Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Data analysis
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(7):1942-1952. doi:10.7150/thno.16236 This issue Cite
Research Paper
Mouse IP-10 Gene Delivered by Folate-modified Chitosan Nanoparticles and Dendritic/tumor Cells Fusion Vaccine Effectively Inhibit the Growth of Hepatocellular Carcinoma in Mice
Zixi Hu1*, Jiaojiao Chen1*, Sufang Zhou1*, Nuo Yang1, Siliang Duan1, Zhenghua Zhang1, Jing Su1, Jian He1, Zhiyong Zhang1, 2 ![]() , Xiaoling Lu1
, Xiaoling Lu1 ![]() , Yongxiang Zhao1
, Yongxiang Zhao1 ![]()
1. National Center for International Research of Biological Targeting Diagnosis and Therapy, Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research, Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China;
2. Department of Surgery, Robert-Wood-Johnson Medical School University Hospital, Rutgers University, The State University of New Jersey, New Brunswick, New Jersey 08901, USA.
* These authors contributed equally to this work.
Received 2016-5-19; Accepted 2017-2-8; Published 2017-5-2
Abstract

Dendritic cells (DC) and tumor cell fusion vaccine (DC/tumor cell fusion vaccine) is considered an effective approach in cancer biotherapy. However, its therapeutic effects in early clinical trials have been suboptimal partially due to the immunosuppressive tumor environment. In this study, we used nanoparticles of folate (FA)-modified chitosan, a non-viral vector capable of targeting tumor cells with high expression of FA receptors. FA-chitosan nanoparticles were used as biological carriers for the expression plasmid of the mouse interferon-induced protein-10 (mIP-10) gene, a potent chemoattractant for cytotoxic T cells. The combination of FA-chitosan/mIP-10 and DC/tumor cell fusion vaccine against hepatocellular carcinoma (HCC) effectively inhibited the growth of implanted HCC tumors and prolonged the survival of mice. The combination therapy significantly reduced myeloid-derived suppressor cells (MDSC) in mouse spleen, local tumor, and bone marrow while increasing tumor-specific IFN-γ responses. Furthermore, the combination therapy significantly inhibited tumor cell proliferation while promoting their apoptosis. Taken together, our data illustrate that the mIP-10 enhances the anti-tumor effect of DC/tumor cell fusion vaccine by alleviating the immunosuppressive tumor environment.
Keywords: DC/tumor fusion cell vaccines, interferon-induced protein-10, folic acid, chitosan.
Introduction
Hepatocellular carcinoma (HCC) has a high prevalence in many countries and is a serious threat to human health world-wide. The efficacy of currently available therapeutic strategies, such as surgery, chemotherapy, and radiotherapy, is limited [1, 2]. Recently, immunotherapy has shown great promise in prolonging the survival of patients with HCC [3, 4]. Dendritic cells (DCs) are professional antigen-presenting cells which play a major role in anti-tumor immunotherapy [5-7]. With advances in DC isolation and fusion techniques, DC/tumor cell fusion vaccine has become a key strategy because of its ability to induce specific cytotoxic T lymphocytes (CTLs) for killing tumor cells in vivo. However, the fused cells as a vaccine for clinical treatment are still inadequate in inducing potent anti-tumor immunity in cancer patients. The failure of DC/tumor fusion vaccine may be attributed to the tumor microenvironment that suppresses tumor-specific CTL responses induced by DC/tumor fusion vaccine and impedes CTL infiltration into the tumor site. Therefore, it is important to explore new strategies to enhance the effectiveness of DC/tumor fusion vaccine.
Interferon-induced protein-10 (IP-10) is a potent chemokine which can efficiently attract T cells to tumor tissues and is produced by activated T cells, fibroblasts, endothelial cells, epithelial cells, keratinocytes, and monocytes. IP-10 can bind to its CXCR3 receptor on activated T cells and induce chemotaxis of T lymphocytes to the local tumor [8, 9]. IP-10 can also inhibit tumor growth and angiogenesis [10, 11]. Therefore, IP-10 is a potential candidate chemokine to enhance anti-tumor immunotherapies in the clinic [12, 13].
The half-life of recombinant IP-10 in vivo may be insufficient to provide a long-lasting effect. This limitation can be overcome by delivery of IP-10 expression plasmid to induce IP-10 expression in vivo. Chitosan is a natural cationic polymer, and has good compatibility, low immunogenicity, and a unique capability to transport across cell membranes [14, 15]. Chitosan has been demonstrated to be an effective non-viral vehicle to deliver plasmid DNA. Furthermore, compared to normal cells, there is a higher density of Folate (FA) receptors on tumor cells. In our previous study, we have reported that treatment with a mixture of FA-chitosan with mouse IP-10 plasmid (FA-chitosan/mIP-10) inhibited tumor growth and angiogenesis, and promoted apoptosis of tumor cells in mice [8]. Based on these findings, we hypothesized that a more potent antitumor-specific CTL response should be induced when the tumor is treated with both FA-chitosan/mIP-10 and DC/tumor fusion vaccine.
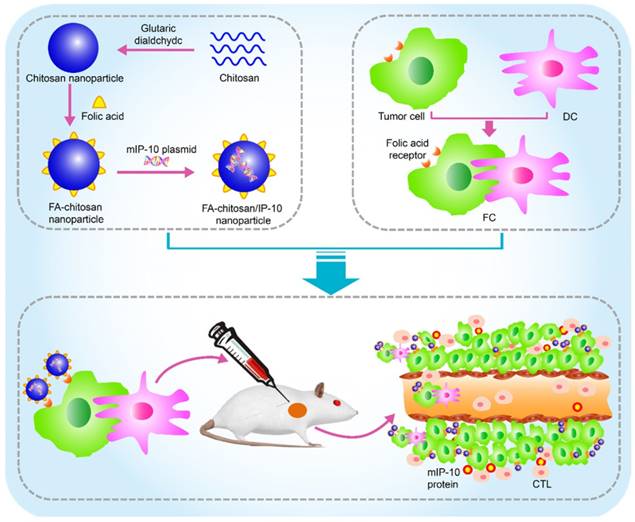
In this study, we prepared FA-chitosan/mIP-10 nanoparticles and DC/tumor fusion vaccine against HCC and tested the therapeutic effect of treatment with the nanoparticles with or without the fusion vaccine on inhibiting the growth of HCC. We also explored the underlying mechanisms of this combined approach in tumor-bearing mice (Figure 1).
Schematic of in vivo therapy using FA-chitosan/mIP-10 nanoparticles and DC/tumor fusion vaccine: FA-chitosan/mIP-10 nanoparticles are prepared by complex coacervation process and DC fusion vaccine (DC/tumor fusion cells, FC) is generated; then nanoparticles of FA-chitosan/mIP-10 and FC fusion vaccine are delivered into tumors. FA-chitosan/mIP-10 nanoparticles targeted folate receptors express and release the mIP-10 protein. Finally, mIP-10 attracts tumor-specific CTLs to kill tumor cells at the tumor site.
Materials and Methods
Materials
Chitosan (deacetylation degree ≥ 90%) was purchased from Solarbio Company (Beijing, China). Folic acid was purchased from Sigma-Aldrich (Sigma, USA). The recombinant IP-10 plasmid was constructed by cloning the mouse IP-10 cDNA fragment into the N-terminal of pReceiver-MO3, which contained the enhanced green fluorescent protein (EGFP) [8]. Monoclonal anti-mouse CD11b-FITC (m1-70) and Ly6G-PE (RB6-8C5), CD80-FITC (16-10A1), CD86-FITC (GL1), MHCII-FITC (NIMR-4), CXCR3-FITC, CD8-PE, and anti-mouse Ki67 were purchased from eBiosciences (San Diego, USA). Chitosanase was purchased from Calbiochem (Dames, Germany). Monoclonal rabbit anti-mouse IP-10 was acquired from Boster (Wuhan, China). Fluorescent apoptosis detection kit was obtained from Roche (Roche, Switzerland). Mouse IL-12 ELISA kit was from Peprotech (Peprotech, USA) and INF-γ ELISPOT assay kit was purchased from Dakewe (Shenzhen, China).
Cell culture and animals
Mouse HCC Hepa1-6 cells were purchased from Chinese Academy of Science (Shanghai, China). Mesocricetus auratus kidney BSR and liver non-tumor BNL.CL.2 cells were obtained from American Type Culture Collection (ATCC, Manassas, USA). Murine dendritic DC2.4 cells were obtained from Xiangya School of Medicine, Central South University (Changsha, China), in DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin at 37℃, 5% CO2 incubator.
Female C57BL/6 mice (H-2b) at 4-6 weeks of age were purchased from Vital River (Beijing, China) and housed in a specific pathogen-free facility. All experiments were performed according to the guidelines of European Federation of Laboratory Animal Science Association. The experimental protocols were approved by the Animal Ethics Committee of Guangxi Medical University (Guangxi, China).
Preparation and transfection of FA-chitosan/mIP-10
FA-chitosan/mIP-10 nanoparticles were prepared as described previously [8]. Briefly, the FA-chitosan in distilled water and the IP-10 plasmid in 25 mM Na2SO4 solution (1 mg/ml) were heated in a 55℃ water bath for 10 min, mixed, vortexed and kept at room temperature for 30 min, followed by centrifugation at 12,000 x g at 4℃ for 20 min. The pellets were re-suspended in phosphate buffer to form FA-chitosan/mIP-10 suspension. Then the prepared nanoparticles were electrophoresed in a 1% agarose gel to detect the chitosan wrapped plasmids. The folate modified-chitosan with control plasmid (FA-chitosan/CP) was prepared with the same method of FA-chitosan/mIP-10.
Hepa1-6 cells were cultured in 24 well-plates and the cells were transfected with 2 μg mIP-10 plasmid, chitosan/mIP-10, or FA-chitosan/mIP-10 for 48 h. The EGFP expression in different groups of cells was observed under a microscope.
Preparation and fluorescent staining of fusion cells
DC2.4 cells were labeled with CSFE and mixed with irradiated (40 Gy) PKH26-labeled Hepa1-6 cells at a ratio of 2: 1. The cells were centrifuged at 38 x g for 6 min, cell pellets were treated with preheated (40℃) polyethylene glycol (PEG) 1450 (Sigma, USA) for 3 min, and PBS was added followed centrifugation. The cells were then washed and stimulated with 50 ng/ml TNF-α in DMEM for 24 h. Subsequently, the cells were stained with DAPI and observed under a fluorescent microscope. Furthermore, the fusion cells were stained with CD80-FITC, CD86-FITC, and MHCII-FITC (eBiosciences, San Diego, USA) as well as isotype control antibodies. The levels of CD80, CD86 and MHCII on the surface of DC2.4/Hepa1-6 cells (from now on referred to as FC) were determined by flow cytometry.
Enzyme-linked immunosorbent assay (ELISA)
The FCs were cultured at a density of 2 × 105 cells/well for 48 h. The levels of IL-12 in the supernatants of cultured cells were determined using a mouse IL-12 detection kit, according to the manufacturers' instruction (Peprotech, USA). The experiments were performed in triplicate and the absorbance at 450 nm was measured in a microplate reader.
Establishment of tumor-bearing mouse model and treatment
Female C57BL/6 mice were injected subcutaneously with 5 × 106 Hepa1-6 cells (100 µl) into their left groins. The mice were randomized and injected intratumorally with control PBS, FA-chitosan/mIP-10 (40 µg mIP-10 in 200 µl), FC (5 × 106 cells in 200 µl), or combination of FA-chitosan/mIP-10 and FC (n = 8/each) weekly beginning at day 5 post inoculation. The growth of implanted tumors was monitored every 5 days with a caliper and the tumor volume was calculated by the formula: L1 × (L2)2 × 0.5 (L1: the maximum diameter of the tumor, L2: the shortest diameter of the tumor). The growth of tumors and survival of tumor-bearing mice were measured.
Flow cytometry analysis
Spleen, bone marrow, and tumor cells were isolated from tumor-bearing mice at day 25 post inoculation. Bone marrow and splenic cells were stained with CD11b-FITC and Ly6G-PE. Tumor cells were stained with CXCR3-FITC and CD8-PE. The isotype antibodies served as controls. The samples were detected by flow cytometry. (Beckman Coulter, Brea, CA, USA).
ELISPOT assay
The frequency of IFN-γ-secreting splenic T cells was determined by ELISPOT assay. Briefly, splenic T cells were isolated from different groups of mice by negative selection and stimulated with the fused cells at a ratio of 10:1 in 96-well ELISPOT plates that had been pre-coated with anti-IFN-γ. The captured cytokine was probed with biotinylated anti-IFN-γ and detected with horseradish peroxidase (HRP)-streptavidin, followed by visualization with diaminobenzidine (DAB). The number of spots from clones was counted on a CTL counter (CTL, Shaker Height, USA).
Histology and Immunohistochemistry
The tumor-bearing mice at 25 days post inoculation were sacrificed and their tumor tissues were dissected out and fixed in 4 % formalin for 24 h. The tumor tissue sections (4 µm) were stained with anti-Ki67 (1: 200 dilution) or anti-mIP-10 antibodies (1: 100 dilution) at 4℃ for 20 h. The bound antibody was detected with HRP-conjugated rabbit anti-rat IgG or goat anti-rabbit IgG and DAB kit (Zhongshan Golden Bridge, Beijing, China), followed by counterstaining with hematoxylin.
The tumor tissue sections were also subjected to TUNEL analysis for determining the frequency of apoptotic cells using a fluorescent apoptosis detection kit, according to the manufacturers' instruction. All images were taken under a fluorescence microscope (Nikon, Japan). Also, the heart, liver, spleen, lung, and kidney tissue sections as well as the tumor tissue sections were stained with H&E.
Data analysis
Data were presented as mean ± standard deviation and the difference among groups was analyzed by analysis of variance (ANOVA) with Bonferroni's post hoc test using the GraphPad Prism 6.0 software. A P-value of < 0.05 was considered statistically significant.
Results
Characterization of nanoparticles
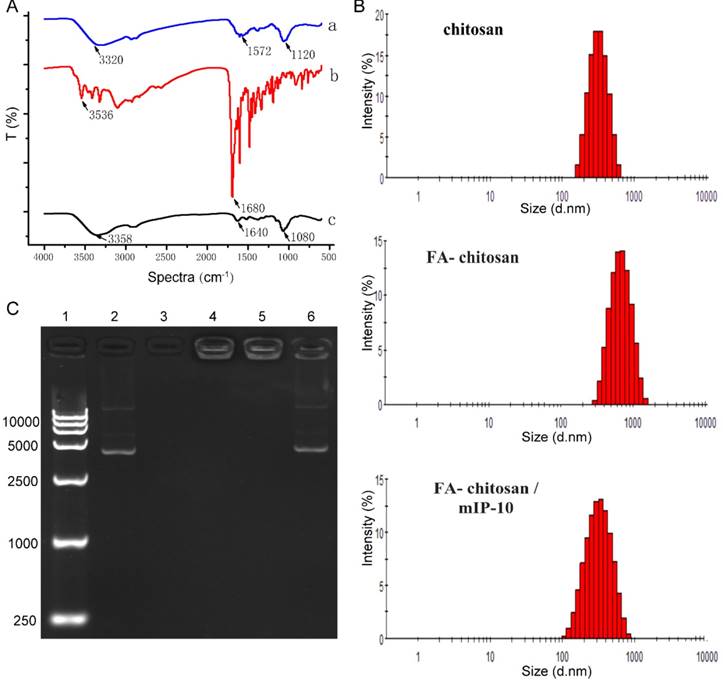
We first characterized the nanoparticles by infrared spectroscopy (FTIR). As shown in Figure 2A , the -OH, -NH2 and C-O-C groups on chitosan (a) had three characteristic peaks at 3320 cm-1, 1572 cm-1, 1120 cm-1; FA (b) had two peaks at 3536 cm-1, 1680 cm-1; The peaks at 1572 cm-1 representative of -NH2 group in chitosan and 1680 cm-1 representative of C=O bond absorption of folate in FA modified chitosan (c) disappeared. It was substituted by amide bond at 1640 cm-1 absorption peak significantly, indicating a successful FA modified chitosan. A laser particle size analyzer was used for the analysis of particle sizes of chitosan. As shown in Figure 2B, the particle size distribution of chitosan, FA-chitosan, and FA-chitosan/mIP-10 was fairly narrow indicating relatively uniform particles. The average particle diameters of chitosan, FA-chitosan, FA-chitosan/mIP-10 nanoparticles were 264.0 nm, 414.4 nm, and 315.5 nm, respectively. Corresponding bands of mIP-10 plasmid were detected using 1% agarose gel electrophoresis. However, the chitosan band was not obvious as it carries a positive charge while chitosan/mIP-10 and FA-chitosan/mIP-10 did not show the mIP-10 plasmid band or only a weak band because mIP-10 plasmid was wrapped. However, the mIP-10 plasmid band became obvious when FA-chitosan/IP-10 was treated with chitonase to destroy the nanoparticle. Results of the gel retardation assay demonstrated that FA-chitosan/mIP-10 successfully wrapped IP-10 plasmid (Figure 2C).
Characterization of chitosan nanoparticles. (A) Infrared spectroscopy results of nanoparticles. (B) Size distribution of nanoparticles. (C) Gel retardation assay. Lane 1: Marker; lane 2: mIP-10 plasmid; lane 3: chitosan; lane 4: chitosan/m IP-10; lane 5: FA-chitosan/mIP-10; 6: chitosanase destroyed FA-chitosan/mIP-10.
Characterization of the DC/HCC fusion vaccine
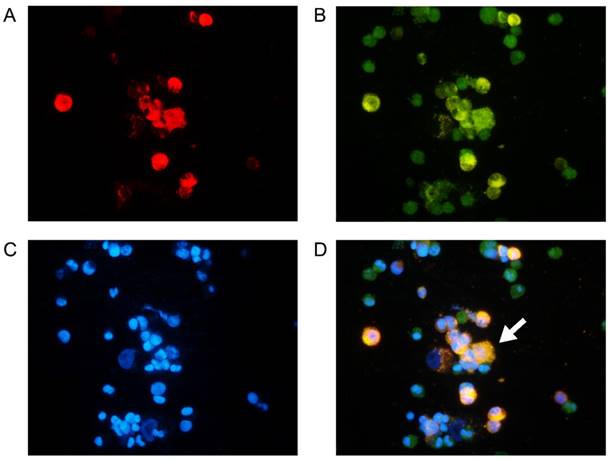
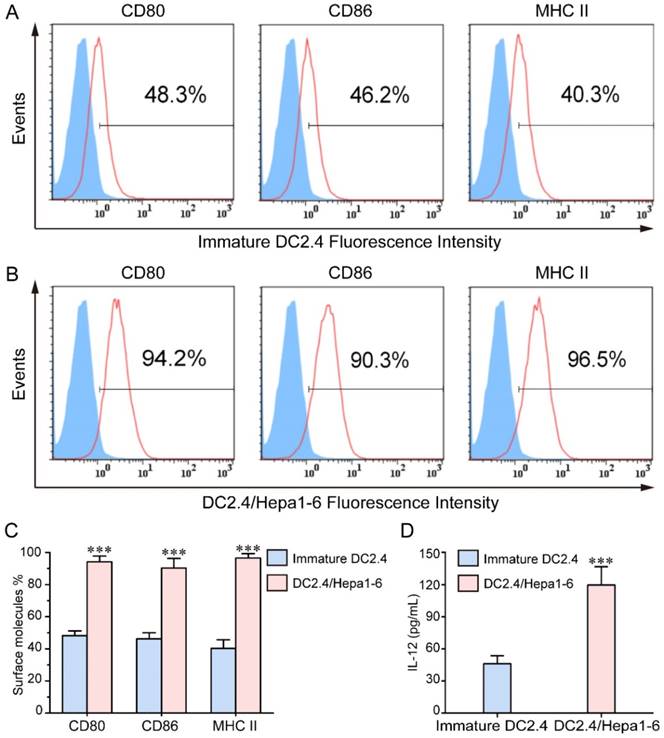
To generate the DC/tumor fusion vaccine, DC2.4 cells were fused with or without irradiated PKH26-labeled Hepa1-6 cells using PEG1450. As shown in Figure 3, we have developed DC2.4/Hepa1-6 fusion vaccine which had double-nuclei or were multi-nucleated with larger cell volumes. As compared with the DC2.4 cells, FC had a high expression of CD80, CD86 and MHCII molecules following TNF-α stimulation (Figure 4A-C), indicating that FC retained the capacity of DC2.4 in response to stimulators and might have a high antigen-presenting ability. The FC secreted significantly higher levels of IL-12 than DC2.4 cells (Figure 4D) which should drive the functional development of IFN-γ-secreting T cells.
Identification of DC2.4/Hepa1-6 fusion cells: DC2.4 and Hepa1-6 cells were labeled with CSFE (green) and PKH26 (red), respectively, followed by fusion with PEG1450. Subsequently, the cells were stained with DAPI (blue) and observed under a fluorescent microscope. Data are representative images from three separate experiments. (A) PKH26 staining Hepa1-6 cells; (B) CFSE staining DC2.4 cells; (C) DAPI staining nuclei; (D) fused DC2.4/Hepa1-6 cells. The white arrow indicates fused cells. Original magnification, ×400.
Original magnification, ×400.
FCs express higher levels of CD80, CD86 and MHCII: The levels of CD80, CD86 and MHCII expression were determined by flow cytometry and the level of IL-12 in the supernatants of cultured cells was determined by ELISA. Data are presented as histograms and expressed as the mean ± SD of individual groups of cells from three separate experiments. (A-B) Flow cytometry analysis. (C) Quantitative analysis. (D) The levels of IL-12. *** P < 0.001.
Combination of FA-chitosan/mIP-10 and FC vaccine effectively inhibits the growth of HCC in mice
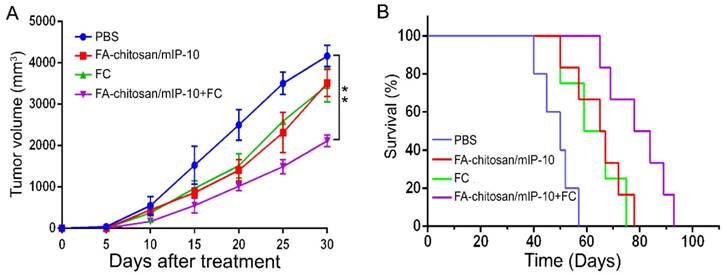
The transfection efficiency of FA-chitosan/mIP-10 was higher than that of chitosan/mIP-10 (Figure S1A-D). We tested the effect of treatment with FA-chitosan/mIP-10 and/or FC vaccine on the growth of implanted Hepa1-6 tumors in mice as well as on the survival of mice. Compared with controls, treatment with either FA-chitosan/mIP-10 or FC vaccine alone significantly reduced the tumor volumes (P < 0.01, Figure 5A) and also prolonged the survival of mice (P < 0.05, Figure 5B). More importantly, upon treatment with both FA-chitosan/mIP-10 and FC vaccine, there was significant inhibition of growth of implanted HCC with an even further reduction in tumor volumes (P < 0.01) and prolonged survival of the tumor-bearing mice (P < 0.05 vs. the control, P < 0.05 vs. the monotherapy).
Treatment with FA-chitosan/mIP-10 and FC vaccine effectively inhibits the growth of implanted HCC and prolongs the survival of tumor-bearing mice. C57BL/6 mice were inoculated with Hepa1-6 cells and five days later, the mice were randomized and treated with PBS, FA-chitosan/mIP-10 and/or FC vaccine weekly for five weeks. (A) Tumor volumes. (B) Survival of tumor-bearing mice. Data are expressed as mean ± SD of the tumor volumes of each group or the percent survival of each group of mice (n = 8 mice per group). ** P < 0.01 vs. the control.
Immunohistochemistry analysis indicated that varying levels of mIP-10 protein were expressed in tumor tissues (Figure S2A-E). The intensity of anti-mIP-10 staining in the tumor tissues from the FA-chitosan/mIP-10 treated mice was significantly higher than that in the controls. Moreover, the intensity of anti-mIP-10 staining was further elevated in the tumor tissues from the mice treated with both FA-chitosan/mIP-10 and FC (Figure S2F). These data indicated that FA-chitosan/mIP-10 effectively delivered mIP-10 plasmid in vivo and induced its expression in the tumor tissues.
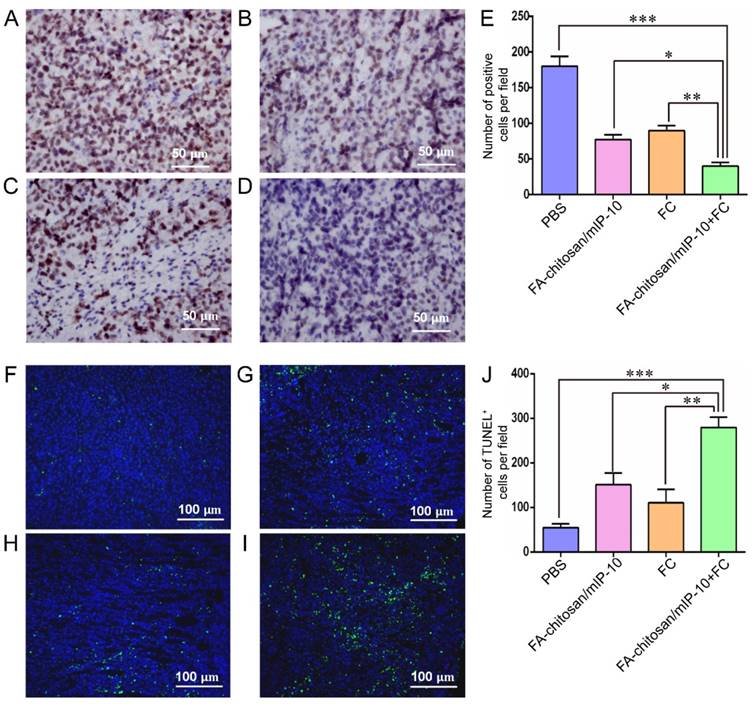
As shown in Figure 6A-E, immunohistochemistry analysis also showed significantly fewer Ki67+ proliferative tumor cells in the tumor tissues from the mice treated with FA-chitosan/mIP-10 or FC vaccine than that in the controls. The numbers of Ki67+ proliferative cells were further reduced when the mice were treated with both FA-chitosan/mIP-10 together with FC vaccine. Similarly, the TUNEL assay revealed that FA-chitosan/mIP-10 or FC vaccine caused a significant increase in the number of apoptotic cells in the tumor tissues as compared to controls (Figure 6F-J), which was further enhanced by the combined treatment with both FA-chitosan/mIP-10 and FC vaccine. Thus our results demonstrated that treatment with both FA-chitosan/mIP-10 and FC vaccine efficiently inhibited the proliferation of tumor cells and triggered their apoptosis in mice.
Treatment with FA-chitosan/mIP-10 and FC vaccine decreases the proliferation of tumor cells and increases their apoptosis in mice: At 25 days post inoculation, the mice were sacrificed and their tumor tissue sections were subjected to immunohistochemistry analysis with anti-Ki67 staining and TUNEL assays. Data are presented as representative images or expressed as the mean ± SD of each group of tumors. Immunohistochemistry analysis of Ki67+ proliferative tumor cells: (A) PBS; (B) FA-chitosan/mIP-10; (C) FC; (D) FA-chitosan/mIP-10 + FC; (E) Quantitative analysis. TUNEL assay of apoptotic tumor cells: (F) PBS; (G) FA-chitosan/mIP-10; (H) FC; (I) FA-chitosan/mIP-10 + FC; (J) Quantitative analysis. * P < 0.05; ** P < 0.01; *** P < 0.001.
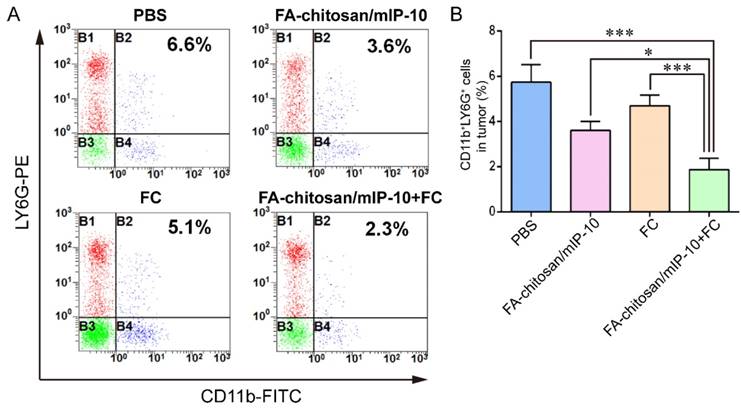
Treatment with FA-chitosan/mIP-10 and FC vaccine significantly reduces the frequency of CD11b+LY6G+ granulocytic cells in mice
To understand mechanisms underlying the action of FA-chitosan/mIP-10 and FC vaccine treatment in vivo, the frequency of CD11b+LY6G+ granulocytic cells in splenocytes, bone marrow cells (Figure S3A and C) and tumor infiltrates from different groups of tumor-bearing mice was assessed by flow cytometry (Figure 7A). The frequency of CD11b+LY6G+ granulocytic cells in tumor infiltrates from the mice treated with FA-chitosan/mIP-10 or FC vaccine was significantly lower than that in the PBS control but higher than that in the mice treated with both FA-chitosan/mIP-10 and FC vaccine (Figure 7B). A similar pattern of the frequency of CD11b+LY6G+ granulocytic cells was detected in splenocytes and bone marrow cells from different groups of mice (Figure S3B and D). Thus, treatment with FA-chitosan/mIP-10 and FC vaccine significantly reduced the frequency of CD11b+LY6G+ granulocytic cells in tumor-bearing mice.
Treatment with FA-chitosan/mIP-10 and FC vaccine decreases the frequency of CD11b+LY6G+ granulocytic tumor infiltrates in mice: Tumor infiltrates were isolated from different groups of tumors and stained with PE-anti-LY6G and FITC-anti-CD11b. The frequency of CD11b+LY6G+ granulocytes in the tumor infiltrates was determined by flow cytometry. Data are presented as representative flow cytometry charts or expressed as the mean ± SD of each group (n = 5) of cells from three separate experiments. * P < 0.05; *** P < 0.001.
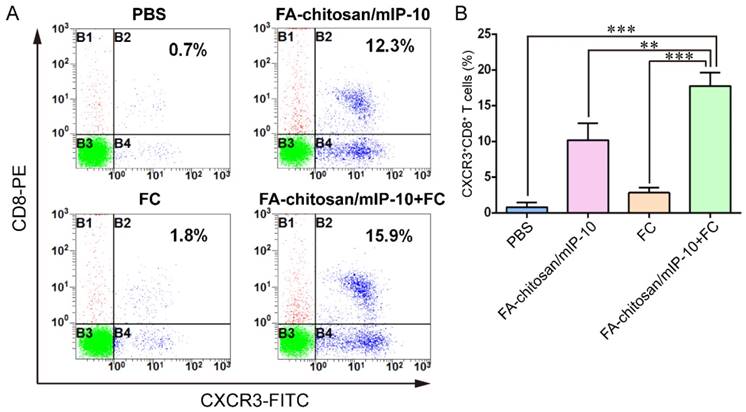
Additional flow cytometry analysis revealed that the frequency of CXCR3+CD8+ T cell in the tumors from the mice that had been treated with FA-chitosan/mIP-10 or FC vaccine was significantly higher than that in the controls (Figure 8). The combined treatment with FA-chitosan/mIP-10 and FC vaccine further increased the frequency of CXCR3+CD8+ T cell in the tumors.
Treatment with FA-chitosan/mIP-10 and FC vaccine increases the frequency of tumor infiltrating CXCR3+CD8+ T cell in mice. The frequency of CXCR3+CD8+ T cells were determined by flow cytometry. Data are presented as representative images or expressed as mean ± SD of each group (n = 5) of cells from three separate experiments. (A) Flow cytometry analysis. (B) Quantitative analysis. ** P < 0.01; *** P < 0.001.
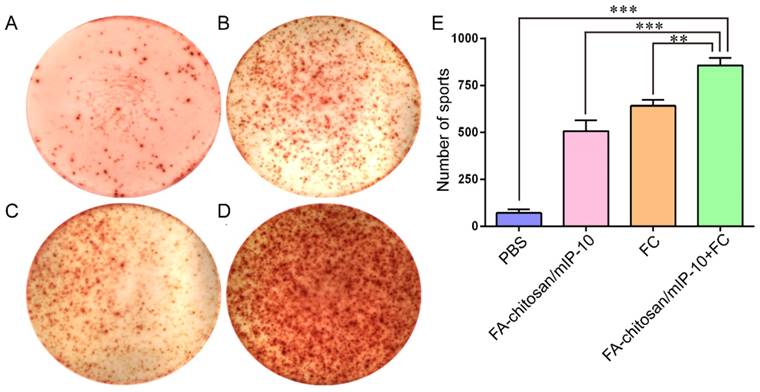
Finally, functional analysis by ELISPOT revealed a significant increase in the numbers of splenic IFN-γ-secreting spot forming cell clones in the mice treated with FA-chitosan/mIP-10 or FC vaccine compared with the controls (Figure 9A-C and E), but significantly less than that in the mice treated with FA-chitosan/mIP-10 and FC vaccine (Figure 9D and E). Therefore, treatment with FA-chitosan/mIP-10 and FC vaccine significantly enhanced tumor-specific T cell-mediated IFN-γ responses in tumor-bearing mice.
ELISPOT analysis of the numbers of tumor-specific IFN-γ-secreting T cells in mice: The numbers of tumor-specific IFN-γ-secreting T cells were determined by ELISPOT. Data are presented as representative images or expressed as the mean ± SD of the numbers of spot forming clones from each group of mice (n = 5) from three separate experiments. (A) PBS; (B) FA-chitosan/mIP-10; (C) FC; (D) FA-chitosan/mIP-10 + FC; (E) Quantitative analysis. ** P < 0.01; *** P < 0.001.
Discussion
In this study, chitosan nanoparticles, which are natural materials derived from chitin, were conjugated with folic acid and used as a biological carrier for the mIP-10 plasmid (FA-chitosan/mIP-10). The FTIR, particle size distribution, and gel retardation assay analyses confirmed the successful synthesis of chitosan particles carrying mIP-10 plasmid. Immunohistochemistry and flow cytometry results showed that FA-chitosan/mIP nanoparticles effectively targeted tumor tissues where they sustained local IP-10 expression, reduced the number of MDSCs, and attracted CXCR3+CD8+ T cells to the tumor thus inducing an effective cellular immune response. We also synthesized fusion cell vaccine of DC2.4/Hepa1-6 cells, which expressed high levels of MHCII and co-stimulatory molecules and had strong antigen presentation ability following stimulation with TNF-α. This novel combination of FA-chitosan/mIP-10 and FC vaccine efficiently inhibited the growth of implanted HCC and prolonged the survival of tumor-bearing mice. Thus our data suggest that this therapeutic strategy has potential for treatment of HCC in the clinic.
IP-10 is a potent chemokine for CXCR3+ activated T cells, NK cells, and macrophages. After binding to its receptor, IP-10 can regulate the function of activated T cells [9, 16, 17]. Expression of IP-10 in the tumor recruited more CXCR3+ CTL cells and enhanced the effects of DC2.4/Hepa1-6 fusion vaccine in the mouse model of HCC used in our study. These findings extended the previous observation that local vaccination with IP-10 plasmid DNA augmented the anti-tumor effect of glioma-specific DC in a mouse model of glioma [18]. Thus, our results support the notion that IP-10 is valuable for enhancing the effectiveness of anti-tumor immunotherapy.
Tumor cells secrete soluble immunosuppressive factors that inhibit the cytotoxicity of NK and CTL cells [19, 20] creating an immune suppressive environment to evade the attack of the immune system. MDSCs are one such class of cells that can secrete inhibitory cytokines [21, 22] and can differentiate into different heterogeneous populations of myeloid cells that inhibit immune function [23]. Previous studies have shown that MDSCs can inhibit the differentiation of effector Th1 cells resulting in immune incompetence and contributing to the development and progression of tumors [24, 25]. In the late stage of tumor progression, MDSCs are capable of suppressing Th1 responses [22, 26, 27]. On the contrary, decreased numbers of peripheral blood, tumor, and bone marrow MDSCs are associated with a reduction in the tumor volume in cancer patients following surgery or molecular targeted therapy [28]. Our present study clearly illustrated that combination of FA-chitosan/mIP-10 and FC vaccine effectively reduced the number of MDSCs in the spleen, bone marrow, and tumor tissues of tumor-bearing mice while increasing antigen-specific Th1 responses. It is possible that vaccination, together with local tumor expression of IP-10, significantly enhanced anti-tumor Th1 and CTL responses which inhibited the development of MDSCs.
It is well established that defects in cell cycle and apoptosis underlie the loss of control of cell growth leading to the development and progression of tumors [29, 30]. In the present study, the combination of FA-chitosan/mIP-10 and FC vaccine significantly reduced the number of Ki67+ tumor cells but increased the number of apoptotic cells in the tumor tissues which was consistent with the previous reports [31, 32, 33]. These data suggest that Th1 and CTL cells induced by combination therapy inhibit the proliferation of tumor cells and trigger apoptosis by direct cell-cell contact or indirectly by soluble mediators.
In summary, our data indicated that combination of FA-chitosan/mIP-10 and fusion cell vaccine efficiently enhanced anti-HCC effects in mice by inhibiting tumor cell proliferation and triggering their apoptosis. The combined treatment also increased the numbers of CTL infiltrates and antigen-specific IFN-γ response, but reduced the numbers of MDSCs. Therefore, the combination of FA-chitosan/mIP-10 and fusion cell vaccine may have potential clinical value for the treatment of HCC.
Abbreviations
FA: folate; mIP-10: mouse interferon-induced protein-10 gene; HCC: hepatocellular carcinoma; MDSC: myeloid-derived suppressor cell; DC: dendritic cell; CTL: cytotoxic T lymphocytes; PEG: polyethylene glycol; FTIR: fourier transform infrared spectroscopy; ELISPOT: enzyme-linked immunospot assay; IFN-γ: interferon-γ; ELISA: enzyme-linked immunosorbent assay; FC: the fused DC2.4/Hepa1-6 cells; DAPI: 4',6-diamidino-2-phenylindole; TUNEL: terminal-deoxynucleotidyl transferase mediated nick end labeling.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported, in part, by grants from Programs for Changjiang Scholars and Innovative Research Team in University (No.IRT_15R13); National Natural Scientific Foundation of China (Nos. 81430055 and 81372452); International Cooperation Project of the Ministry of Science and Technology of China (No. 2015DFA31320); Project for Innovative Research Team in Guangxi Natural Science Foundation (No.2015GXNSFFA-139001); Project of Science and Technology of Guangxi (Nos. 14125008-2-12, 1599005-2-10); Science Fund for Distinguished Young Scholars of Guangxi (No. 2012GXNSFFA-060006).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ingle PV, Samsudin SZ, Chan P. et al. Development and novel therapeutics in hepatocellular carcinoma: a review. Ther Clin Risk Manag. 2016;12:445-55
2. Liu D, Staveley-O'Carroll KF, Li G. Immune-based Therapy Clinical Trials in Hepatocellular Carcinoma. J Clin Cell Immunol. 2015;6:376
3. Bachy E, Coiffier B. Time has come for immunotherapy in PTCL. Blood. 2014;123:3059-60
4. Hutchings M. Targeted immunotherapy in Hodgkin lymphoma. Blood. 2015;125:3967-8
5. Anguille S, Van Acker HH, Van den Bergh J. et al. Interleukin-15 Dendritic Cells Harness NK Cell Cytotoxic Effector Function in a Contact- and IL-15-Dependent Manner. PLoS One. 2015;10:e0123340
6. Dong MB, Rahman MJ, Tarbell KV. Flow cytometric gating for spleen monocyte and DC subsets: differences in autoimmune NOD mice and with acute inflammation. J Immunol Methods. 2016;432:4-12
7. Vandenberk L, Garg AD, Verschuere T. et al. Irradiation of necrotic cancer cells, employed for pulsing dendritic cells (DCs), potentiates DC vaccine-induced antitumor immunity against high-grade glioma. Oncoimmunology. 2015;5:e1083669
8. Lai C, Yu X, Zhuo H. et al. Anti-tumor immune response of folate-conjugated chitosan nanoparticles containing the IP-10 gene in mice with hepatocellular carcinoma. J Biomed Nanotechnol. 2014;10:3576-89
9. Zhang M, He J, Hou J. et al. The immunosuppressant Protosappanin A diminished recipient T cell migration into allograft via inhibition of IP-10 in rat heart transplant. PLoS One. 2014;9:e96138
10. Jiang X, Lu X, Hu P. et al. Improved therapeutic efficacy using vaccination with glioma lysate-pulsed dendritic cells combined with IP-10 in murine glioma. Vaccine. 2009;27:6210-6
11. Zhuo H, Peng Y, Yao Q. et al. Tumor imaging and interferon-γ-inducible protein-10 gene transfer using a highly efficient transferrin-conjugated liposome system in mice. Clin Cancer Res. 2013;19:4206-17
12. Taslimi Y, Zahedifard F, Habibzadeh S. et al. Antitumor Effect of IP-10 by Using Two Different Approaches: Live Delivery System and Gene Therapy. J Breast Cancer. 2016;19:34-44
13. He J, Duan S, Yu X. et al. Folate-modified Chitosan Nanoparticles Containing the IP-10 Gene Enhance Melanoma-specific Cytotoxic CD8(+)CD28(+) T Lymphocyte Responses. Theranostics. 2016;6:752-61
14. Lai C, Ye B, Hou X. et al. Anti-Tumor effect of folic acid-conjugated chitosan nanoparticles containing IL-33 gene in hepatocellular carcinoma. Cell Communications. 2014;1:30-40
15. Kudryashova EV, Suhoverkov KV, Sokolov NN. PEG-chitosan branched copolymers to improve the biocatalytic properties of Erwinia carotovora recombinant L-asparaginase. Biomed Khim. 2015;61:480-7
16. Lunardi S, Jamieson NB, Lim SY. et al. IP-10/CXCL10 induction in human pancreatic cancer stroma influences lymphocytes recruitment and correlates with poor survival. Oncotarget. 2014;5:11064-80
17. Ramirez LA, Arango TA, Thompson E. et al. High IP-10 levels decrease T cell function in HIV-1-infected individuals on ART. J Leukoc Biol. 2014;96:1055-63
18. Enderlin M, Kleinmann EV, Struyf S. et al. TNF-alpha and the IFN-gamma-inducible protein 10 (IP-10/CXCL-10) delivered by parvoviral vectors act in synergy to induce antitumor effects in mouse glioblastoma. Cancer Gene Ther. 2009;16:149-60
19. Gleason MK, Ross JA, Warlick ED. et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood. 2014;123:3016-26
20. Wang J, Gong L, Zhu S. et al. The Human Homolog of Drosophila Headcase Acts as a Tumor Suppressor through Its Blocking Effect on the Cell Cycle in Hepatocellular Carcinoma. PLoS One. 2015;10:e0137579
21. Pinton L, Solito S, Damuzzo V. et al. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget. 2016;7:1168-84
22. Zhang H, Maric I, DiPrima MJ. et al. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. Blood. 2013;122(7):1105-13
23. Liechtenstein T, Perez-Janices N, Gato M. et al. A highly efficient tumor-infiltrating MDSC differentiation system for discovery of anti-neoplastic targets, which circumvents the need for tumor establishment in mice. Oncotarget. 2014;5:7843-57
24. De Vries CR, Monken CE, Lattime EC. The addition of recombinant vaccinia HER2/neu to oncolytic vaccinia-GMCSF given into the tumor microenvironment overcomes MDSC-mediated immune escape and systemic anergy. Cancer Gene Ther. 2015;22:154-62
25. Danelli L, Frossi B, Pucillo CE. Mast cell/MDSC a liaison immunosuppressive for tumor microenvironment. Oncoimmunology. 2015;4:e1001232
26. Valanparambil RM, Tam M, Jardim A. et al. Primary Heligmosomoides polygyrus bakeri infection induces myeloid-derived suppressor cells that suppress CD4+Th2 responses and promote chronic infection. Mucosal Immunol. 2016 [Epub ahead of print]
27. Moses K, Brandau S. Human neutrophils: Their role in cancer and relation to myeloid-derived suppressor cells. Semi n Immunol. 2016 [Epub ahead of print]
28. Noman MZ, Desantis G, Janji B. et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781-90
29. Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclinD1 ablation. Nature. 2001;411:1017-21
30. Wang Y, Wang X, Flores ER. et al. Dysfunctional telomeres induce p53-dependent and independent apoptosis to compromise cellular proliferation and inhibit tumor formation. Aging Cell. 2016 [Epub ahead of print]
31. Liang SN, Huang YJ, Liu LL. et al. Study on the correlation between the expression of Ki67 and FasL and prognosis of cervical carcinoma. Genet Mol Res. 2015;14:8634-9
32. Wilkinson RD, Young A, Burden RE. et al. A bioavailable cathepsin S nitrile inhibitor abrogates tumor development. Mol Cancer. 2016;15:29
33. Bekker-Méndez C, Guzmán-Aguilar RM, Hernández-Cueto MA. et al. TUNEL-positive cells in the surgical border of an amputation due to infected diabetic foot. Mol Med Rep. 2012;5:363-72
Author contact
![]() Corresponding authors: Yongxiang Zhao, M.D., Ph.D., Professor, National Center for International Research of Biological Targeting Diagnosis and Therapy / Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research / Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China. Tel: (86) 771-5630 196; Fax: (86) 771-5630 196; E-mail: yongxiang_zhaocom. Other corresponding authors: Xiaoling Lu, Ph.D., Professor, National Center for International Research of Biological Targeting Diagnosis and Therapy / Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research / Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China. Tel: (86) 771-5317 061; Fax: (86) 771-5317 061; Email: luwuliucom. Zhiyong Zhang, Ph.D., Professor, National Center for International Research of Biological Targeting Diagnosis and Therapy / Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research / Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China. Department of Surgery, Robert-Wood-Johnson Medical School University Hospital, Rutgers University, The State University of New Jersey, New Brunswick, New Jersey 08901, USA. Tel: (86) 771-5359 200; Fax: (86) 771-5317 061; Email: zhiyongzhangcom.
Corresponding authors: Yongxiang Zhao, M.D., Ph.D., Professor, National Center for International Research of Biological Targeting Diagnosis and Therapy / Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research / Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China. Tel: (86) 771-5630 196; Fax: (86) 771-5630 196; E-mail: yongxiang_zhaocom. Other corresponding authors: Xiaoling Lu, Ph.D., Professor, National Center for International Research of Biological Targeting Diagnosis and Therapy / Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research / Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China. Tel: (86) 771-5317 061; Fax: (86) 771-5317 061; Email: luwuliucom. Zhiyong Zhang, Ph.D., Professor, National Center for International Research of Biological Targeting Diagnosis and Therapy / Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research / Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China. Department of Surgery, Robert-Wood-Johnson Medical School University Hospital, Rutgers University, The State University of New Jersey, New Brunswick, New Jersey 08901, USA. Tel: (86) 771-5359 200; Fax: (86) 771-5317 061; Email: zhiyongzhangcom.