Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(6):1447-1462. doi:10.7150/thno.17451 This issue Cite
Research Paper
The antihelminthic drug niclosamide effectively inhibits the malignant phenotypes of uveal melanoma in vitro and in vivo
Jingfeng Zhou, Bei Jin, Yanli Jin, Yizhi Liu, Jingxuan Pan ![]()
State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, 54 S. Xianlie Road, Guangzhou, 510060, China.
Received 2016-9-3; Accepted 2017-1-22; Published 2017-4-3
Abstract

Uveal melanoma (UM) is a lethal intraocular malignancy with an average survival of only 2~8 months in patients with hepatic metastasis. Currently, there is no effective therapy for metastatic UM. Here, we reported that niclosamide, an effective repellence of tapeworm that has been approved for use in patients for approximately 50 years, exhibited strong antitumor activity in UM cells in vitro and in vivo. We showed that niclosamide potently inhibited UM cell proliferation, induced apoptosis and reduced migration and invasion. p-Niclosamide, a water-soluble niclosamide, exerted potent in vivo antitumor activity in a UM xenograft mouse model. Mechanistically, niclosamide abrogated the activation of the NF-κB pathway induced by tumor necrosis factor α (TNFα) in UM cells, while niclosamide elevated the levels of intracellullar and mitochondrial reactive oxygen species (ROS) in UM cells. Quenching ROS by N-acetylcysteine (NAC) weakened the ability of niclosamide-mediated apoptosis. Matrix metalloproteinase 9 (MMP-9) knockdown by shRNA potentiated, while ectopic expression of MMP-9 rescued, the niclosamide-attenuated invasion, implying that MMP-9 is pivotal for invasion blockage by niclosamide in UM cells. Furthermore, our results showed that niclosamide eliminated cancer stem-like cells (CSCs) as reflected by a decrease in the Aldefluor+ percentage and serial re-plating melanosphere formation, and these phenotypes were associated with the suppressed Wnt/β-catenin pathway by niclosamide in UM. Niclosamide caused a dose- and time-dependent reduction of β-catenin and the key components [e.g., DVLs, phospho-GSK3β (S9), c-Myc and Cyclin D1] in the canonical Wnt/β-catenin pathway. Additionally, niclosamide treatment in UM cells reduced ATP and cAMP contents, and decreased PKA-dependent phosphorylation of β-catenin at S552 and S675 which determine the stability of β-catenin protein, suggesting that niclosamide may work as a mitochondrial un-coupler. Taken together, our results shed light on the mechanism of antitumor action of niclosamide and warrant clinical trial for treatment of UM patients.
Keywords: niclosamide, uveal melanoma, migration, invasion, cancer stem cells
Introduction
Uveal melanoma (UM), the most common intraocular malignant tumor in adults, accounts for ~3% of all melanomas [1]. Enucleation of the affected eye is a conventional approach for the treatment of primary lesions. More conservative treatment options (e.g. proton-beam radiotherapy, plaque radiotherapy, transpupillary thermotherapy, and systemic chemotherapy) to desire vision preservation are becoming a tendency. Despite these multimodal approaches, the event-free survival of patients with UM has not been improved in the past decades [2, 3]. In particular, ~50% patients with UM develop metastasis, predominantly hepatic metastasis [1, 2], which accounts for significant death in UM patients. Patients with metastatic UM display poor prognosis with an average survival of 2 to 8 months [4, 5].
Continuous efforts were made to develop medicines for the treatment of metastatic UM. Although mutations in GNAQ and GNA11 were observed in ~80% of patients with UM, which may drive the growth of the primary lesion, little is known about drivers of UM metastasis [6]. Furthermore, directly targeting mutant GNAQ and GNA11 has been demonstrated to be structurally challenging [7]. Targeting protein kinase C (PKC) and mitogen-activated protein kinase (MAPK), the downstream components of GNAQ and GNA11, are being evaluated in clinical trial and the curative options on metastatic UM need a further investigation [8]. Given the predominant hepatic metastasis of UM cells, disruption of the interaction between UM cells and liver microenvironment was examined [3]. For instance, based on the rationale of binding membrane receptor tyrosine kinase c-Met on UM cells by the secreted hepatocyte growth factor (HGF) from hepatocytes, a clinical trial of targeting c-Met is undergoing [9].
More recently, treatment of patients with advanced, unresectable cutaneous melanoma by immunotherapy including ipilimumab [a human monoclonal antibody that blocks the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4)] and pembrolizumab [fully human monoclonal antibodies targeting the programmed cell death 1 (PD-1) receptor] has been obtained inspiring clinical treatment efficacy [8, 10]. However, probably due to vitally distinct molecular genetics of UM from that of cutaneous melanoma [5], the clinical activity of ipilimumab in patients with metastatic UM is limited and the clinical efficacy of pembrolizumab remains to be evaluated [8]. Therefore, novel therapeutic drugs for effective treatment of patients with UM, especially for the patients with metastatic UM, are urgently needed.
Niclosamide is a cell-permeable salicylanilide well known for its antihelmintic efficacy, and has been approved by FDA for application in humans for approximately 50 years [11]. Recent studies from us and others have demonstrated that niclosamide has antitumor activity in multiple types of cancers (e.g., leukemia, colorectal and prostate cancer) [12-17], and that the underlying mechanisms involve the inhibition of NF-κB pathway, the increase in reactive oxygen species (ROS) and the blockage of Wnt/β-catenin pathway.
In this study, our purpose was to determine whether niclosamide was active against UM. Our results showed that niclosamide potently inhibited cell proliferation, induced apoptosis in UM cells, reduced migration and invasion, and eradicated CSCs of UM. These effects of niclosamide on UM involved the inhibition of the NF-κB pathway, MMP-9 and Wnt/β-catenin pathways, and the elevation of ROS. Our study may provide the first pre-clinical evidence for further testing of niclosamide for its efficacy in patients with this ocular disease.
Materials and Methods
Chemicals
Niclosamide (2′, 5-dichloro-4′-nitrosalicylanilide, purity > 95%, HPLC), Annexin V-FITC, propidium iodide (PI), and N-acetylcysteine (NAC) were purchased from Sigma-Aldrich (Shanghai, China). p-Niclosamide was synthesized in our lab, as described previously [12]. Niclosamide was dissolved in DMSO at 10 mmol/L and stored at -20 °C. 4′, 6-diamidino-2-phenylindole (DAPI) and CM-H2DCF-DA were purchased from Invitrogen (Shanghai, China). Dibutyryl-cAMP sodium salt (dbcAMP) was from Selleck (Shanghai, China).
Cell lines and culture
The UM cells 92.1, Mel270, Omm1, and Omm2.3 were grown in RPMI1640 medium (Invitrogen, Shanghai, China) with 10% fetal bovine serum (Biological Industries, Kibbutz Beit Haemek, Israel) and 2 mmol/L glutamine and incubated at 37 °C humidified incubator containing 5% CO2 as described previously [18]. The cells were routinely confirmed to be mycoplasma-free.
Cell viability assay and protein biomass assay
Cell viability was determined using MTS assay (CellTiter 96 Aqueous One Solution reagent, Promega), while the protein biomass of UM cells was examined by sulforhodamine B (Sigma-Aldrich, Shanghai, China) assay as described previously [18, 19]. Briefly, the UM cells seeded overnight in 96-well plates (5,000 cells/well) were exposed to varying concentrations of niclosamide for 72 h. For the MTS assay, MTS (20 μL/well) was added to the culture at the end of treatment, and optical density was read with a wave length of 490 nm. IC50 value was determined by curve fitting of the sigmoidal dose-response curve. For the sulforhodamine B protein biomass assay, 50 μL of cold 30% trichloroacetic acid (Sigma-Aldrich, Shanghai, China) buffer per well were added to the medium and fixed at 4 °C for 1 h. After fixation, the supernatants were removed and washed 5 times with deionized water and dried at room temperature. Seventy microliter of 0.4% sulforhodamine B (w/v, in 1% acetic acid) per well was added and stained for 30 min at room temperature. Plates were then washed with 1% acetic acid and dried at room temperature. The bound sulforhodamine B in cells was dissolved in 200 μL of 10 mM Tris base (pH 10.5) per well and the plate was read at 560 nm.
pH stress experiments in uveal melanoma cell lines
The different pH level of the RPMI1640 medium was adjusted by ethanesulfonic acid (for acidic pH, pH=6.0, pH=6.5, pH=7.0), NaHCO3 (for physiologic pH, pH=7.4) or HEPES (for alkaline pH, pH=8.0, pH=8.5) [20].
Colony-formation assay
The colony-forming capacity of UM cells was analyzed by use of agarose as described [21, 22]. Briefly, 400 μL of complete RPMI1640 medium containing 1% agarose (Invitrogen, Guangzhou, China) was added to each well of 24-well plates. The UM cells that were pretreated with niclosamide and resuspended in 400 μL complete RPMI1640 medium containing 0.5% agarose were plated over the layer of the agarose-containing medium. These plates were kept at 4 °C for 10 min to allow solidification, and then incubated at 37 °C in a humidified atmosphere containing 5% CO2 for 2 weeks. Colonies consisting of ≥ 50 cells were counted under an inverted phase-contrast microscope.
Western blot analysis
Cell lysates were prepared using the radioimmunoprecipitation assay buffer (1 × PBS, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with freshly added 10 mmol/L β-glycerophosphate, 1 mmol/L sodium orthovanadate, 10 mmol/L NaF, 1 mmol/L phenylmethylsulfonyl fluoride, and 1 × Roche complete Mini protease inhibitor cocktail (Roche, Indianapolis, IN) [18]. Cytosolic fractionation for cytochrome c detection was prepared using the digitonin extraction buffer (10 mM PIPES, 0.015% digitonin, 300 mM sucrose, 100 mM NaCl, 3 mM MgCl2, 5 mM EDTA, and 1 mM phenylmethylsulfonyl fluroride) [18]. Cytoplasmic and nuclear fractionations for detection of subcellular distribution of NF-κB components were prepared using the methods previously described [12]. Antibodies against caspase-3, PARP (clone 4C10-5), XIAP, Bcl-2, cytochrome c (clone 6H2.B4) and phospho-GSK3β (Y216) were obtained from BD Biosciences (San Jose, CA). Antibodies against p65, IκBα, phospho-GSK3β (S9), DVL2, phospho-β-catenin (S552), phospho-β-catenin (S675), proliferating cell nuclear antigen (PCNA), Bcl-XL, phospho-IKKα (S180)/IKKβ (S181), phospho-IκBα (S32), and MMP-9 were purchased from Cell Signaling Technology (Beverly, MA). Antibodies against β-actin, Active caspase-3 and Flag were from Sigma-Aldrich (Shanghai, China). Anti-mouse and anti-rabbit immunoglobulin G fluorescent-conjugated secondary antibodies were from LI-COR Biotechnology (Nebraska, USA). The NC membranes were scanned using the Odyssey infrared imaging system (LI-COR).
Measurement of mitochondrial transmembrane potential
The UM cells were collected after incubation with niclosamide at the indicated times and stained with MitoTracker probes (CMXRos and MTGreen, Invitrogen, Shanghai, China) at 37 °C for 1 h. The cells were washed with PBS and resuspended in serum-free RPMI1640 medium, and the inner mitochondrial transmembrane potential (△Ψm) was measured by flow cytometry as described previously [18].
Immunofluorescence staining
Immunofluorescence staining was performed as previously described [12]. In brief, after the cells attached on the coverslips in the 6-well plates, the medium was replaced with the serum-free RPMI1640 medium in the presence or absence of niclosamide for 24 h, followed by addition of TNFα (20 ng/mL) for 15 min. After fixation with 3% paraformaldehyde for 20 min, the cells were permeabilized with 1% Triton X-100 and 0.5% NP-40 for 10 min and blocked with 5% bovine serum albumin (BSA) for 1 h at room temperature. They were then washed with 0.1% NP-40 and incubated with anti-p65 (1:100) overnight at 4 °C followed by the corresponding secondary antibody for 1 h at room temperature. DAPI was applied to stain nucleus, immunofluorescence analysis of p65 localization was performed by confocal microscope.
Luciferase assay
Cells (5×104/well) were seeded in the 24-well plates, and then co-transfected with the promoter reporter constructs encoding NF-κB-TATA-Luc (0.5 μg/well) and pEFRenilla-Luc (10 ng/well) as previously described [12, 23]. Twenty-four hours after transfection, the cells were exposed to serum-free medium with or without niclosamide for 16 h, followed by treatment with TNFα (20 ng/mL) for 8 h. The cells were harvested for luciferase activity using the dual-luciferase assay kits [12].
Measurement of intracellular and mitochondria reactive oxygen species
The intracellular and mitochondria specific ROS were determined using CM-H2DCF-DA and MitoSOX Red Reagent (Invitrogen, Shanghai, China), respectively [18, 24]. After cells were treated with niclosamide at the indicated times, CM-H2DCF-DA (3 μmol/L) or MitoSOX Red (5 μmol/L) was added to the culture 1 h prior to the termination of treatment. The cells were harvested and washed twice with PBS at room temperature, and were analyzed by flow cytometry.
Wound-healing scratch assay
UM cells were grown to confluence on 6-well plates, and then each plate was scratched using a 200 μL sterile micropipette tip. The cells were washed with PBS and replaced with fresh RPMI1640 containing 10% FBS, and incubated in the presence or absence of niclosamide (2 μmol/L) for the indicated times. The same wounded area of microscopic field was recorded using an inverted phase-contrast microscope [18].
In vitro migration and invasion assay
The in vitro migration and invasion assays were performed as previously described [25]. For the migration assay, 92.1 and Omm2.3 cells were pretreated with niclosamide for 48 h. After washing with PBS, 5,000 cells were resuspended in 200 μL serum-free RPMI1640 medium, and seeded in the upper chamber (inserts); for invasion assay, 4×104 cells were placed in matrigel (Corning Incorporated) coated bottom chambers. RPMI1640 medium plus 10% FBS was added in the lower chamber as the chemo-attractant. Forty-eight hours later, the inserts were removed, and the cells on the surface of the bottom chambers were fixed with 3% paraformaldehyde, followed by staining with 0.5% violet. The cells in 3 random microscopic fields were counted and photographed using an inverted phase-contrast microscope.
DNA constructs and lentiviral transduction in uveal melanoma cells
Human matrix metalloproteinase 9 (MMP-9, NCBI Reference Sequence ID: NM_004994.2) were cloned into the pTSB-CMV-MCS-SBP-3Flag-GFP-F2A-PuroR (lentivirus) construct (Transheep, Shanghai, China) using ClonExpress MultiS One Step Cloning Kit (Vazyme, Nanjing, China). Scramble (pLKO.1-puro-Non-target shRNA) or human MMP-9 specific target shRNA (pLKO.1-puro-hMMP-9-target shRNA) were from Sigma-Aldrich (Shanghai, China) (Supplementary Table S1). Lentivirus supernatants were produced by 293T cells with pCMV-dR8.2 (the packing construct) and the pCMV-VSVG (envelope construct) lentivirus packing system as described previously [26]. Briefly, the control or target lentivirus plasmid together with pCMV-dR8.2 and pCMV-VSVG were transfected into 293T cells using polyethyleneimine (Polysciences, Inc., Warrington, PA). Forty-eight or 72 h after transfection, virus-containing supernatants were collected and purified with 0.45 μm filter. Two rounds of addition of lentivirus supernatants into the growing 92.1 cells were conducted; the transduced cells were then incubated with puromycin (1 μg/mL) for approximately 5 days to establish stable clones to overexpress or silence MMP-9.
Melanosphere culture
UM cells treated with niclosamide (2 μmol/L) were washed with PBS, and then 5,000 cells were resuspended in DMEM/F-12 medium (HyClone, containing B27: 1 mL, bFGF: 10 ng/mL and EGF: 20 ng/mL) and plated into each well of 24-well CorningTM Ultra-Low Attachment Plates (Thermo Fisher Scientific Inc, Waltham, MA) [18]. After 7-day incubation, melanospheres (cells ≥ 50) were counted. The cultures were then harvested, and re-plated (5,000 cells/well) for the secondary, tertiary and fourth rounds of culture, respectively. Melanospheres were counted on day 7 following each round of re-plating [18].
Aldehyde dehydrogenase assay
Aldehyde dehydrogenase (ALDH) was assayed using the ALDEFLUORTM kit (Stem Cell Technologies, Vancouver, BC, Canada). Briefly, the ALDEFLUORTM reagent was added and mixed, followed by transferring 0.5 mL of the mixture aliquote to a fresh tube containing 5 μL of ALDEFLUORTM DEAB reagent. After incubation at 37 °C for 30 ~ 60 min, the samples were centrifuged and supernatant was discarded. Cell pellets were resuspended in 0.5 mL of ALDEFLUORTM assay buffer and then analyzed by FACS LSR Fortessa flow cytometer [18].
ATP content measurement
ATP levels in uveal melanoma cells were measured using a luciferase-based ATP measurement kit (ThermoFisher, Shanghai, China) [27]. Briefly, the UM cells seeded overnight in 6-well plates were exposed to varying concentrations of niclosamide for 3 h. After harvested and washed with cold PBS, the cells (2 ×105) were resuspended in 100 μL ATP extaction buffer (100 mM Tris, 4 mM EDTA, pH 7.75) and then incubated in 100 °C water bath for 90 s. The samples were centrifuged at 10, 000 g for 30 s and supernatants were transferred to another fresh tube. Five microliter of supernatant was added into 95 μL fresh reaction buffer and the luciferase activity was measured by the Multiscan Spectrum (Bio-Tek, Winooski, VT).
cAMP measurement
Cytoplasmic cAMP levels in uveal melanoma cells were detected with the Cyclic AMP EIA kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer's instructions. In brief, Mel270 and Omm1 cells seeded overnight in 6-well plates were exposed to escalating concentrations of niclosamide or 10 μmol/L forskolin for 3 h. The culture medium was then aspirated and 1 mL of 0.1 M HCl was added and incubated at room temperature for another 20 min. The cells were scraped, homogenized by pipetting up and down and transferred to a fresh centrifuge tube. The samples were centrifuged at 1,000 g for 10 min and the supernatant was decanted into a clean test tube. Add 50 μL samples or cAMP ELISA standard to each 96 wells (mouse anti-Rabbit IgG coated plate, Cayman). Each plate was incubated for 18 h at 4 °C. The wells were rinsed 5 times with wash buffer. The Ellman's Reagent (200 μL per well) was added and the plates were incubated in dark for 90~120 min, and then read at a wavelength of 412 nm. The concentrations of cAMP were calculated according to the cAMP standard curve.
Animal experiments
Omm1 cells (3 × 106 cells in 200 μL PBS/mouse) were subcutaneously inoculated into the flanks of male (4 to 6-week-old) NOD/SCID mice (Vitalriver, Beijing, China). Tumor volumes were measured with calipers every other day, and calculated using the following formula: a2 × b × 0.4, where a refers to the smallest diameter and b is the diameter perpendicular to a. When the tumors reached ~200 mm3, mice were randomly divided to control or experimental groups, and treated daily with intraperitoneal injection of either vehicle (PBS) or p-niclosamide (25 mg/kg) for 2 weeks. Animals were then euthanized and the xenografts were dissected and weighed. For immunohistochemistry staining of the xenografts, specimens were fixed, embedded, and sectioned (4 μm), and then immunostained using the MaxVision kit (Maixin Biol, Fuzhou, China) following the manufacturer's instructions [28]. The animal studies were approved by the Sun Yat-sen University Institutional Animal Care and Use Committee.
Statistical analysis
All experiments were repeated three times, and data were expressed as mean ± standard deviation (SD). Comparisons between 2 groups were analyzed by 2-tailed Student's t test; differences among multiple groups were analyzed by one-way ANOVA with post hoc comparison by the Dunmett's test, unless otherwise stated. GraphPad Prism 6.02 software (GraphPad, San Diego, CA) was used for statistical analysis. p < 0.05 was considered statistically significant.
Results
Niclosamide counteracts proliferation of uveal melanoma cells
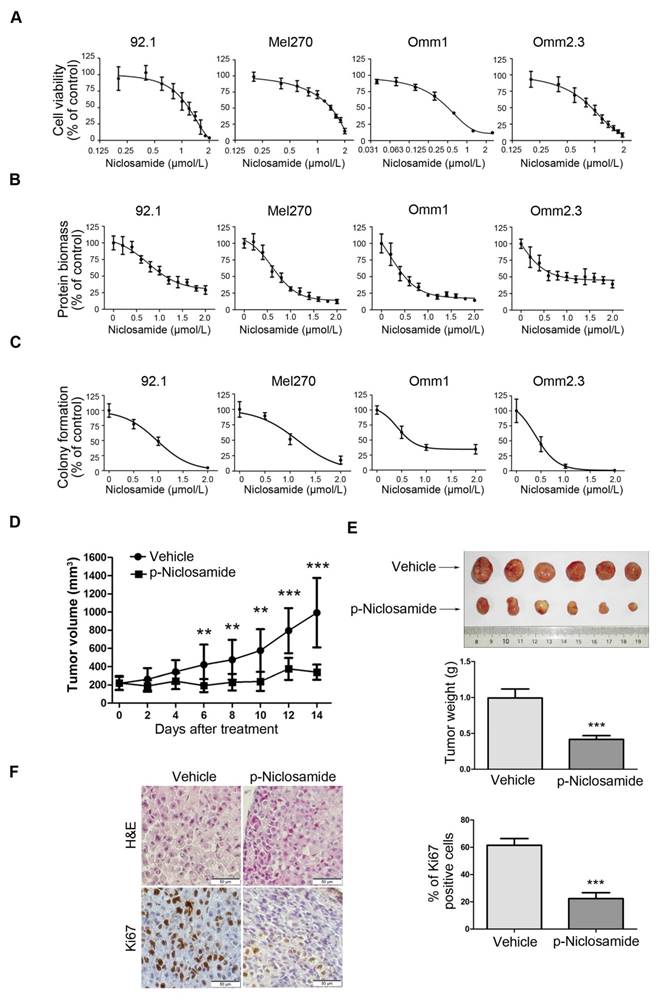
We first determined the effect of niclosamide on growth of UM cells. 92.1, Mel270, Omm1, and Omm2.3 cells were exposed to escalating concentrations of niclosamide for 72 h, and cell viability was determined by MTS assay. As shown in Figure 1A, niclosamide inhibited the cell viability in a dose-dependent manner, with IC50 values of 1.18 μmol/L, 1.35 μmol/L, 0.58 μmol/L, 1.0 μmol/L in 92.1, Mel270, Omm1, and Omm2.3 cells, respectively. The inhibitory effect of niclosamide was further cross-validated by sulforhodamine B protein biomass assay, with IC50 values of 1.29 μmol/L, 0.84 μmol/L, 0.62 μmol/L, 0.91 μmol/L in 92.1, Mel270, Omm1, and Omm2.3 cells, respectively (Figure 1B). In addition, similar inhibitory effect of niclosamide on the ability of UM cells to form colony in agarose was observed (Figure 1C).
Niclosamide inhibits the proliferation of uveal melanoma cells in vitro and in vivo. (A) UM cells (92.1, Mel270, Omm1 and Omm2.3) were exposed to escalating concentrations of niclosamide for 72 h, and the cell viability was determined by MTS assay. (B) UM cells (92.1, Mel270, Omm1 and Omm2.3) were exposed to escalating concentrations of niclosamide for 72 h, and protein biomass was determined by sulforhodamine B assay. (C) UM cells were seeded in drug-free agarose culture and incubated for 14 days, colonies were counted. Clonogenicity was expressed by normalizing to control. (D) NOD/SCID mice bearing palpable Omm1 xenografted tumors were treated with vehicle (PBS) or p-niclosamide (25 mg/kg, i.p.) daily. The tumor volumes measured every other day versus time were plotted. Error bars, SD. **, p< 0.01; ***, p< 0.001, Student's t test. (E) Weights of tumors dissected on day 14 after treatment with p-niclosamide. Representative tumors are shown (top). Data (bottom) shown are the mean ± SD of tumor weights from each group. Vehicle (n=6), p-niclosamide (n=8). ***, p< 0.001, Student's t test. (F) Hematoxylin-eosin (H&E) staining and immnunohistochemical analysis with anti-Ki67 of xenograft tissues from mice sacrificed 14 days after p-niclosamide treatment (left). The total number of Ki67 positive cells (brown-stained nuclei, regardless of staining intensity were counted as positive) in 3 random microscopic fields were counted by Image-Pro Plus 6.0. Data (right) shown are the mean ± SD of Ki67 index from 3 random microscopic fields. ***, p< 0.001, Student's t test.
Niclosamide inhibits the cell viability of uveal melanoma cells in a pH-dependent manner
Given that the acidification niche of tumor (pH 6.5-6.9) which usually derives from the Warburg effect and hypoxia may facilitate the malignant and aggressive phenotype of cancer [20], we next asked whether extracellular acidification would affect the toxicity of niclosamide on uveal melanoma cells. We exposed the UM cells to niclosamide dissolved in escalating pH levels of culture medium from 6.0 to 8.5 for 72 h. The results showed that niclosamide exerted its antitumor activity against all the tested 4 lines of UM cells in a pH-dependent manner (Supplementary Figure S1), which is consistent with previous reports [20].
Niclosamide suppresses growth of the human uveal melanoma xenografts in NOD/SCID mice
The in vivo antitumor activity of p-niclosamide [12], a water-soluble form of niclosamide, was examined in the NOD/SCID mice bearing Omm1 UM xenografts. As shown in Figure 1D, tumor-bearing mice treated with p-niclosamide (25 mg/kg/day, i.p.) for 14 days showed significantly smaller tumor volume, as compared with the corresponding vehicle-treated mice. There was a significant difference in the tumor weights between the p-niclosamide-treated and vehicle-treated groups (Figure 1E). The body weights of mice were stable, with no significant difference between the vehicle and treatment groups (data not shown). The immunohistochemistry staining for Ki67, a cell proliferation marker, was also decreased in the tumor xenografts of the p-niclosamide-treated mice (Figure 1F).
Niclosamide elicits apoptosis in uveal melanoma cells
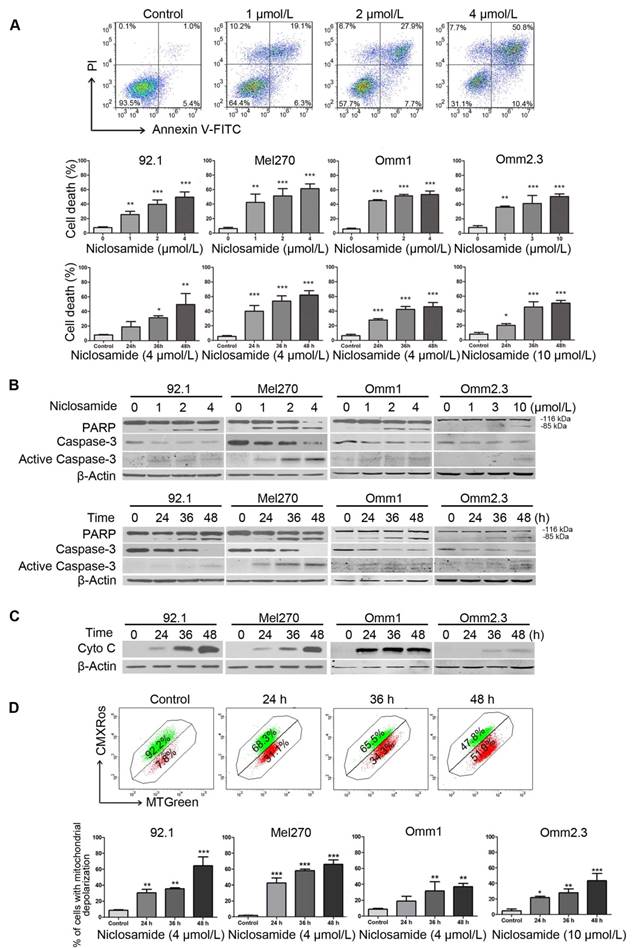
We next determined the effect of niclosamide on apoptosis in UM cells. We observed a dose- and time-dependent induction of apoptosis in the tumor cells treated with niclosamide, as determined by flow cytometric analysis of Annexin V-FITC/PI staining (Figure 2A). Occurrence of apoptosis in the niclosamide-treated UM cells was also evidenced by dose- and time-dependent increases of cleaved PARP and caspase-3 (Figure 2B), as well as a time-dependent release of cytochrome c into the cytosol (Figure 2C). The effect of niclosamide on mitochondria was further evidenced by significant elevations of the cell population with loss of mitochondrial trans-membrane potential (ΔΨm), as measured by flow cytometric analysis of CMXRos and MTGreen dual staining (Figure 2D). No apparent alteration of Bcl-2 family members (e.g., Bcl-2, Bcl-XL, Bax) and IAP (inhibitor apoptosis protein) family proteins (e.g., XIAP, Survivin) was observed in UM cells treated with niclosamide for 48 h (Supplementary Figure S2).
Niclosamide induces apoptosis in uveal melanoma cells. (A) UM cells were exposed to increasing concentrations of niclosamide for 48 h, or a fixed concentration (4 μmol/L or 10 μmol/L) of niclosamide for different time-periods, apoptosis was analyzed by flow cytometry after Annexin V-FITC/PI dual staining. Representative flow cytometry dot plots for Mel270 were shown (top); Bar graphs: mean ± SD from 3 independent experiments (middle & bottom). *, p< 0.05; **, p< 0.01; ***, p< 0.001, one-way ANOVA, post hoc comparisons, Dunnett's test. (B) Dose- (48 h) and time-dependent (4 μmol/L for 92.1, Mel270 and Omm1; 10 μmol/L for Omm2.3) cleavages of PARP and caspase-3 were determined by Western blot after treatment with niclosamide in UM cells. (C) UM cells were exposed to niclosamide for 48 h, Western blot analysis of cytochrome c in the cytosolic extracts. (D) Mitochondrial potential of UM cells exposed to niclosamide was measured by flow cytometry after staining with CMXRos and MTGreen. Representative fluorescence histograms from three independent experiments (92.1) (top); Results for 3 independent experiments are shown (bottom). *, p< 0.05; **, p< 0.01; ***, p< 0.001, one-way ANOVA, post hoc comparisons, Dunnett's test.
Niclosamide inhibits TNFα-induced activation of the NF-κB pathway
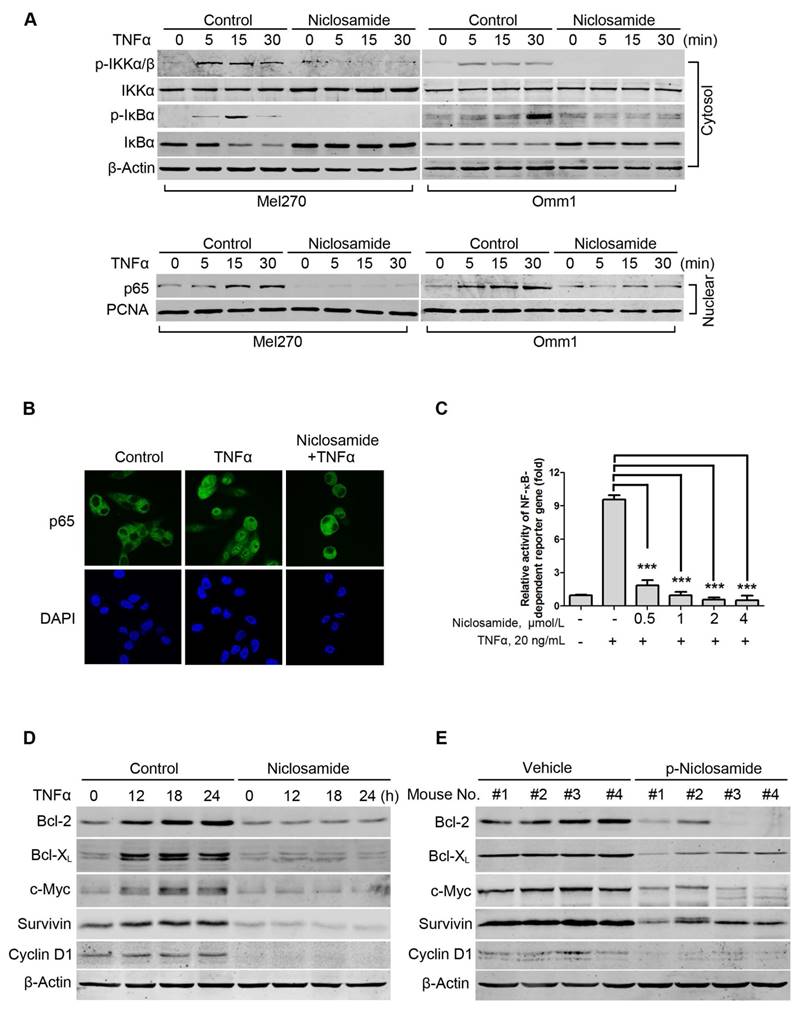
As constitutive activation of NF-κB is critical in primary and metastatic UM cells [29], we next determined the effect of niclosamide on the activity of NF-κB in the tumor cells. We first examined whether niclosamide modulated IκBα phosphorylation and p65 nucleus translocation. Mel270 or Omm1 cells were pretreated with vehicle or niclosamide for 24 h, and then incubated with TNFα for different periods. The results showed that the treatment with TNFα resulted in an increase in levels of phospho-IKKα/β and phospho-IκBα and a decrease in IκBα in the cytoplasmic extracts from control cells which were not observed in the cells pre-treated with niclosamide (Figure 3A). Similarly, the effect of TNFα-induced increase of p65 in the nuclear fraction was blocked by niclosamide (Figure 3A). The blockage of the TNFα-induced nuclear translocation of p65 by niclosamide was further verified by immunofluorescence microscopy examination (Figure 3B). In Mel270 cells transiently co-transfected with NF-κB-TATA-Luc and Renilla luciferase reporter constructs, we demonstrated that niclosamide treatment led to significant suppression of the TNFα-induced NF-κB reporter activity (Figure 3C), suggesting that this drug can inhibit TNFα-induced transcription of the NF-κB-dependent genes. Moreover, pre-treatment of UM cells with niclosamide abrogated the TNFα-induced expression of Bcl-2, Bcl-XL, c-Myc, Survivin and Cyclin D1 proteins involved in cell survival (Figure 3D). Additionally, the NF-κB-dependent proteins were dramatically decreased in the tumor xenografts of the p-niclosamide-treated mice (Figure 3E).
Niclosamide abrogates TNFα-induced NF-κB activation in uveal melanoma cells. (A) Mel270 and Omm1 cells were pre-incubated with 4 μmol/L niclosamide for 16 h, followed by treatment with TNFα (10 ng/mL) for the indicated times. Cytoplasmic and nuclear extracts were analyzed by Western blot using specific antibodies for p-IKKα/β, IKKα/β, p-IκBα, IκBα and p65. (B) Mel270 cells were pre-treated with niclosamide (4 μmol/L) for 24 h and then incubated with TNFα (20 ng/mL) for 15 min. After fixation, immunofluorescence analysis of p65 localization was performed by confocal microscope. DAPI (4', 6-diamidino -2-phenylindole) was applied to stain nuclei. (C) Mel270 cells were transiently co-transfected with NF-κB-TATA-Luc reporter constructs (0.5 μg) and Renilla luciferase reporter constructs (10 ng). Twenty-four hours later, equal numbers of the cells were exposed to different concentrations of niclosamide for 16 h, followed by addition of TNFα (20 ng/mL) to the culture for 8 h. Luciferase activity was detected at the end of incubation. Results are fold change ± SD. ***, p< 0.001, one-way ANOVA, post hoc comparisons, Tukey's test. (D) Mel270 cells were pre-treated with 4 μmol/L niclosamide for 24 h and then incubated with TNFα (20 ng/mL) for the indicated times. The expression of NF-κB-dependent proteins was analyzed by Western blot. (E) Western blot of NF-κB-dependent proteins in NF-κB pathway were analyzed from xenografted tumor tissues of vehicle- or p-niclosamide-treated mice.
Niclosamide increases intracellular and mitochondrial reactive oxygen species in uveal melanoma cells
We previously reported that niclosamide can increase the level of ROS in acute myeloid leukemia cells [12]. We therefore determined intracellular ROS in the niclosamide-treated UM cells. After Mel270 cells were exposed to 4 μmol/L niclosamide, the intracellular ROS level based on CM-H2DCF-DA started to rise at 2 h, and peaked at 16 h following treatment (Figure 4A, top). Given that mitochondria serve as the major intracellular source of ROS [30], we also detected the mitochondria-specific ROS by using MitoSOX Red probe. As anticipated, the mitochondrial ROS levels were elevated in the UM cells treated with niclosamide (Figure 4A, bottom). The increased production of ROS appeared to be, at least in part, responsible for induction of apoptosis, since pre-treatment of the UM cells with the ROS scavenger, N-acetylcysteine (NAC), weakened the effect of niclosamide-mediated apoptosis (Figure 4B).
Niclosamide increases intracellular and mitochondria specific ROS in uveal melanoma cells. (A) Mel270 cells were treated with niclosamide (4 μmol/L) for indicated times, and then stained with CM-H2DCF-DA (for measurement of intracellular ROS) or MitoSOX Red Reagent (for measurement of mitochondria specific ROS) for 1 h. ROS was determined by flow cytometer. The median intensity of DCF-DA (top) or MitoSOX Red (bottom) is shown. (B) Mel270 cells were pre-incubated with NAC (20 mmol/L) for 1 h, and then exposed to niclosamide (4 μmol/L) for 24 h. ROS and apoptosis were evaluated by flow cytometry (left). Mean ± SD of apoptotic cells from 3 independent experiments are shown (right). *, p< 0.05,one-way ANOVA, post hoc comparisons, Tukey's test. (C) Mel270 cells were exposed to niclosamide (4 μmol/L) for 24 h with or without NAC (20 mmol/L), and then stimulated with TNFα (10 ng/mL) for 30 min. Cytoplasmic and nuclear extracts were analyzed by Western blot for p-IκBα, IκBα (cytosol) and p65 (nuclear). (D) Mel270 cells were transiently co-transfected with NF-κB-TATA-Luc reporter constructs (0.5 μg) and Renilla luciferase reporter constructs (10 ng). Twenty-four hours later, the cells were exposed to different concentrations of niclosamide in the absence or presence of NAC for 16 h, followed by stimulation with TNFα (20 ng/mL) for 8 h. Luciferase activity was detected at the end of treatment. Results are fold change ± SD. n.s., no significant difference, one-way ANOVA, post hoc comparisons, Tukey's test. (E) Mel270 cells were treated with niclosamide and TNFα, and then ROS was detected by flow cytometry.
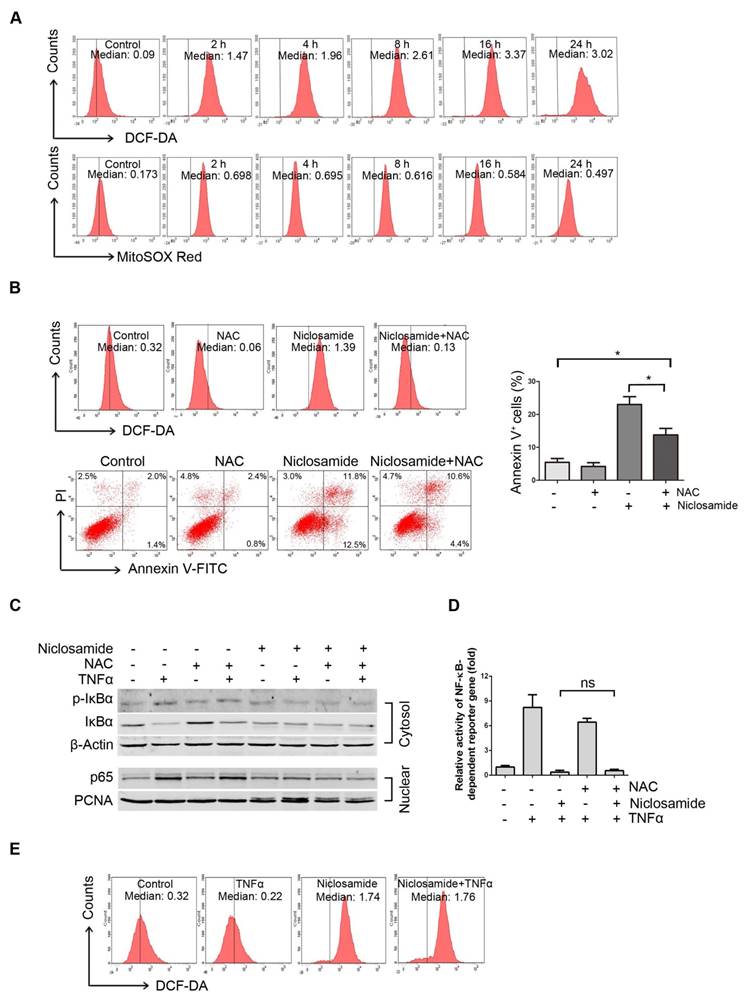
As certain NF-κB-regulated genes play a role in regulating the amount of intracellular ROS, and ROS has either inhibitory or stimulatory roles in the NF-κB signaling pathway [12], we examined whether ROS affected the inactivation of NF-κB pathway in the niclosamide-treated UM cells. NAC did not attenuate the phosphorylation and degradation of IκBα and nuclear translocation of p65 (Figure 4C), and did not alter the transcription of TNFα-induced NF-κB-dependent genes (Figure 4D). On the other hand, the TNFα-induced NF-κB activation did not influence the niclosamide-induced ROS elevation (Figure 4E). These results imply that niclosamide-induced elevation of ROS and inhibition of the NF-κB pathway appears to be independent mechanisms in UM cells.
Niclosamide represses migration and invasion of uveal melanoma cells in vitro
Dissemination of tumor cells is one of the malignant features of UM [5, 31]. We thus determined the effects of niclosamide on migration and invasion of UM cells. Wound healing experiment was used to evaluate the effect of niclosamide on migration of tumor cells. The results showed that the would-healing ability of 92.1 or Omm2.3 cells was dramatically inhibited by niclosamide treatment (Figure 5A). Similar results were obtained by quantitative measurement of migration ability using transwell assay (Figure 5B). Niclosamide treatment also significantly reduced the invasion capacity of UM cells (Figure 5B), as assessed by matrigel invasion assay. Consistently, the expression of MMP-9, an invasion-associated protein, was dramatically reduced in UM cells treated with niclosamide (Figure 5C). These results demonstrated that niclosamide had a striking inhibitory effect on migration and invasion in UM cells.
Niclosamide suppresses migration and invasion of uveal melanoma cells. (A) 92.1 and Omm2.3 were cultured in 6-well plates and scratches were created using sterile pipette tips after cells reached confluence. Records were taken at the indicated times after niclosamide (2 μmol/L) treatment. Representative scratches for 92.1 and Omm2.3 (left); the breadth of scratches was measured and bar graphs (right) were the quantitative analysis of percentage of wound closure relative to the breadth of beginning from 3 random microscopic fields at the indicated time points. Data represent mean ± SD. *, p< 0.05; **, p< 0.01; ***, p< 0.001, Student's t test. (B) 92.1 and Omm2.3 cells were incubated in the presence or absence of niclosamide for 48 h, and then subjected to transwell or matrigel invasion chamber assays. The migrated or invaded cells were counted in 3 random fields. Data represent mean ± SD. *, p< 0.05; **, p< 0.01; ***, p< 0.001, Student's t test. (C) The migration and invasion related protein MMP-9 in the whole cell lysates from the 92.1 and Omm2.3 cells treated with niclosamide was detected by Western blot. (D) 92.1 cells were transduced with either lentivirus MMP-9 cDNA or MMP-9-shRNA constructs, and treated with or without niclosamide for 36 h, and then subjected to matrigel invasion chamber assays. The invaded cells were counted in 3 random fields. Data represent mean ± SD. *, p< 0.05; **, p< 0.01; ***, p< 0.001; ns, no significance, Student's t test.
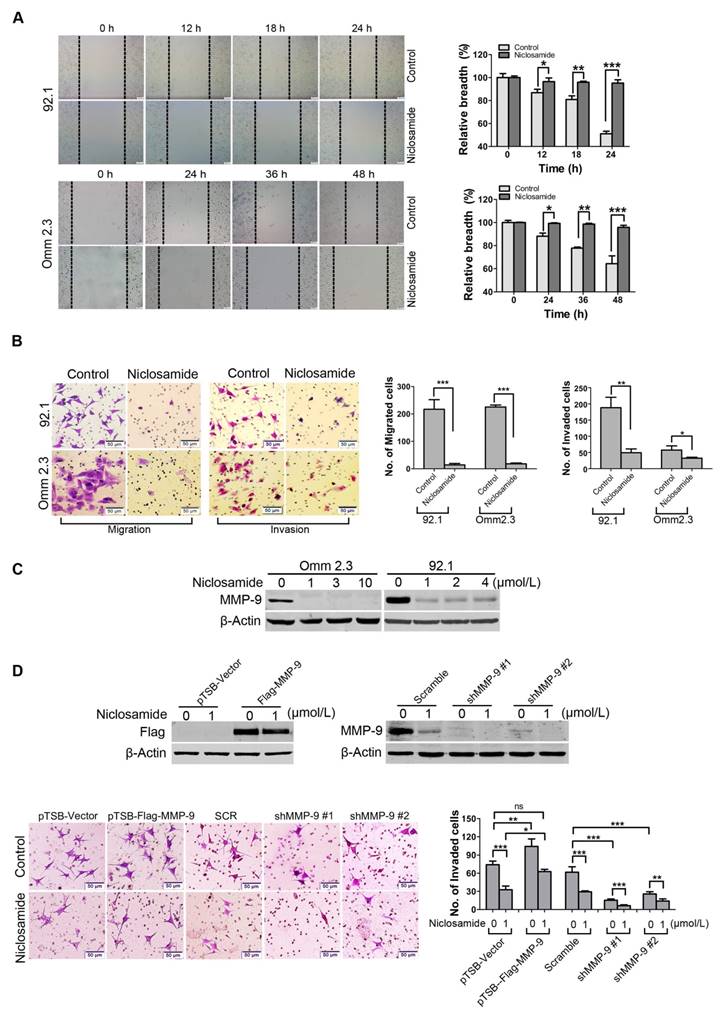
MMP-9 is pivotal for invasion blockage by niclosamide in uveal melanoma cells
To investigate the role of MMP-9 in UM cells, 92.1 cells were transduced with lentivirus constructs encoding human MMP-9, and then incubated in presence or absence of niclosamide for 36 h. The data showed that ectopic overexpression of MMP-9 boosted the invasion capacity of UM cells compared with the cells transduced with the lentiviral empty vector. Conversely, knockdown of MMP-9 by lentiviral shRNA significantly weakened the invasion capacity of UM cells compared with the cells transfected with Control shRNA (Figure 5D). Furthermore, the abrogation of invasion mediated by niclosamide was reversed by ectopic overexpression of MMP-9, but enhanced by knockdown of MMP-9-shRNA (Figure 5D). Taken together, these data indicate the importance of MMP-9 in niclosamide-mediated inhibitory invasion.
Niclosamide eliminates cancer stem-like cells in uveal melanoma
Traits of CSCs such as self-renewal capacity are believed to confer tumor metastasis, drug resistance and cancer relapse [32, 33]. We tested the effect of niclosamide on self-renewal capacity of UM cells. UM cells were exposed to niclosamide (2 μmol/L) for 48 h, and then cultured for melanosphere in the drug-free DMEM/F-12 medium. As shown in Figure 6A, niclosamide dramatically reduced the size and number of melanospheres of UM cells. Four rounds of re-plating experiments showed that the serially re-plating capacity was also declined in the niclosamide-treated UM cells (Figure 6A).
Niclosamide eliminates uveal melanoma cancer stem-like cells with Wnt/β-catenin signaling pathway blocked. (A) The UM cells were incubated with niclosamide (2 μmol/L) for 48 h, and then inoculated in the ultralow-attachment 24-well plates filled with tumorsphere culture medium. Melanospheres were counted on day 7. The cells were then harvested and re-plated for the secondary, tertiary and quaternary rounds of melanosphere culture, respectively. Error bars, SD. **, p< 0.01; ***, p< 0.001, Student's t test. (B) Mel270 and Omm2.3 cells were treated with niclosamide (1 μmol/L) for 48 h, and the percentage of Aldefluor+ cells was determined by flow cytometry. Representative flow cytometry data and bar graph with SD from 3 independent experiments are shown. **, p< 0.01; ***, p< 0.001, Student's t test. (C) Mel270 and Omm1 cells were exposed to increasing concentrations of niclosamide for 48 h. Cell lysates were harvested and detected by Western blots with the indicated antibodies. (D) Mel270 and Omm1 cells were exposed to varying concentrations of niclosamide for 3 h, the cells were harvested for ATP measurement assay. Data were normalized to control. Results for 3 independent experiments are shown. ***, p< 0.001, one-way ANOVA, post hoc comparisons, Dunnett's test. (E) Mel270 and Omm1 cells seeded overnight in 6-well plates were exposed to increasing concentrations of niclosamide or 10 μmol/L forskolin for 3 h and cAMP level was measured. Data were normalized to control. Results for 3 independent experiments are shown. *, p< 0.05; **, p< 0.01; ***, p< 0.001, one-way ANOVA, post hoc comparisons, Tukey's test. (F) The effect of dbcAMP (dibutyryl-cAMP sodium salt) on niclosamide-mediated down-regulation of phospho-β-catenin (S552 and S675) and total level of β-catenin. Mel270 and Omm1 cells were treated with niclosamide (1 μmol/L) in the presence or absence of dbcAMP (500 μmol/L). The phospho-β-catenin (S552 and S675) and total level of β-catenin in the whole cell lysates from Mel270 and Omm1 cells treated with niclosamide (1 μmol/L) in the presence or absence of dbcAMP (500 μmol/L) were detected by Western blot.
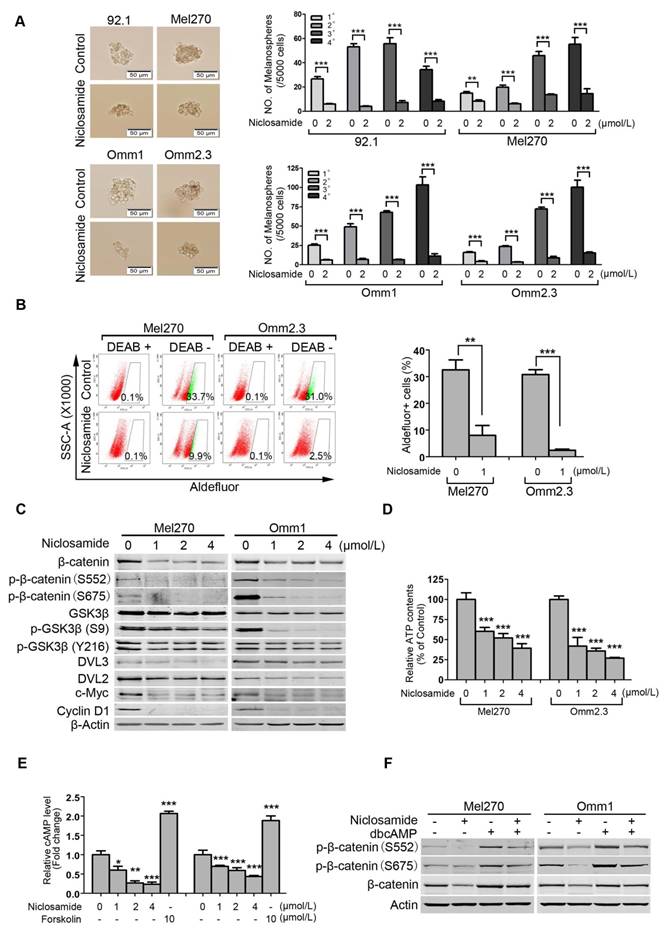
ALDH has been widely used as a putative universal marker for CSCs in multiple types of cancer [34-36]. We thus evaluated the effect of niclosamide on Aldefluor+ cells using flow cytometry. The results showed that niclosamide significantly decreased the percentage of Aldefluor+ cells in Mel270 and Omm2.3 (Figure 6B). In support of the roles of niclosamide in eradicating CSCs, we demonstrated that this agent caused a dose- and time-dependent reduction of β-catenin in Mel270 and Omm1 cells (Figure 6C and Supplementary Figure S3). c-Myc and Cyclin D1, two important downstream proteins in the Wnt/β-catenin pathway, were also decreased in the niclosamide-treated UM cells (Figure 6C). Additionally, the protein levels of DVL2 and DVL3, the positive regulators of β-catenin, and phospho-GSK3β (S9) but not phospho-GSK3β (Y216), were decreased in UM cells treated with niclosamide (Figure 6C). The levels of the key components [e.g. DVLs, phospho-GSK3β (S9), c-Myc and Cyclin D1] in the canonical Wnt/β-catenin pathway were also found to be decreased in the tumor xenografts of the p-niclosamide-treated mice (Supplementary Figure S4 and Figure 3E).
Since niclosamide has the capacity to uncouple mitochondria [37, 38], we measured intracellular ATP and cAMP content. As anticipated, the ATP and cAMP contents in UM cells were significantly decreased after the treatment with niclosamide (Figure 6D and Figure 6E). Consistently, the protein kinase A (PKA)-dependent phosphorylation of β-catenin at S552 and S675 which can facilitate stability of β-catenin protein [38, 39] were reduced in niclosamide-treated UM cells (Figure 6C). Likewise, addition of PKA activator dbcAMP into culture medium resulted in phosphorylation of β-catenin at S552 and S675 in UM cells (Figure 6F). The pre-treatment with dbcAMP abolished the effect of niclosamide-mediated down-regulation in phospho-β-catenin (S552 and S675) and total level of β-catenin (Figure 6F). These results suggested that niclosamide may work as a mitochondrial un-coupler thus reducing PKA-β-catenin signaling in UM cells [38].
Discussion
In the present study, we demonstrate that the antihelminthic drug, niclosamide, is highly effective in suppressing the growth, migration and invasion, and in eradicating CSCs of UM. Importantly, p-niclosamide shows strong antitumor activity in a UM xenograft mice model. As niclosamide has been safely used in patients with infection of certain types of worm for decades, based on the results reported here, this drug may be further exploited as an antineoplastic therapy in UM.
Disclosing new indication and application of old drugs could be more economic and rapid than discovering new drugs from scratch [11]. Here, we found that niclosamide which has been approved by FDA and safely used in human for over 50 years, displayed potent antitumor activity in UM cells. Pharmacokinetics study showed that Cmax (maximal plasma concentration), which reached 71.972 μmol/L after a single 25mg/kg oral administration of p-niclosamide in rats (data not shown), are much higher than those concentrations that reached its in vitro anti-tumor activity. In addition, niclosamide displayed minimal cytotoxicity in normal cells in vitro [12, 13] and in vivo (oral LD50 in rats, >5, 000 mg/kg) [40].
Inflammation has been regarded as one of the most important hallmarks of cancer [41]. Although the eye is an immune privileged site, the inflammation environment can be established when ocular tumor occurs [42, 43]. Such aberrant ocular niches likely lead to deregulated secretion of cytokines [44, 45] and may lead to active NF-κB pathway [46-49]. Indeed, NF-κB is constitutively activated in primary and metastatic UM [29]. Studies have shown that the NF-κB pathway regulates expression of multiple genes that are associated with cell proliferation (e.g. Cyclin D1), anti-apoptosis (e.g. Bcl-2, Bcl-XL, Mcl-1 and Survivin), migration and invasion (e.g., MMP-9) [48, 50, 51]. Consistent with the reported studies [12], we found that niclosamide has an inhibitory effect on the NF-κB pathway.
It has been reported that excess amounts of ROS is toxic to cells [52]. Consistent with our previous observation in leukemia cells [12], here we showed that niclosamide increased ROS levels in UM cells, nevertheless, we noticed that increased ROS caused by niclosamide treatment only partially contributes to the onset of apoptosis in UM cells. Furthermore, our experiments showed quenching of ROS by NAC had no effect on the niclosamide-mediated inhibition of the NF-κB pathway, and vice versa, suggesting that the elevation of ROS and blocking of NF-κB in the UM cells are independent mechanisms of niclosamide.
MMP-9, an important member of matrix metalloproteinases, has the ability to degrade basement membranes in the extracellular matrix, boosting cellular invasion [53]. Accumulating evidence indicates that MMP-9 positively correlated with aggressiveness and metastasis in several types of cancer including breast cancer and glioblastoma [53, 54]. MMP-9 was reported to be predominantly expressed in epithelioid uveal melanomas (a more aggressive type) or epithelioid regions of mixed tumors, a negatively prognostic factor in UM patients [55, 56], the role of MMP-9 in the invasion of UM cells remains to largely unknown. To our knowledge, this is the first time to delineate the fuction of MMP-9 in UM cells invasion. Our data showed that MMP-9 promoted the invasion capacity of UM cells and played pivotal role in the blockage of invasion of UM cells mediated by niclosamide (Figure 5D).
CSCs are believed to contribute to tumor metastasis, drug resistance, and the disease relapse [32, 33]. Promotion of cancer metastasis by CSCs involves multiple mechanisms such as epithelial mesenchymal transition (EMT), angiogenesis and lymphangiogenesis [57]. Therefore, targeting CSCs may be an alternative strategy for the prevention and treatment of UM patients with metastasis. Since the biomarkers of UM CSCs remain elusive, ALDH activity and serial melanosphere formation assays were employed to evaluate the CSCs function. Our results demonstrated here that niclosamide remarkably decreased the percentage of Aldefluor+ cells and the serially re-plating ability of UM cells to form melanospheres, indicating that niclosamide is capable of eliminating CSCs and impairing their self-renewal trait in UM. Consistently, the effect of niclosamide on CSCs has been observed in other types of cancer such as leukemias and breast cancers [12, 17, 58]. More recently, it was reported that a niclosamide loaded rigid core mixed micelle could selectively reduce the CD44+ CSC population in cutaneous melanoma cells [59].
Wnt/β-catenin is one of the intrinsic regulators to control the destiny of CSCs [60, 61]. Our data revealed that niclosamide down-regulates the protein level of β-catenin in vitro and in vivo. Because GSK3β phosphorylate β-catenin at S33, S37 and T41 which results in its subsequent proteasome-ubiquitination [62], reduction in phospho-GSK3β (S9) by niclosamide may be helpful in explaining the reduced β-catenin. DVL is a crucial component of Wnt/β-catenin signaling pathway that have the ability to recruit the Axin thereby deconstructing the degradation complex and resulting in stabilization of β-catenin protein [61]. Lowered DVL mediated by niclosamide is likely an important reason of decline in β-catenin.
Our results showed that the decreased levels of phospho-β-catenin at S552 and S675 in the niclosamide-treated UM cells may reflect the repressed PKA activity because PKA is the major kinase that phosphorylate β-catenin on the sites of S552 and S675 to maintain the stability of β-catenin [38, 39]. This reasoning appeared to be supported by the niclosamide-mediated reduction of cAMP which is an endogenous activator of PKA. Indeed, exogenous PKA activator dbcAMP abrogated the effect of niclosamide on phospho-β-catenin (S552 and S675) and β-catenin. It is plausible that niclosamide may also work as a mitochondrial un-coupler thus reducing PKA-β-catenin signaling. Taken together, decreased β-catenin by niclosamide was likely to be caused through targeting multiple knot regulators in the Wnt/β-catenin pathway.
In conclusion, niclosamide possesses strong in vitro and in vivo antitumor activity in UM. Niclosamide inhibited the NF-κB pathway activation while elevated the levels of intracellullar and mitochondrial ROS in UM cells. MMP-9 was pivotal for invasion blockage by niclosamide in UM cells. Niclosamide eliminated CSCs associated with the suppressed canonical Wnt/β-catenin pathway and PKA-β-catenin pathway in UM. Our results shed light on the mechanism of antitumor action of niclosamide and warrant clinical trial for treatment of UM patients with metastasis.
Supplementary Material
Supplementary figures and table.
Abbreviations
UM: uveal melanoma; TNFα: tumor necrosis factor α; ROS: reactive oxygen species; PKC: protein kinase C; MAPK: mitogen-activated protein kinase; HGF: hepatocyte growth factor; CTLA-4: cytotoxic T-lymphocyte-associated antigen 4; PD-1: programmed cell death 1; CSCs: cancer stem-like cells; NAC: N-acetylcysteine; PI: propidium iodide; ALDH: aldehyde dehydrogenase; BSA: bovine serum albumin; MMP-9: matrix metalloproteinase 9; PKA: protein kinase A; dbcAMP: dibutyryl-cAMP sodium salt; EMT: epithelial mesenchymal transition.
Acknowledgements
This study was supported by grants from National Natural Science Funds (no. U1301226, no. 81373434, no. 81025021, and no. 91213304 to J. Pan), the National Basic Research Program of China (973 Program grant no. 2009CB825506 to J. Pan), the Research Foundation of Education Bureau of Guangdong Province, China (Grant cxzd1103 to J. Pan), and Natural Science Foundation of Guangdong province (Grant 2015A030312014 to J. Pan). The authors thank Dr. T.Z. Liu (Division of Oncology Research Mayo Clinic, Rochester, MN, USA) for a critical reading of the manuscript. Present address of JFZ and YLJ is Jinan University Institute of Tumor Pharmacology, College of Pharmacy, Jinan University, Guangzhou, China.
Authorship Contributions
JFZ designed, performed experiments, analyzed data and drafted the manuscript; BJ and YLJ performed experiments; YZL analyzed data; JXP designed, guided research, analyzed data, and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schoenfield L. Uveal melanoma: A pathologist's perspective and review of translational developments. Adv Anat Pathol. 2014;21:138-43
2. Singh AD, Turell ME, Topham AK. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 2011;118:1881-5
3. Woodman SE. Metastatic uveal melanoma: biology and emerging treatments. Cancer J. 2012;18:148-52
4. Singh AD, Bergman L, Seregard S. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin North Am. 2005;18:75-84
5. Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z. et al. Mutant Gq/11 Promote Uveal Melanoma Tumorigenesis by Activating YAP. Cancer Cell. 2014;25:1-9
6. Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M. et al. Hippo-Independent Activation of YAP by the GNAQ Uveal Melanoma Oncogene through a Trio-Regulated Rho GTPase Signaling Circuitry. Cancer Cell. 2014;25:831-45
7. Cheng H, Terai M, Kageyama K, Ozaki S, McCue PA, Sato T. et al. Paracrine Effect of NRG1 and HGF Drives Resistance to MEK Inhibitors in Metastatic Uveal Melanoma. Cancer Res. 2015;75:2737-48
8. Carvajal RD, Schwartz GK, Tezel T, Marr B, Francis JH, Nathan PD. Metastatic disease from uveal melanoma: treatment options and future prospects. Br J Ophthalmol. 2017;101:38-44
9. Surriga O, Rajasekhar VK, Ambrosini G, Dogan Y, Huang R, Schwartz GK. Crizotinib, a c-Met inhibitor, prevents metastasis in a metastatic uveal melanoma model. Mol Cancer Ther. 2013;12:2817-26
10. Dummer R, Hauschild A, Lindenblatt N, Pentheroudakis G, Keilholz U. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(Suppl 5):v126-32
11. Li Y, Li PK, Roberts MJ, Arend RC, Samant RS, Buchsbaum DJ. Multi-targeted therapy of cancer by niclosamide: A new application for an old drug. Cancer Lett. 2014;349:8-14
12. Jin Y, Lu Z, Ding K, Li J, Du X, Chen C. et al. Antineoplastic Mechanisms of Niclosamide in Acute Myelogenous Leukemia Stem Cells: Inactivation of the NF-κB Pathway and Generation of Reactive Oxygen Species. Cancer Res. 2010;70:2516-27
13. Osada T, Chen M, Yang XY, Spasojevic I, Vandeusen JB, Hsu D. et al. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011;71:4172-82
14. Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP. et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res. 2014;20:3198-210
15. Wieland A, Trageser D, Gogolok S, Reinartz R, Hofer H, Keller M. et al. Anticancer effects of niclosamide in human glioblastoma. Clin Cancer Res. 2013;19:4124-36
16. Londono-Joshi AI, Arend RC, Aristizabal L, Lu W, Samant RS, Metge BJ. et al. Effect of niclosamide on basal-like breast cancers. Mol Cancer Ther. 2014;13:800-11
17. Jin B, Wang C, Li J, Du X, Ding K, Pan J. Anthelmintic niclosamide disrupts the interplay of p65 and FOXM1/beta-catenin and eradicates leukemia stem cells in chronic myelogenous leukemia. Clin Cancer Res. 2017;23:789-803
18. Dai W, Zhou J, Jin B, Pan J. Class III-specific HDAC inhibitor Tenovin-6 induces apoptosis, suppresses migration and eliminates cancer stem cells in uveal melanoma. Sci Rep. 2016;6:1-14
19. Shi X, Wang D, Ding K, Lu Z, Jin Y, Zhang J. et al. GDP366, a novel small molecule dual inhibitor of survivin and Op18, induces cell growth inhibition, cellular senescence and mitotic catastrophe in human cancer cells. Cancer Biol Ther. 2010;9:640-50
20. Ippolito JE, Brandenburg MW, Ge X, Crowley JR, Kirmess KM, Som A. et al. Extracellular pH Modulates Neuroendocrine Prostate Cancer Cell Metabolism and Susceptibility to the Mitochondrial Inhibitor Niclosamide. PLoS One. 2016;11:e0159675
21. Reuben Lotan TS, Dafna Lotan. Isolation and Analysis of Melanoma Cell Mutants Resistant to the Antiproliferative Action of Retinole Acid. Cancer Res. 1983;43:2868-75
22. Onken MD, Ehlers JP, Worley LA, Makita J, Yokota Y, Harbour JW. Functional gene expression analysis uncovers phenotypic switch in aggressive uveal melanomas. Cancer Res. 2006;66:4602-9
23. Wang F, Ma J, Wang KS, Mi C, Lee JJ, Jin X. Blockade of TNF-alpha-induced NF-kappaB signaling pathway and anti-cancer therapeutic response of dihydrotanshinone I. Int Immunopharmacol. 2015;28:764-72
24. Yoo JY, Jaime-Ramirez AC, Bolyard C, Dai H, Nallanagulagari T, Wojton J. et al. Bortezomib Treatment Sensitizes Oncolytic HSV-1-Treated Tumors to NK Cell Immunotherapy. Clin Cancer Res. 2016;22:5265-76
25. Yuan R, Wang K, Hu J, Yan C, Li M, Yu X. et al. Ubiquitin-like protein FAT10 promotes the invasion and metastasis of hepatocellular carcinoma by modifying beta-catenin degradation. Cancer Res. 2014;74:5287-300
26. Jin Y, Yao Y, Chen L, Zhu X, Jin B, Shen Y. et al. Depletion of gamma-catenin by Histone Deacetylase Inhibition Confers Elimination of CML Stem Cells in Combination with Imatinib. Theranostics. 2016;6:1947-62
27. Sun D, Li B, Qiu R, Fang H, Lyu J. Cell Type-Specific Modulation of Respiratory Chain Supercomplex Organization. Int J Mol Sci. 2016;17:1-14
28. Wu Y, Chen C, Sun X, Shi X, Jin B, Ding K. et al. Cyclin-dependent kinase 7/9 inhibitor SNS-032 abrogates FIP1-like-1 platelet-derived growth factor receptor alpha and bcr-abl oncogene addiction in malignant hematologic cells. Clin Cancer Res. 2012;18:1966-78
29. Dror R, Lederman M, Umezawa K, Barak V, Pe'er J, Chowers I. Characterizing the involvement of the nuclear factor-kappa B (NF kappa B) transcription factor in uveal melanoma. Invest Ophthalmol Vis Sci. 2010;51:1811-6
30. Yue C, Yang Y, Zhang C, Alfranca G, Cheng S, Ma L. et al. ROS-Responsive Mitochondria-Targeting Blended Nanoparticles: Chemo- and Photodynamic Synergistic Therapy for Lung Cancer with On-Demand Drug Release upon Irradiation with a Single Light Source. Theranostics. 2016;6:2352-66
31. Ambrosini G, Khanin R, Carvajal RD, Schwartz GK. Overexpression of DDX43 mediates MEK inhibitor resistance through RAS Upregulation in uveal melanoma cells. Mol Cancer Ther. 2014;13:2073-80
32. Adorno-Cruz V, Kibria G, Liu X, Doherty M, Junk DJ, Guan D. et al. Cancer stem cells: targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res. 2015;75:924-9
33. Rycaj K, Tang DG. Cell-of-Origin of Cancer versus Cancer Stem Cells: Assays and Interpretations. Cancer Res. 2015;75:4003-11
34. Serrano D, Bleau AM, Fernandez-Garcia I, Fernandez-Marcelo T, Iniesta P, Ortiz-de-Solorzano C. et al. Inhibition of telomerase activity preferentially targets aldehyde dehydrogenase-positive cancer stem-like cells in lung cancer. Mol Cancer. 2011;10:1-15
35. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M. et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555-67
36. Jiang F, Qiu Q, Khanna A, Todd NW, Deepak J, Xing L. et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol Cancer Res. 2009;7:330-8
37. Tao H, Zhang Y, Zeng X, Shulman GI, Jin S. Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat Med. 2014;20:1263-9
38. Chowdhury MK, Wu LE, Coleman JL, Smith NJ, Morris MJ, Shepherd PR. et al. Niclosamide blocks glucagon phosphorylation of Ser552 on beta-catenin in primary rat hepatocytes via PKA signalling. Biochem J. 2016;473:1247-55
39. Hino S, Tanji C, Nakayama KI, Kikuchi A. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase stabilizes beta-catenin through inhibition of its ubiquitination. Mol Cell Biol. 2005;25:9063-72
40. Merschjohann K, Steverding D. In vitro trypanocidal activity of the anti-helminthic drug niclosamide. Exp Parasitol. 2008;118:637-40
41. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
42. Bronkhorst IH, Jager MJ. Uveal melanoma: the inflammatory microenvironment. J Innate Immun. 2012;4:454-62
43. McKenna KC, Chen PW. Influence of immune privilege on ocular tumor development. Ocul Immunol Inflamm. 2010;18:80-90
44. Ly LV, Bronkhorst IH, van Beelen E, Vrolijk J, Taylor AW, Versluis M. et al. Inflammatory cytokines in eyes with uveal melanoma and relation with macrophage infiltration. Invest Ophthalmol Vis Sci. 2010;51:5445-51
45. Bronkhorst IH, Jager MJ. Inflammation in uveal melanoma. Eye (Lond). 2013;27:217-23
46. Tsimberidou AM, Estey E, Wen S, Pierce S, Kantarjian H, Albitar M. et al. The prognostic significance of cytokine levels in newly diagnosed acute myeloid leukemia and high-risk myelodysplastic syndromes. Cancer. 2008;113:1605-13
47. Zheng Y, Yang W, Aldape K, He J, Lu Z. Epidermal growth factor (EGF)-enhanced vascular cell adhesion molecule-1 (VCAM-1) expression promotes macrophage and glioblastoma cell interaction and tumor cell invasion. J Biol Chem. 2013;288:31488-95
48. Zeng Q, Li S, Zhou Y, Ou W, Cai X, Zhang L. et al. Interleukin-32 contributes to invasion and metastasis of primary lung adenocarcinoma via NF-kappaB induced matrix metalloproteinases 2 and 9 expression. Cytokine. 2014;65:24-32
49. Guo Z, Xing Z, Cheng X, Fang Z, Jiang C, Su J. et al. Somatostatin Derivate (smsDX) Attenuates the TAM-Stimulated Proliferation, Migration and Invasion of Prostate Cancer via NF-kappaB Regulation. PLoS One. 2015;10:1-15
50. Sethi G, Ahn KS, Pandey MK, Aggarwal BB. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB activation. Blood. 2007;109:2727-35
51. Amin H, Wani NA, Farooq S, Nayak D, Chakraborty S, Shankar S. et al. Inhibition of Invasion in Pancreatic Cancer Cells by Conjugate of EPA with beta(3,3)-Pip-OH via PI3K/Akt/NF-kB Pathway. ACS Med Chem Lett. 2015;6:1071-4
52. Shi X, Zhang Y, Zheng J, Pan J. Reactive Oxygen Species in Cancer Stem Cells. Antioxid Redox Signaling. 2012;16:1215-28
53. Mehner C, Hockla A, Miller E, Ran S, Radisky DC, Radisky ES. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget. 2014;5:2736-49
54. Song H, Li Y, Lee J, Schwartz AL, Bu G. Low-density lipoprotein receptor-related protein 1 promotes cancer cell migration and invasion by inducing the expression of matrix metalloproteinases 2 and 9. Cancer Res. 2009;69:879-86
55. El-Shabrawi Y, Ardjomand N, Radner H, Ardjomand N. MMP-9 is predominantly expressed in epithelioid and not spindle cell uveal melanoma. J Pathol. 2001;194:201-6
56. Sahin A, Kiratli H, Soylemezoglu F, Tezel GG, Bilgic S, Saracbasi O. Expression of vascular endothelial growth factor-A, matrix metalloproteinase-9, and extravascular matrix patterns and their correlations with clinicopathologic parameters in posterior uveal melanomas. Jpn J Ophthalmol. 2007;51:325-31
57. Li S, Li Q. Cancer stem cells and tumor metastasis (Review). Int J Oncol. 2014;44:1806-12
58. Wang YC, Chao TK, Chang CC, Yo YT, Yu MH, Lai HC. Drug screening identifies niclosamide as an inhibitor of breast cancer stem-like cells. PLoS One. 2013;8:1-10
59. Misra SK, Jensen TW, Pan D. Enriched inhibition of cancer and stem-like cancer cells via STAT-3 modulating niclocelles. Nanoscale. 2015;7:7127-32
60. Duchartre Y, Kim YM, Kahn M. The Wnt signaling pathway in cancer. Crit Rev Oncol Hematol. 2015:1-9
61. Kahn M. Wnt Signaling in Stem Cells and Tumor Stem Cells. Semin Reprod Med. 2015;33:317-25
62. Jin Y, Nie D, Li J, Du X, Lu Y, Li Y. et al. Gas6/AXL signaling regulates self-renewal of chronic myelogenous leukemia stem cells by stabilizing beta-catenin. Clin Cancer Res. 2016
Author contact
![]() Corresponding author: Jingxuan Pan, M.D., Ph.D., State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, 54 S. Xianlie Road, Guangzhou 510060, People's Republic of China, Phone: +86-20-37628262, E-mail: panjx2sysu.edu.cn
Corresponding author: Jingxuan Pan, M.D., Ph.D., State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, 54 S. Xianlie Road, Guangzhou 510060, People's Republic of China, Phone: +86-20-37628262, E-mail: panjx2sysu.edu.cn