAs including miR-205, miR-663, miR-675, and hcmv-miRUL112-1 [36, 58]. Interestingly, miR-205 reportedly interacts with IL-32 in prostate cancer cells [36] and was identified as a key player in EC inflammation, AAA, and atherogenesis [33, 34, 59]. In addition, previous studies demonstrated that increased arterial miR-205 expression enhances EC inflammation, AAA, and atherosclerosis by suppressing Timp3 and Reck, leading to MMP and ADAM activation [33, 34]. However, the correlation between IL-32α and miR-205 in vascular inflammation and atherosclerosis has not previously been studied. In the current study, we showed that hIL-32α overexpression significantly decreased endogenous miR-205 expression, although the biological function of an exogenous miR-205 mimic was unaltered by hIL-32α expression. We further found that miR-205 was processed via a Rprd2-Dgcr8/Ddx5-Dicer1 axis-dependent canonical miRNA biogenesis pathway, which was effectively suppressed by IL-32α in inflamed ECs. Moreover, we demonstrated that IL-32α suppressed Rprd2-dependent miR-205 biogenesis and thereby increased Timp3 and Reck expression. Thus, these findings strongly suggested that IL-32α preserves Timp3 and Reck expression and functions by inhibiting miR-205 expression by regulating the Rprd2-Dgcr8/Ddx5-Dicer1 axis-dependent miRNA biogenesis pathway.
Intriguingly, although IL-32 has been identified as a pro-inflammatory cytokine [60], its anti-inflammatory function in diverse diseases has been reported with some conflicting findings [18, 19, 23, 61-63]. In previous studies, we also demonstrated an inhibitory role of the IL-32 isoforms (α, β, γ) in inflammatory diseases [19, 21], cancer [61, 62, 64, 65], and liver damage [18, 23]. Interestingly, a previous study suggested a paradoxical dual function of IL-32γ in colitis [66]. Similarly, although IL-32β and IL-32γ have been proposed as a pro-atherogenic cytokine [16, 26], our current study provides strong evidence that IL-32α is an anti-inflammatory and vascular-protective cytokine that exerts inhibitory effects on EC inflammation, arterial wall thickening, and atherosclerosis by reducing miR-205 biogenesis through regulating miRNA biogenesis pathways, which in turn protects Timp3 and Reck expression. In addition, IL-32α inhibits the proliferation and migration of VSMCs, contributing to the prevention of atherosclerosis. These findings thereby provide novel mechanistic insights into the inhibitory role of IL-32α on vascular inflammation and atherosclerosis and demonstrate for the first time that IL-32α has a regulatory role in miRNA biogenesis. Therefore, the knowledge gained from this work could be translatable to humans, providing potential clinical relevance for IL-32α as a diagnostic and therapeutic protein in atherosclerosis.
Abbreviations
IL: interleukin; Tg: transgenic; EC: endothelial cells; VSMC: vascular smooth muscle cell; IFN: interferon; TNF: tumor necrosis factor; CCL2: C-C motif ligand 2; VCAM-1: vascular cell adhesion molecule-1; MMP: matrix metalloproteinases; PMA: phorbol 12-myristate 13-acetate; Timp3: tissue inhibitor of metalloproteinase 3; Reck: reversion-inducing cysteine-rich protein with Kazal motifs; iMAEC: immortalized mouse arterial endothelial cell; ICAM-1: intracellular adhesion molecule-1; HUVEC: human umbilical vein endothelial cell; BrdU: bromodeoxyuridine; PDGF: platelet-derived growth factor; RCA: right common carotid artery; LCA: left common carotid artery; OPC: oligomeric procyanidins; eNOS: endothelial constitutive nitric oxide synthase; ADAM: a disintegrin and metalloprotease; AAA: abdominal aortic aneurysm; GO: gene ontology; ECM: extracellular matrix
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by a funding from the MSIP and Basic Programs of the National Research Foundation (NRF) of Korea (grant nos. MRC, 2008-0062275 and 2015R1A2A2A09001137). We thank Drs. Hanjoong Jo and Sandeep Kumar at the Georgia Institute of Technology and Emory University (Atlanta, GA, USA) for providing Dual-luciferase reporter constructs containing the 3′UTR of Timp3 and Reck.
Contributions
D.J.S. and Y.Y.J. conducted most of the experiments, performed data analysis, generated most of the experimental mice, and were the primary writers of the manuscript. Y.S.S. performed in vitro monocyte-EC adhesion and western blotting assays for cultured iMAECs and HUVECs samples. D.H.L. assisted in the generation of ApoE-/-/hIL-32α-Tg mice and was involved in the qPCR and western blotting analyses. S.H.K. performed the bioinformatic analysis. D.Y.Y. provided IL-32α isoforms expressing plasmid vectors and IL-32 antibody. H.P., Y.Y.J., Y.S.R., S.B.H., and D.Y.Y. provided advice throughout the project. J.T.H. supervised the entire project and had major roles in experimental design, data interpretation, and writing the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135-43
2. Ross R. Atherosclerosis-an inflammatory disease. The New England journal of medicine. 1999;340:115-26
3. Sima AV, Stancu CS, Simionescu M. Vascular endothelium in atherosclerosis. Cell Tissue Res. 2009;335:191-203
4. Michiels C. Endothelial cell functions. J Cell Physiol. 2003;196:430-43
5. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115:1285-95
6. Davies PF. Endothelial mechanisms of flow-mediated athero-protection and susceptibility. Circulation research. 2007;101:10-2
7. Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204-12
8. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317-25
9. Mangge H, Hubmann H, Pilz S, Schauenstein K, Renner W, Marz W. Beyond cholesterol-inflammatory cytokines, the key mediators in atherosclerosis. Clin Chem Lab Med. 2004;42:467-74
10. Ramji DP, Davies TS. Cytokines in atherosclerosis: Key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev. 2015;26:673-85
11. Aukrust P, Halvorsen B, Yndestad A, Ueland T, Oie E, Otterdal K. et al. Chemokines and cardiovascular risk. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:1909-19
12. Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovascular research. 2008;79:360-76
13. Nold-Petry CA, Nold MF, Zepp JA, Kim SH, Voelkel NF, Dinarello CA. IL-32-dependent effects of IL-1beta on endothelial cell functions. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3883-8
14. Kobayashi H, Lin PC. Molecular characterization of IL-32 in human endothelial cells. Cytokine. 2009;46:351-8
15. Kobayashi H, Yazlovitskaya EM, Lin PC. Interleukin-32 positively regulates radiation-induced vascular inflammation. Int J Radiat Oncol Biol Phys. 2009;74:1573-9
16. Heinhuis B, Popa CD, van Tits BL, Kim SH, Zeeuwen PL, van den Berg WB. et al. Towards a role of interleukin-32 in atherosclerosis. Cytokine. 2013;64:433-40
17. Jhun H, Choi J, Hong J, Lee S, Kwak A, Kim E. et al. IL-32gamma overexpression accelerates streptozotocin (STZ)-induced type 1 diabetes. Cytokine. 2014;69:1-5
18. Lee DH, Kim DH, Hwang CJ, Song S, Han SB, Kim Y. et al. Interleukin-32gamma attenuates ethanol-induced liver injury by the inhibition of cytochrome P450 2E1 expression and inflammatory responses. Clin Sci (Lond). 2015;128:695-706
19. Park MH, Yoon DY, Ban JO, Kim DH, Lee DH, Song S. et al. Decreased severity of collagen antibody and lipopolysaccharide-induced arthritis in human IL-32beta overexpressed transgenic mice. Oncotarget. 2015;6:38566-77
20. Lee EJ, Lee EJ, Chung YH, Song DH, Hong S, Lee CK. et al. High level of interleukin-32 gamma in the joint of ankylosing spondylitis is associated with osteoblast differentiation. Arthritis Res Ther. 2015;17:350
21. Yun J, Gu SM, Yun HM, Son DJ, Park MH, Lee MS. et al. Myelin oligodendrocyte glycoprotein (MOG35-55)-induced experimental autoimmune encephalomyelitis is ameliorated in interleukin-32 alpha transgenic mice. Oncotarget. 2015;6:40452-63
22. Dahl CA, Schall RP, He HL, Cairns JS. Identification of a novel gene expressed in activated natural killer cells and T cells. Journal of immunology. 1992;148:597-603
23. Lee DH, Hong JE, Yun HM, Hwang CJ, Park JH, Han SB. et al. Interleukin-32beta ameliorates metabolic disorder and liver damage in mice fed high-fat diet. Obesity (Silver Spring). 2015;23:615-22
24. Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA. Interleukin-32: a cytokine and inducer of TNFalpha. Immunity. 2005;22:131-42
25. Kang JW, Park YS, Lee DH, Kim MS, Bak Y, Ham SY. et al. Interaction network mapping among IL-32 isoforms. Biochimie. 2014;101:248-51
26. Kobayashi H, Huang J, Ye F, Shyr Y, Blackwell TS, Lin PC. Interleukin-32beta propagates vascular inflammation and exacerbates sepsis in a mouse model. PloS one. 2010;5:e9458
27. Kang J-W, Park YS, Lee DH, Kim J-h, Kim MS, Bak Y. et al. Intracellular Interaction of Interleukin (IL)-32α with Protein Kinase Cϵ (PKCϵ) and STAT3 Protein Augments IL-6 Production in THP-1 Promonocytic Cells. The Journal of biological chemistry. 2012;287:35556-64
28. Raines EW. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004;15:237-54
29. Newby AC. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovascular research. 2006;69:614-24
30. Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A. et al. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. American journal of physiology Heart and circulatory physiology. 2009;297:H1535-43
31. Ni CW, Qiu H, Rezvan A, Kwon K, Nam D, Son DJ. et al. Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood. 2010;116:e66-73
32. Garcia-Conesa MT, Tribolo S, Guyot S, Tomas-Barberan FA, Kroon PA. Oligomeric procyanidins inhibit cell migration and modulate the expression of migration and proliferation associated genes in human umbilical vascular endothelial cells. Molecular nutrition & food research. 2009;53:266-76
33. Kim CW, Kumar S, Son DJ, Jang IH, Griendling KK, Jo H. Prevention of abdominal aortic aneurysm by anti-microRNA-712 or anti-microRNA-205 in angiotensin II-infused mice. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:1412-21
34. Son DJ, Kumar S, Takabe W, Kim CW, Ni CW, Alberts-Grill N. et al. The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nature communications. 2013;4:3000
35. Kheirolomoom A, Kim CW, Seo JW, Kumar S, Son DJ, Gagnon MK. et al. Multifunctional Nanoparticles Facilitate Molecular Targeting and miRNA Delivery to Inhibit Atherosclerosis in ApoE(-/-) Mice. ACS Nano. 2015;9:8885-97
36. Majid S, Dar AA, Saini S, Yamamura S, Hirata H, Tanaka Y. et al. MicroRNA-205-directed transcriptional activation of tumor suppressor genes in prostate cancer. Cancer. 2010;116:5637-49
37. Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N. et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235-40
38. Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J. et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415-9
39. Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A. et al. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature. 2009;459:1010-4
40. Trabucchi M, Briata P, Filipowicz W, Ramos A, Gherzi R, Rosenfeld MG. KSRP promotes the maturation of a group of miRNA precursors. Adv Exp Med Biol. 2010;700:36-42
41. Ni Z, Xu C, Guo X, Hunter GO, Kuznetsova OV, Tempel W. et al. RPRD1A and RPRD1B are human RNA polymerase II C-terminal domain scaffolds for Ser5 dephosphorylation. Nat Struct Mol Biol. 2014;21:686-95
42. Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15:321-33
43. Ni Z, Olsen JB, Guo X, Zhong G, Ruan ED, Marcon E. et al. Control of the RNA polymerase II phosphorylation state in promoter regions by CTD interaction domain-containing proteins RPRD1A and RPRD1B. Transcription. 2011;2:237-42
44. Alberts-Grill N, Rezvan A, Son DJ, Qiu H, Kim CW, Kemp ML. et al. Dynamic immune cell accumulation during flow-induced atherogenesis in mouse carotid artery: an expanded flow cytometry method. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:623-32
45. Cardellini M, Menghini R, Martelli E, Casagrande V, Marino A, Rizza S. et al. TIMP3 is reduced in atherosclerotic plaques from subjects with type 2 diabetes and increased by SirT1. Diabetes. 2009;58:2396-401
46. Knox JB, Sukhova GK, Whittemore AD, Libby P. Evidence for altered balance between matrix metalloproteinases and their inhibitors in human aortic diseases. Circulation. 1997;95:205-12
47. Meng N, Li Y, Zhang H, Sun XF. RECK, a novel matrix metalloproteinase regulator. Histol Histopathol. 2008;23:1003-10
48. Ikonomidis JS, Jones JA, Barbour JR, Stroud RE, Clark LL, Kaplan BS. et al. Expression of matrix metalloproteinases and endogenous inhibitors within ascending aortic aneurysms of patients with Marfan syndrome. Circulation. 2006;114:I365-70
49. Fabunmi RP, Sukhova GK, Sugiyama S, Libby P. Expression of tissue inhibitor of metalloproteinases-3 in human atheroma and regulation in lesion-associated cells: a potential protective mechanism in plaque stability. Circulation research. 1998;83:270-8
50. Di Gregoli K, Mohamad Anuar NN, Bianco R, White SJ, Newby AC, George SJ. et al. MicroRNA-181b Controls Atherosclerosis and Aneurysms Through Regulation of TIMP-3 and Elastin. Circulation research. 2017;120:49-65
51. Muto A, Fitzgerald TN, Pimiento JM, Maloney SP, Teso D, Paszkowiak JJ. et al. Smooth muscle cell signal transduction: implications of vascular biology for vascular surgeons. J Vasc Surg. 2007:45 Suppl A: A15-24
52. Kawai-Kowase K, Owens GK. Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells. Am J Physiol Cell Physiol. 2007;292:C59-69
53. Romaine SP, Tomaszewski M, Condorelli G, Samani NJ. MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart. 2015;101:921-8
54. Calway T, Kim GH. Harnessing the Therapeutic Potential of MicroRNAs for Cardiovascular Disease. J Cardiovasc Pharmacol Ther. 2015;20:131-43
55. Hartmann P, Schober A, Weber C. Chemokines and microRNAs in atherosclerosis. Cell Mol Life Sci. 2015;72:3253-66
56. Alexy T, Rooney K, Weber M, Gray WD, Searles CD. TNF-α alters the release and transfer of microparticle-encapsulated miRNAs from endothelial cells. Physiological Genomics. 2014;46:833-40
57. He PP, Ouyang XP, Tang YY, Liao L, Wang ZB, Lv YC. et al. MicroRNA-590 attenuates lipid accumulation and pro-inflammatory cytokine secretion by targeting lipoprotein lipase gene in human THP-1 macrophages. Biochimie. 2014;106:81-90
58. Alanazi I, Hoffmann P, Adelson DL. MicroRNAs are part of the regulatory network that controls EGF induced apoptosis, including elements of the JAK/STAT pathway, in A431 cells. PloS one. 2015;10:e0120337
59. Afonyushkin T, Oskolkova OV, Bochkov VN. Permissive role of miR-663 in induction of VEGF and activation of the ATF4 branch of unfolded protein response in endothelial cells by oxidized phospholipids. Atherosclerosis. 2012;225:50-5
60. Joosten LA, Netea MG, Kim SH, Yoon DY, Oppers-Walgreen B, Radstake TR. et al. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3298-303
61. Yun HM, Oh JH, Shim JH, Ban JO, Park KR, Kim JH. et al. Antitumor activity of IL-32[beta] through the activation of lymphocytes, and the inactivation of NF-[kappa]B and STAT3 signals. Cell death & disease. 2013;4:e640
62. Oh JH, Cho MC, Kim JH, Lee SY, Kim HJ, Park ES. et al. IL-32[gamma] inhibits cancer cell growth through inactivation of NF-[kappa]B and STAT3 signals. Oncogene. 2011;30:3345-59
63. Bang BR, Kwon HS, Kim SH, Yoon SY, Choi JD, Hong GH. et al. Interleukin-32gamma suppresses allergic airway inflammation in mouse models of asthma. Am J Respir Cell Mol Biol. 2014;50:1021-30
64. Yun HM, Park KR, Kim EC, Han SB, Yoon do Y, Hong JT. IL-32alpha suppresses colorectal cancer development via TNFR1-mediated death signaling. Oncotarget. 2015;6:9061-72
65. Park MH, Song MJ, Cho MC, Moon DC, Yoon do Y, Han SB. et al. Interleukin-32 enhances cytotoxic effect of natural killer cells to cancer cells via activation of death receptor 3. Immunology. 2012;135:63-72
66. Choi J, Bae S, Hong J, Ryoo S, Jhun H, Hong K. et al. Paradoxical effects of constitutive human IL-32{gamma} in transgenic mice during experimental colitis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21082-6
Author contact
![]() Corresponding authors: Jin Tae Hong, PhD; Professor, College of Pharmacy & Medical Research Center, Chungbuk National University, 194-31 Osongsaengmyeong 1-ro, Osong-Biocampus Building Room 303, Osong-eup, Heungdeok-gu, Cheongju, Chungbuk 28160, Korea, Telephone: 82-43-261-2813, FAX: 82-43-268-2732, E-mail: jinthongac.kr Professor, Do Young Yoon, Department of Bioscience and Biotechnology, Konkuk University, 120 Neungdong-ro, Gwangjin-gu, Seoul 05029, Korea, Telephone: 82-2-444-2188, FAX: 82-2-444-6176, E-mail: ydy4218ac.kr
Corresponding authors: Jin Tae Hong, PhD; Professor, College of Pharmacy & Medical Research Center, Chungbuk National University, 194-31 Osongsaengmyeong 1-ro, Osong-Biocampus Building Room 303, Osong-eup, Heungdeok-gu, Cheongju, Chungbuk 28160, Korea, Telephone: 82-43-261-2813, FAX: 82-43-268-2732, E-mail: jinthongac.kr Professor, Do Young Yoon, Department of Bioscience and Biotechnology, Konkuk University, 120 Neungdong-ro, Gwangjin-gu, Seoul 05029, Korea, Telephone: 82-2-444-2188, FAX: 82-2-444-6176, E-mail: ydy4218ac.kr